

Abstract
Deciphering and storing information coded in different firing patterns are important properties of neuronal networks, as they allow organisms to respond and adapt to external and internal events. Here we report that hippocampal CA1 pyramidal neurons respond to brief bursts of high-frequency stimulation (HFS) and θ burst stimulation (TBS) with long-lasting enhanced responses (long-term potentiation [LTP]), albeit by engaging different signaling pathways. TBS induces LTP through calpain-1-mediated suprachiasmatic nucleus circadian oscillatory protein degradation, ERK activation, and actin polymerization, whereas HFS requires adenosine A2 receptors, PKA, and actin polymerization. TBS- but not HFS-induced LTP is impaired in calpain-1 knock-out mice. However, TBS-induced LTP and learning impairment in knock-out mice are restored by activating the HFS pathway. Thus, different patterns of rhythmic activities trigger potentiation by activating different pathways, and cross talks between these can be used to restore LTP and learning when elements of the pathways are impaired.
Keywords: calpain, ERK, hippocampus, learning, long-term potentiation, PKA
Introduction
Rhythmic activities in the θ (5–10 Hz) and γ (40–120 Hz) frequencies and their interactions are critical for coding and storing information in brain (Lisman and Jensen, 2013). θ preponderance increases during locomotion and attention and is necessary for memory formation (Winson, 1978); hippocampal γ activity facilitates memory encoding (Jutras et al., 2009; Colgin and Moser, 2010). These two types of oscillatory activity frequently co-occur, and θ-γ coupling is now viewed as a correlate of memory formation (Tort et al., 2009). Therefore, it is likely that these types of oscillatory activities trigger long-lasting synaptic modifications underlying learning and memory. Indeed, a brief train or repeated trains of high-frequency stimulation (HFS) in γ frequency (100 Hz, 1 s) results in long-term potentiation (LTP) (Bliss and Collingridge, 1993), and this protocol has been widely used to elicit LTP in numerous neuronal networks. Similarly, θ burst stimulation (TBS) (trains of brief bursts of high-frequency stimulation, generally at 100 Hz, delivered at θ frequency, 5–7 Hz) also produce LTP (Larson and Lynch, 1986), and this protocol has also been frequently used to induce LTP.
Numerous pathways have been shown to be involved in LTP, although a consensus regarding the events critically involved in linking activation of NMDA receptors, the event common to TBS- and HFS-induced LTP, with long-lasting increases in AMPA receptors and changes in dendritic spine structure, the synaptic modifications considered to underlie LTP maintenance (Morris et al., 1986; Matsuzaki et al., 2004) is still lacking. Because activation of NMDA receptors and the resulting calcium influx are critical for LTP induction by both HFS and TBS, the search for intracellular cascades has focused on calcium-dependent enzymes. CaM kinase II has been consistently shown to participate in LTP induced by either HFS or TBS (Lisman, 1994). The role of PKC and some of its isoforms, including PKMζ, in synaptic plasticity and learning and memory remains much more controversial (Sacktor, 2011; Lee et al., 2013; Volk et al., 2013). Two other protein kinases, cAMP-dependent protein kinase A (PKA) and extracellular regulated kinase (ERK), have also been proposed to link NMDA receptor activation to long-lasting changes in synaptic function (Huang and Kandel, 1994; Korte et al., 1995; Impey et al., 1998). The calcium-dependent protease calpain has also been shown to participate in LTP (Lynch and Baudry, 1984), and we recently discovered that the two major calpain isoforms, calpain-1 and calpain-2, play opposite roles in LTP (Wang et al., 2014).
Although there are multiple forms of LTP (Bliss and Collingridge, 2013; Padamsey and Emptage, 2013; Park et al., 2014), the relationship between stimulation parameters and signaling cascades involved in LTP induction and consolidation remains poorly understood. In the present study, we used pharmacological and genetic manipulations to address this issue in field CA1 of hippocampal slices. Our results indicate that different types of oscillatory activities trigger LTP through different signaling pathways and that cross talks between these can restore LTP when elements of the pathways are impaired.
Materials and Methods
Reagents.
Forskolin (FSK), H89, PD98059, AP5, rolipram, and SCH58261 were purchased from Tocris Bioscience. Rhodamine phalloidin was obtained from Invitrogen. Calpain inhibitor III was obtained from EMD Millipore. TrkB-FC chimera or control IgG-FC (R&D Systems) was prepared in Tris-buffered saline containing 0.1% BSA and diluted to working concentrations in aCSF. All other reagents were from Sigma. Water-insoluble compounds were dissolved in DMSO and the final concentration of DMSO was <0.1%.
Animals.
Animal use in all experiments followed National Institutes of Health guidelines, and all protocols were approved by the Institution Animal Care and Use Committee of Western University of Health Sciences. Calpain-1 knock-out (KO) mice on a C57BL/6 background were obtained from a breeding colony established from breeding pairs generously provided by Dr. Chishti (Tufts University). C57BL/6 mice were purchased from The Jackson Laboratory and were used as the corresponding wild-type (WT).
Acute hippocampal slice preparation.
Hippocampal slice preparation and electrophysiological recording were performed as previously reported (Wang et al., 2014). Adult male mice (3- to 4-month-old) were anesthetized with halothane and decapitated. Brains were quickly removed and transferred to oxygenated, ice-cold cutting medium (in mm) as follows: 124 NaCl, 26 NaHCO3, 10 glucose, 3 KCl, 1.25 KH2PO4, 5 MgSO4, and 3.4 CaCl2. Hippocampal transversal slices (350 μm thick) were prepared using a McIlwain-type tissue chopper and transferred to an interface recording chamber and exposed to a warm, humidified atmosphere of 95% O2/5% CO2 and continuously perfused with oxygenated and preheated (33 ± 0.5°C) aCSF (in mm) as follows: 110 NaCl, 5 KCl, 2.5 CaCl2, 1.5 MgSO4, 1.24 KH2PO4, 10 d-glucose, 27.4 NaHCO3, at a speed of 1.4 ml/min.
Electrophysiology.
After 2 h of incubation at 33.0 ± 0.5°C in recording chamber, a single glass pipette filled with 2 m NaCl was used to record field EPSPs (fEPSPs) elicited by stimulation of the Schaffer collateral pathway with twisted nichrome wires (single bare wire diameter, 50 μm) placed in CA1 stratum radiatum. Responses were recorded through a differential amplifier (DAM 50, World Precision Instruments) with a 10 kHz low-pass and 0.1 Hz high-pass filter. Before each experiment, the input–output relation was examined by varying the intensity of the stimulation. Paired-pulse facilitation was tested at 20–300 ms interval. LTP was induced by three different paradigms: high-frequency stimulation (HFS, 100 Hz, 1 s), TBS (10 bursts of 4 pulses at 100 Hz delivered at 5 Hz), and θ frequency stimulation (5 Hz, 30 s). For LTP and paired-pulse facilitation experiments, stimulation intensity was adjusted to elicit 40% of the maximal EPSP slope, except when otherwise indicated. Responses during baseline and after tetanus stimulation were recorded every 20 s. Data were collected and digitized by Clampex, and the slope of fEPSP was analyzed. LTP level was normalized to the average slope of responses recorded during the 10 min baseline, and responses were recorded for at least 40 min after LTP induction.
Western blotting.
To analyze activity-dependent regulation of different proteins, CA1 mini-slices were obtained by dissecting out the CA1 region before transferring them to the recording chamber, as previously reported (Wang et al., 2014). After 2 h incubation in aCSF, slices were subjected to different stimulations. Ten minutes after stimulation, slices were collected for Western blotting assay. Slices were lysed and protein concentrations were measured using the BCA protein assay kit (Thermo Scientific). Equal amounts of proteins were processed for SDS-PAGE and Western blot. The primary antibodies used were suprachiasmatic nucleus circadian oscillatory protein (SCOP) (1:1000, Millipore), actin (1:10,000, Millipore), phospho-ERK (1:3000, Cell Signaling Technology), ERK (1:3000, Cell Signaling Technology), phospho-AKT S473 (1:2000, Cell Signaling Technology), AKT (1:2000, Cell Signaling Technology), Calpain-1 (1:1000, Cell Signaling Technology), Calpain-2 (1:1000, Millipore), PKA (1:2000, Cell Signaling Technology), and phospho-PKA (Ser/Thr) (1:2000, Cell Signaling Technology).
Immunohistochemistry.
For immunohistochemistry, hippocampal slices were fixed in 4% PFA for 1 h and cryoprotected in 30% sucrose for 1 h at 4°C, and sectioned on a freezing microtome at 20 μm. Sections were blocked in 0.1 m PBS containing 10% goat serum and 0.4% Triton X-100, and then incubated with primary antibodies, including rabbit anti-spectrin breakdown products (1:1000), mouse anti-PSD95 (1:500, MA1-045, Thermo Scientific), or rabbit anti-pERK (1:500, 4370, Cell Signaling Technology) in 0.1 m PBS containing 5% goat serum and 0.4% Triton X-100 overnight at 4°C. Sections were washed 3 times (10 min each) in PBS and incubated in AlexaFluor-594 goat anti-rabbit IgG (A-11037, Invitrogen) and AlexaFluor-488 goat anti-mouse IgG (A-11001, Invitrogen) for 2 h at room temperature. Localization of the stimulation and recording electrodes was evident in the sections, and we were able to reproducibly analyze mean fluorescence intensity (MFI) of various markers (PSD95, spectrin breakdown product [SBP], p-ERK) in a 500 μm2 area between these electrodes. For puncta analysis, a Nikon C1 confocal laser-scanning microscope 100× objective was used.
In situ phalloidin labeling.
Methods for analyzing actin polymerization were slightly modified from those described previously (Kramár et al., 2006). Rhodamine-phalloidin (6 μm) was applied topically from a micropipette every 5 min for 20 min in slices that received low-frequency stimulation in the presence or absence of drugs or 25 min after the delivery of HFS or TBS. Slices were then collected and fixed in 4% PFA for 1 h, cryoprotected in 30% sucrose for 1 h at 4°C, and sectioned on a freezing microtome at 20 μm. Labeling was examined using a Nikon C1 confocal laser-scanning microscope (60×). Identification and measurement of labeled spines were performed on a 500 μm2 sampling area within the zone of physiological recording as described previously (Kramár et al., 2006). Spine numbers in each image were analyzed with the ImageJ software. The threshold was set to count the puncta numbers. Particles with sizes from 20 to 100 pixels were counted in each field.
Behavioral tests: Novel Object Recognition task.
The Novel Object Recognition task was conducted in an open field arena with two different kinds of objects. Both objects were consistent in height and volume but were different in shape and appearance. During habituation, animals were allowed to explore an empty arena. Twenty-four hours after habituation, animals were exposed to the familiar arena with two identical objects added. The next day, mice were allowed to explore the open field in the presence of one familiar object and a novel object to test long-term recognition memory. Time spent exploring each object was measured and a discrimination index percentage was calculated as Tnew/(Tnew + Told) × 100%.
Fear conditioning.
Mice were housed individually with normal 12/12 h daylight cycle. They were handled daily for 5 d before training. On training day, mice were first injected intraperitoneally with rolipram (1 mg/kg body weight). Thirty minutes later, they were placed in the fear-conditioning chamber (H10–11M-TC, Coulbourn Instruments) located in the center of a sound-attenuating cubicle (Coulbourn Instruments). The conditioning chamber was cleaned with 10% ethanol to provide a background odor. A ventilation fan provided a background noise at ∼55 dB. After a 2 min exploration period, one or three tone-footshock pairings separated by 1 min intervals were delivered. The 85 dB 2 kHz tone lasted for 30 s, and the footshocks were 0.75 mA and lasted for 2 s. Footshocks coterminated with the tone. Mice remained in the training chamber for another 30 s before being returned to their home cages. Context test was performed 1 d after training. Mice were placed back into the original conditioning chamber, and their behaviors were recorded for 5 min. On day 3, animals were subjected to cue/tone test. The same conditioning chamber was modified by changing its metal grid floor to a plastic sheet, white metal walls to plastic walls gridded with red tapes, and odor from ethanol to acetic acid. The ventilation fan was turned off to reduce background noise, and the ceiling light was changed from yellow to white. Mice were placed in the altered chamber for 5 min to measure freezing level in the altered context; and after this 5 min period, a tone (85 dB, 2 kHz) was delivered for 1 min to measure freezing to tone. Mice behavior was recorded with the Freezeframe software and analyzed with Freezeview software (Coulbourn Instruments). Motionless bouts lasting >1 s were considered as freezing. The percentage of time animal froze was calculated, and the group means with SEM and accumulative distribution of percentage freeze were analyzed. All experiments were performed blind, as the experimenter had no knowledge of the treatment (saline or drugs).
Statistical analyses.
Data are presented as means ± SEM. For experiments where only two groups were compared, two-tailed t test was used for determining statistical significance. When more than two groups were compared, we used one-way or two-way ANOVA followed by Bonferroni test to determine statistical significance. p values <0.05 were considered statistically significant.
Results
TBS-, but not HFS-induced, LTP is impaired in calpain-1 KO mice
Several studies have shown that calpain is involved in LTP induction (Denny et al., 1990; del Cerro et al., 1990). We first used calpain-1 KO mice to confirm the specific role of calpain-1 in LTP. Calpain-1 was not detectable in hippocampus and cortex from calpain-1 KO mice, whereas levels of calpain-2 were not altered (Fig. 1A). We then analyzed basal synaptic transmission at the Shaffer collateral to CA1 synapses, including input–output responses and paired-pulse facilitation in calpain-1 KO mice. The input–output curve was shifted downward in mutant compared with WT mice (Fig. 1B). On the other hand, paired-pulse facilitation was not affected (Fig. 1C). Next, we induced LTP using different stimulation paradigms. TBS (10 bursts of 4 pulses at 100 Hz delivered at 5 Hz) induced LTP in WT mice (Fig. 1D). However, in slices from calpain-1 KO mice, synaptic responses returned to basal levels 40 min after TBS (EPSP slope values at 40 min: 140 ± 4% of baseline response in WT vs 105 ± 6% in calpain-1 KO mice) (Fig. 1D). In contrast, HFS (100 Hz, 1 s) induced identical LTP in slices from calpain-1 KO and WT mice (Fig. 1E). A third stimulation paradigm, θ frequency stimulation (5 Hz for 30 s), which has previously been shown to induce a slowly developing LTP in hippocampal slices from rats and mice (Winder et al., 1999), induced LTP in slices from WT, but not calpain-1 KO, mice (Fig. 1F). As input–output responses in calpain-1 KO mice were shifted downward, compared with WT mice, we determined whether impaired LTP in calpain-1 KO mice was due to smaller EPSP responses. We first decreased stimulation intensity in WT mice to produce 20% of the maximal response. Under this condition, TBS still induced LTP (Fig. 1G). We then increased stimulation intensity in slices from calpain-1 KO mice to produce 65% of the maximal response. Under this condition, LTP was still impaired (Fig. 1H). These data indicate that the impairment in TBS-LTP in calpain-1 KO mice is not due to the downward shift in input–output curve and to smaller responses.
Figure 1.

TBS-, but not HFS-induced LTP, is impaired in hippocampal slices from calpain-1 KO mice. A, Lack of calpain-1, but normal calpain-2, levels in calpain-1 KO mice. B, Input–output response curves for WT and calpain-1 KO mice. Black line indicates significant difference between the two groups (two-tailed Student's t test). C, Paired-pulse facilitation at various time intervals in calpain-1 KO and WT mice. D, TBS (10 bursts, 4 pulses) does not induce LTP in calpain-1 KO mice. E, HFS (1 s, 100 Hz) induces LTP in calpain-1 KO mice. F, Impaired θ frequency stimulation (30 s, 5 Hz) induced LTP in calpain-1 KO mice. G, Stimulation intensity was decreased in slices from WT mice to elicit 20% of the maximal response. Under this condition, LTP was still induced. H, In calpain-1 KO mice, the current was increased to induce 65% of the maximal response. LTP was still impaired. I, A different TBS (TBS′; 10 bursts, 4 pulses, pulse duration 400 μs) induced LTP in calpain-1 KO mice. J, TBS′-induced LTP in calpain-1 KO mice was NMDA receptor-dependent, as it was blocked by AP5 (10 μm, black bar). K, A nonselective calpain inhibitor (calpain inhibitor III, 10 μm, gray bar) did not affect TBS′-LTP in calpain-1 KO mice. In all cases, data are means ± SEM for 6–10 slices from 3–5 mice in each group. Insets, Representative traces from baseline (gray) and 40 min after TBS (black) or HFS. Calibration: 0.5 mV, 10 ms. L, TBS with 400 μs stimulation duration elicited many spikes between bursts.
We previously reported that the same calpain-1 KO mice as the ones used in this study exhibited normal TBS-induced LTP (Grammer et al., 2005). However, while a similar stimulus pattern (10 bursts of 4 pulses at 100 Hz delivered at 5 Hz) was applied, the pulse duration during TBS was much longer (400 μs) than the one used in the present study (100 μs). When we used the same stimulation protocol, similar LTP was induced in hippocampal slices from WT and calpain-1 KO mice (Fig. 1I). Under these conditions, LTP was completely blocked by the NMDA receptor blocker AP5 (10 μm) (Fig. 1J), but calpain inhibitor III (10 μm), which inhibits TBS-induced LTP using the normal stimulation parameters (Wang et al., 2014), did not impair this form of LTP (Fig. 1K). To better understand the differences between these two types of stimulation, we compared the pattern of responses elicited during TBS with 100 and 400 μs duration. Interestingly, TBS with 400 μs stimulation duration elicited many spikes between the bursts in slices from calpain-1 KO mice, indicating that this pattern of stimulation is more closely related to high-frequency than to TBS (Fig. 1L) (a similar pattern was observed in slices from WT mice). These results indicate that calpain-1 is only involved in LTP induced by specific patterns and parameters of synaptic activation.
TBS, but not HFS, triggers calpain activation
As TBS-, but not HFS-induced, LTP was impaired in calpain-1 KO mice, we determined whether TBS or HFS could trigger calpain activation in pyramidal neurons in field CA1. The SBP generated by calpain has been widely used as a marker for calpain activation (Roberts-Lewis et al., 1994), and an antibody specific for SBP provides a useful tool to localize calpain activation under various experimental conditions (Saido et al., 1993). Hippocampal slices from WT mice were stimulated with TBS, HFS, or low-frequency stimulation (LFS, 0.05 Hz, used as control) and fixed 10 min after stimulation. TBS induced significant calpain activation in dendrites around the stimulation electrode 10 min after stimulation (Fig. 2A,B). In contrast, there was no obvious change in SBP around the stimulation electrode when LFS (Ctrl) or HFS (100 Hz, 1 s) was applied (Fig. 2B). We determined the MFI in regions of interest under various experimental conditions to quantitatively analyze calpain activation. Calpain was significantly activated in stratum radiatum (Fig. 2C) and stratum pyramidale (Fig. 2D) 10 min following TBS, but not HFS, compared with LFS. We performed double-staining with PSD95, a postsynaptic marker, to verify that calpain activation was present in dendritic spines. Ten minutes after TBS, the number of SBP-positive puncta was significantly increased, compared with the value found during baseline stimulation (Fig. 2E,F). In addition, the percentage of SBP- and PSD-95-positive puncta was significantly increased after TBS (control: 1.3 ± 0.4%; TBS: 16.0 ± 1.3%) (Fig. 2G). Neither TBS nor HFS increased SBP levels in hippocampal slices from calpain-1 KO mice (Fig. 2B–D).
Figure 2.

TBS, but not HFS, triggers calpain-mediated spectrin degradation. A, Representative image with SBP staining after TBS in hippocampal slices from WT mice. B, Baseline, TBS-, and HFS-induced spectrin truncation in slices from WT (top row) and calpain-1 KO mice (bottom row). Scale bar, 50 μm. sp, Stratum pyramidale; sr, stratum radiatum. C, D, Quantification of SBP mean fluorescence intensity around the stimulus electrode in stratum radiatum and stratum pyramidale of CA1. E, Colocalization of SBP and the postsynaptic marker, PSD-95. Scale bar, 5 μm. F, Quantification of SBP punctas after TBS. G, Quantification of SBP-positive PSD-95 punctas, calculated as percentage of PSD-95 punctas (for quantification description, see Material and Methods). In all cases, data are means ± SEM for 6 slices from 3 animals in each group. *p < 0.05, compared with Ctrl in WT (two-way ANOVA followed by Bonferroni test). #p < 0.05, compared with TBS in WT (two-way ANOVA followed by Bonferroni test). @p < 0.05, compared with AP5 + TBS in WT (two-way ANOVA followed by Bonferroni test).
TBS-induced LTP has been shown to require activation of NMDA receptors (Larson and Lynch, 1988) as well as of the BDNF receptor TrkB (Minichiello et al., 2002). We analyzed calpain activation following blockade of NMDA or BDNF receptors. Whereas blocking NMDA receptors totally prevented spectrin truncation, TrkB-FC (2 μg/ml, pretreatment for at least 1 h) only partially decreased it (Fig. 2C,D). Thus, in agreement with our previous results (Wang et al., 2013), activation of synaptic NMDA receptors results in calpain-1 activation, and BDNF receptor activation also facilitates calpain stimulation (Zadran et al., 2010).
TBS- and HFS-induced LTP rely on different signaling pathways
The contribution of ERK activation in LTP is still debated, as some studies found that LTP was prevented by MAPK inhibition (Winder et al., 1999; Shalin et al., 2006), whereas others did not (Selcher et al., 2003). Therefore, we examined the contribution of ERK activation in TBS- and HFS-induced LTP. Ten minutes following TBS, p-ERK immunostaining was clearly increased around the stimulation electrode, whereas HFS induced only a slight increase in p-ERK staining (Fig. 3A). Quantitative analysis revealed a significant increase in p-ERK levels in CA1 stratum radiatum only with TBS and not with HFS (Fig. 3B). We recently reported that the ERK-negative regulator SCOP was rapidly degraded following TBS in rat hippocampal slices, an effect associated with increased ERK phosphorylation (Wang et al., 2014). We reproduced these results in mouse hippocampal slices and compared the effects of TBS with those of HFS on SCOP, p-ERK, and p-AKT (Fig. 3C–F). Whereas TBS decreased SCOP and increased p-ERK and p-AKT levels, HFS did not modify any of these proteins. In calpain-1 KO mice, neither TBS nor HFS induced ERK phosphorylation (Fig. 3E,F). Consistent with the biochemical data, ERK inhibition impaired TBS-, but not HFS-induced, LTP (Fig. 3G,H). Together, these data indicate that TBS-, but not HFS-induced, LTP is both calpain-1- and ERK-dependent.
Figure 3.
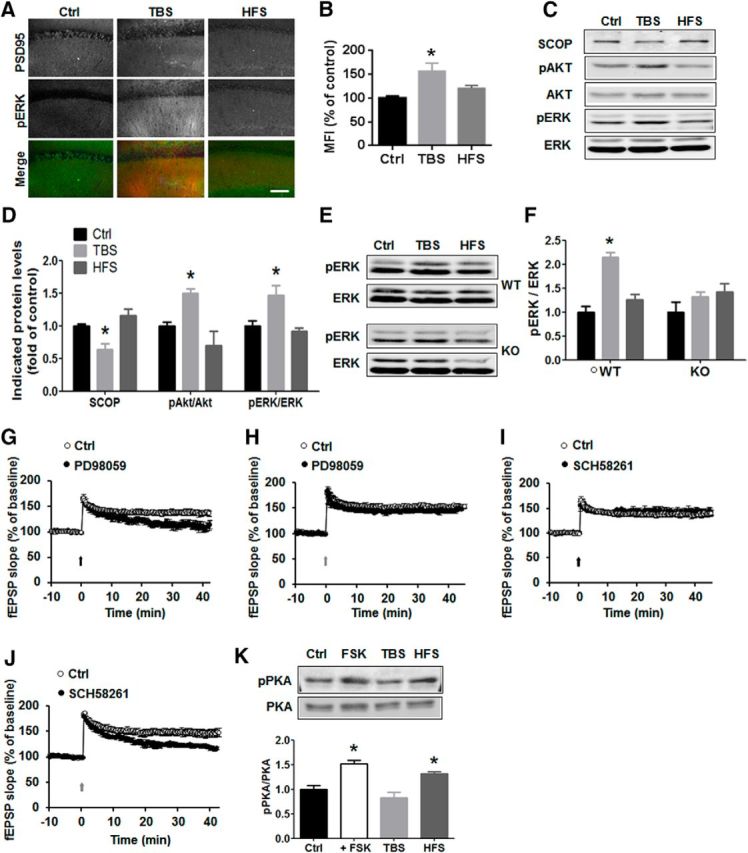
TBS- and HFS activate different signaling pathways. A, Immunohistochemical staining of p-ERK 10 min after TBS and HFS in WT mice. Scale bar, 50 μm. B, Quantification of p-ERK fluorescence intensity in control, TBS, and HFS. n = 4 slices from 3 animals in each group. *p < 0.05, compared with Ctrl (one-way ANOVA followed by Bonferroni test). C, Western blots for SCOP, p-AKT and AKT, and p-ERK and ERK 10 min following TBS or HFS in CA1 mini-slices. D, Quantitative data for SCOP, p-Akt/Akt, and p-ERK/ERK. Data are means ± SEM of 3 experiments. *p < 0.05, compared with Ctrl (one-way ANOVA followed by Bonferroni test). E, TBS, but not HFS, induced ERK activation in WT, but not in calpain-1 KO, mice. F, Quantification data for p-ERK levels. n = 4. *p < 0.05, compared with Ctrl (one-way ANOVA followed by Bonferroni test). G, H, Effect of the MEK inhibitor, PD98059 (50 μm, starting 20 min before TBS, black line) on TBS-LTP (G) and HFS-LTP (H) in WT mice. I, J, Effect of the adenosine A2 receptor blocker, SCH58261 (50 nm) (starting 20 min before TBS, black line) on TBS-LTP (I) and HFS-LTP (J). K, Effect of FSK, TBS, and HFS on PKA activation/phosphorylation. Top, Representative Western blot. Bottom, Quantitative analysis of Western blots. Data are means ± SEM for 3 slices from 3 animals in each group. *p < 0.05, compared with LFS control (one-way ANOVA followed by Bonferroni test). PD98059 or SCH58261 was applied 30 min before stimulation.
We next addressed the question of the molecular mechanism underlying HFS-induced LTP. The adenosine A2A receptor has been shown to be involved in HFS-induced LTP (Costenla et al., 2011). Although PKA has also been repeatedly shown to be involved in LTP, its role appears to be dependent on the hippocampus subfield and the stimulation parameters used to induce LTP (Huang and Kandel, 1994; Huang et al., 1994). When a selective adenosine A2A receptor antagonist (SCH 58261, 50 nm) was applied 30 min before stimulation, HFS-, but not TBS-induced, LTP was impaired (Fig. 3I,J). A2A receptor activation results in adenylate cyclase stimulation, cAMP synthesis, and PKA signaling activation (Higley and Sabatini, 2010), and we verified, using CA1 mini-slices, that HFS, but not TBS, increased PKA phosphorylation (Fig. 3K). As a positive control, FSK treatment (10 μm for 20 min) also increased p-PKA levels (Fig. 3K).
These results confirm that HFS does not trigger calpain activation but stimulates PKA, whereas TBS activates calpain but not PKA. As both HFS and TBS produce NMDA receptor activation (because both HFS- and TBS-induced LTP are blocked by AP5), resulting in calcium influx, the lack of calpain activation following HFS was surprising. We and others have previously reported that calpain-2 could be activated by phosphorylation at serine 50 by ERK, and inactivated by phosphorylation at serine369/threonine370 by PKA (Shiraha et al., 2002; Glading et al., 2004; Zadran et al., 2010). As these residues are conserved in other calpain isoforms (Franco and Huttenlocher, 2005), including calpain-1, we postulated that the lack of calpain activation by HFS could be due to PKA-mediated calpain phosphorylation/inactivation. If this assumption were true, delivering TBS in the presence of a PKA activator should prevent calpain activation, whereas HFS delivered in the presence of a PKA inhibitor should trigger calpain activation: both predictions were verified (Fig. 4A,B). Moreover, PKA inhibition did not affect TBS-induced spectrin truncation 10 min after TBS nor did PKA stimulation affect the lack of spectrin truncation after HFS (Fig. 4A,B).
Figure 4.

Effect of PKA activation/inhibition on TBS- or HFS-induced calpain-mediated spectrin truncation. A, Representative images of SBP staining after TBS or HFS delivered in the absence or presence of the PKA activator, FSK (10 μm), or the PKA inhibitor, H89 (10 μm). SCH58261 was applied 30 min before stimulation. FSK was applied 10 min before stimulation for 20 min. Blocking PKA with H89 enabled HFS-induced spectrin truncation but did not affect TBS-induced spectrin truncation, whereas activating PKA with FSK did not affect the lack of HFS-induced calpain activation but blocked TBS-induced spectrin truncation in WT mice. Scale bar, 80 μm. B, Quantification of SBP staining in CA1 region around the stimulating electrode. Data are means ± SEM of 3–5 experiments. *p < 0.05, compared with TBS (one-way ANOVA followed by Bonferroni test). #p < 0.05, compared with HFS (one-way ANOVA followed by Bonferroni test).
TBS and HFS stimulate actin polymerization
Actin polymerization has been shown to be a necessary event for TBS-induced LTP (Lin et al., 2005) as well as for HFS-induced LTP (Fukazawa et al., 2003), and for long-term memory formation (Huang et al., 2013). Using the phalloidin-labeling assay to visualize fibrous actin (F-actin), we observed an increase in actin polymerization 25 min after TBS or HFS in stratum radiatum of WT mice (Fig. 5A). In calpain-1 KO mice, HFS, but not TBS, stimulated actin polymerization, a result consistent with the requirement for actin polymerization in LTP stabilization (Fig. 5A,B). To verify that actin polymerization was the result of the activation of different signaling pathways following TBS or HFS, we evaluated the effects of inhibitors of these pathways on stimulation-induced actin polymerization. Thus, ERK inhibition decreased TBS-, but not HFS-induced, actin polymerization (Fig. 5B). In contrast, PKA inhibition significantly decreased HFS-, but not TBS-induced, actin polymerization (Fig. 5B).
Figure 5.

Effect of TBS and HFS on actin polymerization. A, Representative images of phalloidin staining in slices from WT and calpain-1 KO mice after 10 min baseline stimulation (Ctrl), TBS, and HFS. Scale bar, 15 μm. B, Quantitative analysis of F-actin staining. Data are means ± SEM of three experiments. **p < 0.01, compared with Ctl (two-way ANOVA followed by Bonferroni test). *p < 0.05, compared with Ctrl (two-way ANOVA followed by Bonferroni test). #p < 0.05, compared with corresponding stimulation conditions in WT mice (two-way ANOVA followed by Bonferroni test). n = 5 for each experimental group.
γ wave stimulation restores TBS-LTP in calpain-1 KO mice by switching to the HFS signaling pathway
As shown above, TBS-, but not HFS-induced, LTP was impaired in calpain-1 KO mice. It was therefore of interest to determine whether it was possible to rescue TBS-induced LTP by activating the pathway engaged by HFS (i.e., the PKA pathway). For this purpose, we first applied the PKA activator FSK before delivering TBS in slices from WT or calpain-1 KO mice. Pretreatment with FSK (10 μm, 20 min) did not alter TBS-induced LTP in WT mice (Fig. 6A) but switched the signaling pathway from the ERK pathway to the PKA pathway. Although TBS-LTP in WT mice was impaired by application of an ERK inhibitor (Fig. 3G), but not by a PKA inhibitor (data not shown), TBS-LTP following FSK treatment became ERK-independent but PKA-dependent (Fig. 6A). In calpain-1 KO mice, TBS could induce LTP following pretreatment with FSK (10 μm, 20 min) (Fig. 6B). In this case as well, the signaling pathway mediating LTP was through PKA, but not ERK, as it was blocked by H89 and not by PD98059 (Fig. 6B). It is important to note that the concentrations of FSK or H89 we used did not affect baseline synaptic transmission in either calpain-1 KO or WT mice (Fig. 6C). In contrast, addition of FSK 5 min after TBS had no effect in calpain-1 KO mice (Fig. 6D) but enhanced LTP in WT mice (Fig. 6E). Consistent with previous results (Barad et al., 1998), activating PKA after HFS did not enhance LTP (Fig. 6F). As PKA blockade could partially rescue spectrin breakdown following HFS (Fig. 4), we tested whether calpain activation due to PKA blockade with HFS could trigger TBS-LTP. However, under this condition, LTP was still lower, although an ERK inhibitor caused a further decrease in LTP under this condition (Fig. 6G).
Figure 6.
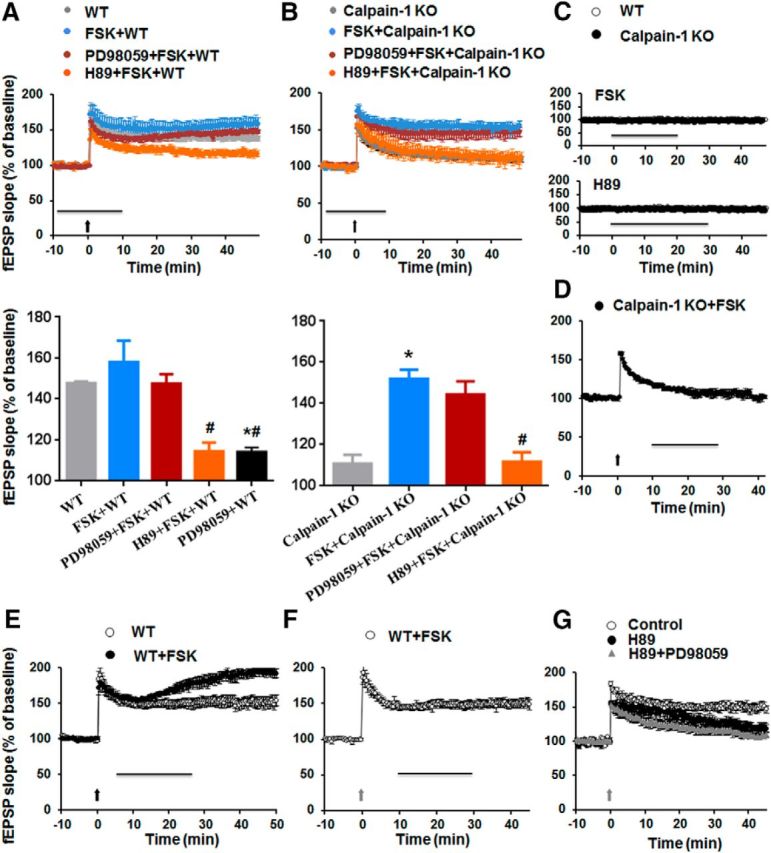
Effect of FSK on TBS-induced LTP in WT and calpain-1 KO mice. A, Effects of FSK (10 μm), PD98059 (10 μm), and H89 (10 μm) on TBS-induced LTP in hippocampal slices from WT mice. Horizontal line indicates FSK application. PD98059 and H89 were applied at least 30 min before TBS (black arrow). LTP magnitude measured 40 min after TBS. Data are percentage of the fEPSP slope normalized to the average value during the 10 min baseline period and expressed as means ± SEM of 5 experiments. B, Effects of FSK (10 μm), PD98059 (10 μm), and H89 (10 μm) on TBS-induced LTP in hippocampal slices from calpain-1 KO mice. Drugs were applied together during the period indicated by the horizontal line. TBS (black arrow) was delivered at t = 0. LTP magnitude 40 min after TBS. Data are percentage of the fEPSP slope normalized to the average value during the 10 min baseline period and expressed as means ± SEM of 5 experiments. *p < 0.05, compared with the groups without drug application (two-way ANOVA followed by Bonferroni test). #p < 0.05, compared with FSK groups (two-way ANOVA followed by Bonferroni test). C, Effect of 10 μm FSK (20 min) and 10 μm H89 (30 min) on baseline synaptic transmission in slices from both WT and calpain-1 KO mice. D, E, Effects of postapplication of FSK (10 μm, 20 min, black line) on TBS-LTP in calpain-1 KO (D) and WT (E) mice. F, Effect of postapplication of FSK (10 μm, 20 min) on HFS-LTP in WT mice. G, Effect of preapplication of H89 (10 μm) on HFS-induced LTP in the absence or presence of PD98059 (10 μm). PD98059 and H89 were applied 30 min before stimulation. Black arrow indicates TBS. Gray arrow indicates HFS.
Alternation between θ and γ rhythms is prominent during successful memory retrieval and electrical stimulation of the fimbria-fornix in amnestic animals patterned accordingly partially rescued memory performance (Shirvalkar et al., 2010). As HFS frequency is in the γ range and a 1 s HFS episode could induce LTP in calpain-1 KO mice, we preapplied this pattern of stimulation, but with different durations, to test the possibility of rescuing TBS-LTP in calpain-1 KO mice (Fig. 7A). We first delivered a 1 s train at 100 Hz (HFS) followed by TBS applied 10 or 60 min later. With the 10 min interval, LTP measured at 50 min was similar in WT and calpain-1 KO mice (Fig. 7B). With the 60 min interval, HFS-induced LTP was similar in both groups at 60 min (Fig. 7C). However, TBS did not induce further enhancement in LTP in calpain-1 KO mice, compared with a large enhancement in WT mice (Fig. 7C). We then decreased the duration of HFS to 0.2 and 0.5 s. These two patterns of HFS did not induce LTP in calpain-1 KO mice (Fig. 7D). However, 0.5 s, but not 0.2 s, HFS restored TBS-induced LTP 10 min later in calpain-1 KO mice (Fig. 7D,E). The restoring effect of the 0.5 s HFS was blocked when slices were incubated with the PKA inhibitor H89 (Fig. 7D,E). These data suggest that TBS-LTP following a brief HFS train in calpain-1 KO mice was now PKA-dependent. Thus, in calpain-1 KO mice, activation of PKA with either FSK or a brief train of HFS (0.5 s) changes the state of synapses such that TBS can induce LTP. We extended this result by using a 1 s train at 50 Hz to test the effects of γ-θ coupling on LTP induction in calpain-1 KO mice. A single train of 1 s at 50 Hz induced only short-term potentiation in calpain-1 KO mice but reestablished LTP induction by TBS delivered 5 min later (Fig. 7F). In WT mice, when TBS was applied first, HFS applied 10 min later elicited a very large LTP, which was even larger than with HFS/TBS (HFS first, TBS 40 min later) (Fig. 7G) (LTP 40 min after last stimulation in TBS/HFS and HFS/TBS groups 222 ± 10% and 184 ± 10%, respectively; p < 0.05).
Figure 7.

Effect of HFS/γ stimulation on TBS-LTP in calpain-1 KO mice. A, Schematic illustration of the paradigm used in the experiment. HFS of different durations was applied before TBS. B, Effects of HFS delivered 10 min before TBS on LTP in WT and calpain-1 KO mice. C, Effects of HFS delivered 1 h before TBS on LTP in WT and calpain-1 KO mice. D, Effects of HFS of various durations delivered 10 min before TBS on LTP in calpain-1 KO mice. H89 (10 μm) blocked the effects of 0.5 s HFS. E, LTP magnitudes measured 40 min after TBS and normalized to the values measured 2 min before TBS in calpain-1 KO mice. *p < 0.05, compared with 0.5 s HFS group (two-way ANOVA followed by Bonferroni test). n = 4–8 for each experimental group. F, Effects of a 1 s 50 Hz HFS on TBS-LTP in calpain-1 KO mice. *p < 0.05, compared with 0.5 s HFS group (two-tailed Student's t test). n = 4–7 for each experimental group. Black arrow indicates TBS. Gray arrow indicates HFS. G, Effect of HFS 10 min after TBS in WT mice.
Rolipram rescues learning and memory impairment in calpain-1 KO mice
Based on LTP results, a brief period of γ activity or preapplication of FSK could restore TBS-LTP in calpain-1 KO mice, presumably by activating the pathway normally activated by HFS (i.e., the PKA pathway). We therefore tested the hypothesis that similar manipulations before learning could rescue learning impairment in calpain-1 KO mice. We first determined that calpain-1 KO mice exhibited deficits in novel object recognition task (Fig. 8A) and, in fear conditioning, when animals were trained with a single footshock pairing (Fig. 8B). We then used systemic injection of the phosphodiesterase 4 inhibitor, rolipram, to activate PKA, as previously reported (Barad et al., 1998), 30 min before training mice in the novel object recognition test or the fear conditioning protocol. Pretraining injection of rolipram (1 mg/kg, i.p.) reversed learning and memory deficits in calpain-1 KO mice in both tasks (Fig. 8A,B). In contrast, post-training injection of the same dose of rolipram did not have any significant effect in the novel recognition task in calpain-1 KO mice (Fig. 8A). On the other hand, post-training, but not pretraining, injection of rolipram in WT mice enhanced learning in the novel object recognition task (Fig. 8A). There was no significant difference between groups in the cue test for fear conditioning (Fig. 8C). We also confirmed our previous findings that, when calpain-1 KO mice were trained with three footshock pairings instead of one, performances in both context and tone protocols were similar to those of WT mice (Fig. 8D) (Grammer et al., 2005).
Figure 8.

Effects of pretraining and post-training injections of rolipram on learning in calpain-1 KO and WT mice. A, Effects of pretraining or post-training injection of rolipram (1 mg/kg, i.p.) on novel object recognition learning in WT and calpain-1 KO mice. *p < 0.05, compared with saline in calpain-1 KO mice (two-way ANOVA followed by Bonferroni test). #p < 0.05, compared with saline after in WT mice (two-way ANOVA followed by Bonferroni test). n = 6–8 animals for each experimental group. B, Effects of pretraining injection of rolipram on context fear conditioning 24 h after one footshock pairing in WT and calpain-1 KO mice. Data are means ± SEM of 6–8 animals. **p < 0.01, compared with calpain-1 KO saline before group (two-way ANOVA followed by Bonferroni test). *p < 0.05, compared with calpain-1 KO saline before group (two-way ANOVA followed by Bonferroni test). C, Effects of pretraining injection of rolipram on tone fear conditioning 24 h after one footshock pairing in WT and calpain-1 KO mice. D, Context and tone fear conditioning are not affected in calpain-1 KO mice 24 h after training with 3 footshock pairings. Data are means ± SEM of 4 animals in each group.
Discussion
Since its discovery in 1966, hippocampal LTP has been the most attractive cellular mechanism underlying certain forms of learning and memory, although the roles of various intracellular pathways engaged by various LTP-inducing patterns of electrical stimulation are still debated. In this study, we focused on two major signaling pathways: calpain and ERK activation, and adenosine A2A receptor stimulation followed by adenylate cyclase activation and PKA stimulation.
TBS and HFS use different signaling pathways to produce actin polymerization and LTP
Our results clearly indicate that TBS-, but not HFS-induced, LTP requires calpain-1 activation. Thus, TBS-induced LTP as well as LTP induced by a long train of stimulation at θ frequency (30 s at 5 Hz) were impaired in calpain-1 KO mice. Whereas the input–output response curve was shifted downward in slices from calpain-1 KO mice, paired-pulse facilitation and burst responses were not modified, suggesting that presynaptic function and NMDA receptor-mediated responses were not affected. Moreover, increasing stimulus intensity did not elicit LTP, indicating that the lack of LTP was not related to understimulation in calpain-1 KO mice. These modifications are very similar to those observed in calpain-4 KO mice (these mice lack both calpain-1 and calpain-2 activity because calpain-4 is the small subunit common to both calpain-1 and calpain-2) (Amini et al., 2013). We also replicated our previous results showing that TBS with a longer stimulus duration during TBS (400 μs stimulus duration instead of 100 μs) induced LTP in calpain-1 KO mice. However, this type of LTP was calpain-independent, although still NMDA receptor-dependent. Moreover, examination of the responses during the bursts under these conditions indicated that the stimulation pattern was more closely related to high frequency than to TBS, therefore accounting for the differences in LTP induction by these two protocols in calpain-1 KO mice.
In our original hypothesis (Lynch and Baudry, 1984), we postulated that calpain-mediated truncation of spectrin was a critical step to modify dendritic spine structure. The present results clearly indicate that calpain-mediated spectrin truncation is associated with TBS-, but not HFS-, LTP. Whereas calpain-1 is likely stimulated by the calcium influx resulting from NMDA receptor activation, calpain-2 could be activated by ERK-mediated phosphorylation resulting from BDNF release after TBS (Briz et al., 2013). When BDNF signaling was blocked by application of the FC fragment of TrkB antibodies, TBS-induced spectrin truncation was significantly decreased, a result supporting this idea.
That HFS, which clearly activates NMDA receptors (as HFS-induced LTP is blocked by AP5), does not produce spectrin breakdown is paradoxical. Our results clearly showed that PKA-mediated phosphorylation and inactivation of calpain are responsible for this paradoxical effect. Indeed, preapplication of a PKA inhibitor resulted in HFS-mediated spectrin breakdown, whereas preapplication of the PKA activator, FSK, completely blocked TBS-induced spectrin breakdown. Importantly, FSK applied after TBS produced an enhancement of LTP in hippocampal slices from WT mice very similar to that produced by calpain-2 inhibition (Wang et al., 2014).
The Ras/MAP kinase pathway has been shown to be involved in both LTP and long-term memory. For example, knocking out the kinase suppressor of ras 1 (KSR1) impaired TBS-, but not HFS-induced, LTP (Shalin et al., 2006). In agreement with previous studies (Winder et al., 1999; Selcher et al., 2003), we found that ERK activation is not necessary for HFS-induced LTP in mice, whereas TBS-induced LTP requires ERK activation. Immunohistochemical staining as well as Western blot analysis showed that TBS, but not HFS, activated ERK in dendrites and dendritic spines in the area surrounding the stimulation electrode. Based on the available data (Kang et al., 1997; Winder et al., 1999; Selcher et al., 2003) and our own results, we conclude that HFS-induced LTP does not involve calpain, TrkB, and ERK activation. On the other hand, PKA was clearly activated following a 1 s train at 100 Hz (Kim et al., 2010), and an A2A receptor antagonist, which decreases PKA activation, reduced HFS-induced LTP, but had no effect on TBS-induced LTP. Moreover, adenosine has been shown to be released following HFS, but not TBS, and to activate adenosine A2A receptors, which facilitate LTP induction (Cunha et al., 1996). Although adenosine receptors are located both presynaptically and postsynaptically in CA1 pyramidal neurons, postsynaptic A2A receptors play critical roles in HFS-induced LTP (Kotaleski and Blackwell, 2010). Although A1 receptors could also play important roles in LTP, we focused our studies on A2A receptors because their activation stimulates the PKA pathway. Previous studies have shown that PKA activation was required for LTP elicited by repeated trains of HFS (Huang and Kandel, 1994). Under our experimental conditions, one episode of HFS was sufficient to induce PKA-dependent LTP. In contrast, Bortolotto and Collingridge (2000) showed that a single HFS was sufficient to induce a long-lasting, PKA-independent form of LTP. The reason for these discrepancies are not clear and could involve differences in animal strains, chamber temperature, and type of stimulating electrode; it should also be noted that PKA activation was clearly detected after one train of stimulation in our and other studies (Kim et al., 2010).
Enlargement of dendritic spines is a critical process for LTP maintenance in CA1 region of hippocampus, and actin polymerization participates in spine enlargement during LTP consolidation (Matsuzaki et al., 2004; Kramár et al., 2006). In agreement with previous studies, we found increased actin polymerization following TBS in the dendritic field in slices from WT but not calpain-1 KO mice. HFS also resulted in actin polymerization in both WT and calpain-1 KO mice. These results indicate that actin polymerization is a common event involved in LTP after TBS or HFS. Numerous pathways are involved in actin polymerization, and it is therefore not surprising that both the ERK pathway and the PKA pathway can trigger it (Huang et al., 2007; Nadella et al., 2009).
Interactions between TBS- and HFS-induced LTP and implications for learning and memory impairment
TBS-induced LTP triggers a refractory period during which a second TBS delivered to the same pathway cannot induce further potentiation for ∼1 h (Kramár et al., 2012; Lynch et al., 2013; but for a different result, see Volianskis and Jensen, 2003). In contrast, there is no such refractory period with HFS-induced LTP (data not shown). Interestingly, HFS delivered 10 min after TBS resulted in the highest level of potentiation. These results also support the idea that TBS and HFS trigger different and nonoverlapping signaling pathways to induce potentiation. In contrast, when TBS was delivered 10 min after HFS, it elicited a very weak potentiation effect. As discussed above, this could be due to the fact that the initial HFS activated PKA, thereby inhibiting calpain-1 and reducing TBS-induced LTP.
Although TBS-induced LTP was impaired in calpain-1 KO mice, HFS-induced LTP was normal. Because TBS could induce LTP following FSK pretreatment in calpain-1 KO mice and switched TBS-LTP to the HFS pathway-mediated LTP in WT mice, these two pathways probably interact at several downstream levels, and defects in some elements of these pathways could be compensated by modulating critical nodes along these pathways. In particular, a brief, repeated, swimming period could rescue TBS-induced LTP impairment caused by Aβ42 through the PKA pathway-mediated “non–TBS-induced LTP” (Li et al., 2007). Similarly, TBS induced-LTP was also impaired in hippocampal slices prepared from HPC-1/Syntaxin 1A KO mice, and this impairment was also rescued by FSK application (Mishima et al., 2012). Thus, in a number of different situations in which TBS-induced LTP is impaired, it is possible to elicit LTP following TBS by activating the PKA pathway, which can under these conditions trigger LTP by the same pathway used for HFS-induced LTP. Interestingly, TBS could induce LTP following a short burst of γ frequency activity (100 Hz, 0.5 s; 50 Hz, 1 s) in calpain-1 KO mice. Thus, θ-γ coupling might be used to trigger different signaling pathways depending on the genetic background or particular environmental conditions (e.g., low estrogen, high BDNF). Our results suggest that a threshold level of PKA activation following application of a low concentration of FSK, or a short duration of HFS (not sufficient to induce LTP) could switch TBS-LTP to HFS-LTP. In contrast, it does not appear that HFS-LTP could be rescued after PKA inhibition through the TBS-dependent mechanism. Although calpain activation was restored, at least partially, following PKA blockade, PKA blockade did decrease HFS-LTP. It is possible that the degree of calpain activation under these conditions is not sufficient to induce enough ERK activation to activate the TBS pathway.
These interactions at the level of LTP extend at the behavioral level, as shown by the rescue of the learning impairment in calpain-1 KO mice provided by rolipram administered before but not after training. In contrast, rolipram had the opposite effects in WT mice, as it had no effect on learning when administered before training, whereas it enhanced learning when administered after learning, as previously reported by Barad et al. (1998) in the fear conditioning protocol. These findings are strikingly similar to the opposite effects of FSK on TBS-induced LTP in calpain-1 KO and WT mice. Interestingly, pretreatment with rolipram was able to enhance LTP elicited by a single HFS but had no effect when delivered after HFS (Barad et al., 1998). Our results would support the idea that TBS-induced LTP is probably used to modify synaptic efficacy and to store some forms of information under certain training conditions. Whether the differences in learning observed with one pairing versus 3 pairings in calpain-1 KO mice reflect the same differences observed between TBS-LTP and HFS-LTP is currently unknown. Nevertheless, there is also evidence that HFS-LTP can mimic and/or occlude certain forms of learning and memory (Pastalkova et al., 2006; Whitlock et al., 2006; Nabavi et al., 2014). As indicated by our results, the existence of cross talks between the two types of protocols (HFS and TBS) suggests that, depending on the conditions, one pathway or the other is the predominant pathway to produce the long-lasting modifications of synaptic efficacy underlying information storage.
In conclusion, in this study, we clearly demonstrated that TBS and HFS, as well as certain stimulation conditions, engage different signaling pathways to induce LTP. TBS-induced LTP requires calpain activation, SCOP degradation, and ERK activation, followed by actin polymerization. In contrast, HFS requires adenosine A2A receptor activation, PKA activation, and actin polymerization. These results account for some of the controversies in the LTP field and underscore the role of oscillatory activity in coding and storing information. Finally, some manipulations can reestablish TBS-LTP and learning in calpain-1 KO mice, thus suggesting new avenues to reverse learning and memory impairments present in a variety of neurological or neuropsychiatric disorders.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant P01NS045260-01 and Grant R01NS057128 to M.B., X.B. is also supported by funds from the Daljit and Elaine Sarkaria Chair. We thank Dr. A. Chishti (Tufts University) for providing us breeding pairs of calpain-1 KO mice to generate the mice used in this study; and Dr. Takaomi Saido for the generous gift of the SBP antibody.
The authors declare no competing financial interests.
References
- Amini M, Ma CL, Farazifard R, Zhu G, Zhang Y, Vanderluit J, Zoltewicz JS, Hage F, Savitt JM, Lagace DC, Slack RS, Beique JC, Baudry M, Greer PA, Bergeron R, Park DS. Conditional disruption of calpain in the CNS alters dendrite morphology, impairs LTP, and promotes neuronal survival following injury. J Neurosci. 2013;33:5773–5784. doi: 10.1523/JNEUROSCI.4247-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad M, Bourtchouladze R, Winder DG, Golan H, Kandel E. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci U S A. 1998;95:15020–15025. doi: 10.1073/pnas.95.25.15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Mol Brain. 2013;6:5. doi: 10.1186/1756-6606-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolotto ZA, Collingridge GL. A role for protein kinase C in a form of metaplasticity that regulates the induction of long-term potentiation at CA1 synapses of the adult rat hippocampus. Eur J Neurosci. 2000;12:4055–4062. doi: 10.1046/j.1460-9568.2000.00291.x. [DOI] [PubMed] [Google Scholar]
- Briz V, Hsu YT, Li Y, Lee E, Bi X, Baudry M. Calpain-2-mediated PTEN degradation contributes to BDNF-induced stimulation of dendritic protein synthesis. J Neurosci. 2013;33:4317–4328. doi: 10.1523/JNEUROSCI.4907-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin LL, Moser EI. Gamma oscillations in the hippocampus. Physiology. 2010;25:319–329. doi: 10.1152/physiol.00021.2010. [DOI] [PubMed] [Google Scholar]
- Costenla AR, Diógenes MJ, Canas PM, Rodrigues RJ, Nogueira C, Maroco J, Agostinho PM, Ribeiro JA, Cunha RA, de Mendonça A. Enhanced role of adenosine A(2A) receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur J Neurosci. 2011;34:12–21. doi: 10.1111/j.1460-9568.2011.07719.x. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Vizi ES, Ribeiro JA, Sebastião AM. Preferential release of ATP and its extracellular catabolism as a source of adenosine upon high- but not low-frequency stimulation of rat hippocampal slices. J Neurochem. 1996;67:2180–2187. doi: 10.1046/j.1471-4159.1996.67052180.x. [DOI] [PubMed] [Google Scholar]
- del Cerro S, Larson J, Oliver MW, Lynch G. Development of hippocampal long-term potentiation is reduced by recently introduced calpain inhibitors. Brain Res. 1990;530:91–95. doi: 10.1016/0006-8993(90)90660-4. [DOI] [PubMed] [Google Scholar]
- Denny JB, Polan-Curtain J, Ghuman A, Wayner MJ, Armstrong DL. Calpain inhibitors block long-term potentiation. Brain Res. 1990;534:317–320. doi: 10.1016/0006-8993(90)90148-5. [DOI] [PubMed] [Google Scholar]
- Franco SJ, Huttenlocher A. Regulating cell migration: calpains make the cut. J Cell Sci. 2005;118:3829–3838. doi: 10.1242/jcs.02562. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/S0896-6273(03)00206-X. [DOI] [PubMed] [Google Scholar]
- Glading A, Bodnar RJ, Reynolds IJ, Shiraha H, Satish L, Potter DA, Blair HC, Wells A. Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol. 2004;24:2499–2512. doi: 10.1128/MCB.24.6.2499-2512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammer M, Kuchay S, Chishti A, Baudry M. Lack of phenotype for LTP and fear conditioning learning in calpain 1 knock-out mice. Neurobiol Learn Mem. 2005;84:222–227. doi: 10.1016/j.nlm.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13:958–966. doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Chotiner JK, Steward O. Actin polymerization and ERK phosphorylation are required for Arc/Arg3.1 mRNA targeting to activated synaptic sites on dendrites. J Neurosci. 2007;27:9054–9067. doi: 10.1523/JNEUROSCI.2410-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Zhu PJ, Zhang S, Zhou H, Stoica L, Galiano M, Krnjeviæ K, Roman G, Costa-Mattioli M. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat Neurosci. 2013;16:441–448. doi: 10.1038/nn.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem. 1994;1:74–82. doi: 10.1101/lm.1.1.74. [DOI] [PubMed] [Google Scholar]
- Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/S0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Jutras MJ, Fries P, Buffalo EA. Gamma-band synchronization in the macaque hippocampus and memory formation. J Neurosci. 2009;29:12521–12531. doi: 10.1523/JNEUROSCI.0640-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/S0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Kim M, Huang T, Abel T, Blackwell KT. Temporal sensitivity of protein kinase A activation in late-phase long term potentiation. PLoS Comput Biol. 2010;6:e1000691. doi: 10.1371/journal.pcbi.1000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotaleski JH, Blackwell KT. Modelling the molecular mechanisms of synaptic plasticity using systems biology approaches. Nat Rev Neurosci. 2010;11:239–251. doi: 10.1038/nrn2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramár EA, Lin B, Rex CS, Gall CM, Lynch G. Integrin-driven actin polymerization consolidates long-term potentiation. Proc Natl Acad Sci U S A. 2006;103:5579–5584. doi: 10.1073/pnas.0601354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramár EA, Babayan AH, Gavin CF, Cox CD, Jafari M, Gall CM, Rumbaugh G, Lynch G. Synaptic evidence for the efficacy of spaced learning. Proc Natl Acad Sci U S A. 2012;109:5121–5126. doi: 10.1073/pnas.1120700109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Lynch G. Induction of synaptic potentiation in hippocampus by patterned stimulation involves two events. Science. 1986;232:985–988. doi: 10.1126/science.3704635. [DOI] [PubMed] [Google Scholar]
- Larson J, Lynch G. Role of N-methyl-d-aspartate receptors in the induction of synaptic potentiation by burst stimulation patterned after the hippocampal θ-rhythm. Brain Res. 1988;441:111–118. doi: 10.1016/0006-8993(88)91388-1. [DOI] [PubMed] [Google Scholar]
- Lee AM, Kanter BR, Wang D, Lim JP, Zou ME, Qiu C, McMahon T, Dadgar J, Fischbach-Weiss SC, Messing RO. Prkcz null mice show normal learning and memory. Nature. 2013;493:416–419. doi: 10.1038/nature11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Feig LA, Hartley DM. A brief, but repeated, swimming protocol is sufficient to overcome amyloid beta-protein inhibition of hippocampal long-term potentiation. Eur J Neurosci. 2007;26:1289–1298. doi: 10.1111/j.1460-9568.2007.05760.x. [DOI] [PubMed] [Google Scholar]
- Lin B, Kramár EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–2069. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J. The CaM kinase II hypothesis for the storage of synaptic memory. Trends Neurosci. 1994;17:406–412. doi: 10.1016/0166-2236(94)90014-0. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Jensen O. The θ-γ neural code. Neuron. 2013;77:1002–1016. doi: 10.1016/j.neuron.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Baudry M. The biochemistry of memory: a new and specific hypothesis. Science. 1984;224:1057–1063. doi: 10.1126/science.6144182. [DOI] [PubMed] [Google Scholar]
- Lynch G, Kramár EA, Babayan AH, Rumbaugh G, Gall CM. Differences between synaptic plasticity thresholds result in new timing rules for maximizing long-term potentiation. Neuropharmacology. 2013;64:27–36. doi: 10.1016/j.neuropharm.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/S0896-6273(02)00942-X. [DOI] [PubMed] [Google Scholar]
- Mishima T, Fujiwara T, Kofuji T, Akagawa K. Impairment of catecholamine systems during induction of long-term potentiation at hippocampal CA1 synapses in HPC-1/syntaxin 1A knock-out mice. J Neurosci. 2012;32:381–389. doi: 10.1523/JNEUROSCI.2911-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R. Engineering a memory with LTD and LTP. Nature. 2014;511:348–352. doi: 10.1038/nature13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadella KS, Saji M, Jacob NK, Pavel E, Ringel MD, Kirschner LS. Regulation of actin function by protein kinase A-mediated phosphorylation of Limk1. EMBO Rep. 2009;10:599–605. doi: 10.1038/embor.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padamsey Z, Emptage N. Two sides to long-term potentiation: a view towards reconciliation. Philos Trans R Soc Lond B Biol Sci. 2013;369:20130154. doi: 10.1098/rstb.2013.0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P, Volianskis A, Sanderson TM, Bortolotto ZA, Jane DE, Zhuo M, Kaang BK, Collingridge GL. NMDA receptor-dependent long-term potentiation comprises a family of temporally overlapping forms of synaptic plasticity that are induced by different patterns of stimulation. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130131. doi: 10.1098/rstb.2013.0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R. Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci. 1994;14:3934–3944. doi: 10.1523/JNEUROSCI.14-06-03934.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC. How does PKMzeta maintain long-term memory? Nat Rev Neurosci. 2011;12:9–15. doi: 10.1038/nrn2949. [DOI] [PubMed] [Google Scholar]
- Saido TC, Yokota M, Nagao S, Yamaura I, Tani E, Tsuchiya T, Suzuki K, Kawashima S. Spatial resolution of fodrin proteolysis in postischemic brain. J Biol Chem. 1993;268:25239–25243. [PubMed] [Google Scholar]
- Selcher JC, Weeber EJ, Christian J, Nekrasova T, Landreth GE, Sweatt JD. A role for ERK MAP kinase in physiologic temporal integration in hippocampal area CA1. Learn Mem. 2003;10:26–39. doi: 10.1101/lm.51103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD. Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron. 2006;50:765–779. doi: 10.1016/j.neuron.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Shiraha H, Glading A, Chou J, Jia Z, Wells A. Activation of m-calpain (calpain II) by epidermal growth factor is limited by protein kinase A phosphorylation of m-calpain. Mol Cell Biol. 2002;22:2716–2727. doi: 10.1128/MCB.22.8.2716-2727.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirvalkar PR, Rapp PR, Shapiro ML. Bidirectional changes to hippocampal θ-γ comodulation predict memory for recent spatial episodes. Proc Natl Acad Sci U S A. 2010;107:7054–7059. doi: 10.1073/pnas.0911184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tort AB, Komorowski RW, Manns JR, Kopell NJ, Eichenbaum H. Theta-γ coupling increases during the learning of item-context associations. Proc Natl Acad Sci U S A. 2009;106:20942–20947. doi: 10.1073/pnas.0911331106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volianskis A, Jensen MS. Transient and sustained types of long-term potentiation in the CA1 area of the rat hippocampus. J Physiol 550. 2003;2:459–492. doi: 10.1113/jphysiol.2003.044214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. PKM-zeta is not required for hippocampal synaptic plasticity, learning and memory. Nature. 2013;493:420–423. doi: 10.1038/nature11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Briz V, Chishti A, Bi X, Baudry M. Distinct roles for mu-calpain and m-calpain in synaptic NMDAR-mediated neuroprotection and extrasynaptic NMDAR-mediated neurodegeneration. J Neurosci. 2013;33:18880–18892. doi: 10.1523/JNEUROSCI.3293-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhu G, Briz V, Hsu YT, Bi X, Baudry M. A molecular brake controls the magnitude of long-term potentiation. Nat Commun. 2014;5:3051. doi: 10.1038/ncomms4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by θ frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/S0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- Winson J. Loss of hippocampal θ rhythm results in spatial memory deficit in the rat. Science. 1978;201:160–163. doi: 10.1126/science.663646. [DOI] [PubMed] [Google Scholar]
- Zadran S, Jourdi H, Rostamiani K, Qin Q, Bi X, Baudry M. Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J Neurosci. 2010;30:1086–1095. doi: 10.1523/JNEUROSCI.5120-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]