

Abstract
Acute insults produce hyperalgesic priming, a neuroplastic change in nociceptors that markedly prolongs inflammatory mediator-induced hyperalgesia. After an acute initiating insult, there is a 72 h delay to the onset of priming, for which the underlying mechanism is unknown. We hypothesized that the delay is due to the time required for a signal to travel from the peripheral terminal to the cell body followed by a return signal to the peripheral terminal. We report that when an inducer of hyperalgesic priming (monocyte chemotactic protein 1) is administered at the spinal cord of Sprague Dawley rats, priming is detected at the peripheral terminal with a delay significantly shorter than when applied peripherally. Spinally induced priming is detected not only when prostaglandin E2 (PGE2) is presented to the peripheral nociceptor terminals, but also when it is presented intrathecally to the central terminals in the spinal cord. Furthermore, when an inducer of priming is administered in the paw, priming can be detected in spinal cord (as prolonged hyperalgesia induced by intrathecal PGE2), but only when the mechanical stimulus is presented to the paw on the side where the priming inducer was administered. Both spinally and peripherally induced priming is prevented by intrathecal oligodeoxynucleotide antisense to the nuclear transcription factor CREB mRNA. Finally, the inhibitor of protein translation reversed hyperalgesic priming only when injected at the site where PGE2 was administered, suggesting that the signal transmitted from the cell body to the peripheral terminal is not a newly translated protein, but possibly a newly expressed mRNA.
Introduction
Hyperalgesic priming is a preclinical model of the transition from acute to chronic pain. It can be produced by inflammatory or neuropathic insults, and is expressed as a long-lasting prolongation in the response to low levels of hyperalgesic agents such as prostaglandin E2 (PGE2), adenosine, and serotonin, with normal nociceptive threshold in the absence of these agents (Aley et al., 2000; Parada et al., 2003a; Reichling and Levine, 2009). While in naive control rats PGE2-induced hyperalgesia is of short duration (∼1 h) and is dependent on stimulatory G-protein activation of protein kinase A (Aley and Levine, 1999), in the primed state PGE2-induced hyperalgesia is markedly prolonged, lasting >24 h (Aley et al., 2000), with the prolongation phase (>1 h) mediated by a signaling pathway involving an inhibitory G-protein, phospholipase C β3 and protein kinase C ε (PKCε) (Aley et al., 2000; Parada et al., 2005; Joseph et al., 2007; Khasar et al., 2008; Dina et al., 2009; Reichling and Levine, 2009; Ferrari et al., 2013a). Importantly, the plasticity observed in hyperalgesic priming has been shown to be restricted to the nonpeptidergic, isolectin B4 (IB4)-positive nociceptors (Joseph and Levine, 2010), which express the extracellular matrix molecule versican (Bogen et al., 2005).
The induction of priming is triggered by pronociceptive mediators that transiently activate the calcium independent novel ε isoform of protein kinase C (PKCε; Aley et al., 2000; Parada et al., 2003a) or molecules downstream of PKCε such as α calmodulin-dependent protein kinase II and the ryanodine receptor (Ferrari et al., 2013b). In turn, cytoplasmic polyadenylation element-binding protein-dependent translation of mRNAs in the peripheral terminal of the nociceptor (Bogen et al., 2012; Ferrari et al., 2013c) contributes to the long-term maintenance of the primed state (Bogen et al., 2012; Ferrari et al., 2013c), which has been shown to persist unattenuated for at least 2 months (Aley et al., 2000; Parada et al., 2005).
A distinctive feature of hyperalgesic priming, whose underlying mechanism remains to be elucidated, is the very prolonged (∼72 h) delay from the acute painful event that induces priming, to the development of the long-lasting primed state (Bogen et al., 2012). In the present study, we explored the hypothesis that this delay is due, in part, to the time required for a signal to be generated in the peripheral terminal of the nociceptor and transported or transmitted to the cell body, where the regulation of transcription occurs, which, in turn, produces a signal that travels to the peripheral terminal of the nociceptor where hyperalgesic priming is expressed.
Materials and Methods
Animals.
The experiments were performed on adult male Sprague Dawley rats (220–400 g; Charles River Laboratories). Animals were housed, three per cage, under a 12 h light/dark cycle in a temperature- and humidity-controlled room in the animal care facility of the University of California, San Francisco. Food and water were available ad libitum. All nociceptive testing was done between 10:00 A.M. and 5:00 P.M., and the experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco, and adhered to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. A concerted effort was made to minimize the number of animals used and their suffering.
Testing mechanical nociceptive threshold.
The mechanical nociceptive threshold was quantified using an Ugo Basile Analgesymeter (Randall-Selitto paw-withdrawal test; Stoelting), a device that applies a linearly increasing mechanical force to the dorsum of the rat hindpaw, as previously described (Taiwo and Levine, 1989; Taiwo et al., 1989; Reichling and Levine, 2009). The nociceptive threshold was defined as the force, reported in grams, at which the rat withdrew its paw, and the baseline paw pressure threshold was defined as the mean of three readings taken before a test agent was administered; the response endpoint was an all-or-none reaction (i.e., either the animal withdraws its paw from the stimulus or does not). Each paw was treated as an independent measure, and each experiment was performed on a separate group of rats. Nociceptive thresholds and experimental treatments were not performed blinded. Data are presented as the mean change from baseline mechanical nociceptive threshold.
Drugs and method of administration.
The drugs used in this study were as follows: the direct-acting hyperalgesic inflammatory mediator PGE2, nerve growth factor (NGF), the protein translation inhibitor cordycepin from Cordyceps militaris, rat recombinant monocyte chemotactic protein 1 (MCP-1), and the adenosine deaminase inhibitor pentostatin, all from Sigma-Aldrich; the PKCε-specific translocation inhibitor peptide PKCεV1–2, (PKCε-I; Calbiochem), the PKCε activator ψεRACK (Biomatik), rat recombinant interleukin-6 (IL-6; PeproTech), and rat recombinant tumor necrosis factor α (TNFα; R&D Systems). The selection of the drug doses was based on our previous studies (Taiwo et al., 1990; Ouseph et al., 1995; Khasar et al., 1999; Aley et al., 2000; Parada et al., 2005; Ferrari et al., 2013c).
Stock solutions of PGE2 in absolute ethanol (1 μg/μl) were further diluted in 0.9% NaCl (1:50; final concentration, 0.02 μg/μl) immediately before injection. The ethanol concentration of the final PGE2 solution was ∼2%, and the injection volume was 5 μl. Stock solutions of cordycepin (1 μg/μl, dissolved in distilled water), prepared and further diluted in 0.9% NaCl, were used for intradermal injection into the dorsum of the hindpaw; for systemic treatment with cordycepin, a dose of 5 mg/kg, dissolved in distilled water, was administered intravenously in the tail vein. Because cordycepin is an adenosine analog that can be converted to an inactive metabolite by the enzyme adenosine deaminase (Tsai et al., 2010; Dalla Rosa et al., 2013), it must be administered with an adenosine deaminase inhibitor such as pentostatin (Johnston, 2011) to be effective (Koç and McCaffrey, 1995; Sugar and McCaffrey, 1998; Foss, 2000; Kodama et al., 2000; Yoshikawa et al., 2007). Pentostatin (1 mg/kg, dissolved in distilled water) was injected intravenously before cordycepin, in the same syringe, separated by an air bubble, to avoid mixing of the drugs. Control groups received vehicle or pentostatin plus vehicle. Before removal of the injection needle, administration of the drugs was followed by a bolus injection of an equal volume of saline solution.
Intradermal injections into the dorsum of the hindpaw were performed using a beveled 30 gauge hypodermic needle attached to a microsyringe by a short length of polyethylene (PE-10) tubing. The administration of cordycepin, PKCε-I, and ψεRACK was preceded by distilled water (2 μl in the same syringe as the drug, separated by a bubble of air to avoid mixing) to induce hypotonic shock, which facilitates cell permeability to these membrane-impermeant agents (Borle and Snowdowne, 1982; Burch and Axelrod, 1987).
For intrathecal drug administration, rats were briefly anesthetized with 2.5% isoflurane (Phoenix Pharmaceuticals) in 95% O2. Then, a 30 gauge hypodermic needle was inserted into the subarachnoid space, on the midline, between the L4 and L5 vertebrae, and the injection was performed (20 μl). Animals regained consciousness ∼1 min after the injection. Intrathecal injection was confirmed by a tail-flick reflex evoked by subarachnoid space access and bolus injection (Mestre et al., 1994).
Hyperalgesic priming.
Hyperalgesic priming, a model of the transition from acute to chronic pain, was produced using a previously described procedure (Aley et al., 2000; Parada et al., 2003a; Reichling and Levine, 2009): to prime the sensory neuron, ψεRACK was injected into the dorsum of the hindpaw, at the site of nociceptive testing. This induces mechanical hyperalgesia that resolves 3–5 d later, following which intradermal injection of PGE2 at the same site produces enhanced and prolonged mechanical hyperalgesia that lasts >4 h and is still significant at 24 h, as opposed to the effect of intradermal injection of PGE2 in a nonprimed paw, which lasts ∼1 h. To induce priming at the spinal terminals of the primary afferent, we used intrathecal injection of TNFα, IL-6, NGF, or MCP-1. That TNFα, IL-6, and NGF induce priming when injected into the dorsum of the hindpaw has been shown previously (Parada et al., 2003b; Dina et al., 2008; Ferrari et al., 2010). MCP-1, which we have observed to induce priming after peripheral injection (Alvarez et al., 2014), was also included in the present experiments because it is a pronociceptive mediator that acts selectively on IB4-positive nociceptors (Bogen et al., 2009), the subtype of neurons shown to be involved in priming (Joseph and Levine, 2010). Thus, in the studies using spinal intrathecal drug administration, MCP-1 provides a more specific target, namely, the central terminals of those primary afferent nociceptors that mediate hyperalgesic priming (Joseph and Levine, 2010).
Oligodeoxynucleotide antisense to CREB mRNA.
The oligodeoxynucleotide (ODN) antisense (AS) sequence for CREB mRNA, 5′-TGG TCA TCT AGT CAC CGG TG-3′, synthesized by Invitrogen, was directed against a unique region of the rat mRNA sequence, which blocks the translation of both known isoforms of CREB (Widnell et al., 1996; Lane-Ladd et al., 1997; Ma et al., 2003). That this ODN AS sequence can be used to downregulate the expression of CREB has been shown previously (Widnell et al., 1996; Lane-Ladd et al., 1997; Ma et al., 2003). The ODN missense sequence 5′-GAC CTC AGG TAG TCG TCG TT-3′ is a scramble of the AS sequence.
Before use, the ODNs were lyophilized and reconstituted in 0.9% NaCl to a concentration of 2 μg/μl. They were injected, as described above, by the spinal intrathecal route. A total of 40 μg of ODN in a volume of 20 μl was slowly injected. The use of the ODN AS sequence to manipulate the expression of proteins in nociceptors, important for their role in nociceptor sensitization, is well supported by previous studies by us (Parada et al., 2003a; Ferrari et al., 2010, 2012; Bogen et al., 2012) and others (Song et al., 2009; Su et al., 2011; Quanhong et al., 2012; Sun et al., 2013).
Statistics.
In all experiments, the dependent variable was the paw withdrawal threshold, expressed as the percentage change from baseline. The average paw withdrawal thresholds before and after [4 d (experiments shown in Figs. 1, 3, 5, and 6), and 1 week (Fig. 4) or 2 weeks (Fig. 7) later, depending on the stimulus] the injection of the priming stimuli were 119.1 ± 0.5 g and 116.7 ± 0.8 g, respectively. The total number of rats used in this study was 231 (462 paws). The left and right hindpaws of the rats were used and considered as independent subjects. To demonstrate the lack of correlation between the outcomes from paws of the same rat (i.e., the independence between the left and right paws of the same animal), we assessed the correlation between the mechanical hyperalgesia produced by the injection of PGE2 into the dorsum of both hindpaws of a group of six rats (12 paws). The Pearson's correlation coefficient based on these six rats, which was only 0.064, was not significantly different from zero, with a p value of 0.904, supporting the notion that the responses are independent. In addition, the analysis of the difference in the PGE2-induced hyperalgesia between the right and left paws of the same animal with a matched-pair t test, which pays attention to the potential connection between the paws in the same animal, also found no significant difference in the mean scores between the paws, with a p value of 0.696. Furthermore, the difference in the PGE2-induced hyperalgesia between the hindpaws was also examined with an independent-groups t test that treated the left paws as a different independent group from the right paws. The results were identical to the matched-pair analysis, in that there was no difference in the mean scores between right and left paws (p = 0.698). Importantly, no statistically significant difference between paw withdrawal threshold absolute values (compared by paired Student's t test) was observed between groups of rats subsequently injected with PGE2, vehicles, or inhibitors. In the experiments shown in Figure 2 (N = 36 rats, 72 paws), to evaluate the change in the mechanical threshold induced by PGE2, the mechanical threshold evaluated immediately before PGE2 injection (24, 48, or 72 h after injection of MCP-1) was considered the baseline threshold. To compare the percentage change in the hyperalgesia induced by injection of PGE2 in different groups, two-way repeated-measures ANOVA, followed by Bonferroni post-test, was performed. Prism version 5.0 (GraphPad Software) was used to plot the graphics and to perform the statistical analysis; p < 0.05 was considered statistically significant. Data are presented as the mean ± SEM.
Figure 1.

Spinal administration of TNFα, IL-6, NGF, or MCP-1 induces hyperalgesic priming. Different groups of rats received a 20 μl spinal injection of TNFα (C, 20 ng/μl), IL-6 (D, 1 ng/μl), NGF (E, 150 ng/μl), or MCP-1 (F, 20 ng/μl). All four mediators induced mechanical hyperalgesia. Ninety-six hours after injection, the mechanical nociceptive paw withdrawal threshold, evaluated by the Randall-Sellitto method, had returned to values not statistically different from preinjection values (paired Student's t test). Average paw withdrawal threshold values before and 96 h after the spinal injections were as follows: TNFα, 121.0 ± 2.1 and 118.3 ± 2.2 g, respectively (t(5) = 1.061; p = 0.3370); NGF, 123.3 ± 1.6 and 120.0 ± 1.5 g, respectively (t(5) = 2.500; p = 0.0545); IL-6, 123.3 ± 1.7 and 123.0 ± 2.1 g, respectively (t(5) = 0.2548; p = 0.8090); and MCP-1, 123.0 ± 1.7 g and 121.3 ± 2.0 g, respectively (t(11) = 1.131; p = 0.2821). PGE2 (100 ng) was then injected intradermally into the dorsum of the hindpaw, and the mechanical threshold evaluated 30 min and 4 h later. In all cases, PGE2-induced hyperalgesia remained unatennuated at the fourth hour after injection [nonsignificant (NS) in all cases: C, p = 0.6505; D, p = 0.7423; E, p = 0.1592; F, p = 0.1098; two-way repeated-measures ANOVA followed by Bonferroni post-test], demonstrating the presence of hyperalgesic priming (C–E, N = 3 rats, 6 paws per group; F, N = 6 rats, 12 paws per group). A, In the schematic, the red arrow represents the signal triggered by the spinal injection (1, red syringe) of the priming agents (TNFα, IL-6, NGF, or MCP-1), in the central terminal of the nociceptor, directed to the cell body. A signal for the neuroplastic change then spreads to the peripheral terminal in the skin (blue arrow), where the presence of hyperalgesic priming can be detected by evaluating the hyperalgesia induced by the injection of PGE2 (2, blue syringe). B, In control experiments, the hyperalgesia induced by PGE2 in naive rats (nonprimed), no longer present at the fourth hour after injection, is shown (***p < 0.0001, when compared with the hyperalgesia at 30 min).
Figure 3.

Hyperalgesic priming can be detected at the spinal cord when it is induced at the hindpaw. Rats received intradermal injection of the PKCε activator ψεRACK (1 μg, black bars) into the dorsum of the right hindpaw. Vehicle (white bars) was injected into the left paw. Four days later, PGE2 (20 ng/μl, 20 μl) was injected into the spinal cord, and the mechanical nociceptive paw withdrawal thresholds evaluated, 30 min and 4 h after PGE2 injection. Average paw withdrawal thresholds before the injection of ψεRACK or vehicle and immediately before the intrathecal injection of PGE2 (4 d later) were as follows: 119.1 ± 1.2 and 117.5 ± 1.2 g, respectively, for the vehicle group (t(11) = 1.101; p = 0.2945, NS); 119.6 ± 1.8 and 119.5 ± 1.3 g, respectively, for the ψεRACK group (t(11) = 0.1121; p = 0.9127, NS); paired Student's t test showed no significant difference between these two values. We observed a statistically significant increase in the magnitude of the hyperalgesia induced by the injection of PGE2 in the paws previously treated with ψεRACK when compared with the vehicle-treated paws [30 min after PGE2: **p < 0.01; 4 h after PGE2: ***p < 0.001; two-way repeated-measures ANOVA followed by Bonferroni post-test, N = 6 rats (12 paws) per group], showing the presence of hyperalgesic priming (F(1,22) = 35.20, p < 0.0001, when comparing both groups). The schematic on the left shows the signal triggered by the injection of ψεRACK (1, red syringe) at the peripheral terminal of the nociceptor in the skin, directed to the cell body (red arrow), and the signal from the cell body to the central terminal (blue arrow), where the presence of hyperalgesic priming can be detected by evaluating the hyperalgesia evoked by intrathecal injection of PGE2 (2, blue syringe).
Figure 5.
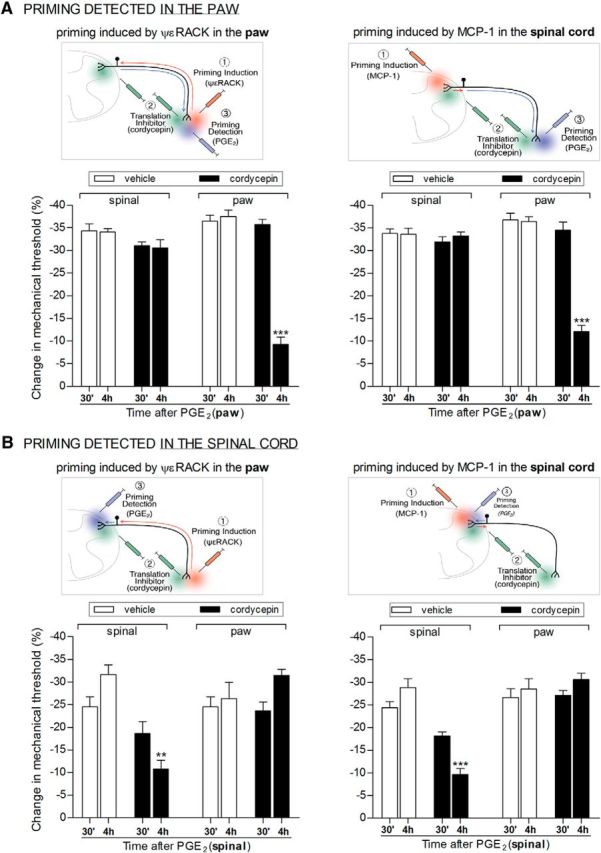
Inhibition of protein translation only reverses priming at the terminal where it is administered. Rats received intradermal injection of the PKCε activator ψεRACK (1 μg) into the dorsum of the hindpaw (lefts panels) or spinal intrathecal injection of MCP-1 (20 ng/μl, 20 μl; right panels). No significant difference between the mechanical thresholds before and 4 d after the injections (when the experiments were performed) was observed (paired Student's t test, data not shown); the average paw withdrawal thresholds before and 4 d after injection of ψεRACK (left panels) were 119.9 ± 0.8 and 119.6 ± 0.7 g, respectively (t(47) = 0.5329; p = 0.5966, NS). For the rats treated with intrathecal injection of MCP-1 (right panels), the average paw withdrawal thresholds before and 4 d after injection were 116.7 ± 0.6 and 116.7 ± 0.7 g, respectively (t(95) = 0.07275; p = 0.9422, NS). Rats were then divided into groups, vehicle (white bars) or the protein translation inhibitor cordycepin (black bars), which were administered intrathecally to the spinal cord (200 ng/μl, 20 μl) or injected into the hindpaw (1 μg). Fifteen minutes later, PGE2 was injected into the dorsum of the hindpaw (100 ng, A) or into the spinal cord (20 ng/μl, 20 μl, B), and the paw withdrawal thresholds were evaluated 30 min and 4 h later. Importantly, cordycepin alone did not affect the mechanical paw withdrawal threshold. However, when it was injected at the same site as PGE2, the PGE2-induced hyperalgesia was significantly attenuated at the 4 h time point (A, left: F(1,10) = 129.42, ***p < 0.001; A, right: F(1,22) = 63.07, ***p < 0.001; B, left: F(1,10) = 28.58, **p = 0.0003; B, right: F(1,22) = 51.52, ***p < 0.001, when the hyperalgesia at 30 min and 4 h are compared, two-way repeated-measures ANOVA followed by Bonferroni post-test). In contrast, when cordycepin was administered at the opposite terminal of the nociceptor from the terminal at which the nociceptive testing was performed, the prolonged PGE2-induced hyperalgesia was not reduced [p > 0.05 in all cases, when comparing vehicle- and cordycepin-treated groups; N = 3 rats (6 paws, ψεRACK treated) or N = 6 rats (12 paws, MCP-1-treated) per group].
Figure 6.

Systemic treatment with protein translation inhibitor reverses hyperalgesic priming in both the paw and spinal cord. Hyperalgesic priming was induced by intradermal injection of the PKCε activator ψεRACK (1 μg) in the paw (A) or by spinal injection of MCP-1 (20 ng/μl, 20 μl; B). Four days later, as indicated by * in the schematics on the top of the figures, when the mechanical nociceptive paw withdrawal thresholds had returned to baseline values (data not shown), different groups of rats received intravenous vehicles (control groups, white bars), pentostatin (1 mg/kg) plus vehicle (gray bars), or pentostatin plus cordycepin (5 mg/kg, black bars), for 4 consecutive days. On the fifth day, PGE2 (100 ng) was injected into the dorsum of the hindpaw, and the mechanical nociceptive paw withdrawal thresholds were evaluated 30 min and 4 h later. There were no significant differences in the average paw withdrawal thresholds before the injection of PGE2 when compared with the thresholds before the injections of the priming stimuli (paired Student's t test): A: vehicle-plus-vehicle group, 111.6 ± 1.6 and 112.3 ± 1.7 g, respectively (t(5) = 0.2454; p = 0.8159, NS); pentostatin-plus-vehicle group, 108.3 ± 3.0 and 111.6 ± 0.9 g, respectively (t(5) = 1.300; p = 0.2504, NS); cordycepin-plus-pentostatin group, 120.0 ± 2.5 and 118.0 ± 2.0 g, respectively (t(5) = 0.7746; p = 0.4736, NS); B: vehicle-plus-vehicle group, 117.6 ± 2.6 and 114.3 ± 2.0 g, respectively (t(5) = 1.976; p = 0.1051, NS); pentostatin-plus-vehicle group, 111.6 ± 1.6 and 111.0 ± 1.3 g, respectively (t(5) = 0.5976; p = 0.5761, NS); cordycepin-plus-pentostatin group, 113.3 ± 2.1 and 112.0 ± 1.0 g, respectively (t(5) = 0.7906; p = 0.4650, NS). Of note, the systemic treatment with the vehicles, pentostatin, or cordycepin (or the combinations), after priming was induced (4 d after injection of ψεRACK or MCP-1) did not induce significant changes on the mechanical thresholds: A: vehicle-plus-vehicle group, t(5) = 1.118; p = 0.3144, NS; pentostatin-plus-vehicle group, t(5) = 0.4152; p = 0.6452, NS; cordycepin-plus-pentostatin group, t(5) = 0.1395; p = 0.8945, NS; B, vehicle-plus-vehicle group, t(5) = 1.400; p = 0.2204, NS; pentostatin-plus-vehicle group, t(5) = 2.739; p = 0.4090, NS; cordycepin-plus-pentostatin group, t(5) = 0.6523; p = 0.5430, NS. Two-way repeated-measures ANOVA followed by Bonferroni post-test showed that in the groups treated with cordycepin plus pentostatin, but not in the controls (vehicles or pentostatin plus vehicle), the hyperalgesia induced by PGE2 was significantly attenuated at the fourth hour [A: ***p < 0.001, when the PGE2-induced hyperalgesia at the 4 h time point in the cordycepin-plus-pentostatin group is compared with the control groups: cordycepin-plus-pentostatin × vehicles group: F(1,50) = 62.66, p < 0.0001; cordycepin-plus-pentostatin × pentostatin-plus-vehicle groups: F(1,50) = 58.22, p < 0.0001; nonsignificant (F(1,50) = 6.45, p > 0.05) difference was observed between vehicles × pentostatin-plus-vehicle groups; B: ***p < 0.001, when the PGE2-induced hyperalgesia at the 4 h time point in the cordycepin-plus-pentostatin is compared with the control groups: cordycepin-plus-pentostatin × vehicles group: F(1,20) = 16.30, p = 0.0024; cordycepin-plus-pentostatin × pentostatin-plus-vehicle groups: F(1,20) = 22.67, p = 0.0008; nonsignificant difference (F(1,20) = 0.01, p = 0.9394) was observed between vehicles × pentostatin-plus-vehicle groups], showing reversal of the ψεRACK- and MCP-1-induced hyperalgesic priming by systemic treatment with cordycepin. In addition, when tested for priming again by intradermal injection of PGE2 20 d later, the hyperalgesia at the fourth hour was still attenuated in the groups previously treated with cordycepin plus pentostatin (A, ***p < 0.001, when compared with the control groups), showing that the reversal of priming was very long lasting [N = 3 rats (6 paws) per group].
Figure 4.
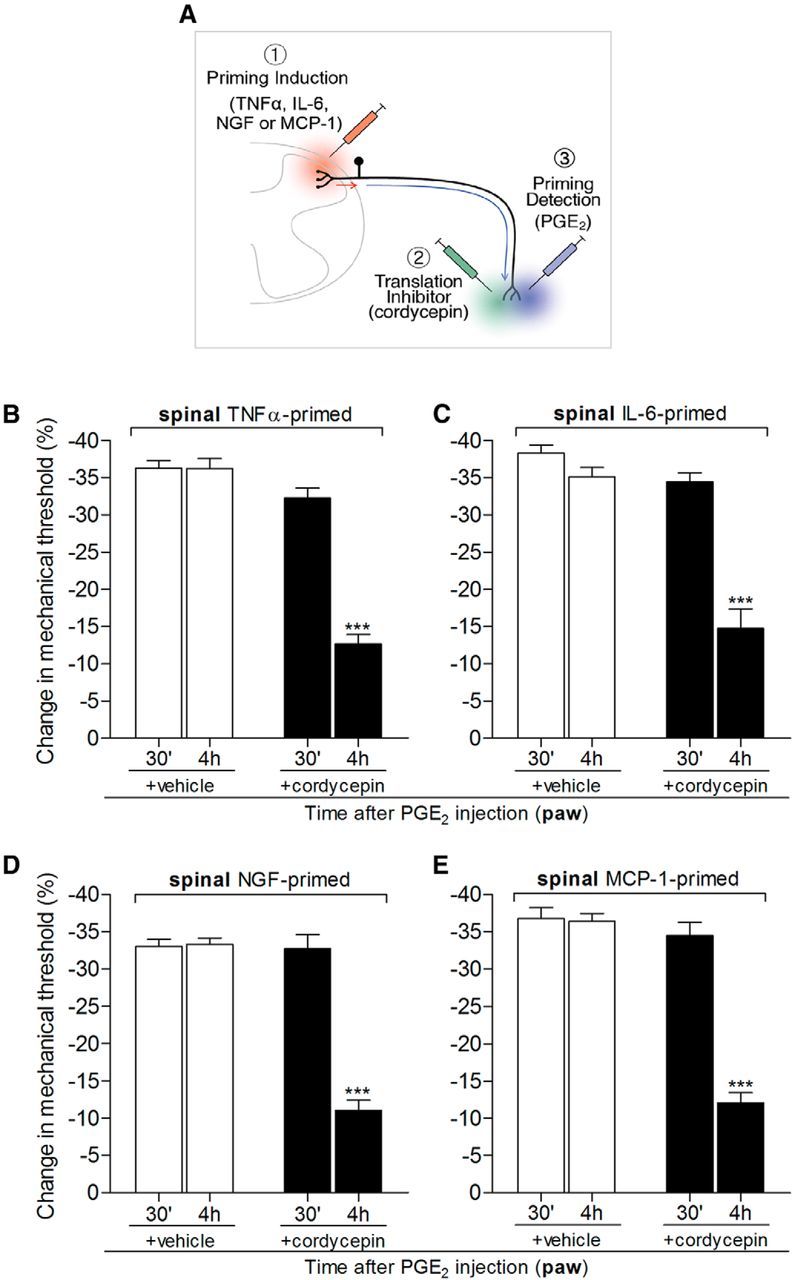
Hyperalgesic priming at the central terminal of the nociceptor, in the spinal cord, depends on local protein translation. Rats that received spinal intrathecal injection of TNFα (B, 20 ng/μl, 20 μl), IL-6 (C, 1 ng/μl, 20 μl), NGF (D, 150 ng/μl, 20 μl), or MCP-1 (E, 20 ng/μl, 20 μl) 1 week before were tested for hyperalgesic priming of the response to PGE2 (100 ng) injected intradermally into the dorsum of the hindpaw, in the presence or absence of the protein translation inhibitor cordycepin (1 μg, injected at the same site as PGE2 15 min before). Average paw withdrawal thresholds before the intrathecal injections of the priming stimuli and before the injection of PGE2 (1 week later) were as follows: TNFα, 120.6 ± 2.7 and 119.0 ± 2.2 g, respectively, for the vehicle-treated group (t(5) = 0.7734; p = 0.4743, NS) and 118.3 ± 1.4 and 118.3 ± 2.0 g, respectively, for the cordycepin-treated group (t(5) = 0.000; p = 1.000, NS); IL-6, 120.6 ± 1.8 and 120.0 ± 1.2 g, respectively, for the vehicle-treated group (t(5) = 0.3953; p = 0.7089, NS) and 122.6 ± 1.6 and 121.3 ± 2.9 g, respectively, for the cordycepin-treated group (t(5) = 0.0984; p = 0.5160, NS); NGF, 121.6 ± 2.2 and 119.6 ± 1.4 g, respectively, for the vehicle-treated group (t(5) = 1.369; p = 0.2292, NS) and 119.0 ± 1.8 and 118.6 ± 0.8 g, respectively, for the cordycepin-treated group (t(5) = 0.1465; p = 0.8893, NS); MCP-1, 118.6 ± 0.8 and 117.3 ± 1.2 g, respectively, for the vehicle-treated group (t(11) = 0.9834; p = 0.3466, NS) and 120.6 ± 1.0 and 119.6 ± 0.9 g, respectively, for the cordycepin-treated group (t(11) = 0.9199; p = 0.3774, NS); paired Student's t test showed no significant difference between these two values. The nociceptive mechanical paw withdrawal threshold was evaluated 30 min and 4 h after PGE2 injection. Two-way repeated-measures ANOVA followed by Bonferroni post-test showed significant attenuation of PGE2-induced hyperalgesia at the fourth hour after injection in the groups pretreated with cordycepin (***p < 0.001 in all cases, when vehicle- and cordycepin-treated groups are compared at the fourth hour; B, F(1,10) = 126.19; C, F(1,10) = 53.36; D, F(1,10) = 60.63; E, F(1,22) = 63.07; p < 0.0001 for all cases, when vehicle and cordycepin groups are compared), indicating a role of local protein translation in hyperalgesic priming induced by spinal injection of TNFα, IL-6, NGF, or MCP-1 [N = 3 rats (6 paws, B–D) or N = 6 rats (12 paws, E) per group]. A, In the schematic, the red arrow represents the signal triggered by the spinal injection (red syringe) of the priming agents (1, TNFα, IL-6, NGF, and MCP-1) originating in the central terminal of the nociceptor and the subsequent signal (blue arrow) to the peripheral nociceptor terminal (in skin) where hyperalgesic priming is detected by the prolongation of hyperalgesia evoked by PGE2 injected in the paw (2, blue syringe). The green syringe represents the site where cordycepin (or vehicle) was injected.
Figure 7.

Knockdown of CREB prevents (A), but does not reverse (B), hyperalgesic priming. A, Rats were treated with daily spinal intrathecal injections of ODN AS (black bars) for CREB mRNA, for 3 consecutive days, to decrease its levels in the sensory neuron and prevent its activation by the priming inducers ψεRACK (1 μg, injected intradermally into the dorsum of the hindpaws; A, left) or MCP-1 (20 ng/μl; 20 μl, injected by the intrathecal route; A, right), injected on the fourth day. Control animals were treated, following the same protocol, with missense (MS; white bars). To prevent further activation of a signaling pathway that will ultimately produce priming during the acute effect of ψεRACK or MCP-1, the ODN treatments continued until the return of the mechanical nociceptive paw withdrawal thresholds to baseline values, when hyperalgesic priming was assessed by the administration of PGE2 (100 ng). PGE2 was injected intradermally into the dorsum of the hindpaw, and the mechanical threshold was evaluated 30 min and 4 h later. Average paw withdrawal thresholds before the injections of the priming stimuli and before the injection of PGE2 (8 d later) were as follows: ψεRACK in the paw, left, 121.3 ± 1.9 and 121.9 ± 1.4 g, respectively, for the CREB MS-treated group (t(11) = 0.000; p = 1.0000, NS), and 125.9 ± 1.7 and 125.5 ± 1.1 g, respectively, for the AS-treated group (t(11) = 0.1461; p = 0.8864, NS); MCP-1, intrathecally, right, 123.3 ± 2.5 and 120.5 ± 1.7 g, respectively, for the MS-treated group (t(11) = 1.787; p = 0.1014, NS) and 120.6 ± 2.3 and 116.5 ± 1.4 g, respectively, for the AS-treated group (t(11) = 1.948; p = 0.0773, NS). Paired Student's t test showed no significant difference between these two values. Two-way repeated-measures ANOVA followed by Bonferroni post-test showed significant mechanical hyperalgesia induced by PGE2 30 min after the injection. However, while in the MS-treated groups the magnitude of PGE2 hyperalgesia was still significant at the fourth hour, in the AS-treated groups it was strongly attenuated in both the ψεRACK- and the MCP-1-primed group (***p < 0.001 when the MS- and the AS-treated groups are compared). When tested again for priming with PGE2 1 week after the last treatment with ODN AS or MS, a time point when the levels of CREB had returned to pre-AS levels, the prolongation of PGE2-induced hyperalgesia was still attenuated (at the 4 h time point) in the ODN AS-treated groups, but not in the MS-treated groups, indicating a role of CREB in the induction of hyperalgesic priming [***p < 0.001 when the MS- and the AS-treated groups are compared; A, N = 6 rats (12 paws) per group]. Of note, no difference in the mechanical thresholds was observed at this time point, when compared with pre-priming stimuli thresholds: ψεRACK, left, t(11) = 0.8710; p = 0.4024 (NS) for the CREB MS-treated group, and t(11) = 1.497; p = 0.1624 (NS) for the AS-treated group; MCP-1, right, t(11) = 2.482; p = 0.305 (NS) for the MS-treated group, and t(11) = 1.915; p = 0.0819 (NS) for the AS-treated group (paired Student's t test). B, Rats that were treated with intradermal injection of the PKCε activator ψεRACK (1 μg) into the dorsum of the hindpaw (B, left) or spinal intrathecal injection of MCP-1 (20 ng/μl, 20 μl; B, right) 2 weeks before were treated with ODN AS (black bars) for CREB mRNA for 3 consecutive days to decrease the levels of CREB in the nociceptor. Control animals were treated, following the same protocol, with MS (white bars). On the fourth day, PGE2 (100 ng) was injected intradermally into the dorsum of the hindpaws, and the mechanical thresholds were evaluated 30 min and 4 h later. Average paw withdrawal thresholds before the injections of ψεRACK or MCP-1 and before the injection of PGE2 were as follows: ψεRACK, left, 121.6 ± 2.7 and 123.3 ± 1.6 g, respectively, for the CREB MS-treated group (t(11) = 0.8752; p = 0.4002, NS), and 113.3 ± 1.8 and 112.6 ± 2.1 g, respectively, for the AS-treated group (t(11) = 0.4838; p = 0.6380, NS); MCP-1, right, 114.1 ± 1.4 and 114.6 ± 1.2 g, respectively, for the MS-treated group (t(11) = 0.2536; p = 0.8045, NS) and 117.5 ± 1.7 and 117.5 ± 1.4 g, respectively, for the AS-treated group (t(11) = 0.0000; p = 1.0000, NS). Paired Student's t test showed no significant difference between these two values. Two-way repeated-measures ANOVA followed by Bonferroni post-test showed no difference in the magnitude of the PGE2-induced hyperalgesia at 30 min and 4 h in both the ODN AS- and MS-treated groups (rats primed with ψεRACK: F(1,22) = 1.47, p = 0.2378; rats primed with MCP-1: F(1,22) = 1.39, p = 0.2511), indicating that CREB does not play a role in the maintenance of hyperalgesic priming [B: N = 6 rats (12 paws) per group].
Figure 2.

Time course of development of hyperalgesic priming induced by intradermal (paw) or intrathecal spinal injection of MCP-1. Different groups of rats received injections of MCP-1 intradermally into the dorsum of the hindpaw (100 ng; A) or intrathecally in the spinal cord (20 ng/μl, 20 μl; B). PGE2 (100 ng) was injected into the dorsum of the hindpaw 24 h (left panels), 48 h (middle panels), or 72 h (right panels) later, and, the mechanical hyperalgesia, evaluated by the Randall-Selitto paw withdrawal test, 30 min and 4 h later. Average paw withdrawal thresholds before, and 24, 48, and 72 h after intradermal injection of MCP-1 (A) were as follows: 120.0 ± 1.4 and 107.5 ± 1.8 g, respectively (after 24 h, t(11) = 10.56; p < 0.0001; paired Student's t test); 119.6 ± 1.4 and 116.1 ± 0.9 g, respectively (after 48 h, t(11) = 2.782; p = 0.1790, NS); and 118.6 ± 1.1 and 114.1 ± 1.3 g, respectively (after 72 h, t(11) = 2.279; p = 0.4360, NS). For the groups treated with intrathecal injection of MCP-1 (B), the average paw withdrawal thresholds before, and 24, 48, and 72 h after injections were as follows: 120.1 ± 1.5 and 98.0 ± 2.0 g, respectively (after 24 h, t(11) = 8.071; p < 0.0001; paired Student's t test); 116.8 ± 2.1 and 110.5 ± 1.2 g, respectively (after 48 h, t(11) = 2.588; p = 0.2502, NS); and 120.3 ± 1.5 and 116.6 ± 1.9 g, respectively (after 72 h, t(11) = 1.910; p = 0.0826, NS). In the groups treated with MCP-1 into the dorsum of the hindpaw (A), hyperalgesic priming was not established until 72 h [24 and 48 h panels: ***p < 0.0001 when comparing the PGE2-induced hyperalgesia at 30 min and 4 h; 72 h panel: p = 0.9404 (NS); two-way repeated-measures ANOVA followed by Bonferroni post-test, N = 6 rats (12 paws) per group]. On the other hand, in the group treated with spinal MCP-1 (B), although the injection of PGE2 on the next day induced short-lasting hyperalgesia, it was no longer present at the fourth hour time point (***p < 0.0001 when comparing both time points); when PGE2 was injected 48 or 72 h after spinal MCP-1, the hyperalgesia remained statistically unattenuated at the fourth hour [NS in both cases: 48 h panel, p = 0.1864; 72 h panel, p = 0.3635; two-way repeated-measures ANOVA followed by Bonferroni post-test, N = 6 rats (12 paws) per group], demonstrating that hyperalgesic priming had already developed by 48 h. Of note, since the hyperalgesia induced by MCP-1 was still significant 24 h after its administration (A and B, both p < 0.0001, paired Student's t test), the baseline used to evaluate the effect induced by PGE2 at the 30 min time point was already decreased. For that reason, the change in the mechanical threshold induced by PGE2 injection appears smaller (left graphics). The schematic in A shows the hypothetical pathway of a signal triggered by the injection of the priming agent MCP-1 (1, red syringe) in the peripheral terminal of the nociceptor (in the skin), directed to the cell body (red arrow), which induces changes in the peripheral terminal (blue arrow) detected by evaluating the hyperalgesia evoked by the injection of PGE2 (2, blue syringe). In B, the signal that triggers hyperalgesic priming (1, red syringe) is spinal injection of MCP-1, and directed to the cell body (red arrow), from which the message for the neuroplastic change will spread (blue arrow) to the peripheral terminal (in skin) of the nociceptor, detected by the prolongation of the hyperalgesia evoked by injection of PGE2 in the paw (2, blue syringe).
Results
Inducing priming by stimulating the central terminal of the nociceptor
Given the marked difference in the length of the central and peripheral branches of nociceptors innervating the hindpaw, we tested whether the delay to onset of the primed state would be shorter if the agent that induces priming was administered to the central branch in the spinal cord. Pronociceptive mediators (TNFα, IL-6, NGF, and MCP-1) that induce priming when administered in peripheral tissue were injected intrathecally into the spinal cord in separate groups of rats. That TNFα, IL-6, and NGF induce priming when injected into the dorsum of the hindpaw has been shown previously (Parada et al., 2003b; Dina et al., 2008; Ferrari et al., 2010). MCP-1, which we have observed to induce priming after peripheral injection (Alvarez et al., 2014), was also included in the present experiment because it is a pronociceptive mediator that acts on IB4-positive, but not IB4-negative, nociceptors (Bogen et al., 2009). To address the role of the cell body in hyperalgesic priming, we first tested whether administration of the inducers of hyperalgesic priming intrathecally in the spinal cord can induce hyperalgesic priming in the paw at the peripheral terminal of the nociceptor. As shown in Figure 1, intrathecal injection of each agent induced peripheral hyperalgesic priming, manifested as prolongation of mechanical hyperalgesia induced by PGE2 injected in the paw (measured 4 h after the injection of PGE2, a time at which PGE2-induced hyperalgesia would be fully resolved in a naive rat; Aley and Levine, 1999). Importantly, although the mechanical hyperalgesia induced by the injection of these agents had different time courses (Fig. 1C–F, first four bars), we waited 96 h (when the mechanical thresholds in all groups had returned to baseline values) to test for the presence of priming (Fig. 1C–F, two bars on the right). The choice of 96 h is based on the observation that the establishment of priming takes ∼72 h after injection of the priming stimulus (Bogen et al., 2012). However, as seen in Figure 2, we have further observed that the delay to the induction of priming was ∼48 h when the inducer was injected intrathecally into the spinal cord (compared with ∼72 h when the inducing agent was injected into the paw).
Induction of hyperalgesic priming in the paw also induces priming at the central terminal of the nociceptor
Following administration of inducers of hyperalgesic priming into the paw, hyperalgesic priming in the spinal cord was detected as a prolongation of mechanical hyperalgesia induced by PGE2 injected intrathecally (Fig. 3). Of note, the intrathecal injection of PGE2 in nonprimed rats, in a concentration similar to the dose injected into the dorsum of the hindpaw (20 ng/μl), but diluted by the larger volume of the CSF, induced less intense hyperalgesia when compared with the hyperalgesia induced by the injection of PGE2 into the paw. We observed that, similar to the hyperalgesia induced by injection of PGE2 into the paw of primed rats, the hyperalgesia induced by the intrathecally injected PGE2 was significantly prolonged.
We have shown previously that hyperalgesic priming in the paw is reversed when protein translation is inhibited locally in the paw (Ferrari et al., 2013c). Hyperalgesic priming induced in the spinal cord is also mediated by a similar mechanism, since the administration of the translation inhibitor cordycepin in the same site in the paw as PGE2, in rats previously primed by intrathecal injection of TNFα, IL-6, NGF, or MCP-1, significantly attenuated hyperalgesic priming (Fig. 4). Consistent with a need for only local protein translation, spinal administration of a translation inhibitor did not affect the prolonged response to PGE2 injected in the paw (Fig. 5A). Similarly, a translation inhibitor administered in the paw had no effect on the prolonged hyperalgesia induced by intrathecally injected PGE2 (Fig. 5B).
Thus, agents that induce priming when administered to the peripheral terminal of the nociceptor also induce priming at the central terminal, suggesting that signals initiated at either the central or peripheral terminal of a nociceptor induce the neuroplastic changes that underlie hyperalgesic priming throughout the axon branches of the sensory neuron (where ongoing local protein translation is critical to maintain the primed state).
Systemic administration of a translation inhibitor reverses central and peripheral priming
Since systemically active translation inhibitors have been developed for clinical use to treat cancer (Behbakht et al., 2011; Demetri et al., 2013; Ma et al., 2013; Maj-Hes et al., 2013; Ogawa et al., 2013), we tested the hypothesis that systemic administration of such drugs would reverse priming both at the central and peripheral terminal of the nociceptor (Fig. 6). Systemic administration of cordycepin (5 mg/kg; and pentostatin, 1 mg/kg, daily intravenous administration for 4 consecutive days, before testing on the fifth day) significantly attenuated hyperalgesic priming measured as the prolongation of the hyperalgesia induced by PGE2 challenge in the paw [which was induced by administration of ψεRACK (1 μg, Fig. 6A) in the paw or MCP-1 (20 ng/μl; 20 μl, Fig. 6B) in the spinal cord], indicating that systemic treatment with protein translation inhibitors can be effective to reverse chronic pain syndromes where hyperalgesic priming plays a role.
Nuclear mechanism in priming
Finally, we tested the hypothesis that the signal traveling along the axon of the sensory neuron from the central or peripheral terminal toward the cell body, required to induce priming at both terminals, engages CREB, a nuclear transcription factor implicated in neuroplasticity. CREB is a cellular transcription factor that binds to nuclear DNA sequences to increase or decrease the transcription of many genes (Mayr and Montminy, 2001), and CREB-related changes in gene expression in the neuronal nucleus have been implicated in long-term sensitization and neuronal regeneration (Herdegen et al., 1992; Fields et al., 1997; Teng and Tang, 2006). We tested the hypothesis that CREB plays a role in the induction, but not necessarily the maintenance, of hyperalgesic priming. In support of this hypothesis, we found that intrathecal administration of an ODN antisense to CREB mRNA prevented (but did not reverse) priming induced either by activation of PKCε in the paw or by intrathecal injection of MCP-1 (Fig. 7). The ODN missense control had no significant effect. In Figure 8, the proposed axonal and nuclear mechanisms underlying hyperalgesic priming are illustrated.
Figure 8.

Schematic of the proposed axonal and nuclear mechanisms underlying hyperalgesic priming. Our experiments support the hypothesis that an inflammatory event that activates PKCε in nonpeptidergic nociceptors (A) triggers gene transcription in the cell body that is dependent on CREB (B, red arrows). The resultant mRNA species will then be transported to the terminals of the nociceptor (C, blue arrows), where translation mechanisms will take place (D) and, as a consequence, produce hyperalgesic priming. This phenomenon will then be maintained, independently of CREB, by local protein translation at the central and peripheral terminals of the nociceptor (E). The time for the development of priming will depend on the transport of the newly formed mRNA to the peripheral or central terminals of the nociceptor. F, The expression of this neuroplasticity, an increased response to pronociceptive inflammatory cytokines, can be verified by the prolongation of the mechanical hyperalgesia induced by the injection of PGE2 (F).
Discussion
The delay to induction of hyperalgesic priming
The 72 h delay in the induction of hyperalgesic priming has been an unexplained feature of this phenomenon. However, since other forms of long-term neuronal plasticity involve changes of gene expression (Lee et al., 2008; Knöll and Nordheim, 2009; Bengtson and Bading, 2012; Kim et al., 2013), we hypothesized that this could play a role in priming, and that the time required for a signal to be carried by the sensory axon from the nerve ending to the nucleus, transcription in the cell body, and a return signal from the nucleus to the peripheral nerve ending followed by peripheral translation (Ferrari et al., 2013c) contribute to this delay. We found that intrathecal administration of a pronociceptive mediator that induces priming when administered at the peripheral terminal, induces priming in the central terminal of the nociceptor (Fig. 1), with a markedly shorter latency to onset (Fig. 2), implicating axoplasmic transport of a signaling molecule from the nerve terminal to the cell body. Since some part of the delay to onset of the primed state is due to nuclear and peripheral translation mechanisms, which adds an unknown additional delay, and the molecule transmitted to the cell body is likely different from the one transmitted from the cell body back to the terminal, we are not yet in a position to implicate a specific mechanism for the relevant signals to move between the terminals and the cell body.
Hyperalgesic priming at the central terminal of the primary afferent neuron
The observation that hyperalgesic priming in the paw can be induced by the administration of inducing agents in the spinal cord raised the possibility that priming might also be expressed in the spinal cord as an increase in the response to inflammatory mediators at the central terminal of the primary afferent. Consistent with hyperalgesic priming occurring at the central terminal, we found that intradermal as well as intrathecal administration of priming stimuli caused a prolongation of mechanical hyperalgesia induced by intrathecally administered PGE2. While the prolonged hyperalgesia induced by peripheral administration of PGE2 is mediated by sensitization of the nociceptor (Aley et al., 2000; Parada et al., 2005; Reichling and Levine, 2009), the mechanism for prolongation of mechanical hyperalgesia induced by intrathecal PGE2 is currently unknown. Because PGE2 can enhance neurotransmitter release (Osaka, 2008; Fehrenbacher et al., 2009; Ma, 2010; Anneken et al., 2013; Lindgren et al., 2013), one possibility is enhanced neurotransmitter release from the central terminal of the nociceptor. A contribution from postsynaptic mechanisms cannot, however, be excluded since prostaglandins also have effects on neurons in the CNS (Hori et al., 2000; Sang and Chen, 2006; Ohinata and Yoshikawa, 2008; O'Banion, 2010).
Trans-terminal priming suggests participation of the sensory neuron cell body
We hypothesized that the induction of priming involves a retrograde signal from the cell body back to nerve endings. The observation that hyperalgesic priming can also be expressed at the central terminal of the nociceptor, when priming was induced by an agent administered at the peripheral terminal, raised the possibility that the retrograde priming signal sent from the cell body to the peripheral terminal might also travel along the central branch of the axon to induce priming at the spinal terminal. Therefore, we tested whether mediators that produce hyperalgesic priming of the nociceptor peripheral terminal, when injected at the peripheral terminal, also induce priming of the peripheral terminal when injected intrathecally to the central terminal. We found that intrathecal injection of these mediators produces hyperalgesic priming, a prolonged response to PGE2 injected into the paw. We also found that injection of an inducing agent into the paw caused hyperalgesic priming at the central terminal. Furthermore, as predicted by the hypothesis that the delay in the induction of priming is dependent on axon length, the latency to the onset of the primed state in both cases was markedly shortened (Devor, 1999). This might explain the distinct time course for priming establishment, depending on the site where the inducer is administered, since the neuroplastic change observed in primed nociceptors is dependent on the transport of nuclear transcripted mRNAs to the terminals where they will be translated (Ferrari et al., 2013c). Moreover, the transmission of the priming signal between the two branches of the sensory nerve axon on the central and peripheral sides of the cell body suggested that mechanisms in the dorsal root ganglion (DRG) could play a crucial role in the induction of priming. However, this finding alone cannot rule out the possibility of a direct transmission of a signal from one branch of the bipolar axon to another, without any involvement of the cell body.
A nuclear mechanism in hyperalgesic priming
To begin to address the question of whether the signal transmitted from the nociceptor terminals along the length of the axon involves a nuclear event in the cell body, we administered an ODN antisense to CREB mRNA (Widnell et al., 1996; Lane-Ladd et al., 1997; Ma et al., 2003), a nuclear transcription factor (Montminy et al., 1990; Montminy, 1997; Shaywitz and Greenberg, 1999) that plays a role in the neuroplasticity of learning and memory (Montminy et al., 1990; Montminy, 1997; Shaywitz and Greenberg, 1999; Kandel, 2012). CREB antisense prevented priming, which is compatible with a role of a nuclear transcription mechanism. CREB antisense, however, did not reverse priming, suggesting that CREB-dependent patterns of gene expression in the nucleus play a critical role in its induction, but is not necessary to maintain the primed state. Therefore, processes that maintain hyperalgesic priming may be entirely localized in the nerve ending, consistent with our previous finding that peripheral administration of a protein translation inhibitor can reverse established hyperalgesic priming.
While the precise mechanism by which CREB contributes to hyperalgesic priming is currently unknown, its function in the DRG neuron has been extensively studied. We have previously shown that repeated administration of the μ-opioid receptor agonist morphine at the peripheral terminal of the nociceptor induces hyperalgesic priming (Joseph et al., 2010), and, while it has yet to be shown that intrathecal opioids induce priming, they are well established to produce analgesia by action at the central terminal of the nociceptor. Of note in this regard, Ma et al. (2001) have shown that repeated morphine exposure increased the number of DRG neurons expressing phosphorylated extracellular signal-regulated protein kinase, also implicated in hyperalgesic priming (Dina et al., 2003; Ferrari et al., 2014), as well as those expressing phosphorylated CREB (pCREB; Ma et al., 2001). In neurons that are selectively deficient in axonal CREB transcripts, increases in nuclear pCREB, CRE-mediated transcription, and neuronal survival elicited by axonal application of NGF (which also produces hyperalgesic priming; Ferrari et al., 2010) are abolished, indicating a signaling function for axon-synthesized CREB (Cox et al., 2008). Furthermore, IL-6, which also produces hyperalgesic priming, engages a similar signaling pathway (Eyre and Baune, 2012; Yang et al., 2012; Chou et al., 2013). Of note, protein synthesis inhibitors prevent NGF-induced maintenance of axonal CREB, implying that NGF promotes the translation of CREB mRNA in the axon (Cox et al., 2008). Exactly how this contributes to the induction of hyperalgesic priming remains to be established.
Cordycepin
An ultimate goal in studying the mechanism of hyperalgesic priming is to create a foundation for novel treatments of chronic pain. In most cases, such treatments would be used after the condition is already established. This study has shown that hyperalgesic priming is maintained independent of the nucleus, by protein translation occurring in both nerve endings (Ferrari et al., 2013b,c), suggesting that a successful clinical intervention would optimally target both sites. Cordycepin is a clinically approved protein translation inhibitor that is systemically administered and appears to cross the blood–brain barrier (Rottenberg et al., 2005). Our demonstration that systemic cordycepin administration can reverse priming produced by stimuli in both the spinal cord and periphery is a preliminary indication that this drug might be useful as a treatment for chronic pain. Our findings that cordycepin attenuated priming only when injected at the same site where PGE2 was administered (Fig. 5), and that the induction, but not maintenance, of priming, depends on CREB-activated nuclear transcription (Fig. 6) suggest that the signal transmitted to the terminals of the nociceptor is not a newly translated protein, but possibly a newly expressed mRNA.
Footnotes
This study was funded by the National Institutes of Health.
The authors declare no competing financial interests.
References
- Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci. 1999;19:2181–2186. doi: 10.1523/JNEUROSCI.19-06-02181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20:4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez P, Green PG, Levine JD. Role for monocyte chemoattractant protein-1 in the induction of chronic muscle pain in the rat. Pain. 2014;155:1161–1167. doi: 10.1016/j.pain.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anneken JH, Cunningham JI, Collins SA, Yamamoto BK, Gudelsky GA. MDMA increases glutamate release and reduces parvalbumin-positive GABAergic cells in the dorsal hippocampus of the rat: role of cyclooxygenase. J Neuroimmune Pharmacol. 2013;8:58–65. doi: 10.1007/s11481-012-9420-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behbakht K, Sill MW, Darcy KM, Rubin SC, Mannel RS, Waggoner S, Schilder RJ, Cai KQ, Godwin AK, Alpaugh RK. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: a Gynecologic Oncology Group study. Gynecol Oncol. 2011;123:19–26. doi: 10.1016/j.ygyno.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson CP, Bading H. Nuclear calcium signaling. Adv Exp Med Biol. 2012;970:377–405. doi: 10.1007/978-3-7091-0932-8_17. [DOI] [PubMed] [Google Scholar]
- Bogen O, Dreger M, Gillen C, Schröder W, Hucho F. Identification of versican as an isolectin B4-binding glycoprotein from mammalian spinal cord tissue. FEBS J. 2005;272:1090–1102. doi: 10.1111/j.1742-4658.2005.04543.x. [DOI] [PubMed] [Google Scholar]
- Bogen O, Dina OA, Gear RW, Levine JD. Dependence of monocyte chemoattractant protein 1 induced hyperalgesia on the isolectin B4-binding protein versican. Neuroscience. 2009;159:780–786. doi: 10.1016/j.neuroscience.2008.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogen O, Alessandri-Haber N, Chu C, Gear RW, Levine JD. Generation of a pain memory in the primary afferent nociceptor triggered by PKCε activation of CPEB. J Neurosci. 2012;32:2018–2026. doi: 10.1523/JNEUROSCI.5138-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borle AB, Snowdowne KW. Measurement of intracellular free calcium in monkey kidney cells with aequorin. Science. 1982;217:252–254. doi: 10.1126/science.6806904. [DOI] [PubMed] [Google Scholar]
- Burch RM, Axelrod J. Dissociation of bradykinin-induced prostaglandin formation from phosphatidylinositol turnover in Swiss 3T3 fibroblasts: evidence for G-protein regulation of phospholipase A2. Proc Natl Acad Sci U S A. 1987;84:6374–6378. doi: 10.1073/pnas.84.18.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CH, Lai SL, Chen CN, Lee PH, Peng FC, Kuo ML, Lai HS. IL-6 regulates Mcl-1L expression through the JAK/PI3K/Akt/CREB signaling pathway in hepatocytes: implication of an anti-apoptotic role during liver regeneration. PLoS One. 2013;8:e66268. doi: 10.1371/journal.pone.0066268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LJ, Hengst U, Gurskaya NG, Lukyanov KA, Jaffrey SR. Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat Cell Biol. 2008;10:149–159. doi: 10.1038/ncb1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla Rosa L, da Silva AS, Gressler LT, Oliveira CB, Dambrs MG, Miletti LC, França RT, Lopes ST, Samara YN, da Veiga ML, Monteiro SG. Cordycepin (3′-deoxyadenosine) pentostatin (deoxycoformycin) combination treatment of mice experimentally infected with Trypanosoma evansi. Parasitology. 2013;140:663–671. doi: 10.1017/S0031182012001990. [DOI] [PubMed] [Google Scholar]
- Demetri GD, Chawla SP, Ray-Coquard I, Le Cesne A, Staddon AP, Milhem MM, Penel N, Riedel RF, Bui-Nguyen B, Cranmer LD, Reichardt P, Bompas E, Alcindor T, Rushing D, Song Y, Lee RM, Ebbinghaus S, Eid JE, Loewy JW, Haluska FG, et al. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol. 2013;31:2485–2492. doi: 10.1200/JCO.2012.45.5766. [DOI] [PubMed] [Google Scholar]
- Devor M. Unexplained peculiarities of the dorsal root ganglion. Pain. 1999;6(Suppl):S27–S35. doi: 10.1016/S0304-3959(99)00135-9. [DOI] [PubMed] [Google Scholar]
- Dina OA, McCarter GC, de Coupade C, Levine JD. Role of the sensory neuron cytoskeleton in second messenger signaling for inflammatory pain. Neuron. 2003;39:613–624. doi: 10.1016/S0896-6273(03)00473-2. [DOI] [PubMed] [Google Scholar]
- Dina OA, Green PG, Levine JD. Role of interleukin-6 in chronic muscle hyperalgesic priming. Neuroscience. 2008;152:521–525. doi: 10.1016/j.neuroscience.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina OA, Khasar SG, Gear RW, Levine JD. Activation of Gi induces mechanical hyperalgesia poststress or inflammation. Neuroscience. 2009;160:501–507. doi: 10.1016/j.neuroscience.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre H, Baune BT. Neuroplastic changes in depression: a role for the immune system. Psychoneuroendocrinology. 2012;37:1397–1416. doi: 10.1016/j.psyneuen.2012.03.019. [DOI] [PubMed] [Google Scholar]
- Fehrenbacher JC, Sun XX, Locke EE, Henry MA, Hargreaves KM. Capsaicin-evoked iCGRP release from human dental pulp: a model system for the study of peripheral neuropeptide secretion in normal healthy tissue. Pain. 2009;144:253–261. doi: 10.1016/j.pain.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Levine JD. Nociceptor subpopulations involved in hyperalgesic priming. Neuroscience. 2010;165:896–901. doi: 10.1016/j.neuroscience.2009.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Alessandri-Haber N, Levine E, Gear RW, Levine JD. Transient decrease in nociceptor GRK2 expression produces long-term enhancement in inflammatory pain. Neuroscience. 2012;222:392–403. doi: 10.1016/j.neuroscience.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Levine E, Levine JD. Role of a novel nociceptor autocrine mechanism in chronic pain. Eur J Neurosci. 2013a;37:1705–1713. doi: 10.1111/ejn.12145. [DOI] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Levine JD. Role of nociceptor αCaMKII in transition from acute to chronic pain (hyperalgesic priming) in male and female rats. J Neurosci. 2013b;33:11002–11011. doi: 10.1523/JNEUROSCI.1785-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Chu C, Levine JD. Peripheral administration of translation inhibitors reverses increased hyperalgesia in a model of chronic pain in the rat. J Pain. 2013c;14:731–738. doi: 10.1016/j.jpain.2013.01.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Levine JD. Second messengers mediating the expression of neuroplasticity in a model of chronic pain in the rat. J Pain. 2014;15:312–320. doi: 10.1016/j.jpain.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Eshete F, Stevens B, Itoh K. Action potential-dependent regulation of gene expression: temporal specificity in Ca2+, cAMP-responsive element binding proteins, and mitogen-activated protein kinase signaling. J Neurosci. 1997;17:7252–7266. doi: 10.1523/JNEUROSCI.17-19-07252.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss FM. Combination therapy with purine nucleoside analogs. Oncology (Williston Park) 2000;14(6 Suppl 2):31–35. [PubMed] [Google Scholar]
- Herdegen T, Fiallos-Estrada CE, Schmid W, Bravo R, Zimmermann M. The transcription factors c-JUN, JUN D and CREB, but not FOS and KROX-24, are differentially regulated in axotomized neurons following transection of rat sciatic nerve. Brain Res Mol Brain Res. 1992;14:155–165. doi: 10.1016/0169-328X(92)90170-G. [DOI] [PubMed] [Google Scholar]
- Hori T, Oka T, Hosoi M, Abe M, Oka K. Hypothalamic mechanisms of pain modulatory actions of cytokines and prostaglandin E2. Ann N Y Acad Sci. 2000;917:106–120. doi: 10.1111/j.1749-6632.2000.tb05375.x. [DOI] [PubMed] [Google Scholar]
- Johnston JB. Mechanism of action of pentostatin and cladribine in hairy cell leukemia. Leuk Lymphoma. 2011;52(Suppl 2):43–45. doi: 10.3109/10428194.2011.570394. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Hyperalgesic priming is restricted to isolectin B4-positive nociceptors. Neuroscience. 2010;169:431–435. doi: 10.1016/j.neuroscience.2010.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Bogen O, Alessandri-Haber N, Levine JD. PLC-beta 3 signals upstream of PKC epsilon in acute and chronic inflammatory hyperalgesia. Pain. 2007;132:67–73. doi: 10.1016/j.pain.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Reichling DB, Levine JD. Shared mechanisms for opioid tolerance and a transition to chronic pain. J Neurosci. 2010;30:4660–4666. doi: 10.1523/JNEUROSCI.5530-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012;5:14. doi: 10.1186/1756-6606-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/S0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Burkham J, Dina OA, Brown AS, Bogen O, Alessandri-Haber N, Green PG, Reichling DB, Levine JD. Stress induces a switch of intracellular signaling in sensory neurons in a model of generalized pain. J Neurosci. 2008;28:5721–5730. doi: 10.1523/JNEUROSCI.0256-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kwon JT, Kim HS, Han JH. CREB and neuronal selection for memory trace. Front Neural Circuits. 2013;7:44. doi: 10.3389/fncir.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöll B, Nordheim A. Functional versatility of transcription factors in the nervous system: the SRF paradigm. Trends Neurosci. 2009;32:432–442. doi: 10.1016/j.tins.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Koç Y, McCaffrey R. 2′,3′-Dideoxyadenosine killing of TdT-positive cells is due to a trace contaminant. Leukemia. 1995;9:53–57. [PubMed] [Google Scholar]
- Kodama EN, McCaffrey RP, Yusa K, Mitsuya H. Antileukemic activity and mechanism of action of cordycepin against terminal deoxynucleotidyl transferase-positive (TdT+) leukemic cells. Biochem Pharmacol. 2000;59:273–281. doi: 10.1016/S0006-2952(99)00325-1. [DOI] [PubMed] [Google Scholar]
- Lane-Ladd SB, Pineda J, Boundy VA, Pfeuffer T, Krupinski J, Aghajanian GK, Nestler EJ. CREB (cAMP response element-binding protein) in the locus coeruleus: biochemical, physiological, and behavioral evidence for a role in opiate dependence. J Neurosci. 1997;17:7890–7901. doi: 10.1523/JNEUROSCI.17-20-07890.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Bailey CH, Kandel ER, Kaang BK. Transcriptional regulation of long-term memory in the marine snail Aplysia. Mol Brain. 2008;1:3. doi: 10.1186/1756-6606-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren CA, Newman ZL, Morford JJ, Ryan SB, Battani KA, Su Z. Cyclooxygenase-2, prostaglandin E2 glycerol ester and nitric oxide are involved in muscarine-induced presynaptic enhancement at the vertebrate neuromuscular junction. J Physiol. 2013;591:4749–4764. doi: 10.1113/jphysiol.2013.256727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CX, Suman VJ, Goetz M, Haluska P, Moynihan T, Nanda R, Olopade O, Pluard T, Guo Z, Chen HX, Erlichman C, Ellis MJ, Fleming GF. A phase I trial of the IGF-1R antibody Cixutumumab in combination with temsirolimus in patients with metastatic breast cancer. Breast Cancer Res Treat. 2013;139:145–153. doi: 10.1007/s10549-013-2528-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W. Chronic prostaglandin E2 treatment induces the synthesis of the pain-related peptide substance P and calcitonin gene-related peptide in cultured sensory ganglion explants. J Neurochem. 2010;115:363–372. doi: 10.1111/j.1471-4159.2010.06927.x. [DOI] [PubMed] [Google Scholar]
- Ma W, Zheng WH, Powell K, Jhamandas K, Quirion R. Chronic morphine exposure increases the phosphorylation of MAP kinases and the transcription factor CREB in dorsal root ganglion neurons: an in vitro and in vivo study. Eur J Neurosci. 2001;14:1091–1104. doi: 10.1046/j.0953-816x.2001.01731.x. [DOI] [PubMed] [Google Scholar]
- Ma W, Hatzis C, Eisenach JC. Intrathecal injection of cAMP response element binding protein (CREB) antisense oligonucleotide attenuates tactile allodynia caused by partial sciatic nerve ligation. Brain Res. 2003;988:97–104. doi: 10.1016/S0006-8993(03)03348-1. [DOI] [PubMed] [Google Scholar]
- Maj-Hes A, Medioni J, Scotte F, Schmidinger M, Kramer G, Combe P, Gornadha Y, Elaidi R, Oudard S. Rechallenge with mTOR inhibitors in metastatic renal cell carcinoma patients who progressed on previous mTOR inhibitor therapy. Oncology. 2013;85:8–13. doi: 10.1159/000350005. [DOI] [PubMed] [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- Mestre C, Pélissier T, Fialip J, Wilcox G, Eschalier A. A method to perform direct transcutaneous intrathecal injection in rats. J Pharmacol Toxicol Methods. 1994;32:197–200. doi: 10.1016/1056-8719(94)90087-6. [DOI] [PubMed] [Google Scholar]
- Montminy M. Transcriptional regulation by cyclic AMP. Annu Rev Biochem. 1997;66:807–822. doi: 10.1146/annurev.biochem.66.1.807. [DOI] [PubMed] [Google Scholar]
- Montminy MR, Gonzalez GA, Yamamoto KK. Regulation of cAMP-inducible genes by CREB. Trends Neurosci. 1990;13:184–188. doi: 10.1016/0166-2236(90)90045-C. [DOI] [PubMed] [Google Scholar]
- O'Banion MK. Prostaglandin E2 synthases in neurologic homeostasis and disease. Prostaglandins Other Lipid Mediat. 2010;91:113–117. doi: 10.1016/j.prostaglandins.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa E, Furusyo N, Kajiwara E, Takahashi K, Nomura H, Maruyama T, Tanabe Y, Satoh T, Nakamuta M, Kotoh K, Azuma K, Dohmen K, Shimoda S, Hayashi J. Efficacy of pegylated interferon alpha-2b and ribavirin treatment on the risk of hepatocellular carcinoma in patients with chronic hepatitis C: a prospective, multicenter study. J Hepatol. 2013;58:495–501. doi: 10.1016/j.jhep.2012.10.017. [DOI] [PubMed] [Google Scholar]
- Ohinata K, Yoshikawa M. Central prostaglandins in food intake regulation. Nutrition. 2008;24:798–801. doi: 10.1016/j.nut.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Osaka T. Prostaglandin E(2) fever mediated by inhibition of the GABAergic transmission in the region immediately adjacent to the organum vasculosum of the lamina terminalis. Pflugers Arch. 2008;456:837–846. doi: 10.1007/s00424-007-0443-8. [DOI] [PubMed] [Google Scholar]
- Ouseph AK, Khasar SG, Levine JD. Multiple second messenger systems act sequentially to mediate rolipram-induced prolongation of prostaglandin E2-induced mechanical hyperalgesia in the rat. Neuroscience. 1995;64:769–776. doi: 10.1016/0306-4522(94)00397-N. [DOI] [PubMed] [Google Scholar]
- Parada CA, Yeh JJ, Reichling DB, Levine JD. Transient attenuation of protein kinase Cepsilon can terminate a chronic hyperalgesic state in the rat. Neuroscience. 2003a;120:219–226. doi: 10.1016/S0306-4522(03)00267-7. [DOI] [PubMed] [Google Scholar]
- Parada CA, Yeh JJ, Joseph EK, Levine JD. Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur J Neurosci. 2003b;17:1847–1852. doi: 10.1046/j.1460-9568.2003.02626.x. [DOI] [PubMed] [Google Scholar]
- Parada CA, Reichling DB, Levine JD. Chronic hyperalgesic priming in the rat involves a novel interaction between cAMP and PKCepsilon second messenger pathways. Pain. 2005;113:185–190. doi: 10.1016/j.pain.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Quanhong Z, Ying X, Moxi C, Tao X, Jing W, Xin Z, Li W, Derong C, Xiaoli Z, Wei J. Intrathecal PLC(beta3) oligodeoxynucleotides antisense potentiates acute morphine efficacy and attenuates chronic morphine tolerance. Brain Res. 2012;1472:38–44. doi: 10.1016/j.brainres.2012.06.030. [DOI] [PubMed] [Google Scholar]
- Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32:611–618. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg ME, Masocha W, Ferella M, Petitto-Assis F, Goto H, Kristensson K, McCaffrey R, Wigzell H. Treatment of African trypanosomiasis with cordycepin and adenosine deaminase inhibitors in a mouse model. J Infect Dis. 2005;192:1658–1665. doi: 10.1086/496896. [DOI] [PubMed] [Google Scholar]
- Sang N, Chen C. Lipid signaling and synaptic plasticity. Neuroscientist. 2006;12:425–434. doi: 10.1177/1073858406290794. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Song MJ, Wang YQ, Wu GC. Additive anti-hyperalgesia of electroacupuncture and intrathecal antisense oligodeoxynucleotide to interleukin-1 receptor type I on carrageenan-induced inflammatory pain in rats. Brain Res Bull. 2009;78:335–341. doi: 10.1016/j.brainresbull.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Su L, Wang C, Yu YH, Ren YY, Xie KL, Wang GL. Role of TRPM8 in dorsal root ganglion in nerve injury-induced chronic pain. BMC Neurosci. 2011;12:120. doi: 10.1186/1471-2202-12-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugar AM, McCaffrey RP. Antifungal activity of 3′-deoxyadenosine (cordycepin) Antimicrob Agents Chemother. 1998;42:1424–1427. doi: 10.1128/aac.42.6.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JL, Xiao C, Lu B, Zhang J, Yuan XZ, Chen W, Yu LN, Zhang FJ, Chen G, Yan M. CX3CL1/CX3CR1 regulates nerve injury-induced pain hypersensitivity through the ERK5 signaling pathway. J Neurosci Res. 2013;91:545–553. doi: 10.1002/jnr.23168. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Prostaglandin effects after elimination of indirect hyperalgesic mechanisms in the skin of the rat. Brain Res. 1989;492:397–399. doi: 10.1016/0006-8993(89)90928-1. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Coderre TJ, Levine JD. The contribution of training to sensitivity in the nociceptive paw-withdrawal test. Brain Res. 1989;487:148–151. doi: 10.1016/0006-8993(89)90950-5. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Heller PH, Levine JD. Characterization of distinct phospholipases mediating bradykinin and noradrenaline hyperalgesia. Neuroscience. 1990;39:523–531. doi: 10.1016/0306-4522(90)90288-F. [DOI] [PubMed] [Google Scholar]
- Teng FY, Tang BL. Axonal regeneration in adult CNS neurons—signaling molecules and pathways. J Neurochem. 2006;96:1501–1508. doi: 10.1111/j.1471-4159.2006.03663.x. [DOI] [PubMed] [Google Scholar]
- Tsai YJ, Lin LC, Tsai TH. Pharmacokinetics of adenosine and cordycepin, a bioactive constituent of Cordyceps sinensis in rat. J Agric Food Chem. 2010;58:4638–4643. doi: 10.1021/jf100269g. [DOI] [PubMed] [Google Scholar]
- Widnell KL, Self DW, Lane SB, Russell DS, Vaidya VA, Miserendino MJ, Rubin CS, Duman RS, Nestler EJ. Regulation of CREB expression: in vivo evidence for a functional role in morphine action in the nucleus accumbens. J Pharmacol Exp Ther. 1996;276:306–315. [PubMed] [Google Scholar]
- Yang Q, Mattick JS, Orman MA, Nguyen TT, Ierapetritou MG, Berthiaume F, Androulakis IP. Dynamics of hepatic gene expression profile in a rat cecal ligation and puncture model. J Surg Res. 2012;176:583–600. doi: 10.1016/j.jss.2011.11.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa N, Nakamura K, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Reinforcement of antitumor effect of Cordyceps sinensis by 2′-deoxycoformycin, an adenosine deaminase inhibitor. In Vivo. 2007;21:291–295. [PubMed] [Google Scholar]