

Abstract
Phosphorylation and dephosphorylation of AMPA-type ionotropic glutamate receptors (AMPARs) by kinases and phosphatases and interactions with scaffold proteins play essential roles in regulating channel biophysical properties and trafficking events that control synaptic strength during NMDA receptor-dependent synaptic plasticity, such as LTP and LTD. We previously demonstrated that palmitoylation of the AMPAR-linked scaffold protein A-kinase anchoring protein (AKAP) 79/150 is required for its targeting to recycling endosomes in dendrites, where it regulates exocytosis from these compartments that is required for LTP-stimulated enlargement of postsynaptic dendritic spines, delivery of AMPARs to the plasma membrane, and maintenance of synaptic potentiation. Here, we report that the recycling endosome-resident palmitoyl acyltransferase DHHC2 interacts with and palmitoylates AKAP79/150 to regulate these plasticity signaling mechanisms. In particular, RNAi-mediated knockdown of DHHC2 expression in rat hippocampal neurons disrupted stimulation of exocytosis from recycling endosomes, enlargement of dendritic spines, AKAP recruitment to spines, and potentiation of AMPAR-mediated synaptic currents that occur during LTP. Importantly, expression of a palmitoylation-independent lipidated AKAP mutant in DHHC2-deficient neurons largely restored normal plasticity regulation. Thus, we conclude that DHHC2-AKAP79/150 signaling is an essential regulator of dendritic recycling endosome exocytosis that controls both structural and functional plasticity at excitatory synapses.
Keywords: A-kinase anchoring protein, AMPAR, LTP, palmitoylation, recycling endosome, trafficking
Introduction
Hebbian tuning of synaptic efficacy is essential for information storage in neural networks. Such modulation of excitatory synaptic strength is frequently achieved through regulated trafficking and postsynaptic positioning of AMPARs (Anggono and Huganir, 2012). Indeed, AMPAR synaptic recruitment (in LTP) and loss (in LTD) are thought to underlie higher level brain functions including learning and memory (Malenka and Bear, 2004). Crucial to these plasticity phenomena is AMPAR phosphorylation regulation by kinases and phosphatases, which control both AMPAR channel activity and trafficking (Kessels and Malinow, 2009). In turn, kinases and phosphatases are targeted to AMPARs and other substrates in postsynaptic nanodomains by a highly organized scaffolding protein network (Sheng and Hoogenraad, 2007). AKAP79/150 (human79/rodent150) is a prototypic multivalent scaffold that anchors the cAMP-dependent protein kinase (PKA), PKC, and the Ca2+-dependent protein phosphatase-2B/calcineurin (CaN) to control receptor phosphorylation and trafficking in complexes with PSD-95 family scaffold proteins (Colledge et al., 2000; Tavalin et al., 2002; Lu et al., 2007; Bhattacharyya et al., 2009; Robertson et al., 2009; Jurado et al., 2010; Sanderson and Dell'Acqua, 2011; Sanderson et al., 2012). Nanodomains important for AMPAR trafficking include not only the PSD, but also perisynaptic and dendritic recycling endosomes (REs), which are key repositories for AMPARs that can be rapidly exocytosed near synapses to support potentiation (Kennedy and Ehlers, 2011). We recently described AKAP79/150 localization in REs, where it regulates exocytosis and facilitates LTP/LTD signaling (Keith et al., 2012). However, the upstream mechanisms regulating AKAP79/150 localization and signaling in REs remain largely unexplored.
Cotranslational and post-translational modification by lipidation is a powerful means of controlling protein interactions with plasma and endomembranes. S-palmitoylation, the attachment of a C-16 fatty acid to cysteine (Cys) residues via thioester linkage, is unique among the major types of lipidation in that it is reversible. Increasingly, palmitoylation is recognized as a critical modification controlling targeting of ion channels, receptors, enzymes, and scaffolds (Linder and Deschenes, 2007; Fukata and Fukata, 2010). Importantly, AKAP79/150 localization to REs, but not the plasma membrane, requires its palmitoylation on two Cys residues within its N-terminal membrane-targeting domain (Keith et al., 2012).
Two classes of enzymes are responsible for palmitoylation regulation: DHHC-motif palmitoyl acyltransferases (PATs) catalyzing palmitate addition (Linder and Jennings, 2013) and protein acylthioesterases catalyzing palmitate removal (Zeidman et al., 2009). Due to the promiscuous nature of PATs and the apparent lack of consensus substrate recognition sequences, identifying specific PAT/substrate pairs has been challenging. In the last few years, important roles for palmitoylation in controlling the trafficking of prominent synaptic scaffold proteins were revealed, and functionally relevant cognate DHHC-family PATs were recently identified for PSD-95 (Noritake et al., 2009), GRIP1b (Thomas et al., 2012), and δ-catenin (Brigidi et al., 2014).
Here we identify DHHC2 as a regulator of AKAP79/150 palmitoylation and signaling that is crucial for postsynaptic regulation of RE function. Specifically, we show that DHHC2 interacts and colocalizes with AKAP79/150 in REs and is critical for regulation of AKAP79/150 postsynaptic recruitment, dendritic spine enlargement, recycling endosome exocytosis, and AMPAR synaptic potentiation.
Materials and Methods
Animal care and use.
Animal procedures were performed in compliance with NIH United States Public Health Service guidelines and were approved by the University of Colorado Denver Institutional Animal Care and Use Committee.
Cell culture.
COS7 cells were seeded on 18 mm or 25 mm coverslips in DMEM +10% FBS media and grown until >80% confluence before transfection with plasmid cDNA via calcium phosphate. Primary dissociated neuron cultures were prepared from both male and female P0–P1 Sprague Dawley rats as previously described (Robertson et al., 2009). Neurons were plated on poly-d-lysine-coated, 12 mm, 18 mm, or 25 mm round no. 1 glass coverslips at a density of 100,000 or 450,000 cells per coverslip. Plasmid DNA was introduced into neurons using Lipofectamine 2000 (Life Technologies) at 10–12 DIV. Cells were fixed, imaged, or recorded from on 12–14 DIV (see below for more details). Plasmids for expressing micro-interfering RNA to DHHC2 (miDHHC2), HA-DHHC2WT, and HA-DHHC2CS were generously provided by Masaki Fukata (National Institute for Physiological Sciences, Japan). Plasmids encoding myc-tagged DHHC2, DHHC3, DHHC 5, DHHC 8, and DHHC 15 were kindly supplied by Shernaz Bamji (University of British Columbia). Superecliptic pHluorin-tagged transferrin receptor was a gift from Michael Ehlers (Duke University, Durham, NC/Pfizer). HA-tagged DHHC5 and DHHC8 plasmids were generously provided by Gareth Thomas (Temple University, Philadelphia, PA).
Immunoprecipitation.
HEK293 or COS7 cells were transfected by calcium phosphate precipitation. At 48 h post transfection cells were lysed in ice-cold lysis buffer (20 mm Tris, pH7.4, 150 mm NaCl, 1% Triton X-100, 1% NP-40, 5 mm EDTA, 5 mm NaF, 2 mg/ml leupeptin, 2 mg/ml pepstatin, and 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride), sonicated (eight 2 s bursts), and centrifuged at 20,800 × g and then supernatants were transferred to fresh Microfuge tubes. Five percent of lysates were set aside for input loading. Lysates were split in half and tumbled overnight at 4°C with 4 μg of rabbit polyclonal antibodies (Ab) to GFP (Life Technologies) or nonimmune rabbit IgG (negative control). Lysates + Ab were then tumbled with 60 μl of Protein A agarose beads at 4°C for 1–2 h. Beads were subsequently pelleted and washed ×3 in lysis buffer. Samples were resolved on Tris-SDS gels and transferred to PVDF membrane, blocked in 3% BSA TBS + 0.1% Tween, and probed with the following mouse monoclonal Abs overnight at 4°C–c-Myc (1:1000; Santa Cruz Biotechnology), HA (1:1000; Covance), and GFP (1:500; Abcam)—and visualized by ECL (Fisher-Pierce) using a Fluorochem gel documentation system (Alpha Innotech).
Immunocytochemistry.
COS7 cells and neurons were processed for immunostaining as previously described (Robertson et al., 2009; Keith et al., 2012). Briefly, cells were washed in PBS, fixed with 3.6% paraformaldehyde in PBS, and permeabilized with PBS + 0.2% Triton X-100. Following overnight block in PBS + 3% BSA, cells were incubated with primary Abs anti-HA (1:1000; Covance), anti-myc (1:1000; Santa Cruz Biotechnology), and anti-PSD-95 (1:500; NeuroMab) for 2 h, washed three times, and incubated with secondary Ab (Alexa 647; 1:500; Life Technologies) for 1 h. After three PBS washes, cells were mounted with Prolong Gold antifade (Life Technologies).
Fluorescence imaging.
Fixed cells were imaged using an Axiovert 200M microscope (Zeiss) with a 63× objective (1.4 NA; Plan Apo) and a CoolSNAP2 (Photometrics) CCD camera. Acquisition and off-line processing were conducted using SlideBook 5.5 (Intelligent Imaging Innovations). Focal plane z-stacks (spaced 0.5 μm apart) were acquired and deconvolved to discard out-of-focus light. Z-stacks of neurons were collapsed using maximum-intensity projections. Images of live neurons (Fig. 4) were acquired using the same microscope in ACSF [containing the following (in mm): 130 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 11 glucose] at 33°C. Baseline images were acquired before and 25 min following a 10 min chemical LTP (cLTP) treatment (ACSF + 0 μm Mg2+ + 200 μm glycine and 50 μm picrotoxin), delivered by peristaltic pump (Harvard Apparatus).
Figure 4.
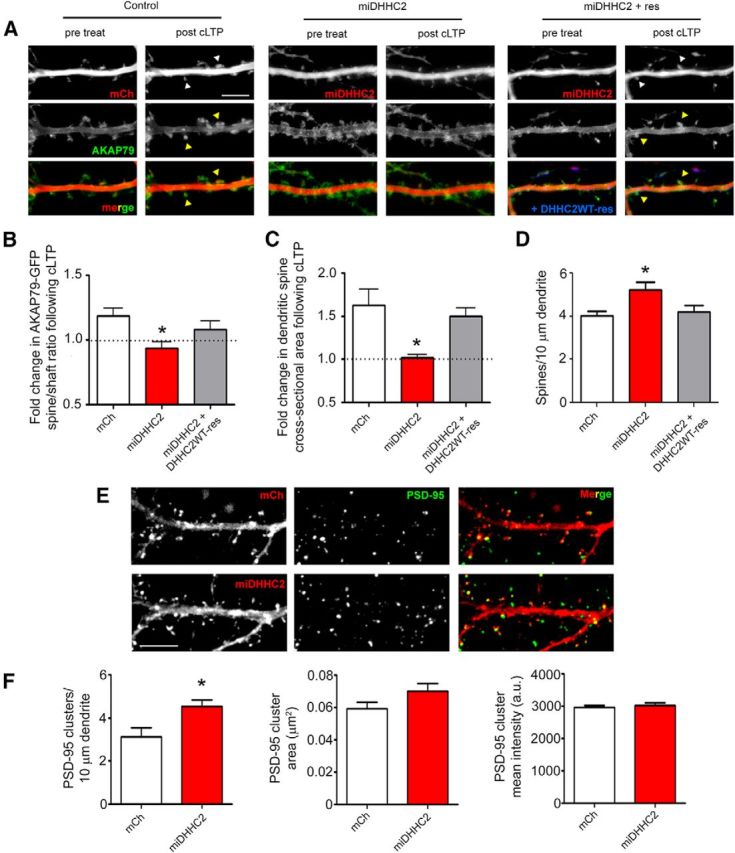
DHHC2 knockdown prevents AKAP79 recruitment to dendritic spines and spine enlargement during synaptic potentiation. A, Live hippocampal neurons (14 DIV) expressing AKAP79-GFP (green) and mCh (red; Control), miDHHC2 (red), or miDHHC2 (red) + DHHC2WT-CFP (blue) were imaged before and 25 min after cLTP treatment (10 min in 0 Mg2+, 200 μm glycine, and 50 μm picrotoxin). White arrowheads identify spines showing enlargement and yellow arrowheads denote spines showing AKAP79 enrichment after cLTP (n = 10–17). B, Spine-to-shaft ratios of AKAP79-GFP fluorescence were quantified before and after cLTP treatment showing that DHHC2 knockdown significantly reduces cLTP-induced AKAP79 spine enrichment, an effect rescued by RNAi-insensitive CFP-DHHC2WT expression. C, cLTP treatment is accompanied by a significant increase in spine size in control cells, but not in DHHC2 knockdown neurons. Spine enlargement following cLTP is restored in miDHHC2 knockdown cells by RNAi-insensitive CFP-DHHC2WT. D, Dendritic spine density is increased basally in cells expressing miDHHC2 relative to control and knockdown-rescue cells (n = 7–16 cells per condition). E, Fixed 14 DIV hippocampal neurons expressing either mCh or miDHHC2 were stained for endogenous PSD-95. F, DHHC2 knockdown results in an increase in the linear density of PSD-95-immunoreactive puncta but no change in mean puncta size or intensity (n = 18–21 cells per condition; all data are represented as mean ± SEM; *p < 0.05, one-way ANOVA, Dunnett's post hoc test). Scale bar, 5 μm.
Live-cell spinning-disk confocal imaging of superecliptic pHluorin (SEP)-TfR exocytosis (Figs. 5, 7) was performed as previously described (Keith et al., 2012). Neurons were transfected from 12 to 14 DIV with SEP-TfR and mCherry (mCh) or miDHHC2 or miDHHC2 + CFP-DHHC2 rescue constructs using Lipofectamine 2000 as above. Images were acquired on an Axio Observer microscope (Zeiss) with a 63× Plan Apo/1.4 NA objective, using 440, 488, and 561 nm laser excitation and a CSU-XI spinning-disk confocal scan head (Yokogawa) coupled to an Evolve 512 EM-CCD camera (Photometrics) driven by SlideBook 5.5. Before imaging, neurons were incubated in ACSF plus 1 mm MgCl2 for 30 min and were maintained during imaging at 33−35°C in a perfusion chamber (Warner Instruments). Baseline rates of SEP-TfR exocytic events were determined by acquiring z-stacks of 10–14 optical sections (1.0 μm spacing) every 7 s for 4 min. For cLTP stimulation of neurons, the bath solution was exchanged by peristaltic pump (Harvard Apparatus) perfusion with ACSF without Mg2+ + 200 μm glycine and 50 μm picrotoxin during 4 min of additional imaging.
Figure 5.

DHHC2 knockdown blocks LTP-induced RE exocytosis. A, Images of 14 DIV neurons transfected with SEP-TfR (green) and mCh or miDHHC2 (red). B, Time-composite images of SEP-TfR signal (green) before (0–4 min) and after (4–8 min) cLTP treatment. Expression of an RNAi-insensitive construct CFP-DHHC2WT was used to rescue DHHC2 knockdown (image not shown). C, 3D time-composite plots showing integrated SEP-TfR intensity on the z-axis in pseudocolor (low, blue; red, high) before (0–4 min) and after (4–8 min) cLTP treatment. D, Quantification of fold increase in SEP-TfR exocytosis in response to cLTP treatment reveals an increase in exocytic events in control neurons but not in miDHHC2 knockdown neurons. This inhibitory effect of miDHHC2 on cLTP-stimulated exocytosis is restored CFP-DHHC2WT expression (n = 7–12 cells/condition; all data are presented as mean ± SEM; *p < 0.05, one-way ANOVA, Dunnett's post hoc test).
Figure 7.

Myristoylated AKAP79 restores LTP-stimulated RE exocytosis in DHHC2-deficient neurons. A, Live images of 14 DIV hippocampal neurons expressing SEP-TfR (green) and mCh (control; red), miDHHC2 (knockdown; red), or miDHHC2 (red) + myr-AKAP79WT (blue). 3D time-composite plots showing integrated SEP-TfR intensity on the z-axis in pseudocolor (low, blue; red, high) depicting 0–4 min baseline (top) and 4–8 min post-cLTP (bottom) imaging periods. B, Quantification of the fold increase in RE exocytosis elicited by cLTP treatment showing that miDHHC2-mediated knockdown blocks increased RE exocytosis while coexpression of myr-AKAP79WT recovers cLTP stimulation. C, Quantification of basal SEP-TfR exocytic rates showing that miDHHC2 elevates basal RE exocytosis relative to mCH controls and that myr-AKAP79WT or myr-AKAP79CS, but not AKAP79CS, significantly suppress this elevated RE exocytosis (n = 7–10 cells/condition; all data are presented as mean ± SEM; *p < 0.05; t test with Bonferroni's correction for multiple comparisons).
Image analysis.
Protein colocalization, spine/shaft ratios, spine cross-sectional area, and RE exocytosis were quantified essential as previously described (Gomez et al., 2002; Horne and Dell'Acqua, 2007; Robertson et al., 2009; Keith et al., 2012). For colocalization analysis, masks for each channel/fluorophore were created using a threshold of 2–2.5× the mean fluorescence intensity. A colocalization mask representing only pixels with superthreshold intensities in both channels was then generated. Ratios between the integrated intensity in the colocalization mask and the total integrated intensity for each channel were then calculated, yielding the proportion/percentage overlap values for each channel that are graphed in the figures or reported in the Results text (Gomez et al., 2002; Horne and Dell'Acqua, 2007). For spine/shaft ratio and spine area changes in living neurons, spines and underlying dendritic shafts from cells expressing mCh as a cell fill were masked. The same spines were analyzed before and after treatment. Fold change in mean intensity in the spine/shaft ratio was quantified along with changes in spine cross-sectional area. Analysis of 4D spinning-disk movies was conducted using SlideBook 5.5 by generating time-lapse series of 2D maximum-intensity projection images from the 3D image z-stacks for each time point and then the series were divided into 4 min pretreatment and post-treatment sequences. Masks defining SEP-TfR membrane fusion events were then created at 2.5× mean dendrite fluorescence intensity in the 488 nm channel. The basal rate of exocytosis was calculated as the number of events/min/100 μm length of dendrite. The total number of events detected 4–8 min following cLTP treatment was divided by the total number of events detected during the 0–4 min baseline period to calculate the fold increase in exocytosis in response to treatment. Time-composite images of the 488 nm (SEP-TfR) channel were generated to visualize the accumulated intensity from the transient events occurring over the entire 4 min periods. These time-composite images were in turn used to generate 3D intensity time-composite plots of the 488 nm (SEP-TfR) channel displayed in pseudocolor on a relative scale of low (blue) to high (red) fluorescence intensity such that exocytic events appear as discrete vertical peaks.
BMCC-biotin palmitoylation assay.
COS7 cells were grown on 10 cm cell-culture dishes and transfected with AKAP79WT, AKAP79CS, mCh, mCh-DHHC2, and mCh-DHHC2CS plasmids via calcium phosphate precipitation. At 48 h post transfection, AKAP79 palmitoylation state was assessed by a BMCC biotinylation assay as described previously (Keith et al., 2012). Briefly, cells were lysed in pH 7.4 lysis buffer + 50 mm N-ethylmaleimide (NEM), permanently blocking sulfhydryls. Samples were split into input (5%), hydroxylamine (HAM)+ and HAM− groups. GFP-tagged AKAP79 was then immunoprecipitated with GFP Ab overnight as described above, maintaining 50 mm NEM in the immunoprecipitation (IP) buffer. Following binding to Protein A agarose beads, the AKAP79/bead complex was incubated with 0.5 m HAM in buffer, pH 7.2, for 45 min at room temperature. Beads were then rinsed with buffer, pH 6.2, and incubated with 10 μm biotin-BMCC reagent (Thermo Scientific) for 1 h at 4°C. Proteins were separated via SDS-PAGE and palmitoylation was detected via HRP-conjugated streptavidin labeling and ECL visualization. For quantification, palmitoylation signal was normalized to total protein signal using a Fluorochem gel documentation system (Alpha Innotech).
Electrophysiology.
Whole-cell voltage-clamp recordings were conducted at room temperature with an Axopatch 200B amplifier (Molecular Devices). Cells were identified by infrared-differential contrast microscopy. Microelectrodes had resistances of 3–6 MΩ. AMPAR mEPSCs were recorded from 12 DIV hippocampal pyramidal neurons (2 d following transfection on 10 DIV) in ACSF [containing the following (in mm): 130 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 11 glucose (osmolarity ∼300 mOsm)] at a holding potential of −70 mV using a potassium gluconate internal solution containing the following (in mm): 135 K-gluconate, 5 KCl, 10 HEPES, 0.2 EGTA, 4.6 MgCl2, 4 Na2ATP, and 0.4 NaGTP, pH 7.3 (Forti et al., 2006). Access resistances were between 15 and 35 MΩ; if the access resistance changed >20%, the recording was discarded. For cLTP, neurons were treated with 200 μm glycine and 50 μm picrotoxin in Mg2+-free ACSF for 10 min at 37°C and then recovered in normal ACSF for 25 min at 37°C before recording. Data were analyzed using the Mini Analysis Program (Synaptosoft). Statistical analyses were performed using GraphPad Prism.
Results
Identification of DHHC2 as a regulator of AKAP79 palmitoylation
We know that palmitoylation of AKAP79/150 regulates NMDAR-dependent excitatory synaptic potentiation (Keith et al., 2012), but the enzyme(s) responsible for AKAP palmitoylation are not yet known. The mammalian PATs are composed of a family of 23 evolutionarily related integral membrane proteins (Fukata et al., 2004; Linder and Jennings, 2013). These enzymes have short intracellular N termini; four trans-membrane domains; a conserved DHHC enzymatic motif located in the sole cytoplasmic loop between trans-membrane domains 2 and 3; and diverse C-terminal tails that can regulate intracellular localization and, in some cases, substrate binding. Rather than screen all 23 of these enzymes as potential regulators of AKAP79 palmitoylation, we screened AKAP79 for interactions with a subset of DHHC proteins (DHHC2, DHHC3, DHHC5, DHHC8, and DHHC15) that are known to palmitoylate other postsynaptic scaffold proteins and be localized to neuronal dendrites (Fukata et al., 2004; Noritake et al., 2009; Fukata and Fukata, 2010; Thomas et al., 2012, 2013; Brigidi et al., 2014). In coimmunoprecipitation (co-IP) experiments using HEK293 cells expressing exogenous myc-tagged DHHC constructs with AKAP79-GFP, we found that myc-DHHC2 robustly precipitated with AKAP79-GFP, while DHHC3, DHHC5, DHHC8, and DHHC15 displayed much weaker precipitation (Fig. 1A). The results of this interaction screen led us to focus on DHHC2 as a potential regulator of AKAP79 in subsequent experiments.
Figure 1.

AKAP79 preferentially interacts with and is palmitoylated by DHHC2. A, Anti-GFP co-IP of the indicated myc-tagged DHHC-family proteins with AKAP79-GFP in HEK293 cells; myc-DHHC2 precipitates robustly with AKAP79-GFP. B, The N-terminal basic polybasic membrane targeting region of AKAP79 (1–153) is sufficient for co-IP with DHHC2 and the C36,129S mutation prevents this interaction. C, Both HA-DHHC2WT and catalytically inactive HA-DHHC2CS co-IP with AKAP79WT-GFP. D, AKAP79 palmitoylation is increased by DHHC2WT but not DHHC2CS in COS7 cells as revealed by the BMCC-biotin palmitoylation assay. E, Quantification reveals higher levels of palmitoylated AKAP79-GFP when mCh-tagged DHHC2WT but not catalytically inactive DHHC2CS is coexpressed normalized to control conditions expressing AKAP79-GFP and mCH alone. AKAP79CS serves as negative control for assay specificity in D and E (n = 5; data represented as mean ± SEM; *p < 0.05, one-way ANOVA, Dunnett's post hoc test).
We further explored the interaction between DHHC2 and AKAP79 by first identifying the region of AKAP79 that interacts with DHHC2. Importantly, an AKAP79 (1–153) N-terminal fragment, which contains the polybasic membrane targeting domain and palmitoylated residues C36 and C129 (Dell'Acqua et al., 1998; Keith et al., 2012), was sufficient for co-IP with myc-DHHC2 (Fig. 1B). Previous studies demonstrated that the palmitoylation-deficient AKAP79C36,129S mutant is excluded from lipid rafts and REs but not nonraft domains in the plasma membrane (Delint-Martinez et al., 2011; Keith et al., 2012). Interestingly, the C36,129S mutation prevented co-IP of DHHC2, in the contexts of both full-length AKAP79 and the 1–153 fragment, suggesting that the Cys substrate sites in the AKAP N-terminal targeting domain are required for interaction with DHHC2 (Fig. 1B). In contrast, a catalytically inactive DHHC2CS mutant still precipitated with AKAP79, indicating that DHHC2 enzymatic activity is not necessary for this interaction (Fig. 1C).
We next examined whether DHHC2 could regulate palmitoylation of AKAP79 by coexpressing these proteins in COS7 cells and detecting the level of palmitoylated AKAP79 using a permutation of the acyl-biotin exchange method (Kang et al., 2008). In this assay, AKAP79-GFP was first precipitated and then palmitoylated Cys residue was detected by biotin-switch labeling with a BMCC-biotin reagent (Keith et al., 2012). DHHC2WT expression significantly increased AKAP79 palmitoylation (∼51%) relative to levels observed in mCh-expressing control cells (Fig. 1D,E). In contrast, the catalytically inactive DHHC2CS mutant did not increase AKAP79 palmitoylation above control levels. Coexpression of DHHC2WT with AKAP79C36,129S revealed significantly reduced BMCC-biotin labeling, thus serving as an additional negative control that demonstrates the specificity of this assay for detecting modification of C36 and C129 (Fig. 1D,E). Together, these experiments strongly support a role for DHHC2 in regulating AKAP79 palmitoylation at C36 and C129.
Imaging DHHC2 colocalization in endosomes with AKAP79
We next examined the subcellular localization of DHHC-family proteins relative to AKAP79 in COS7 cells and neurons. Previous work on DHHC localization conducted in cultured cell lines indicates that DHHCs are regionally restricted in their subcellular distribution (Ohno et al., 2006). More recently, it was reported that DHHC2 is targeted to Rab11-positive REs as well as the plasma membrane in PC12 cells (Greaves et al., 2011) and DHHC2, DHHC5, and DHHC8 may all be present to various degrees at both the plasma membrane and in endosomes in neurons (Noritake et al., 2009; Thomas et al., 2012). Due to the importance of REs for synaptic plasticity and our recent findings that AKAP79/150 associates with REs and its trafficking to dendritic spines is regulated by Rab11(Keith et al., 2012), we sought to determine which DHHC family members are most localized to the RE compartment. COS7 cells are an ideal model system for initial interrogation because AKAP79 localizes very prominently to distinct, large, and easily resolved REs in these cells (Keith et al., 2012). Thus, we examined the localization of myc-tagged DHHC2, DHHC5, DHCC8, and DHCC15 in COS7 cells relative to both AKAP79-GFP and an established RE marker, the transferrin receptor tagged with mCh (TfR-mCh; Fig. 2A; Park et al., 2006). Colocalization analysis revealed a high degree of overlap between AKAP79 and DHHC2, which was significantly greater than that observed for the other DHHCs examined (Fig. 2B; Proportion overlap with AKAP79: DHHC2 0.49 ± 0.04, DHHC5 0.31 ± 0.05, DHHC8 0.26 ± 0.07, DHHC15 0.27 ± 0.03). DHHC2 also colocalized extensively with TfR-positive puncta, indicating enrichment in REs. Some DHHC/TfR-mCh overlap was also observed in cells expressing other DHHC isoforms, but this colocalization was significantly less than that seen in cells expressing DHHC2 (Fig. 2C; Proportion overlap with TfR: DHHC2 0.71 ± 0.04, DHHC5 0.56 ± 0.03, DHHC8 0.43 ± 0.04, DHHC15 0.51 ± 0.05). Of note, ∼37% of DHHC2 intensity was found in puncta that were also both AKAP79 and TfR positive, consistent with the presence of an AKAP79/DHHC2 complex in REs (Fig. 2D; Proportion DHHC in AKAP79+REs: DHHC2 0.37 ± 0.04, DHHC5 0.23 ± 0.04, DHHC8 0.16 ± 0.02, DHHC15 0.19 ± 0.03).
Figure 2.
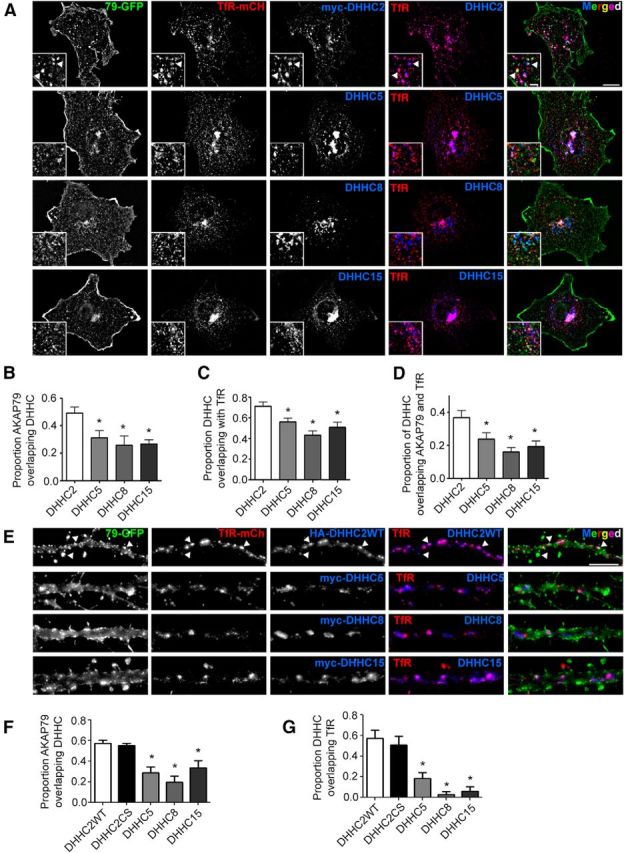
DHHC2 is enriched in REs containing AKAP79 in both COS7 cells and hippocampal neuron dendrites. A, Images of COS7 cells transfected with AKAP79-GFP (green), TfR-mCh (red), and the indicated HA- or myc-tagged DHHC constructs (blue). Arrowheads in insets indicate representative RE puncta that are positive for AKAP79, TfR, and DHHC2 (white in merged image). B, Colocalization analysis in COS7 cells reveals that AKAP79 colocalizes the most with DHHC2 and that DHHC2 (C) is the PAT most enriched in REs (D) containing both AKAP79 and TfR (n = 9 cells/condition). E, Images of 14 DIV hippocampal neuron dendrites expressing AKAP79-GFP (green), TfR-mCh (red), and the indicated HA- or myc-tagged DHHC constructs (blue). Arrowheads indicate representative RE puncta positive for AKAP79, TfR, and DHHC2WT. F, Colocalization analysis in dendrites reveals that AKAP79 colocalizes more with DHHC2 than with DHHC5, DHHC8, and DHHC15. G, DHHC2 also displays the greatest degree of colocalization in dendritic REs with TfR. Disabling DHHC2 enzymatic activity (DHHC2CS) has no effect on its localization (n = 5–10 cells/condition; all data are presented as mean ± SEM; *p < 0.05, one-way ANOVA, Dunnett's post hoc test). Scale bars: A, 10 μm; inset 2.5 μm; B, 5 μm.
Cellular distribution of DHHC family members is less well characterized in neurons, so we conducted additional colocalization experiments in dendrites of 14 DIV cultured hippocampal neurons (Fig. 2E). As in COS7 cells, significantly more AKAP79-GFP colocalized with HA-tagged DHHC2 in dendrites than with myc-tagged DHHC5, DHHC8, and DHCC15 (Fig. 2F; Proportion AKAP79 overlap with DHHCs: DHHC2WT 0.57 ± 0.03, DHHC2CS 0.55 ± 0.02, DHHC5 0.29 ± 0.06, DHHC8 0.20 ± 0.06, DHHC15 0.33 ± 0.07). DHHC2/TfR colocalization in dendrites was even more striking than in COS7 cells and was much higher than observed for the other DHHC enzymes, thus identifying DHHC2 as the PAT most distinctly localized to dendritic REs (Fig. 2G; Proportion overlap with TfR: DHHC2WT 0.57 ± 0.08, DHHC2CS 0.51 ± 0.08, DHHC3 0.14 ± 0.07, DHHC5 0.18 ± 0.06, DHHC8 0.03 ± 0.03, DHHC15 0.06 ± 0.04). Interestingly, the enzymatically dead HA-DHHC2CS mutant was localized to TfR-positive structures to a similar degree as HA-DHHC2WT, indicating that DHHC2 subcellular targeting is independent of its catalytic activity. Together, these findings demonstrate that DHHC2 is enriched in dendritic REs and is ideally positioned to interact with and regulate AKAP79/150 palmitoylation in these compartments.
DHHC2 knockdown interferes with NMDA receptor-dependent synaptic potentiation
Dendritic REs carry important plasticity-related cargo in the form of AMPARs and other proteins (Ehlers, 2000; Park et al., 2004, 2006; Brown et al., 2007; Fernández-Monreal et al., 2012; Keith et al., 2012). Indeed, REs and their cargo are recruited to the vicinity of synapses during potentiation and provide an important pool of AMPARs for synaptic strengthening (Kopec et al., 2006; Brown et al., 2007; Makino and Malinow, 2009; Petrini et al., 2009; Kennedy et al., 2010; Keith et al., 2012). Our previous work demonstrated that the palmitoylation-deficient AKAP79CS mutant increased basal AMPAR synaptic activity but then prevented acute stimulation of RE exocytosis and AMPAR delivery required to maintain synaptic potentiation (Keith et al., 2012). We therefore reasoned that if DHHC2 is the principle PAT mediating AKAP79 palmitoylation in the RE compartment, then loss of DHHC2 should similarly alter synaptic function and plasticity regulation. Accordingly, we recorded AMPAR mEPSCs in 12 DIV cultured hippocampal neurons transfected with a plasmid-encoded miRNA (miDHHC2; coexpresses mCh) that was previously demonstrated to effectively suppress DHHC2 expression in neurons (Noritake et al., 2009; Fig. 3). Compared with control neurons expressing mCh alone, AMPAR mEPSC amplitudes were significantly elevated in miDHHC2-expressing neurons 48 h post transfection, as visualized by both a rightward shift in the cumulative distribution of amplitudes [Kolmogorov–Smirnov (K–S) test, p < 0.0001] and an increase in mean amplitude (∼35%; Fig. 3A,B,D). Interestingly, this increase in mEPSC amplitudes with DHHC2 knockdown is very similar to previous observations mentioned above for AKAP79C36,129S expression (Keith et al., 2012). Also of note was the detection of a significant rightward shift in the interevent interval distribution in miDHHC2-expressing cells relative to controls, indicating a DHHC2 knockdown-induced increase in the number of active synapses (Fig. 3A,C; K–S test, p < 0.0001). Thus, these results demonstrate that loss of DHHC2 expression, like loss of AKAP79 palmitoylation, triggers basal synaptic strengthening over a period of ∼48 h.
Figure 3.

Synaptic potentiation is altered by DHHC2 knockdown in hippocampal neurons. A, Representative traces of mEPSCs recorded from 12 DIV cultured hippocampal neurons transfected as indicated with mCh alone (Control) or miDHHC2 (Knockdown). B, C, Cumulative plots of mEPSC amplitude (2 pA bins) and interevent interval (ms; related to frequency) for recordings as in A. D, Mean (±SEM) mEPSC amplitudes for mCh (control; n = 16) and miDHHC2-expressing neurons (n = 12) with and without cLTP treatment (10 min in 0 Mg2+, 200 μm glycine, and 50 μm picrotoxin; 25 min recovery). miDHHC2 expression increases basal mean mEPSC amplitudes. cLTP treatment potentiates synapses in control cells (n = 18) but depresses synapses in miDHHC2-expressing cells (n = 14). E, Mean (±SEM) mEPSC frequency (Hz) for mCh (control) and miDHHC2-expressing neurons with and without cLTP treatment. Control cells significantly increase and miDHHC2 cells significantly decrease mEPSC frequency with cLTP treatment (*p < 0.05, **p < 0.01, one-way ANOVA, Tukey's post hoc test).
We next determined whether DHHC2 knockdown interferes with acute induction of NMDA-dependent synaptic potentiation by using a well established cLTP protocol where neurons are treated with glycine in Mg2+-free media (Lu et al., 2001; Park et al., 2004; Fortin et al., 2010; Kennedy et al., 2010; Keith et al., 2012). In response to cLTP stimulation, control neurons, as expected, showed a significant increase in mean mEPSC amplitude (∼28%) and a rightward shift in the distribution of amplitudes on cumulative plots (Fig. 3A,B,D; K–S test, p < 0.0001). In contrast, DHHC2-deficient neurons not only failed to undergo further synaptic strengthening but exhibited a significant depression of mean mEPSC amplitude (∼29%) and a leftward shift in the cumulative distribution of amplitudes relative to untreated miDHHC2 neurons (Fig. 3A,B,D; K–S test, p < 0.0001). cLTP stimulation is also known to increase mEPSC frequency due to postsynaptic unsilencing (Lu et al., 2001; Fortin et al., 2010; Keith et al., 2012). Accordingly, a significant increase in AMPAR mean mEPSC frequency (∼96%) and a corresponding leftward shift in the distribution of interevent intervals toward shorter times (K–S test, p < 0.005) was observed following cLTP stimulation in control neurons (Fig. 3A,C,E). However, miDHHC2-expressing cells exhibited reduced mEPSC frequency (∼47%) and a rightward shift in interevent interval distribution toward longer times in response to cLTP treatment (K–S test, p < 0.001), indicating that functional silencing of synapses occurred in response to what should have been a potentiating stimulus. Again, this reversal of the plasticity response in favor of LTD over LTP in DHHC2 knockdown neurons echoes prior experiments on neurons expressing palmitoylation-deficient AKAP79CS, in which inappropriate depression of both mEPSC amplitude and frequency was observed in response to cLTP (Keith et al., 2012). Collectively, these experiments demonstrate that loss of DHHC2 has profound effects on both basal excitatory synapse function and regulation of synaptic potentiation that closely mirror loss of AKAP79 palmitoylation.
DHHC2 controls synaptic recruitment of AKAP79 and dendritic spine enlargement following potentiation
Having established DHHC2 as a critical regulator of AMPAR synaptic function, we next investigated whether DHHC2 also controls spine structural plasticity and AKAP79 recruitment to spines, both of which occur during cLTP and require regulation of RE trafficking and exocytosis (Kopec et al., 2006; Park et al., 2006; Keith et al., 2012). To determine whether DHHC2 is required for cLTP-induced AKAP79 trafficking to spines, we imaged live, AKAP79-GFP-expressing hippocampal neurons before and then 25 min after 10 min of cLTP stimulation. Spine localization of AKAP79-GFP was determined by measuring dendritic spine/shaft fluorescence intensity ratios before and after cLTP for the same neuron (Fig. 4A,B). As expected, mCh-expressing control neurons exhibited an increase in AKAP79 spine localization following cLTP (1.19 ± 0.06-fold increase). However, in cells expressing miDHHC2, AKAP79 did not redistribute to spines (0.93 ± 0.05-fold change). Re-expression of DHHC2WT, using an RNAi-resistant construct, restored AKAP79 redistribution nearly to control levels (1.08 ± 0.06-fold increase). These results demonstrate a requirement for DHHC2 in AKAP79 recruitment to spines following acute synaptic potentiation in hippocampal neurons.
LTP is usually also associated with the enlargement of dendritic spines (Matsuzaki et al., 2001; Okamoto et al., 2004; Kopec et al., 2006), and we previously found that the AKAP79CS mutant prevented cLTP-induced spine enlargement in cultured neurons (Keith et al., 2012). We therefore investigated whether DHHC2 is also required for dendritic spine growth following cLTP treatment. By analyzing the same neurons used above to assess AKAP79 spine localization, we were able to track spine size before and after cLTP treatment by imaging coexpressed mCh fluorescence filling the spine cytoplasm (Fig. 4A,C). Control cells exhibited a significant mean increase in spine size, measured as the cross-sectional area of the spine head, following cLTP. In contrast, miDHHC2-expressing cells failed to exhibit spine enlargement, and this defect was rescued by expression of RNAi-insensitive DHHC2WT (Fold increase in spine area: mCh 1.6 ± 0.2, miDHHC2 1.02 ± 0.04, miDHHC2 + DHHC2WT-res 1.50 ± 0.09). Thus, DHHC2 knockdown prevents both AKAP79 trafficking to spines and dendritic spine enlargement in response to cLTP implicating DHHC2 in the regulation of spine structural plasticity that accompanies acute synaptic potentiation.
Interestingly, in these experiments we found that DHHC2 knockdown also increased basal dendritic spine density by ∼30% (Fig. 4D; Spines/10 μm of dendrite mCh: 4.0 ± 0.2, miDHHC2: 5.2 ± 0.3, miDHHC2 + DHHC2WT-res: 4.2 ± 0.3) but without significantly affecting basal dendritic spine size [Spine area (μm2): mCh 0.42 ± 0.03, miDHHC2 0.41 ± 0.03, miDHHC2 + res 0.35 ± 0.05]. To independently assess whether DHHC2 knockdown increased the number of synapses, we examined localization of the postsynaptic marker PSD-95 in dendritic spines (Fig. 4E), and after DHHC2 knockdown, found a significant increase in PSD-95 cluster density (Fig. 4F; PSD-95 clusters/10 μm of dendrite mCh: 3.1 ± 0.4, miDHHC2: 4.6 ± 0.3). These DHHC2 knockdown-induced increases in both dendritic spine and PSD-95 cluster numbers are consistent with the somewhat increased basal mEPSC frequency found in electrophysiological experiments (Fig. 3A,C; K–S test, p < 0.001) and confirm that there was an increase in the total number of active synapses in response to DHHC2 depletion. Since PSD-95 is an AKAP79/150 binding partner and a DHHC2 substrate and its synaptic clustering is regulated by palmitoylation (Colledge et al., 2000; El-Husseini et al., 2002; Noritake et al., 2009; Robertson et al., 2009), we also determined whether DHHC2 knockdown altered the amount of PSD-95 within dendritic spine clusters. Previous studies knocking down DHHC2 expression for a prolonged period of time (∼10 d; Noritake et al., 2009; Fukata et al., 2013) observed decreased postsynaptic clustering of PSD-95. Thus, somewhat unexpectedly, following more acute knockdown of DHHC2 expression for only 48 h, we found no changes in either PSD-95 mean cluster size [Fig. 4F; PSD-95 cluster area (μm2): mCh 0.059 ± 0.004, miDHHC2 0.070 ± 0.005] or intensity [Fig. 4F; PSD-95 cluster intensity (a.f.u.): mCh 2960 ± 60, miDHHC2 3030 ± 70]. Thus, interestingly, the increases in basal mEPSC amplitude we measured after DHHC2 knockdown (Fig. 3A,B,D) were not accompanied by any increases in the average size or PSD-95 content of individual dendritic spines (Fig. 4D–F).
DHHC2 regulates LTP stimulation of RE exocytosis in dendrites
In the experiments detailed above, we found that DHHC2 is required for rapid changes in postsynaptic structure, protein composition, and AMPAR function during cLTP. Neurons maintain tight control over synaptic strength by regulating the exchange of AMPARs between the synaptic and extrasynaptic plasma membrane and intracellular pools through local endocytosis and RE exocytosis, which occurs near synapses in spines or on the adjacent dendrite shaft. (Makino and Malinow, 2009; Petrini et al., 2009; Kennedy et al., 2010; Opazo et al., 2010). Accordingly, as an RE-resident PAT for AKAP79/150, we reasoned that loss of DHHC2 function may also result in altered regulation of RE exocytosis that is required for LTP (Park et al., 2004; Kennedy and Ehlers, 2011), similar to previous observations with the AKAP79C36,129S mutant (Keith et al., 2012). To test this hypothesis, we used a genetically encoded optical reporter of RE exocytosis, the TfR conjugated to the pH-sensitive GFP variant SEP-TfR (Park et al., 2006; Kennedy et al., 2010; Keith et al., 2012). When this construct is present in REs, the SEP moiety faces the acidic (pH ∼5.5) lumen where its fluorescence is quenched. Following exocytosis, SEP is exposed to the neutral extracellular environment and a flash of green fluorescence is observed. We observed neurons expressing the SEP-TfR reporter and mCh or miDHHC2 (Fig. 5A) for a period of 4 min using a spinning-disk confocal microscope and then perfused cLTP ACSF and continued to image for another 4 min. Time-composite images showing the accumulated SEP-TfR intensity over the corresponding pre- and post-cLTP imaging periods were then generated (Fig. 5B). In addition, because static images are not ideal for conveying transient, rapid exocytic events, we also represented these data using 3D time-composite, cumulative plots that show SEP-TfR exocytic events as fluorescence intensity peaks on the z-axis (Fig. 5C). Quantification of the fold change in exocytic events between pre- and post-cLTP treatment revealed a significant increase in events in control neurons, as expected from previous work (Park et al., 2006; Kennedy et al., 2010; Keith et al., 2012). In contrast, cLTP stimulation did not increase RE exocytic events in DHHC2-deficient neurons (Fig. 5D). Re-expression of CFP-DHHC2WT (CFP channel not shown) rescued this inhibitory effect of DHHC2 knockdown (mCh: 1.4 ± 0.1, miDHHC2: 0.90 ± 0.08, miDHHC2 + DHHC2WT-res: 1.37 ± 0.09). Overall, these experiments reveal that DHHC2 is required for regulation of RE exocytosis associated with LTP.
DHHC2 controls RE exocytosis and synaptic potentiation through AKAP79 palmitoylation
PATs are somewhat promiscuous and modify numerous substrates, and DHHC2 is a prime example, with at least eight described neuronal substrates to date, three of which are synaptic including PSD-95 (Fukata and Fukata, 2010). Though many of the DHHC2 knockdown phenotypes documented above are very similar to those observed with the AKAP79CS mutant (Keith et al., 2012) and we did not detect any decreases in PSD-95 synaptic clustering, it is still possible that the observed effects of DHHC2 knockdown could also be mediated at least in part through altered palmitoylation of other substrate proteins. To address this issue, we tested whether normal synaptic plasticity could be restored in DHHC2 knockdown neurons by engineering an N-terminal myristoylation sequence on AKAP79 to render it constitutively lipidated (Fig. 6A). Importantly, this strategy has been used previously to produce constitutively lipidated forms of other palmitoylated proteins, such as GRIP1b, and circumvent their regulation by depalmitoylation (Thomas and Huganir, 2013). We first determined whether myristoylation could substitute for palmitoylation in targeting AKAP79 to endosomes in COS7 cells (Fig. 6B). As expected from previous work, AKAP79CS failed to associate with REs, while AKAP79WT associated with >30% of REs (Fig. 6C). Myristoylation of AKAP79CS was able to recover approximately half of AKAP79 RE localization, similar to previously seen for AKAP79 mutants lacking one palmitoylation site (Keith et al., 2012). Myr-AKAP79WT did not differ significantly from AKAP79WT in its RE association, despite the potential for three lipid modifications. However, we predicted that overexpression of myr-AKAP79WT might prove effective in restoring functional losses resulting from DHHC2 knockdown, due not only to constitutive myristoylation but perhaps also to being more resistant to depalmitoylation than WT due to stronger membrane association.
Figure 6.
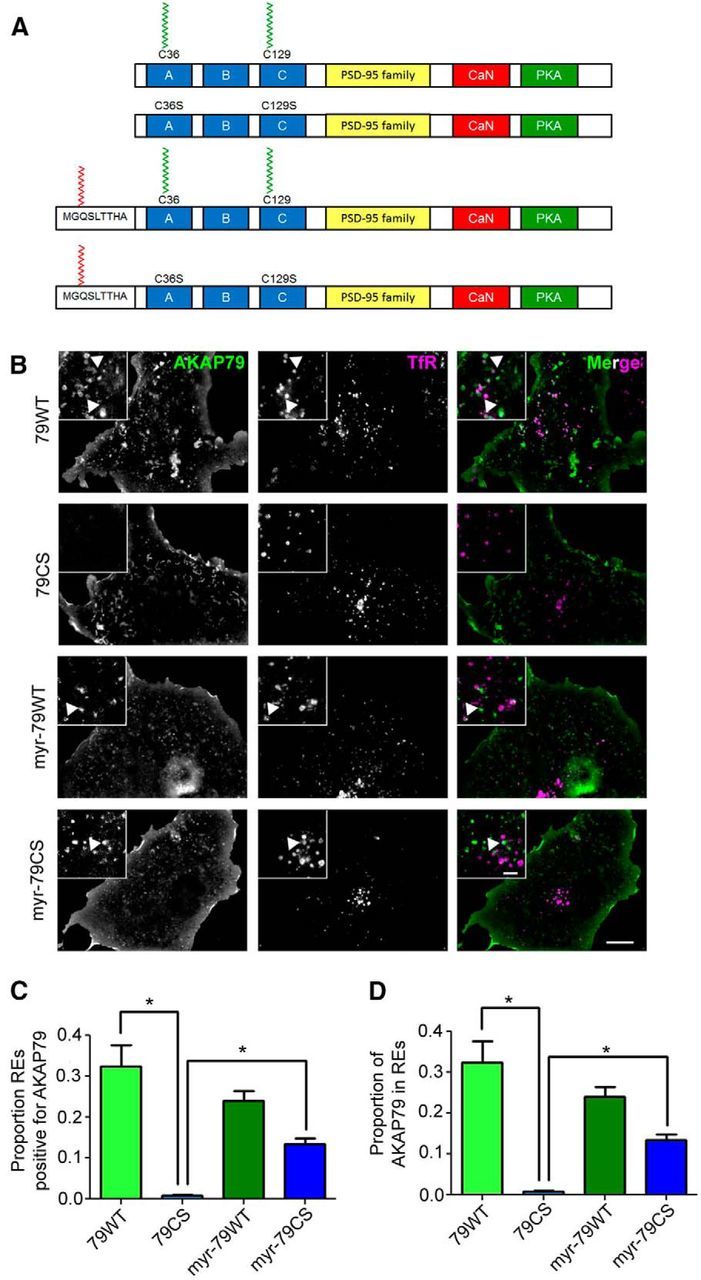
Myristoylation can substitute for palmitoylation in AKAP79 endosomal targeting. A, Structural schematics of AKAP79 lipidation mutants. The N-terminal MGQSLTTHA myristoylation consensus sequence confers cotranslational myristate addition to AKAP79. The locations of the Cys to Ser mutations at positions 36 and 129 that prevent AKAP79 palmitoylation are also indicated. B, Images of COS7 cells coexpressing GFP-tagged AKAP79 lipidation mutants (green) and the RE marker TfR-mCh (magenta). Note: all AKAP79-GFP constructs, including AKAP79CS, display strong plasma membrane localization. C, D, Quantification of colocalization of AKAP79 mutants and TfR-mCh reveals total loss of AKAP79CS but not myr-AKAP79CS from REs (n = 11–18 cells/condition; all data are presented as mean ± SEM; *p < 0.05, one-way ANOVA, Tukey's post hoc test).
The efficacy of myr-AKAP79 in “rescuing” the effects of DHHC2 knockdown was tested first using the RE exocytosis assay in neurons expressing SEP-TfR and miDHHC2 (Fig. 7A–C). Importantly, a robust cLTP-induced increase in RE exocytosis was observed in the miDHHC2 + myr-AKAP79WT condition, indicating restoration of normal RE regulation in DHHC2-deficient neurons (Fig. 7B). Expression of myr-AKAP79CS in the presence of miDHHC2 exhibited a trend toward restoring increased exocytosis in response to cLTP, but this effect was not significantly higher than DHHC2 knockdown alone. In contrast, AKAP79CS was completely incapable of recovering cLTP stimulation of RE exocytosis in DHHC2 knockdown cells (mCh: 1.6 ± 0.1, miDHHC2: 1.17 ± 0.09, miDHHC2+myr-79WT: 2.2 ± 0.4, miDHHC2 + myr-79CS: 1.3 ± 0.3, miDHHC2 + 79CS: 1.0 ± 0.2; Fig. 7C). From these results, we conclude that the myr-AKAP79WT mutant, as predicted, allows DHHC2 knockdown neurons to resist the functional impacts of AKAP79 depalmitoylation on RE exocytosis regulation.
We also assessed the effects of the myr-AKAP79 constructs on basal rates of RE exocytosis measured as the number of exocytic events occurring before cLTP treatment normalized to total dendrite length, and found a significant elevation of basal RE exocytosis in miDHHC2 neurons (Fig. 7C). However, miDHHC2 coexpression with either myr-AKAP79WT or myr-AKAP79CS significantly reduced basal exocytosis relative to miDHHC2 alone (Fig. 7C; Exocytosis events/min/100 μm dendrite mCh: 2.5 ± 0.3, miDHHC2: 4.4 ± 0.6, miDHHC2 + myr-79WT: 1.8 ± 0.4, miDHHC2 + myr79CS 0.9 ± 0.3, miDHHC2 + 79CS: 3.7 ± 1.2). Only AKAP79CS was incapable of reducing basal RE exocytosis in the presence of DHHC2 knockdown, in agreement with our previous findings that AKAP79CS overexpression enhances basal exocytosis compared with WT (Keith et al., 2012). Overall, these findings are consistent with a shared mechanism for RE trafficking dysfunction between DHHC2 knockdown and AKAP79CS expression, which both increase basal RE exocytosis rates but then prevent stimulation by cLTP.
Having established rescue of regulated RE exocytosis by myr-AKAP79WT, we next asked whether this constitutively lipidated mutant could restore normal cLTP-synaptic potentiation in the context of DHHC2 knockdown by recording mEPSCs from neurons expressing miDHHC2 and myr-AKAP79WT (Fig. 8A). While expression of myr-AKAP79 was able to reverse elevated basal mEPSC frequency observed with DHHC2 knockdown (Fig. 8A,C; K–S test, p < 0.0001; E, t test, p < 0.05), it had no significant impact on the increased basal mEPSC amplitudes (Fig. 8A,B,D; p > 0.05). However, while mean mEPSC amplitude was not significantly enhanced by cLTP in neurons expressing miDHHC2 and myr-AKAP79WT (Fig. 8D), a significant rightward shift in cumulative plots of mEPSC amplitudes was observed indicating restoration of a small amount of synaptic potentiation (Fig. 8B; K–S test, p < 0.05). Importantly, these small, positive effects on mEPSC amplitude were very different from those observed in cells expressing miDHHC2 alone, which exhibited a significant reduction in mean amplitude and a leftward shift in amplitude cumulative distribution in response to cLTP (Figs. 3, 8B,D). Concordantly, mEPSC frequency was significantly increased by cLTP treatment in neurons expressing miDHHC2 and myr-AKAP79WT, as shown by analysis of both mean frequency (∼91%) and interevent interval distribution (Fig. 8A,C,E; K–S test, p < 0.001), an effect also opposite to that observed with DHHC2 knockdown alone (Figs. 3, 8C,E). These mEPSC recordings demonstrate that myr-AKAP79 reverses most of the alterations in plasticity regulation that are characteristic of DHHC2 knockdown. Collectively, these data provide strong evidence that AKAP79 is a major target of DHHC2 in the RE compartment and that this signaling pathway is a critical regulator of excitatory postsynaptic plasticity mechanisms that are required for LTP.
Figure 8.

Myristoylated AKAP79 restores normal control of synaptic potentiation in DHHC2-deficient neurons. A, Representative traces of mEPSCs recorded from 12 DIV cultured hippocampal neurons expressing miDHHC2 + myr-AKAP79WT ± cLTP. B, Cumulative plots of mEPSC amplitude (2 pA bins) and interevent interval (ms; related to frequency, C) for recordings as in A. Data from Figure 3B and C for miDHHC2 are reproduced in gray for comparison. D, Quantification of mEPSC mean (±SEM) amplitude. cLTP does not significantly increase mean mEPSC amplitude in cells expressing miDHHC2 + myr-AKAP79WT, but mean amplitude does not decrease as for miDHHC2 alone (miDHHC2 data reproduced from Fig. 3D). E, Quantification of mEPSC mean (±SEM) frequency (Hz). cLTP treatment of neurons coexpressing myr-AKAP79WT and miDHHC2 shows a significant increase in mEPSC frequency, an effect opposite to that seen for miDHHC2 knockdown alone (miDHHC2 data reproduced from Fig. 3E; n = 13–15 cells/condition; *p < 0.05, t test).
Discussion
Precise organization of postsynaptic signaling proteins in multiprotein complexes is critical for localized, rapid signal transduction events required for synaptic plasticity. S-palmitoylation of postsynaptic scaffolds is emerging as an integral mechanism orchestrating the assembly of such signaling complexes. Here, we used biochemical, imaging and electrophysiological approaches to show that AKAP79/150 palmitoylation by DHHC2 is essential for control of dendritic RE exocytosis and AMPAR potentiation.
DHHC2 interacts with and palmitoylates AKAP79/150
Protein palmitoylation is catalyzed by DHHC-family PATs, but despite several large-scale proteomic efforts (Fukata et al., 2004; Roth et al., 2006; Kang et al., 2008; Huang et al., 2009), relatively few specific enzyme/substrate pairs have been described. This limited progress is largely due to the lack of consensus substrate sequences that specify palmitoylation by one PAT versus another. Thus, extensive screening for functional interactions is typically needed to identify cognate PATs for substrates of interest. We used DHHC coimmunoprecipitation as our primary screen, but biotin-switch or 3H-palmitate labeling assays are viable alternatives (Fukata et al., 2004; Brigidi et al., 2014). While DHHC2 exhibited the most pronounced interaction with AKAP79, it is notable that other DHHCs showed weaker, but detectable precipitation with AKAP79 (Fig. 1A). Weak AKAP79 association with other DHHCs is likely due to both the substrate promiscuity and partially overlapping subcellular distributions of these enzymes. These findings also highlight the limitations of using biochemical approaches alone to detect important functional interactions between DHHCs and potential substrates, as subcellular spatial information is lost, and support use of our multimodal approach that included not only biochemistry but also imaging to identify DHHC2 regulation of AKAP79-dependent functions in REs.
How do AKAP79/150 and DHHC2 interact? As mentioned above, DHHC2 is a tetra-spanning integral membrane protein with N-terminal and C-terminal cytosolic tails and a DHHC enzymatic motif located in the lone intracellular loop. Unlike other DHHCs that feature canonical protein–protein interaction domains, such as C-terminal PDZ-ligands in DHHC5 and DHHC8 that bind to PSD-95 and GRIP1b (Li et al., 2010; Thomas et al., 2012), DHHC2 does not have any obvious substrate interaction motifs. AKAP79/150, on the other hand, is a peripheral membrane protein with a polybasic N-terminal membrane targeting domain that binds to phosphoinositide lipids (Dell'Acqua et al., 1998). Since AKAP79/150 is palmitoylated in its N-terminal targeting domain, we reasoned that this domain would interact with DHHC2. Indeed, an AKAP79 (1–153) truncation mutant containing the N-terminal domain was sufficient for co-IP DHHC2. Strikingly, mutation of the C36 and C129 palmitoylation sites within this domain prevented DHHC2 interaction. However, loss of DHHC2 enzymatic activity did not disrupt the DHHC2/AKAP79 complex. Our previous work found that palmitoylation is essential for AKAP79 localization in REs (Keith et al., 2012). Therefore, AKAP79CS may not come into contact with DHHC2 in cells both due to lack of suitable Cys substrate sites as well as exclusion from REs where DHHC2 is enriched. Conversely, the enzymatically dead DHHC2CS mutant retains RE localization (Fig. 2B), making it available to interact with AKAP79 palmitoylated by endogenous PATs in this compartment. However, additional DHHC2 structural determinants that further specify interaction with AKAP79 likely exist.
The C-terminal tails of DHHC family members contain a high degree of sequence variability and are thought to determine subcellular localization of these enzymes (Ohno et al., 2006). Although certain DHHCs are enriched in specific membrane compartments, there is evidence for territorial overlap between species. For example, DHHC2 displays pronounced RE localization (Fig. 2; Greaves et al., 2011), yet some DHHC2 likely also traffics to the plasma membrane upon endosome fusion (Noritake et al., 2009). Similarly, DHHC5 and DHHC8 are expressed prominently on the plasma membrane but also traffic through endosomes (Thomas et al., 2012; Fig. 2). Determining the degree to which these DHHCs are active toward various substrates as they move through different membrane compartments and characterizing the targeting signals that maintain their semidiscrete distributions will be essential for furthering our understanding of how these enzymes regulate neuronal function.
DHHC2 control of synaptic structure and function
Excitatory synapses can undergo multiple types of plasticity defined by timescale, underlying molecular mechanisms, and proposed roles in information processing. Over the last decade, the need for neurons to globally adjust synaptic strength to maintain bounds on action potential firing rates has been recognized and collectively referred to as homeostatic plasticity (Turrigiano, 2012). Specifically, in response to decreases or increases in firing over hours and days, neurons alter many synaptic components in a compensatory manner to either globally enhance or depress AMPAR synaptic strength, respectively (Ehlers, 2003). Importantly, DHHC2 participates in homeostatic synaptic scaling-up in response to decreased firing by increasing PSD-95 palmitoylation and postsynaptic clustering to promote AMPAR synaptic incorporation (Noritake et al., 2009).
More rapid forms of plasticity can also involve changes in postsynaptic palmitoylation. NMDAR activation associated with LTD stimulates depalmitoylation of PSD-95 and AKAP79/150 within minutes and promotes concomitant loss of synaptic AMPARs (El-Husseini et al., 2002; Keith et al., 2012). Conversely, cLTP induction results in rapid AKAP79/150 palmitoylation and synaptic recruitment. Importantly, palmitoylation-deficient AKAP79CS blocks cLTP-induced synaptic potentiation (Keith et al., 2012). In this study, we demonstrate that DHHC2 also regulates this form of acute synaptic potentiation (Fig. 3). Strikingly, DHHC2 knockdown recapitulated effects of AKAP79CS expression on mEPSC amplitude and frequency; both manipulations elevated basal AMPAR activity and then exhibited inappropriate depression of AMPAR activity in response to cLTP.
Despite the striking phenotypic similarities between DHHC2 knockdown and AKAP79CS expression, it was important to confirm the AKAP as a critical substrate of DHHC2 required for cLTP. To address this issue, we used a method previously used to circumvent the need for palmitoylation by engineering a mutant that is alternatively and irreversibly lipidated (El-Husseini et al., 2000; Thomas et al., 2012). Importantly, addition of an N-terminal myristoylation sequence to AKAP79 was able to support RE localization (Fig. 6) and reverse many of the functional impacts of DHHC2 knockdown. In particular, myr-AKAP79WT completely restored cLTP stimulation of RE exocytosis in DHHC2 knockdown neurons (Fig. 7) and for the most part normalized regulation of AMPAR synaptic function, with cLTP inducing net-positive effects on overall mEPSC activity instead of inappropriate reductions as seen with DHHC2 knockdown alone (Fig. 8). However, it should be noted that myr-AKAP79WT failed to reverse the increased basal mEPSC amplitudes in DHHC2 knockdown neurons, and as such, was not capable of restoring significant enhancement of mean mEPSC amplitude following cLTP. But despite these limitations, rescue of DHHC2 loss of function by myr-AKAP79 was surprisingly successful, especially given that C-14 myristate is not as strong a lipid-raft targeting signal as C-16 palmitate and the engineered myristate is located at the N terminus and not within the polybasic domains like C36 and C129. Nonetheless, for a majority of the plasticity-related phenomena assayed, myr-AKAP79 restored WT-like responses in DHHC2-deficient neurons, lending strong support to the idea that DHHC2 acts via AKAP palmitoylation to control synaptic potentiation. Overall, our past and present findings suggest that DHHC2 and its palmitoylation of AKAP79/150 in REs paradoxically limit basal exocytosis and synaptic strength yet promote stimulation of RE exocytosis and AMPAR synaptic incorporation during LTP.
The DHHC2 knockdown-induced increases in basal mEPSC frequency, and accompanying increases in spine and PSD-95 cluster numbers, we report here are not necessarily in conflict with previous studies mentioned above showing DHHC2 knockdown decreases PSD-95 clustering and interferes with homeostatic plasticity (Noritake et al., 2009). Our DHHC2 knockdown was performed for a much shorter period of time to assess the acute effects of DHHC2 loss on LTP mechanisms, while Noritake et al. (2009) used longer term knockdown to assess the impacts of chronic DHHC2 loss on synapse homeostasis. Importantly, our short-term DHHC2 knockdown, which did not decrease PSD-95 clustering as observed with prolonged knockdown in Noritake et al., (2009) helped us characterize AKAP79/150 as a novel DHHC2 substrate that is important for regulation of plasticity mechanisms in a way that is distinct from PSD-95.
DHHC2 and AKAP79 regulation of postsynaptic exocytosis during plasticity
Despite recent advances in uncovering mechanisms underlying plasticity at excitatory synapses, many important questions remain. Basic understanding of recruitment, docking, and NMDAR-triggered fusion of REs in dendrites and spines is still rudimentary. As both a plasma membrane and RE-resident scaffold protein, AKAP79/150 may target regulatory enzymes to important components of the endosome trafficking, docking, and release machinery in these locations. From prior studies we know that AKAP79/150 anchoring of PKA and CaN plays important roles controlling AMPAR phosphorylation and trafficking during LTP and LTD (Lu et al., 2007; Jurado et al., 2010; Sanderson et al., 2012); however, we do not yet know if specific enzymes anchored to this AKAP also directly regulate RE trafficking and exocytosis. Likewise, we do not know if DHHC2 delivery to the dendritic plasma membrane near synapses during LTP could play additional roles by palmitoylating other proteins associated with AKAP79/150, such as PSD-95 or AMPARs, to favor their synaptic incorporation and/or retention. Thus, much remains to be investigated to fully understand the likely complex interplay between DHHC2 and the AKAP79/150 signaling scaffold in control of postsynaptic membrane and protein trafficking during synaptic plasticity.
Footnotes
This research was supported by a grant from National Institutes of Health (NIH)–National Institute of Neurological Disorders and Stroke (NINDS) to M.L.D. (R01NS040701). K.M.W. was supported by NIH–NINDS (T32NS007083) and an American Heart Association Postdoctoral Fellowship. Spinning-disk confocal microscopy was performed in the University of Colorado Anschutz Medical Campus Advanced Light Microscopy Core supported by NIH–NINDS through the Rocky Mountain Neurological Disorders Core Center (P30NS048154) and NIH–NCATS Colorado CTSI (UL1TR001082). We thank Masaki Fukata, Michael Ehlers, Gareth Thomas, Richard Huganir, and Shernaz Bamji for providing reagents. We thank Drs. K. Ulrich Bayer and Matthew Kennedy for critical reading of this manuscript.
The authors declare no competing financial interests.
References
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22:461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigidi GS, Sun Y, Beccano-Kelly D, Pitman K, Mobasser M, Borgland SL, Milnerwood AJ, Bamji SX. Palmitoylation of delta-catenin by DHHC5 mediates activity-induced synapse plasticity. Nat Neurosci. 2014;17:522–532. doi: 10.1038/nn.3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TC, Correia SS, Petrok CN, Esteban JA. Functional compartmentalization of endosomal trafficking for the synaptic delivery of AMPA receptors during long-term potentiation. J Neurosci. 2007;27:13311–13315. doi: 10.1523/JNEUROSCI.4258-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/S0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Delint-Martinez I, Willoughby D, Hammond GV, Ayling LJ, Cooper DM. Palmitoylation targets AKAP79 to lipid rafts and promotes its regulation of the calcium sensitive adenylyl cyclase type 8. J Biol Chem. 2011;286:32962–32975. doi: 10.1074/jbc.M111.243899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua ML, Faux MC, Thorburn J, Thorburn A, Scott JD. Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4, 5- bisphosphate. EMBO J. 1998;17:2246–2260. doi: 10.1093/emboj/17.8.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/S0896-6273(00)00129-X. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Craven SE, Chetkovich DM, Firestein BL, Schnell E, Aoki C, Bredt DS. Dual palmitoylation of PSD-95 mediates its vesiculotubular sorting, postsynaptic targeting, and ion channel clustering. J Cell Biol. 2000;148:159–172. doi: 10.1083/jcb.148.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini Ael-D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/S0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- Fernández-Monreal M, Brown TC, Royo M, Esteban JA. The balance between receptor recycling and trafficking toward lysosomes determines synaptic strength during long-term depression. J Neurosci. 2012;32:13200–13205. doi: 10.1523/JNEUROSCI.0061-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forti L, Cesana E, Mapelli J, D'Angelo E. Ionic mechanisms of autorhythmic firing in rat cerebellar Golgi cells. J Physiol. 2006;574:711–729. doi: 10.1113/jphysiol.2006.110858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, Soderling TR. Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J Neurosci. 2010;30:11565–11575. doi: 10.1523/JNEUROSCI.1746-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Dimitrov A, Boncompain G, Vielemeyer O, Perez F, Fukata M. Local palmitoylation cycles define activity-regulated postsynaptic subdomains. J Cell Biol. 2013;202:145–161. doi: 10.1083/jcb.201302071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci. 2002;22:7027–7044. doi: 10.1523/JNEUROSCI.22-16-07027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Carmichael JA, Chamberlain LH. The palmitoyl transferase DHHC2 targets a dynamic membrane cycling pathway: regulation by a C-terminal domain. Mol Biol Cell. 2011;22:1887–1895. doi: 10.1091/mbc.E10-11-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne EA, Dell'Acqua ML. Phospholipase C is required for changes in postsynaptic structure and function associated with NMDA receptor-dependent long-term depression. J Neurosci. 2007;27:3523–3534. doi: 10.1523/JNEUROSCI.4340-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Sanders S, Singaraja R, Orban P, Cijsouw T, Arstikaitis P, Yanai A, Hayden MR, El-Husseini A. Neuronal palmitoyl acyl transferases exhibit distinct substrate specificity. FASEB J. 2009;23:2605–2615. doi: 10.1096/fj.08-127399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Biou V, Malenka RC. A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat Neurosci. 2010;13:1053–1055. doi: 10.1038/nn.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, Green WN, Yates JR, 3rd, Davis NG, El-Husseini A. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456:904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith DJ, Sanderson JL, Gibson ES, Woolfrey KM, Robertson HR, Olszewski K, Kang R, El-Husseini A, Dell'acqua ML. Palmitoylation of A-kinase anchoring protein 79/150 regulates dendritic endosomal targeting and synaptic plasticity mechanisms. J Neurosci. 2012;32:7119–7136. doi: 10.1523/JNEUROSCI.0784-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Ehlers MD. Mechanisms and function of dendritic exocytosis. Neuron. 2011;69:856–875. doi: 10.1016/j.neuron.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hu J, Höfer K, Wong AM, Cooper JD, Birnbaum SG, Hammer RE, Hofmann SL. DHHC5 interacts with PDZ domain 3 of post-synaptic density-95 (PSD-95) protein and plays a role in learning and memory. J Biol Chem. 2010;285:13022–13031. doi: 10.1074/jbc.M109.079426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- Linder ME, Jennings BC. Mechanism and function of DHHC S-acyltransferases. Biochem Soc Trans. 2013;41:29–34. doi: 10.1042/BST20120328. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/S0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noritake J, Fukata Y, Iwanaga T, Hosomi N, Tsutsumi R, Matsuda N, Tani H, Iwanari H, Mochizuki Y, Kodama T, Matsuura Y, Bredt DS, Hamakubo T, Fukata M. Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J Cell Biol. 2009;186:147–160. doi: 10.1083/jcb.200903101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno Y, Kihara A, Sano T, Igarashi Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim Biophys Acta. 2006;1761:474–483. doi: 10.1016/j.bbalip.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of Stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, Choquet D. Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron. 2009;63:92–105. doi: 10.1016/j.neuron.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HR, Gibson ES, Benke TA, Dell'Acqua ML. Regulation of postsynaptic structure and function by an A-kinase anchoring protein-membrane-associated guanylate kinase scaffolding complex. J Neurosci. 2009;29:7929–7943. doi: 10.1523/JNEUROSCI.6093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Wan J, Bailey AO, Sun B, Kuchar JA, Green WN, Phinney BS, Yates JR, 3rd, Davis NG. Global analysis of protein palmitoylation in yeast. Cell. 2006;125:1003–1013. doi: 10.1016/j.cell.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Dell'Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist. 2011;17:321–336. doi: 10.1177/1073858410384740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, Gibson ES, Lam P, Freund RK, Chick WS, Dell'Acqua ML. AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors. J Neurosci. 2012;32:15036–15052. doi: 10.1523/JNEUROSCI.3326-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J Neurosci. 2002;22:3044–3051. doi: 10.1523/JNEUROSCI.22-08-03044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. Palmitoylation-dependent regulation of glutamate receptors and their PDZ domain-containing partners. Biochem Soc Trans. 2013;41:72–78. doi: 10.1042/BST20120223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Hayashi T, Chiu SL, Chen CM, Huganir RL. Palmitoylation by DHHC5/8 targets GRIP1 to dendritic endosomes to regulate AMPA-R trafficking. Neuron. 2012;73:482–496. doi: 10.1016/j.neuron.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Hayashi T, Huganir RL, Linden DJ. DHHC8-dependent PICK1 palmitoylation is required for induction of cerebellar long-term synaptic depression. J Neurosci. 2013;33:15401–15407. doi: 10.1523/JNEUROSCI.1283-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 2012;4:a005736. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidman R, Jackson CS, Magee AI. Protein acyl thioesterases (review) Mol Membr Biol. 2009;26:32–41. doi: 10.1080/09687680802629329. [DOI] [PubMed] [Google Scholar]