

Abstract
Acute brain lesions induce profound alterations of the peripheral immune response comprising the opposing phenomena of early immune activation and subsequent immunosuppression. The mechanisms underlying this brain-immune signaling are largely unknown. We used animal models for experimental brain ischemia as a paradigm of acute brain lesions and additionally investigated a large cohort of stroke patients. We investigated the inflammatory potency of HMGB1 and its signaling pathways by immunological in vivo and in vitro techniques. Features of the complex behavioral sickness behavior syndrome were characterized by homecage behavior analysis. HMGB1 downstream signaling, particularly with RAGE, was studied in various transgenic animal models and by pharmacological blockade. Our results indicate that HMGB1 was released from the ischemic brain in the hyperacute phase of stroke in mice and patients. Cytokines secreted in the periphery in response to brain injury induced sickness behavior, which could be abrogated by inhibition of the HMGB1-RAGE pathway or direct cytokine neutralization. Subsequently, HMGB1-release induced bone marrow egress and splenic proliferation of bone marrow-derived suppressor cells, inhibiting the adaptive immune responses in vivo and vitro. Furthermore, HMGB1-RAGE signaling resulted in functional exhaustion of mature monocytes and lymphopenia, the hallmarks of immune suppression after extensive ischemia. This study introduces the HMGB1-RAGE-mediated pathway as a key mechanism explaining the complex postischemic brain-immune interactions.
Keywords: alarmins, HMGB1, immunomodulation, myeloid-derived suppressor cell, RAGE, stroke
Introduction
Acute brain injuries, including stroke, induce profound alterations of the peripheral immune system (Chamorro et al., 2012), which contribute substantially to morbidity and mortality of patients. The best described component of these systemic immune alterations is an acquired immune deficiency in the subacute phase after ischemic stroke, which predisposes patients to infections (Meisel et al., 2005). The main features of this syndrome are an inactivation of circulating monocytes and lymphopenia (Liesz et al., 2009; Urra et al., 2009b). On the other hand, activation of the peripheral immune system preceding the immunosuppressive syndrome has also been consistently described (Offner et al., 2006a). This sterile inflammatory response includes the massive release of proinflammatory cytokines from systemic innate immune cells within hours after the brain lesion (An et al., 2014). Yet, the pathophysiological impact of this early immune activation as well as its mediators are unknown.
In view of the extensive impact of the perturbed peripheral immune system on the damaged brain as well as on systemic infectious complications, the identification of the signaling pathways underlying systemic immune modulation is of great translational relevance. Previous reports have suggested a role of the autonomic nervous system in mediating subacute immunosuppression after acute brain injuries because elevated catecholamine levels were observed in some of the previous experimental stroke studies. Blocking of adrenoreceptors reversed aspects of poststroke immunodepression, including susceptibility to bacterial infections in experimental stroke (Prass et al., 2003; Meisel et al., 2005). However, recent clinical (Liesz et al., 2013a) as well experimental (Mracsko et al., 2014) studies have yielded mixed results regarding the implications of catecholamines in immunodepression.
Therefore, we focused in this study on soluble mediators released from the necrotic brain tissue as an alternative pathway in poststroke brain-immune interaction. Previous studies suggested a strong correlation between the volume of damaged brain tissue and the extent of systemic immune alterations, regardless of infarct location (Gendron et al., 2002; Hug et al., 2009). Accordingly, we speculated that soluble mediators would be released from dying neurons in proportion to the extent of necrotic tissue. As a prototypic alarmin, high mobility group box 1 (HMGB1) is a nuclear protein, which is released from necrotic cells with high inflammatory potency (Lotze and Tracey, 2005). As such, HMGB1 can act itself as a cytokine or amplify the binding of cytokines and nucleosomes to Toll-like receptors (TLRs) (Bianchi, 2009). The receptor of advanced glycation end-products (RAGE) has been identified as a key receptor (in addition toTLR2 and TLR4) for HMGB1, mediating activation pathways to a variety of cell types in an organ-specific manner (Lotze and Tracey, 2005).
Our study introduces the signaling of brain-released alarmins, such as HMGB1, through RAGE as an important link between acute brain injury and systemic immune alterations. We demonstrate a biphasic scenario where this signaling cascade initially activates peripheral monocytes and dendritic cells, resulting in a massive release of proinflammatory cytokines. Subsequently, initial (over)activation of the peripheral immune system results in exhaustion of monocytes and recruitment of immature monocytes. Suppressive signaling by the altered monocyte population leads to apoptosis of lymphocytes, the hallmark of poststroke immunosuppression.
Materials and Methods
Animals.
The study was conducted in accordance with national guidelines for the use of experimental animals, and the protocols were approved by the governmental committee (Regierungspraesidium Karlsruhe, Germany). Animal experiments were designed, performed, and data reported according to the ARRIVE guidelines (Kilkenny et al., 2010). We used age-matched, male mice (C57BL/6J, 10–12 weeks, 23–25 g body weight, Charles River Laboratories). RAGE−/− (Liliensiek et al., 2004) and MyD88−/− (Adachi et al., 1998) mice were bred in the core animal facility of the University of Heidelberg under specified pathogen-free conditions.
Patient recruitment and blood sampling.
Blood samples used for analysis in this study were collected during a previous study (Liesz et al., 2013a) conducted between October 2011 until November 2012. In brief, inclusion criteria were study enrollment within 24 h after symptom onset. Exclusion criteria were recent clinical infection (fever, cough, antibiotic treatment, laboratory markers) within 14 d before admission, a chronic immunological disorder, preexisting immunosuppression, cancer, and pregnancy. Blood samples from 104 patients obtained within 24 h after symptom onset were analyzed. Patient characteristics are depicted in Tables 1 and 2. Stroke patients received routine cranial imaging by CT (Somatom Sensation 16) and/or 3T MRI (Trio and/or Verio; all Siemens Medical Systems) to detect ischemic brain lesions. Volume was calculated by integrating areas on each image multiplied by section diameter using a Syngo Multimodality workplace with syngoMMWP-software (version VE31A, Siemens). For the control group, patients were included that presented at the Department of Ophthalmology for presurgical screening for cataract operation. Patients were selected to be in the same range for age and gender of patients with cerebrovascular disorders. All procedures were performed after written informed consent was obtained by the patient or a legal representative.
Table 1.
Patient characteristics of stroke and control patients for HMGB1 blood analysis
| Controls | Stroke patients | p | |
|---|---|---|---|
| Total patients per group | 20 | 104 | |
| Age (years) | 69.3 ± 9.2 | 73.2 ± 10.4 | 0.241 |
| NIHSS on admission | 0 | 11.7 ± 6.4 | — |
| Median stroke volume (ml) | NA | 29.4 | — |
| Laterality (right/left) | NA | 53/51 | — |
| Blood samples (n) obtained within 24 h after symptom onset | NA | 104 | — |
| Blood samples (n) obtained within 72 h after symptom onset | NA | 153a | — |
| Mean time: symptom onset to first blood sampling (hours) | NA | 10.7 ± 5.2 | — |
| Comorbidities | |||
| Hypercholesterolemia | 6 (30%) | 27 (26%) | 0.998 |
| Arterial hypertension | 17 (85%) | 72 (69%) | 0.151 |
| Diabetes | 7 (35%) | 30 (29%) | 0.581 |
| Premedication | |||
| β-AR blocker | 7 (35%) | 39 (38%) | 0.832 |
| Oral anticoagulationb | 4 (20%) | 17 (16%) | 0.689 |
| Antithromboticsc | 8 (27%) | 17 (24%) | 0.167 |
| Sympathicoactive medicationd | 3 (15%) | 7 (7%) | 0.069 |
a Forty-nine patients were measured twice (at hospital admission and at day 3 after stroke onset).
b Phenprocoumon, rivaroxaban.
c Aspirin, aspirin + dipyridamole, clopidogrel.
d Clonidin, oxybutinin, inhalable β2-AR agonists, doxazosin, terazosin.
Table 2.
Patient characteristics of stroke and control patients for MDSC FACS (Fig. 3D)
| Patient | Age (years) | Hypertension | Diabetes | Lesion localization | NIHSS | Lesion volume (cm3) |
|---|---|---|---|---|---|---|
| Control 1 | 67 | + | − | NA | NA | NA |
| Control 2 | 91 | + | − | NA | NA | NA |
| Control 3 | 86 | − | + | NA | NA | NA |
| Control 4 | 84 | + | + | NA | NA | NA |
| Control 5 | 31 | − | − | NA | NA | NA |
| Control 6 | 64 | + | − | NA | NA | NA |
| Stroke 1 | 69 | + | − | MCA, R | 18 | 143 |
| Stroke 2 | 76 | + | + | MCA, R | 33 | 317 |
| Stroke 3 | 87 | + | − | MCA, R | 24 | 214 |
| Stroke 4 | 43 | − | − | ACA + MCA, L | 19 | 293 |
| Stroke 5 | 64 | − | − | MCA, L | 22 | 184 |
| Stroke 6 | 73 | + | + | MCA, R | 18 | 174 |
NA, Not applicable; ACA, anterior cerebral artery; R, right; L, left.
Animal experiments.
Animals received intraperitoneal injection of 100 μg monoclonal anti-HMGB1 antibodies (clone 2g7) (Qin et al., 2006) 30 min before ischemia induction or an isotype control antibody (BioXCell); 100 μg soluble RAGE (full-length recombinant human sRAGE) was injected before middle cerebral artery occlusion (MCAO) induction and 4 h after MCAO. The β2-adrenoreceptor inhibitor ICI 118,551 (Sigma) was administered intraperitoneally at 4 mg/kg body weight before MCAO and at 4, 8, and 12 h after MCAO (Mracsko et al., 2014). Neutralization of circulating cytokines was achieved by intraperitoneal injection of 200 μg each of anti-IL-1beta, anti-TNF-α, or anti-IL-6 antibodies (all from eBioscience, functional grade purified) in a total volume of 200 μl PBS intraperitoneally. Control mice received 300 μg of isotype control antibody. Organ sampling (brain, spleen, thymus, bone marrow, blood from retro-orbital sinus) for further analysis was performed in anesthesia with ketamin/xylazine. Rectal temperature and body weight were measured daily. Neurological deficits were assessed by a modified “Bederson score” (Bederson et al., 1986): 0, no deficit; 1, forelimb flexion; 2, decreased resistance to lateral push; 3, unidirectional circling; 4, longitudinal spinning; 5, no movement. Sensorimotor deficit was measured using the Rotarod test as previously described (Jones and Roberts, 1968; Liesz et al., 2013b). Briefly, mice were trained at first at constant 4 rpm, then three times at acceleration from 4 to 40 rpm in 210 s. Baseline assessment was performed as for training with acceleration and the mean calculated from three runs with a 10 min break in between. Time was stopped if the mouse fell off the beam or passively rotated twice on the beam. Assessment 24 h after MCAO was performed similar to baseline measurement.
Experimental stroke models
Coagulation model.
We used this stroke model for experiments shown in Figure 1A, B. Mice were anesthetized with 1.0%–2.0% halothane in O2/N2O. After making a 1 cm skin incision between the left eye and ear, we drilled a burr hole through the temporal skull. We removed the dura mater and occluded the MCA permanently using a bipolar electrocoagulation forceps (ERBOTOM, Erbe). During the operation, body temperature was kept at 37°C using a feedback controlled heating pad. We sutured the skin lesion and placed the mouse in a cage under an infrared heating lamp until recovery from anesthesia. Sham treatment was given according to the same surgical protocol as for MCAO but without coagulation of the MCA. The overall mortality in the coagulation model was <5% for the 3 d observation period.
Figure 1.

Inhibition of the HMGB1 pathway after stroke improves mortality. A, Representative cresyl-violet-stained brain sections (left) after permanent distal MCA occlusion (C-MCAO), 30 min or 90 min transient proximal filament-MCAO (F30-MCAO and F90-MCAO), and analysis of mean infarct volumes in the respective MCAO model (3 d after MCAO, n = 12–15 per model, C57BL/6J WT mice). B, Serum HMGB1 concentration was measured by ELISA in unoperated naive animals, in the three stroke models, and in respective sham-operated animals (C-sham and F-sham, respectively) at the indicated time points after MCAO (n = 6 per group). *p < 0.05, between naive and the indicated group. Bars represent p < 0.05 between marked groups within one time point. Data are representative for 3 individual experiments per time point. C, Increase of serum HMGB1 in the 90 min filament-occlusion model was verified by Western blot analysis at 2 h after MCAO compared with the sham-operated control mice (n = 4 per group). D, Dot plot analysis of HMGB1 serum concentrations of ischemic stroke patients up to 72 h after stroke onset (n = 153). E, Serum HMGB1 was measured in matched control patients without stroke (n = 20) and in stroke patients. For analysis of stroke patients (n = 104), only blood samples of individual patients (no repetitive measurements) up to 24 h after symptom onset were included and patient groups dichotomized into small/moderate (<30 ml) and severe (>30 ml) lesion volumes. F, Stroke lesion volume was analyzed 3 d after 90 min filament-occlusion MCAO (F-90 min) in control animals (C57BL/6J) and RAGE−/− animals receiving anti-HMGB1 or control (isotype IgG) treatment. Infarct volumes did not significantly differ among groups (n = 18–34 per group, 4 individual experiments, p = 0.21). G, The Bederson score reflecting poststroke motor deficits was not affected by anti-HMGB1 treatment or RAGE deficiency. H, Kaplan–Meier curves of poststroke survival within the first week after F-90 min MCAO in control, anti-HMGB1 treated, and RAGE−/− mice. Overall p value = 0.02: Mantel-Cox test; n = 32(control), 17(RAGE−/−), 22(anti-HMGB1). p value (RAGE−/− vs control) = 0.008. I, Body weight (left) and rectal body temperature (right) were measured before surgery and on poststroke days 1–3 (n = 15–18 per group). *p < 0.05, control versus RAGE−/−. #p < 0.05, control versus anti-HMGB1. F–I, Data are representative of 4 individual experiments.
Filament model.
We used this model for all remaining experiments. Mice were anesthetized with 1.0%–2.0% halothane in O2/N2O-enriched air. We placed the laser Doppler probe over the cortical area supplied by the MCA. Baseline CBF was measured as relative perfusion units and defined as 100% flow. After neck dissection, we made an incision into the left external carotid artery between two ligations and advanced a silicon-covered 8-0 nylon monofilament through the internal carotid artery to occlude the MCA. MCA occlusion was documented as a decrease in relative perfusion values by laser Doppler to <20% of original flow, a less pronounced decrease in CBF was used as an exclusion criterion. We fixed the filament in this position by ligation, closed the neck, removed the Doppler probe, and placed the mouse in its cage. Then, 30 min (only for experiments in Fig. 1A,B) or 90 min, respectively, after filament insertion, the mouse was anesthetized again and the filament removed. After we closed the surgical wound, the mice were transferred to their cages with free access to water and food. During the operations body temperature was kept at 37°C with a feedback-controlled heating pad. We maintained normal body temperature of the mice between operations and until recovery after the procedure by an infrared heating lamp. Sham operation was performed in exactly the same manner as described above, including repeated anesthesia 30 or 90 min after sham surgery, except that the filament was only introduced briefly into the external carotid artery. The overall mortality in the 30 min occlusion model was <5% for the 3 d observation period; mortality for the 90 min occlusion model is reported in Figure 1H.
Assessment of infarct volume.
We anesthetized the mice and perfused them transcardially with 20 ml normal saline. We removed the brains from the skull and froze them immediately in isopentane (−20°C). We cut 20-μm-thick coronal cryosections every 400 μm, stained the sections using a standard cresyl-violet staining protocol, scanned them at 600 dpi, and analyzed the infarct area on each section (ImageJ). The Swanson method was applied to indirectly measure the infarct area and to correct for cortical swelling (Swanson et al., 1990). The total infarct volume was determined by integrating measured areas and distances between sections.
ELISA.
Human and mouse serum and cell culture supernatants were collected and immediately frozen at −80°C until analysis. Commercial ELISA kits were used for the quantitative analysis of HMGB1 (IBL) and cytokine expression of IL-1β, TNF-α, IL-6, and IL-12p40 (all R&D Systems).
Homecage behavior analysis.
Mice were held individually in 37 × 16 × 13 cm transparent plastic cages as homecages. Mice were recorded for 17–24 h for each day/night/d cycle before MCAO and at day 1 and/or day 3 after MCAO using CCD cameras and an infrared illumination system during the dark cycle. The camera angle was set up for recording of the long side of cages to observe horizontal as well as vertical activities, such as rearing and hanging. Ethovision (EthovisionXT, version 4.1, by Noldus Information Technologies) video tracking software was used to digitize the trials at a rate of 12.5 frames/s.
Bacterial culture.
For bacterial blood cultures, 30 μl blood was drawn from each mouse and diluted in 270 μl sterile PBS. For bacterial lung cultures, both lung lobes were homogenized in PBS in a total volume of 300 μl; 50 μl of each blood sample or 100 μl of each lung homogenate was plated on Columbia agar with 5% sheep blood (BD Biosciences). Plates were incubated at 37°C in the presence of 5% CO2 and colony forming units were evaluated after 24 h.
Determination of bacterial DNA in lung homogenates by quantitative PCR.
Bacterial load in lung homogenates was determined applying a quantitative real-time PCR that amplifies 16S ribosomal DNA. Both lung lobes were homogenized, and DNA was extracted using the QIAmpDNA Mini kit (QIAGEN) according to the manufacturer's instructions with an eluate volume of 100 μl. Samples were amplified with primers targeting conserved regions within the 16S rRNA gene (forward, 5′-TGG AGC ATG TGG TTT AAT TCG A-3′; reverse, 5′-TGC GGG ACT TAA CCC AAC A-3′). A defined plasmid standard (16S rRNA amplicon from Escherichia coli, cloned in pCR2.1, Invitrogen; plasmids quantified by spectrophotometry) was used as positive control for qPCR. Samples were assayed in triplicate in a 25 μl reaction mixture containing 2 μl of template DNA, 12.5 μl of 2× Fast SYBR Green Master Mix (Applied Biosystems) and 12.5 pmol of forward primer and reverse primer (MWG). Cycling conditions were as follows: 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min each. For quantification, background levels of the nontemplate control were subtracted from the specific samples.
Western blotting.
Proteins were isolated for HMGB1 and ARG1 Western blotting from samples using ice-cold RIPA buffer as previously reported (Liesz et al., 2013b). Lysates were electrophoresed, transferred to nitrocellulose membranes, and blocked with skim milk. Blocked membranes were probed with rabbit anti-HMGB1 (1:1000) or incubated with sheep anti-ARG1 (1:500; R&D Systems) in TBS-T plus 5% milk overnight at 4°C Peroxidase-conjugated goat anti-rabbit and donkey anti-sheep secondary antibodies were used, respectively, and blots developed by standard chemiluminescence protocols.
Stress mediator analysis.
Serum cortisol and metanephrine/normetanephrine concentrations were measured in the central laboratory of the University Hospital Heidelberg by the same procedures used and validated for routine diagnostic analysis. Cortisol concentrations were measured by chemiluminescence immunoassays, and serum metanephrine/normetanephrine were detected by ELISA.
Magnetic cell sorting.
MACS sorting using magnetic beads was used for isolation of CD11b-, CD11c-, and CD3-positive cells for in vitro studies. Single-cell suspensions were prepared from spleens of the respective naive donor animals or after experimental brain ischemia as indicated in the manuscript. Single-cell suspensions were then processed for further purification of the subpopulation according to the manufacturer's protocol using commercial MACS kits (Miltenyi Biotec) for the purification of the indicated population (total T cells, monocytes, dendritic cells, myeloid-derived suppressor cells).
Real-time PCR.
RNA was isolated from spleens or isolated cells using the RNeasy kit (QIAGEN). Reverse transcription was performed with the High Capacity complementary DNA Archive Kit and real-time PCR with SYBR-Green assays on an ABI7500 real-time PCR system (Applied Biosystems). Primers were purchased as ready-to-use primer sets for each gene (Super Array). All assays were run in duplicate. Expression of each gene was quantified according to the relative standard curve method relative to the expression of the housekeeping gene encoding for β-actin.
In vitro assays
T-cell stimulation assay.
For intracellular cytokine staining of isolated T cells, we plated single-cell suspensions of 300,000 mononuclear cells on 96-well plates (RPMI-1640, + 10% FCS, + glutamine, +HEPES, + penicillin/streptomycin, + mercaptoethanol). Each sample was processed in triplicate. We primed the cells for 48 h with plate-bound anti-mouse CD3e (clone 145-2C11, BD Biosciences) and 2 μg/ml soluble anti-mouse CD28 (clone 37, clone 51, BD Biosciences), followed by restimulation with 500 ng calcimycin (Sigma-Aldrich), 5 ng/ml PMA (Sigma-Aldrich), and protein transport inhibitor (BD GolgiPlug, BD Biosciences) for 5 h.
Dendritic cell stimulation assay.
CD11c+ splenic dendritic cells were isolated by MACS following the manufacturer's protocol (Miltenyi Biotec). CD11c+ cells were cultured at a concentration of 100,000 cells/well for 24 h. Fully phosphorothioate modified CpG-ODN 1668 (TIB Molbiol) was used at a concentration of 100 nm by adding the stimulus directly into the culture medium.
Marrow-derived suppressor cell (MDSC) suppression assay.
CD11b+Ly-6Chigh+ cells were isolated by MACS and cocultered with CD3+ T cells (MACS-isolated) at the indicated ratio with a total cell count of 200,000 cells per well of a 96-well round-bottom plate. T cells were labeled with CFSE (CFSE cell proliferation kit, Invitrogen), and their proliferation was stimulated by plate-bound anti-CD3ε (10 μg/ml) and addition of concavalin A at 2 μg/ml for a culture time of 42 h. Suppression of T-cell proliferation was assessed by measuring the CFSE dilutions and analyzed using Flowjo 4.2 software (TreeStar).
HMGB1 stimulation assay.
For assessment of the cytokine-inducing activity of HMGB1, mixed splenocyte cultures (Fig. 2D) or cocultures of T cells with antigen-presenting cells (APCs) (Fig. 2E) were used. Erythrocyte-depleted primary cell suspensions of spleens from naive animals were used for mixed splenocyte cultures. For cocultures, T cells were isolated by MACS (T cell isolation kit I, Miltenyi) and APCs were isolated using the same kit. APCs were either irradiated or received sham treatment (placement in chamber with no irradiation). Cells were cultured in RPMI-1640 medium, “Cytokine HMGB1”(C23-C45 disulfide bond, purchased from HMGBiotech) was added to the culture medium at the indicated concentrations.
Flow cytometry.
We collected organs (spleen, blood, mesenteric lymph nodes, and bone marrow) after transcardial perfusion with saline at various time points after MCAO for flow cytometric analysis. We stained single-cell suspensions for anti-mouse CD3 (clone 17A2), CD4 (clone RM 4–5), CD8 (clone 53–6.7), B220 (clone RA3–6B2), NK1.1 (clone PK136), Ly-6C (clone HK1.4), Ly-6G (Gr-1; clone RB6–8C5), CD80 (clone 16–10A1), CD86 (clone GL1), CD25 (clone PC61), CD69 (clone H1.2F3), CD11b (clone M1/70), MHC-II (M5/114.15.2), and CD11c (clone N418). Expression of IFN-γ (clone XMG1.2) and IL-4 (clone 11B11) was intracellularly detected after stimulation of T cells by the above published stimulation protocol.
Blood samples from stroke patients for FACS analysis were collected in EDTA tubes and immediately processed. Mononuclear cells were isolated by Ficoll gradient centrifugation and single-cell suspensions stained for anti-human CD11b (clone ICRF44), CD33 (clone P67.6), and HLA-DR (clone L243). We performed flow cytometry on a Becton Dickinson FACS Calibur and analyzed the data by CellQuest Pro software.
Statistical analysis.
Sample size of the experiments was determined by a priori calculation presuming Type I error of 0.05, power of 0.8, and specific variance based on according preexperiments using g* power statistical software. All values in bar graphs are expressed as mean ± SD. We analyzed infarct volumes and functional outcome tests by two-tailed Student's t test after validating the normal distribution of these datasets (Kolmogorov–Smirnov test) or for >2 groups by ANOVA with Tukey post hoc analysis. For the remaining data without normal distribution, we used two-tailed Wilcoxon rank-sum test for two groups or the Kruskal–Wallis test with Dunn's multiple comparison post hoc analysis test for more than two groups, using GraphPad Prism 5 software. p < 0.05 was considered statistically significant.
Results
HMGB1 is released from necrotic brain lesions
We analyzed HMGB1 serum concentrations by ELISA in different experimental stroke models varying by location and volume of the resulting infarct (Fig. 1A) and detected a significant increase only after large infarcts at early time points after stroke (Fig. 1B). This was verified by HMGB1 Western blotting at 2 h after stroke onset (Fig. 1C). We also found massively increased HMGB1 serum concentrations in stroke patients within the first hours after symptom onset (Fig. 1D). To verify HMGB1 release in human stroke, we further evaluated HMGB1 concentrations in blood samples from 104 patients (patient characteristics summarized in Table 1) within 24 h after symptom onset of small or large lesions (dichotomized by the median infarct volume, in this cohort 30 ml) and compared it with control patients (Fig. 1E). Because human and murine results showed a consistent pattern of early HMGB1 release into the circulation after extensive ischemic brain lesions, further experimental studies were performed exclusively in the 90 min transient MCAO model.
We analyzed the impact of blocking HMGB1 by neutralizing antibodies or by elimination of its receptor RAGE in knock-out mice on stroke outcome. Infarct volumes (Fig. 1F) and basic sensory-motor deficits measured by the Bederson score (Fig. 1G) did not differ significantly among anti-HMGB1 treated animals, RAGE−/− mice, and controls in our experiments. Intriguingly, despite the lack of effect on infarct size and neurological deficit, 7 d mortality was substantially reduced in RAGE−/− mice (p = 0.008, n = 22) and tended also to be lower in anti-HMGB1-treated mice (p = 0.07, n = 17) (Fig. 1H). Both HMGB1 neutralization and RAGE deficiency also attenuated loss of body weight and hypothermia after stroke (Fig. 1I). This dissociation between similar lesion size and differing mortality might indicate that HMGB1-RAGE blockade modified important systemic consequences of brain infarction rather than resulting in immediate structural neuroprotection.
The early phase after brain injury is characterized by massive systemic cytokine secretion mediating sickness behavior
To determine the contribution of HMGB1 signaling on sterile systemic inflammation, cytokine concentrations were determined within the acute phase after brain ischemia. Expression levels of the proinflammatory cytokines IL-1β, TNF-α, and IL-6 were markedly increased in the acute phase after stroke onset (Fig. 3A). Antibody-mediated neutralization of HMGB1 significantly attenuated splenic expression and serum concentrations of these proinflammatory cytokines (Fig. 3A,B). Supporting the immune-stimulatory role of HMGB1, in vitro stimulation of splenocyte cultures with the cytokine-stimulating isoform of HMGB1 resulted in increased expression of T cell- (IFN-γ) and monocyte- (IL-12) derived cytokine secretion (Fig. 3C). Moreover, we detected a dose–response relation for HMGB1 concentration and IFN-γ secretion by isolated T cells, which depended on the presence of viable APCs (Fig. 3D).
Figure 3.

HMGB1 signaling induces early innate and adaptive immune activation. A, Cytokine mRNA expression was analyzed in spleens at the indicated time points after MCAO (n = 7 per group and time point). B, Expression levels of IL-1β and TNF-α were verified on protein level in the serum of control mice (isotype IgG) and anti-HMGB1-treated mice without brain lesion (naive) and 24 h after F-90 min MCAO by ELISA (n = 6 per group). A, B, Data are representative of 2 individual experiments per time point. C, The cytokine-inducing function of HMGB1 was tested in vitro. Interferon-γ (left) and IL-12 (right) concentration was analyzed in supernatants of mixed splenocyte cultures stimulated with disulphide-HMGB1 at the indicated concentration and compared with unstimulated samples (control). Bars represent p < 0.05 between indicated groups. Each data point represents one individual experiment. D, Naive CD3+ T cells were cocultured with untreated (vital) or irradiated APCs and stimulated with HMGB1 at the indicated concentration for 24 h. Interferon-γ concentrations were measured in the supernatant by ELISA (data representative of 5 individual experiments). Vital APCs are required for stimulation of IFN-γ production by T cells.
Weight loss and hypothermia, as shown in Figure 1I, are considered as basal markers of sickness in models of immune-mediated diseases (Dantzer, 2006). We further investigated this phenomenon in a homecage behavior analysis setup, which enabled the automated analysis of circadian rhythm and complex mouse behavior (Jhuang et al., 2010; Spulber et al., 2012) (Fig. 4A). Although no behavioral difference was detected between RAGE−/− animals and WT controls at baseline before MCAO (data not shown), we detected a disturbance in circadian rhythm at day 1 after stroke. This was more pronounced in WT compared with RAGE−/− animals with respect to arousal after the start of the day cycle (6 A.M.) and disparity between overall night and day activity (Fig. 4B). Furthermore, mobility and exploratory behavior (total distance moved), targeted movements to reach the water bottle (rearing time and frequency), and challenging physical activities (hanging time) were significantly less affected during the early phase after stroke induction in RAGE−/− compared with WT mice (Fig. 4C).
Figure 4.

RAGE signaling mediates sickness-like behavior in the acute phase after stroke. A, Representative images of the unmodified tracking zones for continuous recording by the homecage behavior system (top). Analysis fields (hanging and rearing zones) and tracking (red lines) of mice in their home cages, indicated for 1 h recording (bottom). B, Brain ischemia by F-90 min MCAO induces a disturbed circadian rhythm with reduced activation response after a shift of night-day cycle at 6 A.M. Data are depicted for total distance moved per 1 h during the 12 h recording period (6 h dark and light cycle, each) and indicate a different degree of disturbed circadian rhythm between RAGE−/− and WT mice at day 1 (d1) after MCAO. C, Results of the homecage behavior analyses for overall mobility (moving distance: left) or motivation to challenging physical activity (rearing time: middle; hanging time: right) shown as cumulative data during the 2 h period from 6 A.M. to 8 A.M. at day 1 and day 3 after MCAO in WT control and RAGE−/− mice. Data were obtained in 3 individual experiments. D, Infarct volumetry 24 h after 90 min transient MCAO in mice receiving neutralizing antibodies against IL-1, TNF-α, and IL-6 (anti-cytokine) or isotype control IgG (control). E, Sensorimotor deficits were determined in the corresponding groups as in D and in a sham-operated group 24 h after F-90 min MCAO and normalized to baseline values. n.s., Not significant. F, The disturbed circadian rhythm after MCAO compared with sham treatment is improved in the anti-cytokine treated group as depicted by the approximation of the activity pattern to one of mice with sham surgery. G, Analysis of the total distance moved within the first 2 h of the light period (6 A.M. to 8 A.M.). D–G, Data were obtained from 3 individual experiments; n(sham) = 9, n(control) = 9, n(anti-cytokine) = 9.
To investigate the role of the peripheral cytokine release in the acute phase after brain injury as the potential link between RAGE deficiency and improved behavior, we antagonized IL-1β, TNF-α, and IL-6 by neutralizing antibodies and analyzed key features of sickness behavior as introduced above. Infarct volumetry revealed similar infarct volumes in control- and anti-cytokine–treated mice (Fig. 4D). Additionally, focal sensorimotor dysfunction analyzed by the Rotarod test did not differ between the treatment groups (Fig. 4E). This demonstrated that peripheral cytokine neutralization had no immediate impact on the primary brain lesion. In contrast, homecage behavior analysis revealed a similar treatment effect of cytokine antagonization as previously seen in RAGE-deficient mice. The cytokine neutralization improved the stroke-induced disturbances in circadian rhythm with reduced variation between light and dark periods (Fig. 4F). It also improved overall motility as depicted by analysis of total distanced moved in the first 2 h of the light period (6 A.M. to 8 A.M.) (Fig. 4G).
RAGE signaling promotes MDSC expansion
Based on the finding that APC viability is critical for the impact of HMGB1 on lymphocyte function (Fig. 3D), we further analyzed the role of monocytes in this model. We found an expansion of a specific immature monocyte subpopulation in the spleen, characterized by the CD11b+Ly-6C+MHC-IIlow expression pattern after stroke (Fig. 5A), which was abrogated by anti-HMGB1 treatment (Fig. 5B). Further analysis of this population in spleen and bone marrow revealed both increased splenic proliferation (marker Ki-67) and a reduced proportion of this subpopulation in the bone marrow (Fig. 5B). These findings are compatible with a combination of local splenic proliferation as well as bone marrow egress of immature monocytes. Moreover, stroke in WT mice resulted in an increase in CD80 expression and a reduction of MHC-II expression on monocytes (Fig. 5C). The stroke-induced rise of immature splenic monocytes compared with sham-operated mice was significantly reduced in RAGE−/− animals 3 d after MCAO, supporting the role of HMGB1-RAGE signaling on immature monocyte recruitment (Fig. 5D).
Figure 5.
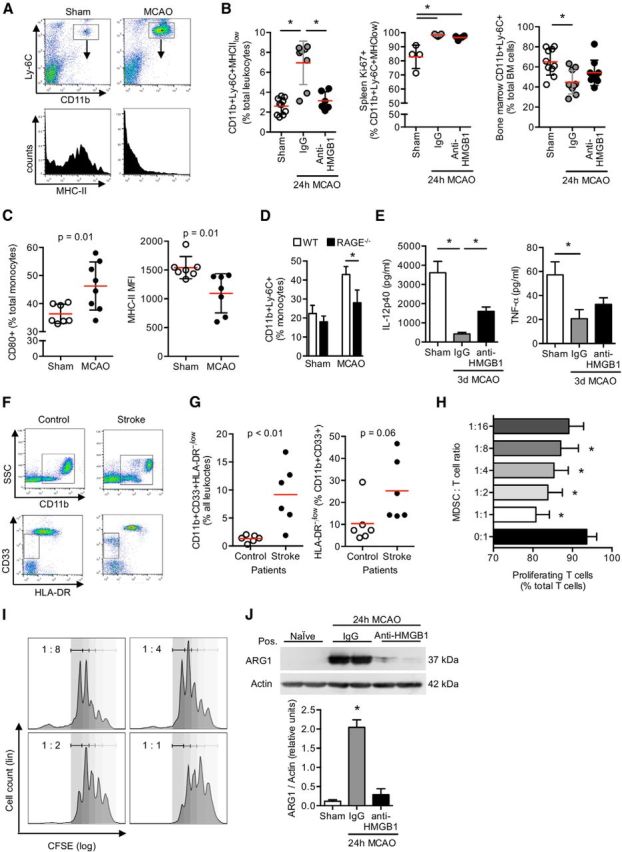
Stroke induces immune exhaustion and expansion of immature monocytes via HMGB1-RAGE signaling in the subacute phase after stroke. A, Representative FACS plots for CD11b/Ly-6C expression of splenic mononuclear cells (CD45+ gated) 3 d after sham or F-90 min MCAO operation and the respective histogram for MHC-II expression of the double-positive cell population. B, Cell count of myeloid-derived progenitor cells (CD11+Ly-6C+MHC-IIlow) and their proliferation (CD11b+Ly-6C+Ki-67+) was analyzed in spleens at 24 h after sham operation or F90 min-MCAO with control (IgG) or anti-HMGB1 treatment (two left panels). Cell count of CD11b+Ly-6C+ cells in the bone marrow was determined in the same groups (right panel). Bars represent p < 0.05 between indicated groups. C, Splenic CD11c+ dendritic cells were analyzed for CD80 and MHC-II expression at 3 d after sham operation (control) or MCAO. D, Frequency of immature CD11+Ly-6C+ monocytes was compared between WT and RAGE−/− mice 3 d after F90 min-MCAO or sham surgery (n = 6 per group). E, Splenic CD11c+ dendritic cells (1 × 105 cells) harvested from sham-operated animals and control antibody (IgG) or anti-HMGB1 treated animals 3 d after MCAO were stimulated with 1 μm CpG-ODN overnight, and IL12p40 and TNF-α cytokine concentrations were measured in the cell culture supernatant (n = 6 per group). B–E, Results are representative of at least two individual experiments per set. F, Representative FACS dot plots for CD11b (top) and CD33/HLA-DR expression (gated for CD11b+, bottom) of total blood leukocytes (CD45+ gated) from control or stroke patients. G, Blood cell counts of immature CD11b+CD33+monocytes (left) and their HLA-DR expression were analyzed in control patients with no neurological disease or in stroke patients at 24 h after extensive brain ischemia. H, Suppression assay analysis was performed to determine the suppressive function of splenic CD11b+Ly-6C+ monocytes harvested from donor animals 24 h after F90 min-MCAO (MDSC). T cells and MDSCs were cocultured at the indicated ratio and the proliferation of CFSE-labeled T cells analyzed by FACS (shown as percentage of total CD3+ T cells). CD11b+Ly-6C cells after MCAO are potent suppressors of T-cell proliferation. Data are representative of 5 individual experiments. I, Representative plots with fitted peaks for cell proliferation analysis by FlowJo software as used for MDSC suppression assay analysis shown before. Labeling in the plot indicates MDSC:T-cell ratio. J, Representative Western blot images (top) and analysis of ARG1 protein expression (bottom). ARG1 expression was measured in isolated splenic CD11b+ cells from sham-operated animals and control antibody (IgG) or anti-HMGB1 treated mice 24 h after F90 min-MCAO. Analysis is depicted as normalized ARG1 density values to actin expression. *p < 0.05 (n = 5 per group, 2 individual experiments).
To assess functional capabilities of splenic monocytes after stroke that were independent of the RAGE signaling pathway, we performed stimulation assays with the TLR9-ligand CpG-ODN. This revealed substantially reduced IL-12 and TNF-α secretion by monocytes 3 d after brain ischemia consistent with a reduced responsiveness of splenic monocytes resulting from functional exhaustion. In vivo anti-HMGB1 treatment partially attenuated monocytic hyporesponsiveness to in vitro stimulation as shown by increased IL-12 secretion in the anti-HMGB1-treated group compared with control mice, although it had no effect on TNF-α production (Fig. 5E). Next, we analyzed the frequency of immature monocytes, delineated by expression of CD11b+CD33+HLA−, in stroke patients with extensive brain lesions (>100 ml lesion volume) and matched control patients without neurological disease (Fig. 5F; Table 2). We detected a significant expansion of immature monocytes also after human stroke (Fig. 5G). We further analyzed the functional properties of the expanded CD11b+Ly-6C+ monocyte population to stimulate or inhibit T cells. Splenic CD11b+ monocytes isolated 3 d after stroke used as APCs in coculture with CD3+ T cells had a suppressive effect on T-cell proliferation (Fig. 5H,I). Additionally, arginase-1 (Arg1) expression, a marker of myeloid-derived suppressor cells (Rodríguez and Ochoa, 2008; Gabrilovich and Nagaraj, 2009), was massively increased in poststroke splenic monocytes, which was prevented by in vivo pretreatment with anti-HMGB1 (Fig. 5J).
Poststroke lymphocytopenia in the subacute phase is partially mediated by HMGB1 signaling
We investigated the role of the HMGB1-RAGE pathway on the previously documented decrease in immune cell cellularity in the subacute phase after stroke (Prass et al., 2003; Offner et al., 2006b; Liesz et al., 2009) (Fig. 6A–D). RAGE deficiency, but not anti-HMGB1 treatment, reduced splenic lymphocyte apoptosis after brain ischemia (Fig. 6A,B). Similarly, the stroke-induced reduction of circulating blood T cells was prevented in RAGE-deficient mice (Fig. 6C). The postischemic effect on B-cell and granulocyte counts, however, was not influenced by either anti-HMGB1 treatment or by RAGE- or MyD88 deficiency, respectively (data not shown). Furthermore, we observed a relative increase of activated T cells (activation markers CD25 and CD69) in RAGE−/− mice compared with WT mice after stroke (Fig. 6D). Because of the incomplete antagonization of the immunosuppressive phenotype by RAGE deficiency, we further investigated the impact of TLR-mediated pathways in stroke-induced immune disturbances using MyD88−/− mice. We detected a similar pattern for T-cell counts in spleen and blood as for RAGE−/− mice after stroke with partial abrogation of lymphopenia in MyD88−/− mice without further augmentation by anti-HMGB1 treatment (Fig. 6E). In summary, these results indicate that the immunosuppressive and lymphotoxic effects of extensive brain ischemia are mediated at least in part by HMGB1 via the RAGE and the TLR-MyD88 pathways.
Figure 6.

HMGB1-RAGE signaling induces profound compromise of the peripheral adaptive immune system in the subacute phase after stroke. A, Representative images, mean weight, and cellularity (±SD) of spleens 3 d after sham operation or F90 min-MCAO in WT mice and RAGE−/− mice receiving control or anti-HMGB1 treatment (n = 5–7 per group). MCAO induces a profound reduction in spleen weight and cell count, which is significantly reversed in RAGE−/− mice. B, Flow cytometric analysis of absolute cell counts of Thelper (CD3+CD4+), Tcytotoxic (CD3+CD8+) cells per spleen and (C) per microliter of whole blood in sham-operated animals and 3 d after F90 min-MCAO in WT and RAGE−/− mice receiving additional control IgG antibodies or anti-HMGB1 treatment. *p < 0.05, between control animals of the same mouse strain and the indicated group. Bars represent p < 0.05 between marked groups within the MCAO+ groups. D, T-cell activation was analyzed (CD69+ right y-axis and CD25+ left y-axis, respectively) in spleens of WT and RAGE−/− mice 3 d after MCAO (n = 6 per group). E, The downstream signaling pathway of TLRs on CD3+ T cells was analyzed in MyD88−/− mice. T-cell counts were measured per total spleen (left) and microliters of whole blood (right) in sham-operated animals and 3 d after F90 min-MCAO in WT and MyD88−/− mice treated with control or anti-HMGB1 antibodies. *p < 0.05, between sham-operated animals of the same mouse strain and the indicated MCAO group. Bars represent p < 0.05 between marked groups within the MCAO groups.
HMGB1-RAGE signaling is a distinct alternative signaling pathway after acute brain injury
To control for potential confounding factors known for the used stroke model of large brain lesion, we performed additional analyses to investigate alternative explanations for the observed effects independent of the HMGB1-RAGE pathway. Previous reports have demonstrated bacterial infections in this model, which could have interfered with the immunological and behavioral readout parameters investigated in this study. However, we did not detect bacteria in lung homogenates or blood of any mice (Fig. 7A). Furthermore, catecholamines released by activation of the sympathetic autonomous nervous system have been reported as mediators of poststroke immunosuppression (Prass et al., 2003; Meisel et al., 2005). Therefore, we measured serum concentrations of glucocorticoid and catecholamine stress mediators in WT control mice, anti-HMGB1 treated, and RAGE-deficient mice after stroke without detecting significant differences among groups (Fig. 7B). Next, we aimed to analyze the functional role of catecholamine signaling on critical parameters affected by the HMGB1-RAGE pathway in the previous experiments, such as cytokine release, spleen cellularity, and MDSC expansion. Therefore, we treated mice either with the β2-adrenoreceptor blocker (b2-INH) ICI 118,551 (Mracsko et al., 2014), sRAGE. or the combination of both. We observed that the inhibition of the RAGE pathway by sRAGE-treatment, but not blockade of the adrenergic b2 receptor by b2-INH, abrogated the increased systemic IL-6 secretion after brain ischemia (Fig. 7C). Both sRAGE treatment and b2-INH injection prevented the expansion of the CD11b+Ly-6C+ monocyte fraction (compare withFig. 5B). However, b2-INH injection also caused a significant reduction in this cell population compared with control-treated mice (Fig. 7D). On the other hand, the marked reduction of spleen cellularity (Fig. 7E) was not affected by b2-INH but significantly abrogated by sRAGE treatment. Interestingly, the combination of the RAGE as well as the catecholamine signaling had an additive effect on this parameter. These results indicate that the HMGB1-RAGE and the stress-mediator pathways might be largely independent but differentially involved in specific signaling cascades, such as acute cytokine release and immunosuppressive pathways.
Figure 7.

The HMGB1-RAGE pathway is distinct from infectious or catecholamine-mediated effects. A, Microbiological analysis of blood samples and lung homogenates 3 d after 90 min Filament-MCAO for presence of bacteria. No significant growth of colony forming units (CFU) was detected after 24 h of incubation time on blood agar plates in any sample (n = 21, 2 individual experiments). B, Serum concentrations of cortisol and the catecholamines metanephrine and normetanephrine were measured 24 h after stroke induction (n = 7 per group, 2 individual experiments). Mice were treated with the β2-adrenoreceptor inhibitor ICI 118,551 (b2-INH) and/or sRAGE as indicated in Materials and Methods. C, sRAGE treatment, but not b2-INH, significantly reduced plasma IL-6 concentrations 24 h after F90 min-MCAO. D, The expansion of CD11b+Ly-6C MDSCs at 24 h after F90 min-MCAO was significantly reduced by sRAGE and, to a lesser extent, by b2-INH and without a measurable additive effect. E, The reduction in spleen cellularity induced by MCAO was abrogated by b2-INH as well sRAGE treatment with an additive effect. C–E, Data were obtained in three individual experiments; n = 5 per group.
Discussion
This study presents data supporting a novel concept of brain-immune interaction after acute brain lesions in which soluble danger signals from the brain mediate a complex immunomodulatory reaction in the peripheral immune system. Previous studies have focused on the role of HMGB1 and other DAMPs on immediate tissue injury within a defined organ, such as the heart and the brain (Andrassy et al., 2008; Muhammad et al., 2008; Shichita et al., 2012). This is the first study uncovering suprasystemic cross signaling via RAGE largely by cytokine-inducing isoforms of the soluble mediator HMGB1 released from the necrotic brain. Our findings provide new insights into the peripheral immune alterations in response to brain injury and propose brain-released alarmins as important mediators of the systemic immune response leading to complex behavioral and metabolic sequelae.
Our results are of clinical relevance for patients presenting with severe acute brain injury because their prognosis is strongly influenced by secondary immune-mediated complications. In the early phase after clinical stroke and traumatic brain injury features of sickness behavior, including sleep disorders, fatigue, and depression, are common (Dantzer, 2006; Crosby et al., 2012), which affect secondary morbidity and physical recovery (Willey et al., 2010; Lerdal and Gay, 2013). The concept of proinflammatory cytokines as the cause of clinical sickness behavior and peripheral inflammation is well established, and IL-1β, IL-6, and TNF-α are seen as the main mediators (Dantzer et al., 2008). These cytokines are also strongly upregulated in the periphery during the acute phase after brain ischemia as shown in the present and previous reports (Offner et al., 2006a; Chamorro et al., 2012), but the underlying signaling and the resulting pathophysiological implications were unknown. In this study, we identified the HMGB1-RAGE signaling pathway as the critical route inducing this overshooting immune reaction after brain injury. Inhibiting this signaling pathway improved the systemic metabolic and behavioral consequences of stroke and reduced mortality but did not affect the size of the brain lesions. Similarly, direct neutralization of the overshooting cytokine-release in the acute phase after MCAO improved similar behavioral parameters, such as circadian rhythm and mobility as for inhibition of the HMGB1-RAGE pathway, supporting the concept of HMGB1-RAGE mediated cytokine-induced behavioral alterations. Because cytokine-induced sickness behavior is still not well established as a distinct syndrome in the clinical setting and also requires more comprehensive characterization in experimental models, future studies will have to further validate this concept. Additionally, we cannot exclude immediate modulatory effects of HMGB1-RAGE signaling on brain structures that are not primarily affected by the brain ischemia (e.g., hippocampus). Also, stress mediators, which are released after acute brain injury, might have a currently unknown direct impact on poststroke behavior mimicking cytokine-induced sickness behavior (see below).
Immunosuppression predisposes patients to bacterial infections, which contribute substantially to poststroke morbidity (Katzan et al., 2003). Based mainly on an experimental landmark study using the β blocker propranolol (Prass et al., 2003), catecholamines have been suggested as the key mediators of poststroke immune suppression and infectious complications (Dirnagl et al., 2007; Iadecola and Anrather, 2011). Furthermore, a previous study reported the modulation of hepatic iNKT cells by noradrenergic signals after experimental stroke (Wong et al., 2011). Indeed, most immune cells express catecholaminergic receptors and may undergo cell death after in vitro stimulation with metanephrines, but blockade of β adrenergic receptor in vivo and in vitro fails to prevent lymphocytopenia, the hallmark of immunodepression (Mracsko et al., 2014). Although increased catecholamine blood levels have been reported after experimental and clinical stroke (Chamorro et al., 2007; Urra et al., 2009b), their correlation with lymphopenia, humoral immunosuppression or bacterial infections was not confirmed in a recent study in stroke patients (Liesz et al., 2013a). Moreover, the catecholaminergic concept of poststroke immune depression suggests the involvement of structures of the autonomic nervous system (e.g., the insula) and a laterality of the observed effect (right vs left hemisphere). However, the evidence from clinical and experimental studies rather points to lesion size instead of lesion location as the primary determinator of immune depression (Gendron et al., 2002; Hug et al., 2009; Liesz et al., 2013a). Therefore, the link between acute ischemic brain injury and the impact on the peripheral immune system remained to be identified. Genetic or pharmacological blockage of pattern recognition receptor signaling via HMGB1 and RAGE in this study abrogated cellular immunosuppression and also restored the activation of lymphocytes in the subacute phase after stroke. We have performed experiments comparing the differential effects of inhibiting the RAGE signaling cascade and beta2-adrenergic signaling on key outcome parameters of the acute immunostimulatory phase (cytokine release) and the subsequent immunosuppressive state in the subacute phase (spleen cellularity and MDSC expansion). Our findings suggest that alarmin-mediated pathways and the poststroke stress-related catecholaminergic effects are largely independent. While brain-released alarmins are released early after brain injury and might cause peripheral immune activation and subsequent immune exhaustion, catecholamines might represent an additional signaling pathway in the subacute phase compromising poststroke immune function (Fig. 8).
Figure 8.
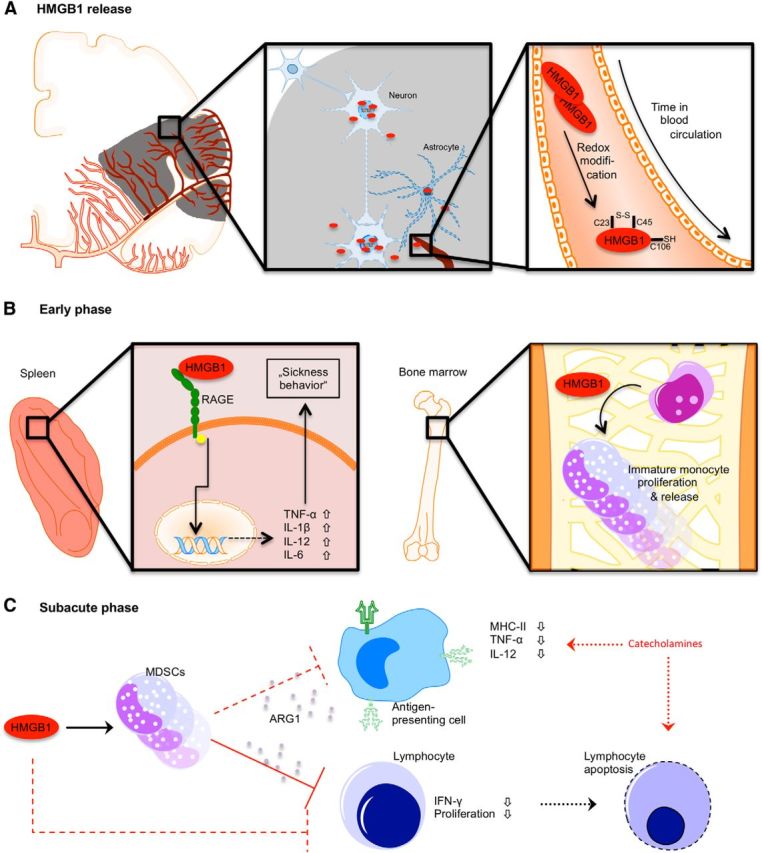
Schematic overview of the proposed mechanisms of HMGB1 release and its biological activity after acute stroke. A, After acute vessel occlusion, cells of the hypoperfused territory become necrotic and release HMGB1 from its nuclear localization. Between 2 and 24 h after ischemia onset, the (C23-C45) disulphide “cytokine HMGB1” becomes the predominant HMGB1 isoform. B, Differential effects of HMGB1 in the spleen (left) and bone marrow (right) in the acute phase (<24 h) after stroke. Splenic monocytes are activated via a RAGE-dependent pathway, producing increased amounts of proinflammatory cytokines. In the bone marrow, HMGB1 induces proliferation of immature monocytes and their release to the circulation. C, The expanded population of (bone marrow-derived) immature monocytes has features of myeloid-derived suppressor cells, which inhibit lymphocyte activation via arginase (ARG1) secretion and might induce lymphocyte apoptosis. Splenic and circulating APCs have an “exhausted” phenotype characterized by reduced MHC-II expression and decreased cytokine production upon stimulation. Inadequate costimualtory signaling to lymphocyte induces T-cell dysfunction and might lead to apoptosis. Additionally, increased catecholamine levels in the subacute phase after stroke result in impaired monocyte function and lymphocyte reduction probably independent of HMGB1-RAGE signaling.
The concept of “stroke-induced immunodepression” (Meisel et al., 2005; Dirnagl et al., 2007) has been perceived as an independent and isolated immunological phenomenon after acute brain injury. However, our results suggest that this immunosuppressive state in the subacute phase after stroke has to be integrated as only one feature of the complex systemic immunomodulation by disturbed brain-immune communication. We detected a marked release of HMGB1 from the ischemic brain in the hyperacute phase after the lesion. Mass spectrometric analysis revealed a “maturation” of the post-translationally unmodified, fully reduced isoform of HMGB1 toward the cytokine-inducing isoform within the acute phase (Fig. 8A). For the cytokine-stimulating function of HMGB1, its residues at C23 and C45 must form a disulfide bond, whereas the unpaired C106 within BoxB must be in the reduced thiol state (Yang et al., 2010). This isoform is a very strong stimulus for various types of immune cells comparable with the stimulatory function of bacterial antigens, such as LPS (Lotze and Tracey, 2005). In this study, we have identified the induction of an “exhausted phenotype” of mature splenic monocytes after the initial (over)activation of the peripheral immune cells by brain-released HMGB1 in the acute phase (Fig. 8B,C). The induction of reduced responsiveness of monocytes/macrophages to endotoxins or bacterial stimuli after a strong inflammatory reaction has been described in immunological conditions, such as sepsis or sterile inflammatory response (Randow et al., 1995; Haupt et al., 1998; Cavaillon et al., 2005). This hyporesponsive state of innate immune cells encompasses the reduced expression of MHC and costimulatory molecules as well as the impaired secretion of proinflammatory cytokines (TNF-α and IL-12). These features have also been reported after brain ischemia (Hug et al., 2009; Urra et al., 2009b; Vogelgesang et al., 2013). Inhibition of the initially overshooting immune activation in the hyperacute phase by neutralizing HMGB1 interaction with its receptors ameliorated also the secondary monocyte exhaustion, indicating a functional link between the distinct and opposing immunological phenotypes observed in the acute and subacute phase after brain lesion.
In addition to the exhaustion of mature monocytes, we describe here for the first time the HMGB1-induced expansion of a monocyte subpopulation after stroke. This subpopulation has features of MDSCs and contributes to the immunosuppressive state in the subacute phase. However, the source of this expanding population could not be determined with certainty. Our findings suggest that it might arise from bone marrow egress, result from peripheral expansion, or represent a mere phenotypical change in the monocyte population. The phenotype of MDSCs was previously identified in several disease models as CD11b+Gr-1+ cells, although distinct functional subsets have meanwhile been characterized in different immunological milieus and disease models (Gabrilovich and Nagaraj, 2009). Although a relative expansion of monocytes after stroke was noted also in previous clinical and experimental studies (Vogelgesang et al., 2008; Liesz et al., 2009, 2013a), the phenotypical and functional characteristics of this expanding population had not been analyzed so far. We detected a strong suppressive function of the expanding CD11+Ly-6C+ population on lymphocytes. Additionally, the expression of Arg1 was strongly increased in the splenic monocyte population. Previous reports suggested a suppressive and even toxic function of MDSCs on lymphocytes by upregulation of Arg1, which induces apoptosis in activated lymphocytes by limiting the essential l-arginine concentration needed for lymphocyte viability (Bronte et al., 2003; Rodríguez and Ochoa, 2008). This novel finding of MDSC expansion and increased Arg1 expression might be one mechanism explaining the induction of lymphocytopenia after stroke, a key feature of poststroke immunosuppression syndrome (Meisel et al., 2005). Indeed, lymphocytopenia has been consistently described in this study and previously in patients and experimental models of extensive brain lesions (Vogelgesang et al., 2008; Klehmet et al., 2009; Urra et al., 2009a) and was found to be an independent predictor of poststroke infections in patients (Liesz et al., 2013a). The above-mentioned characteristics of lymphocyte-suppressing MDSCs after stroke and the necessity of APCs for the stimulation of T cells by HMGB1 demonstrated herein, suggest an interaction of monocytes/APCs with lymphocytes downstream of the HMGB1-mediated pathways. However, the exact molecular pathways underlying this potential monocyte/APC–lymphocyte interaction after brain ischemia require further investigation. Furthermore, also other immunosuppressive cell populations induced by the acute brain injury might be involved in the observed peripheral immune alterations.
In conclusion, our data strongly suggest that HMGB1 release from the ischemic brain and signaling through the pattern recognition receptor RAGE is a key mechanism of the intricate peripheral immune response after brain injury that has profound consequences on clinical outcome. Interfering with this pathway may provide a promising therapeutic target for prevention of immune-mediated complications of acute brain damage.
Footnotes
This work was supported by the German Research Foundation DFG VE196/3-1 to R.V. and the Else-Kröner-Fresenius Foundation 2012/A118 to A.L., P.P.N. received funding by the German research foundation (Deutsche Forschungsgemeinschaft) for the collaborative research clusters (SFB) 1118 and 938. The authors thank the participating patients for their contribution to this study and Simone Karcher, Sabrina Heide, and Rene Karayilan for their technical support.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/S1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- An C, Shi Y, Li P, Hu X, Gan Y, Stetler RA, Leak RK, Gao Y, Sun BL, Zheng P, Chen J. Molecular dialogs between the ischemic brain and the peripheral immune system: dualistic roles in injury and repair. Prog Neurobiol. 2014;115:6–24. doi: 10.1016/j.pneurobio.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrassy M, Volz HC, Igwe JC, Funke B, Eichberger SN, Kaya Z, Buss S, Autschbach F, Pleger ST, Lukic IK, Bea F, Hardt SE, Humpert PM, Bianchi ME, Mairbäurl H, Nawroth PP, Remppis A, Katus HA, Bierhaus A. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008;117:3216–3226. doi: 10.1161/CIRCULATIONAHA.108.769331. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.STR.17.3.472. [DOI] [PubMed] [Google Scholar]
- Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009;86:573–576. doi: 10.1189/jlb.1008585. [DOI] [PubMed] [Google Scholar]
- Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 2003;24:302–306. doi: 10.1016/S1471-4906(03)00132-7. [DOI] [PubMed] [Google Scholar]
- Cavaillon JM, Adrie C, Fitting C, Adib-Conquy M. Reprogramming of circulatory cells in sepsis and SIRS. J Endotoxin Res. 2005;11:311–320. doi: 10.1177/09680519050110050901. [DOI] [PubMed] [Google Scholar]
- Chamorro A, Amaro S, Vargas M, Obach V, Cervera A, Gómez-Choco M, Torres F, Planas AM. Catecholamines, infection, and death in acute ischemic stroke. J Neurol Sci. 2007;252:29–35. doi: 10.1016/j.jns.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Chamorro Á, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8:401–410. doi: 10.1038/nrneurol.2012.98. [DOI] [PubMed] [Google Scholar]
- Crosby GA, Munshi S, Karat AS, Worthington E, Lincoln NB. Fatigue after stroke: frequency and effect on daily life. Disabil Rehabil. 2012;34:633–637. doi: 10.3109/09638288.2011.613517. [DOI] [PubMed] [Google Scholar]
- Dantzer R. Cytokine, sickness behavior, and depression. Neurol Clin. 2006;24:441–460. doi: 10.1016/j.ncl.2006.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T, Prass K, Meisel A. Stroke-induced immunodepression: experimental evidence and clinical relevance. Stroke. 2007;38:770–773. doi: 10.1161/01.STR.0000251441.89665.bc. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron A, Teitelbaum J, Cossette C, Nuara S, Dumont M, Geadah D, du Souich P, Kouassi E. Temporal effects of left versus right middle cerebral artery occlusion on spleen lymphocyte subsets and mitogenic response in Wistar rats. Brain Res. 2002;955:85–97. doi: 10.1016/S0006-8993(02)03368-1. [DOI] [PubMed] [Google Scholar]
- Haupt W, Riese J, Mehler C, Weber K, Zowe M, Hohenberger W. Monocyte function before and after surgical trauma. Dig Surg. 1998;15:102–104. doi: 10.1159/000018601. [DOI] [PubMed] [Google Scholar]
- Hug A, Dalpke A, Wieczorek N, Giese T, Lorenz A, Auffarth G, Liesz A, Veltkamp R. Infarct volume is a major determiner of stroke immune cell function and susceptibility to infection. Stroke. 2009;40:3226–3232. doi: 10.1161/STROKEAHA.109.557967. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhuang H, Garrote E, Mutch J, Yu X, Khilnani V, Poggio T, Steele AD, Serre T. Automated home-cage behavioural phenotyping of mice. Nat Commun. 2010;1:68. doi: 10.1038/ncomms1064. [DOI] [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ. A rotarod suitable for quantitative measurements of motor incoordination in naive mice. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol. 1968;259:211. doi: 10.1007/BF00537801. [DOI] [PubMed] [Google Scholar]
- Katzan IL, Cebul RD, Husak SH, Dawson NV, Baker DW. The effect of pneumonia on mortality among patients hospitalized for acute stroke. Neurology. 2003;60:620–625. doi: 10.1212/01.WNL.0000046586.38284.60. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klehmet J, Harms H, Richter M, Prass K, Volk HD, Dirnagl U, Meisel A, Meisel C. Stroke-induced immunodepression and stroke infections: lessons from the preventive antibacterial therapy in stroke trial. Neuroscience. 2009;158:1184–1193. doi: 10.1016/j.neuroscience.2008.07.044. [DOI] [PubMed] [Google Scholar]
- Lerdal A, Gay CL. Fatigue in the acute phase after first stroke predicts poorer physical health 18 months later. Neurology. 2013;81:1581–1587. doi: 10.1212/WNL.0b013e3182a9f471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L, Hug A, Zorn M, Dalpke A, Nawroth P, Veltkamp R. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke. 2009;40:2849–2858. doi: 10.1161/STROKEAHA.109.549618. [DOI] [PubMed] [Google Scholar]
- Liesz A, Rüger H, Purrucker J, Zorn M, Dalpke A, Möhlenbruch M, Englert S, Nawroth PP, Veltkamp R. Stress mediators and immune dysfunction in patients with acute cerebrovascular diseases. PLoS One. 2013a;8:e74839. doi: 10.1371/journal.pone.0074839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Na SY, Hämmerling GJ, Garbi N, Karcher S, Mracsko E, Backs J, Rivest S, Veltkamp R. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J Neurosci. 2013b;33:17350–17362. doi: 10.1523/JNEUROSCI.4901-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Gröne HJ, Kurschus FC, Schmidt AM, Yan SD, Martin E, Schleicher E, Stern DM, Hämmerling GG, Nawroth PP, Arnold B. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004;113:1641–1650. doi: 10.1172/JCI18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- Mracsko E, Liesz A, Karcher S, Zorn M, Bari F, Veltkamp R. Differential effects of sympathetic nervous system and hypothalamic-pituitary-adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav Immun. 2014;41:200–209. doi: 10.1016/j.bbi.2014.05.015. [DOI] [PubMed] [Google Scholar]
- Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G, Nawroth PP, Bierhaus A, Schwaninger M. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008;28:12023–12031. doi: 10.1523/JNEUROSCI.2435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006a;26:654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, Vandenbark AA, Hurn PD. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006b;176:6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- Prass K, Meisel C, Höflich C, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U, Volk HD, Meisel A. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Wang H, Yuan R, Li H, Ochani M, Ochani K, Rosas-Ballina M, Czura CJ, Huston JM, Miller E, Lin X, Sherry B, Kumar A, Larosa G, Newman W, Tracey KJ, Yang H. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006;203:1637–1642. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randow F, Syrbe U, Meisel C, Krausch D, Zuckermann H, Platzer C, Volk HD. Mechanism of endotoxin desensitization: involvement of interleukin 10 and transforming growth factor beta. J Exp Med. 1995;181:1887–1892. doi: 10.1084/jem.181.5.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–191. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, Sekiya T, Ooboshi H, Kitazono T, Yanagawa T, Ishii T, Takahashi H, Mori S, Nishibori M, Kuroda K, Akira S, Miyake K, Yoshimura A. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012;18:911–917. doi: 10.1038/nm.2749. [DOI] [PubMed] [Google Scholar]
- Spulber S, Edoff K, Hong L, Morisawa S, Shirahata S, Ceccatelli S. Molecular hydrogen reduces LPS-induced neuroinflammation and promotes recovery from sickness behaviour in mice. PLoS One. 2012;7:e42078. doi: 10.1371/journal.pone.0042078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Urra X, Cervera A, Villamor N, Planas AM, Chamorro A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience. 2009a;158:1174–1183. doi: 10.1016/j.neuroscience.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Urra X, Cervera A, Obach V, Climent N, Planas AM, Chamorro A. Monocytes are major players in the prognosis and risk of infection after acute stroke. Stroke. 2009b;40:1262–1268. doi: 10.1161/STROKEAHA.108.532085. [DOI] [PubMed] [Google Scholar]
- Vogelgesang A, Grunwald U, Langner S, Jack R, Bröker BM, Kessler C, Dressel A. Analysis of lymphocyte subsets in patients with stroke and their influence on infection after stroke. Stroke. 2008;39:237–241. doi: 10.1161/STROKEAHA.107.493635. [DOI] [PubMed] [Google Scholar]
- Vogelgesang A, Becker KJ, Dressel A. Immunological consequences of ischemic stroke. Acta Neurol Scand. 2013;129:1–12. doi: 10.1111/ane.12165. [DOI] [PubMed] [Google Scholar]
- Willey JZ, Disla N, Moon YP, Paik MC, Sacco RL, Boden-Albala B, Elkind MS, Wright CB. Early depressed mood after stroke predicts long-term disability: the Northern Manhattan Stroke Study (NOMASS) Stroke. 2010;41:1896–1900. doi: 10.1161/STROKEAHA.110.583997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CH, Jenne CN, Lee WY, Léger C, Kubes P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science. 2011;334:101–105. doi: 10.1126/science.1210301. [DOI] [PubMed] [Google Scholar]
- Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, Akira S, Bierhaus A, Erlandsson-Harris H, Andersson U, Tracey KJ. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]