

Abstract
Severe autonomic dysfunction, including the loss of control of the cardiovascular, respiratory, and gastrointestinal systems, is a common comorbidity of stroke and other bleeding head injuries. Previous studies suggest that this collapse of autonomic control may be caused by thrombin acting on astrocytic protease-activated receptors (PAR1) in the hindbrain. Using calcium imaging and electrophysiological techniques, we evaluated the mechanisms by which astrocytic PAR1s modulate the activity of presynaptic vagal afferent terminals and postsynaptic neurons in the rat nucleus of the solitary tract (NST). Our calcium-imaging data show that astrocytic and neuronal calcium levels increase after brain slices are treated with the PAR1 agonist SFLLRN-NH2. This increase in activity is blocked by pretreating the slices with the glial metabolic blocker fluorocitrate. In addition, PAR1-activated astrocytes communicate directly with NST neurons by releasing glutamate. Calcium responses to SFLLRN-NH2 in the astrocytes and neurons significantly increase after bath application of the excitatory amino acid transporter blocker dl-threo-β-benzyloxyaspartic acid (TBOA) and significantly decrease after bath application of the NMDA receptor antagonist dl-2-amino-5-phosphonopentanoic acid (dl-AP5). Furthermore, astrocytic glutamate activates neuronal GluN2B-containing NMDA receptors. Voltage-clamp recordings of miniature EPSCs (mEPSCs) from NST neurons show that astrocytes control presynaptic vagal afferent excitability directly under resting and activated conditions. Fluorocitrate significantly decreases mEPSC frequency and SFLLRN-NH2 significantly increases mEPSC frequency. These data show that astrocytes act within a tripartite synapse in the NST, controlling the excitability of both postsynaptic NST neurons and presynaptic vagal afferent terminals.
Keywords: calcium imaging, electrophysiology, gliotransmitter, GluN2B, mEPSC, tripartite synapse
Introduction
Gastrointestinal stasis, nausea, emesis, and anorexia are common comorbidities of patients with ischemic or bleeding traumatic brain injury (Young et al., 1992; Lu et al., 1997). Similar complications also can occur after other traumatic bodily injuries, burns, and transplantation surgeries associated with extensive bleeding (Berkowitz et al., 1995; Sodhi et al., 2002). These patients can require aggressive nutritional or gastrointestinal surgical interventions due to gastric feeding intolerance (Akindipe et al., 2000) and prolonged gastric stasis can require intensive management of the airway due to the significant risk of aspiration (Berkowitz et al., 1995). Elevated intracranial pressure cannot fully explain the visceral malaise that occurs after head injury (Larson et al., 1984; Garrick et al., 1988) and is not a factor in gastrointestinal dysfunction after injuries that occur outside of the CNS. One common factor in these injuries producing autonomic failure is the significant increase in the production of the serine protease thrombin (Gingrich and Traynelis, 2000; Junge et al., 2003; Olson et al., 2004; Nicole et al., 2005; Xi et al., 2006; Lavrentieva et al., 2008; Hamill et al., 2009).
Thrombin catalyzes the proteolysis of fibrinogen to enable clot formation and wound healing (Gingrich and Traynelis, 2000) and also activates protease-activated receptors (PARs) found on platelet, endothelial, intestinal, airway, and brain cells (Coughlin, 2000; Traynelis and Trejo, 2007). Four PARs (PAR1–PAR4) have been identified in the mammalian genome and differ in cellular expression and activating protease (Coughlin, 2000; Traynelis and Trejo, 2007). PAR1 is activated by the cleavage of the extracellular N-terminal domain by proteases such as thrombin and plasmin. This cleavage releases a tethered ligand (SFLLRN) that activates the receptor and initiates a signaling cascade through Gαq/11, Gαi/o, or Gα12/13 G-proteins (Coughlin, 2000; Macfarlane et al., 2001; Traynelis and Trejo, 2007). In the medullary hindbrain nucleus of the solitary tract (NST), PAR1 is expressed exclusively in astrocytes (Hermann et al., 2009), but may be expressed in both neurons and astrocytes in the hippocampus, dentate gyrus, basal ganglia, cortex, and amygdala (Weinstein et al., 1995; Niclou et al., 1998; Striggow et al., 2001; Wang et al., 2002; Junge et al., 2004).
Our previous studies have shown that activation of PAR1 evokes a dramatic increase in cytoplasmic calcium in NST astrocytes. This event apparently drives excitatory gliotransmitter release onto NST neurons. In turn, gastric vago-vagal reflexes are invoked, significantly slowing gastric transit (Hermann et al., 2009). Therefore, it is possible that the suppression of gastric motility and other autonomic events caused by bleeding head injuries is a result of thrombin activation of astrocytic PAR1 in the NST. Although it is clear that astrocytic PAR1 activation influences NST neurons, the mechanisms by which astrocytes and PAR1 control the NST have not been evaluated. To address this question, we used live-cell calcium-imaging coupled with whole-cell voltage-clamp recordings to identify the gliotransmitter released by activated NST astrocytes and the receptors that mediate the neuronal calcium response.
Materials and Methods
Animals and procedures.
Long–Evans rats (27 female, 2 male; 130–300 g) were used for these studies. Animals were obtained from the breeding colony at Pennington Biomedical Research Center, were maintained in a room with a 12 h light/dark cycle with constant temperature and humidity, and had access to food and water ad libitum. All experimental protocols were approved by the Institutional Animal Care and Use Committees of Pennington Biomedical Research Center and were performed according to the guidelines determined by the National Institutes of Health.
Direct injection of SR101 and calcium green-1 AM into the NST.
For our live-cell calcium-imaging experiments, the dyes calcium green-1 AM (CAG) and sulforhodamine 101 (SR101) were injected into the NST as described previously (Hermann et al., 2009). CAG is a calcium reporter dye that labels both neurons and astrocytes, whereas SR101 labels only astrocytes (McDougal et al., 2011), allowing us to discriminate between astrocytes and neurons in our imaging recordings. Briefly, animals were deeply anesthetized using ethyl carbamate (urethane; 1.5 g/kg; Sigma); urethane readily washes out of the brain slices (Hara and Harris, 2002). The animals were placed in a stereotaxic frame, the occipital plate of the skull was removed using aseptic technique, and the medullary brainstem was exposed. A micropipette (50 μm tip diameter) filled with 1% CAG (Life Technologies), 0.3% SR101 (Sigma), and 10% pluronic-DMSO (Life Technologies; Tris-PBS buffer, pH 7.2) was directed into the solitary nucleus using a stereotaxic carrier. Three to four injections of the pipette solution (40 nl each) were made unilaterally in the rostrocaudal direction to fill the NST at a depth of 300 μm below the surface. Cellular uptake of these dyes occurred within 30–45 min. The anesthetized rat was decapitated and the brainstem was quickly harvested.
Brainstem slice preparation.
The prelabeled brainstem was glued to the stage of a vibrating microtome (Leica VT1200) and submerged in a cold (4°C) carbogenated (95% O2/5% CO2) cutting solution containing the following (in mm): 92 N-methyl-d-glucamine, 30 NaHCO3, 25 glucose, 20 HEPES, 10 MgSO4-7H2O, 5 sodium ascorbate, 3 sodium pyruvate, 2.5 KCl, 2 thiourea, 1.25 NaH2PO4, and 0.5 CaCl2, titrated to pH 7.4 with HCl (Zhao et al., 2011). The brainstem was cut into coronal sections (300 μm thick), which were incubated at 32–34°C in the cutting solution for 10–15 min. The brainstem sections subsequently were incubated at room temperature (22–24°C) for 1–5 h in carbogenated recording solution containing the following (in mm): 124 NaCl, 25 NaHCO3, 10 glucose, 3 KCl, 2 CaCl2, 1.5 NaH2PO4, and 1 MgSO4-7H2O, which was supplemented with 5 sodium ascorbate, 3 sodium pyruvate, and 2 thiourea and titrated to pH 7.4 with HCl (Zhao et al., 2011).
Live-cell calcium imaging.
Live-cell calcium imaging of prelabeled astrocytes and neurons was performed as described previously (Hermann et al., 2009). Note that our previous studies showed that repeated exposures to SFLLRN-NH2 did not evoke comparable fluorescence responses, so each slice could not serve as its own control (Hermann et al., 2009). Briefly, slices were placed in the chamber of a Nikon F1 fixed stage upright microscope and perfused with carbogenated recording solution containing one of the following pharmacological modulators (33°C; 2.5 ml/min flow rate): control (recording solution alone); suramin (100 μm), a purinergic P2 receptor antagonist (Abcam); dl-threo-β-benzyloxyaspartic acid (TBOA; 100 μm), an excitatory amino acid transporter reuptake inhibitor (Tocris Bioscience); dl-2-amino-5-phosphonopentanoic acid (dl-AP5; 200 μm), an NMDA receptor antagonist (Tocris Bioscience); DNQX (20 μm), an AMPA receptor antagonist (Abcam); dl-2-amino-3-phosphonopropionic acid (dl-AP3; 300 μm), an mGluR antagonist (Tocris Bioscience); ifenprodil (3 μm), a GluN2B NMDA receptor antagonist (Abcam); or TCN 201 (10 μm), a GluN2A NMDA receptor antagonist (Abcam). After pharmacological pretreatment by incubation in the recording chamber for 10 min, the slices were stimulated by an 80 s application of 45 μm SFLLRN-NH2 (Peptides International), a selective PAR1 agonist, and were then washed with recording solution alone.
Selective inhibition of glial metabolism by fluorocitrate (FC) was as described previously (Hassel et al., 1992; Bonansco et al., 2011) to determine whether the SFLLRN-NH2 effects were mediated by astrocytic PAR1. Note that our own, in vivo, neurophysiological work has demonstrated that FC has no independent effect to alter NST neuron activity (Hermann et al., 2014). For those imaging experiments, slices were incubated in 200 μm FC for 20–25 min before being placed in the recording chamber. SFLLRN-NH2 was applied as described above and, after 10 minutes of incubation in normal recording solution, the slice was exposed to 1 mm l-glutamate and 100 μm ATP prepared in recording solution (Fig. 1). The glutamate/ATP challenge was used as a control to confirm that the neurons and astrocytes were capable of producing calcium signals in response to agonists. Only neurons and astrocytes that produced a calcium response after glutamate and ATP application were included in our FC analyses. Similar controls were conducted for the dl-AP5 and presynaptic transmission block experiments.
Figure 1.
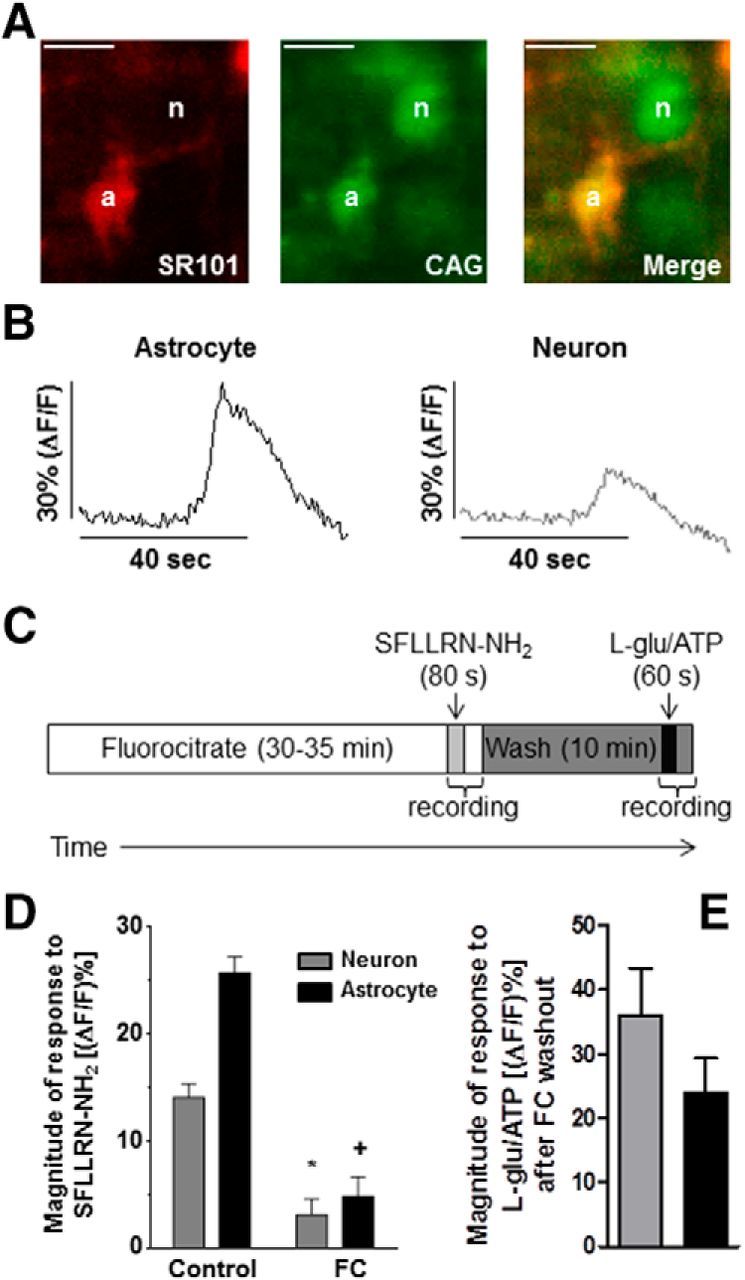
Astrocytic and neuronal responses to SFLLRN-NH2. A, SR101 selectively labels astrocytes (left) and CAG is the calcium reporter dye and labels both astrocytes and neurons (middle). Overlaying the two images allows for identification of neurons and glial cells (right). Scale bar, 8 μm. B, Prelabeled hindbrain slices containing the NST were activated by SFLLRN-NH2, which resulted in an increase in the fluorescence intensity of the CAG signal for both glial and neuronal cells. Responses to SFLLRN-NH2 are represented as (ΔF/F)%. C, Timeline of the fluorocitrate-imaging experiments. Slices were pretreated with FC for 30–35 min and then bath exposed to SFLLRN-NH2 for 80 s. After SFLLRN-NH2 stimulation, the slices were perfused with recording solution alone for 10 min and then stimulated with l-glutamate plus ATP (60 s) to confirm that the astrocytes and neurons were capable of producing agonist-evoked calcium responses after FC treatment. D, Inhibiting astrocytic activity with FC significantly inhibited both the neuronal and astrocytic responses to stimulation with SFLLRN-NH2. Data were analyzed for statistical significance with an unpaired t test. *p < 0.05 compared with the control neuronal response. +p < 0.05 compared with the control glial response. E, After FC washout, stimulation with l-glutamate plus ATP evoked a robust response from both neurons and astrocytes, demonstrating that FC effects are reversible and that these cells were still healthy. Only those astrocytes and neurons that responded to l-glu/ATP after FC exposure were included in our FC analyses (Fig. 1D).
A Prairie Technologies (Bruker) line-scanning laser confocal head equipped with a Photometrics CoolSNAP HQ camera performed time-lapse laser confocal calcium imaging at a rate of three frames per second. The CAG-labeled cells were visualized using a 488 nm excitation/509 nm emission filter and the SR101-labeled astrocytes were visualized using a 568 nm excitation/607 nm emission filter.
Calcium-imaging data analysis.
Nikon Elements AR software was used to analyze the confocal live cell fluorescent signals in the astrocytes and neurons as described previously (Hermann et al., 2009). Individual astrocytes and neurons were designated as regions of interest and their fluorescence signal over time was captured. Background fluorescence was subtracted from the fluorescence signal. The relative changes in cytoplasmic calcium in the cells were expressed as changes in fluorescence [(ΔF/F)%], where F is the intensity of the baseline fluorescence signal before stimulation and ΔF is the difference between the peak fluorescence intensity and the baseline signal. Data were evaluated for significance using the unpaired t test (to compare the responses evoked by SFLLRN-NH2 under control conditions with the responses evoked after FC pretreatment and to compare the responses evoked by SFLLRN-NH2 under control conditions with the responses evoked under presynaptic transmission blockade) or one-way ANOVA and Tukey's post hoc test (for the remaining analyses). Significance was set at p < 0.05. Data are reported as mean ± SEM.
Voltage-clamp recording from neurons in the NTS.
Given that astrocytes are not electrically excitable, prelabeling of the astrocytes and neurons was not necessary; however, all other preparations are as described in the “Brainstem slice preparation” section for our electrophysiological recordings. Slices were placed in the recording chamber of an upright microscope and perfused with normal recording solution supplemented with 0.5 μm TTX to block sodium channels and to prevent action potentials; 10 μm bicuculline was added to inhibit GABA receptors (Lee et al., 2007). Recording electrodes were filled with solution containing the following (in mm): 120 Cs-methanesulfonate, 15 CsCl, 10 tetraethylammonim chloride, 10 HEPES, 8 NaCl, 3 Mg-ATP, 1.5 MgCl2, 0.3 Na-GTP, and 0.2 EGTA at pH 7.3 (Guzman et al., 2009). Recordings of NST mEPSCs (VHOLD = −60 mV) were made using a Multiclamp 700B amplifier (Molecular Devices), filtered at 8 kHz, and digitized at 20 kHz using Axon pClamp10 software.
Electrophysiology data analysis.
Spontaneous miniature EPSCs (mEPSCs) from whole-cell voltage-clamp recordings from NST neurons were analyzed as described previously using the Mini Analysis Program (Synaptosoft; Lee et al., 2007). Only events with amplitudes >3 times the root mean square noise were included in the analysis. Selected mEPSCs from each recording were scaled and averaged, and the deactivation time constants were calculated by fitting the following single exponential equation to the data:
 |
where τ is the deactivation time constant and Amp is the current amplitude of the deactivation component.
Statistical significance of the distribution of mEPSC inter-event intervals was determined using the Kolmogorov–Smirnov nonparametric analysis and the paired t test (for the control vs SFLLRN-NH2 experiments) or one-way ANOVA with repeated measures and Tukey's post hoc test (for the FC and FC plus SFLLRN-NH2 experiments) were used to evaluate amplitudes and deactivation time constants for statistical significance. Significance was set at p < 0.05. Data are reported as mean ± SEM.
Results
Astrocytes mediate the PAR1 response in the NST
Although previous immunohistochemical studies indicated that PAR1 is expressed exclusively on astrocytes in the NST (Hermann et al., 2009), we used live-cell calcium imaging of rat hindbrain slices that contained the NST and pharmacological inhibition of the astrocytes with fluorocitrate to evaluate the specificity of PAR1 activation in the NST. Astrocytes were preloaded with the astrocyte-specific dye SR101 and both the astrocytes and neurons were preloaded with the calcium-sensitive dye CAG (Fig. 1A). An increase in intracellular calcium corresponded to an increase in fluorescence and is indicative of cellular activation. In control experiments, selective activation of PAR1 evoked a 25.7 ± 1.5% increase in astrocytic fluorescence compared with resting baseline levels (n = 121) and the peak neuronal fluorescence was 14.1 ± 1.2% compared with the baseline (n = 39; Fig. 1B,D, Table 1).
Table 1.
PAR1-dependent increases in calcium influx are diminished by inhibiting glial metabolism
| Condition | (ΔF/F)% | n |
|---|---|---|
| Neurons | ||
| Control | 14.1 ± 1.3 | 39 |
| Fluorocitrate | 3.0 ± 1.5* | 30 |
| Astrocytes | ||
| Control | 25.7 ± 1.5 | 121 |
| Fluorocitrate | 4.8 ± 1.8** | 28 |
PAR1 was activated by bath application of SFLLRN-NH2. Peak responses were determined for each cell type. FC significantly inhibited the responses of both neurons and astrocytes to PAR1 activation. (Unpaired t test within each cell type;
* p < 0.05 compared with the control neuronal response;
** p < 0.05 compared with the control glial response.)
Pretreating the slices with FC to block astrocyte metabolism (Bonansco et al., 2011) resulted in significantly reduced responses to SFLLRN-NH2 in both astrocytes and neurons (astrocytes: 4.8 ± 1.8%; n = 28; neurons: 3.0 ± 1.5%; n = 30; unpaired t test; p < 0.05; Fig. 1C,D, Table 1). To confirm that the neurons and astrocytes were still viable after FC treatment, the slices were bathed in normal recording solution for 10 min after SFLLRN-NH2 application, after which the slices were exposed to 1 mm l-glutamate plus 100 μm ATP for 60 s (Fig. 1C,E). The glutamate/ATP challenge evoked robust calcium responses in both astrocytes (23.9 ± 5.4%, n = 28) and neurons (35.9 ± 7.5%, n = 30). Only those astrocytes and neurons that had a calcium response after ATP/glutamate application were included in our FC analyses.
In a separate experiment, we pretreated the slices with FC and then immediately bath applied the glutamate/ATP challenge to evaluate whether the neurons could respond to other agonists while astrocytic metabolism was inhibited. In these experiments, the glutamate/ATP challenge evoked responses in both the astrocytes (20.5 ± 1.2%, n = 39) and the neurons (36.0 ± 2.1%, n = 49). Therefore, astrocytic and neuronal ligand-gated ion channels (i.e., for glutamate and ATP) remain functional while under the influence of FC (Table 2). However, the metabotropic actions of SFLLRN-NH2 on PAR1 receptors and the gliotransmission response to elevated cell calcium are diminished (Fig. 1D), similar to what has been observed with FC treatment in previous studies (Clarke, 1991; Martín et al., 2007; Bonansco et al., 2011; Choe et al., 2012; Tanahashi et al., 2012). Furthermore, these data suggest that astrocytes, but not neurons, are selectively activated by the PAR1 agonist SFLLRN-NH2, which results in gliotransmitter release and neuronal activation.
Table 2.
Astrocytic and neuronal ligand-gated ion channels (e.g., glutamate and ATP) remain functional while under the influence of FC
| Condition | (ΔF/F)% | n |
|---|---|---|
| Neurons | ||
| After FC washout | 35.9 ± 7.5 | 30 |
| During FC | 36.1 ± 2.1 | 39 |
| Astrocytes | ||
| After FC washout | 23.9 ± 5.4 | 28 |
| During FC | 20.5 ± 1.2 | 39 |
Bath application of glutamate/ATP combination evoked the same robust peak responses during exposure to FC and after FC washout in both neurons and astrocytes (unpaired t test; p > 0.05). These data suggest that the effects of the glial metabolic blocker FC specifically interfere with astrocytic ability to respond to metabotropic agonists (e.g., SFLLRN-NH2) and that 200 μm FC is not toxic to either neurons or astrocytes.
NST astrocytes activate NST neurons directly after PAR1 stimulation
The increase in neuronal calcium after astrocytic PAR1 activation may be due to direct gliotransmitter release onto the postsynaptic neurons or could be caused indirectly by astrocytic stimulation of the glutamatergic presynaptic inputs (e.g., from vagal afferents) that synapse upon NST neurons. Therefore, to discount the possibility of action-potential-dependent or spontaneous neuronal glutamate release, TTX (0.5 μm), high Mg2+ (6 mm), and low Ca2+ (0.9 mm) were added to the perfusion bath to inhibit presynaptic glutamate release (Katz et al., 1997). As a demonstration of the ability to impede presynaptic neurotransmitter release via the use of high Mg2+ and low Ca2+ presynaptic transmission blockade, we compared mEPSC frequencies and amplitudes recorded in the control recording solution (0.5 μm TTX, 10 μm bicuculline, 3 mm Ca2+, and 1 mm Mg2+) and under presynaptic block conditions (TTX and bicuculline, 0.9 mm Ca2+, and 6 mm Mg2+). The frequency of mEPSC occurrence decreased from 12.9 ± 4.6 Hz to 5.0 ± 1.6 Hz (n = 4) and the inter-event interval shifted significantly to the right (p < 0.05; Kolmogorov–Smirnov nonparametric analysis; Fig. 2). mEPSC amplitude was not significantly different between the two conditions (p > 0.05; paired t test). These data suggest that presynaptic glutamate release is significantly limited under the presynaptic block conditions, favoring astrocyte-to-neuron communication in these experiments.
Figure 2.
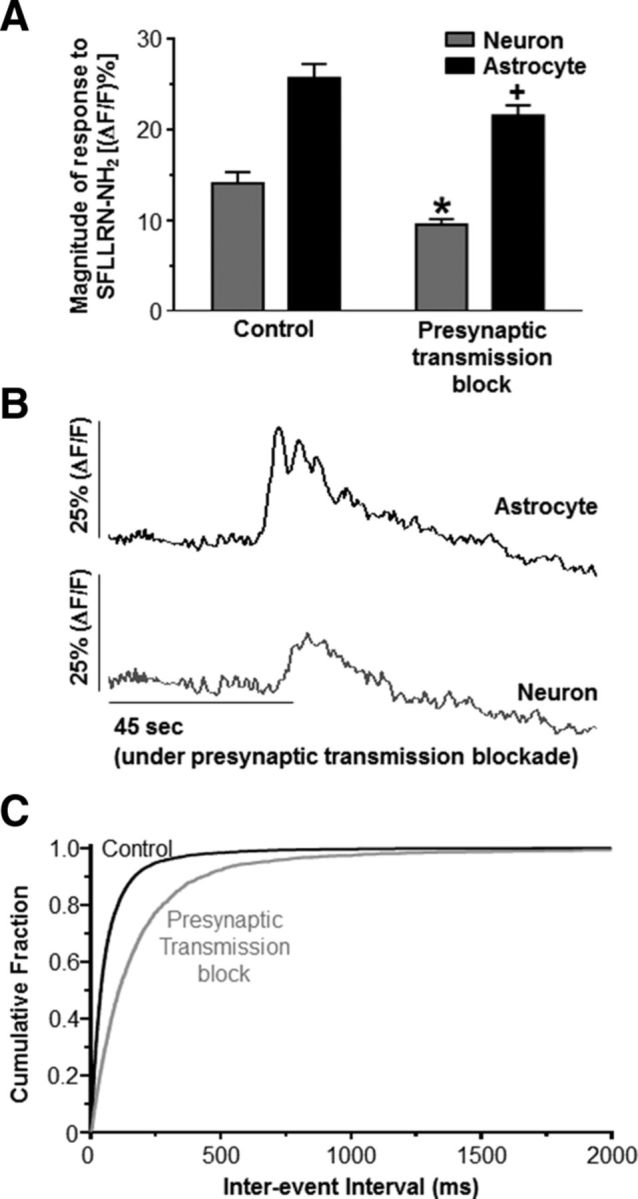
Astrocytes excite neurons directly after PAR1 activation. A, Calcium-imaging experiments with hindbrain slices containing the NST were perfused in the recording chamber with a bath solution containing 0.5 μm TTX, 0.9 mm Ca2+, and 6 mm Mg2+TTX (i.e., “high Mg2+ and low Ca2+”; presynaptic transmission blockade). Even under control conditions, astrocytes are more responsive than neurons to SFLLRN-NH2 challenge (refer to Fig. 1B). Presynaptic transmission block only slightly but significantly reduced the responses of both the neurons and astrocytes to the PAR1 agonist (unpaired t test; *p < 0.05 compared with the control neuronal response; +p < 0.05 compared with the control glial response). These data suggest that the majority of the neuronal response to PAR1 activation is due to direct astrocyte-to-neuron gliotransmission, although there is a small component that may be due to astrocyte-induced glutamate release from other neuronal sources (e.g., afferent terminals). B, Representative responses from an NST-astrocyte and NST-neuron in the same hindbrain slice evoked by bath application of SFLLRN-NH2 under presynaptic transmission blockade conditions (TTX, high Mg2+, and low Ca2+). C, As a demonstration of the ability to impede presynaptic neurotransmitter release via the use of high Mg2+ and low Ca2+ presynaptic transmission blockade, voltage-clamp experiments on NST-neurons recorded mEPSCs under both “control” (0.5 μm TTX, 10 μm bicuculline, 3 mm Ca2+, and 1 mm Mg2+) and “presynaptic block” (TTX and bicuculline, 0.9 mm Ca2+, and 6 mm Mg2+) conditions. The inter-event interval significantly shifted to the right after presynaptic block (p < 0.05; Kolmogorov–Smirnov nonparametric analysis), suggesting that presynaptic glutamate release is significantly inhibited by the “presynaptic block” protocol.
Next, we evaluated whether the neuronal response after bath application of SFLLRN-NH2 was decreased after presynaptic inhibition. Bath application of SFLLRN-NH2 evoked robust, yet significantly reduced, peak response signals in both the astrocytes (21.6 ± 1.1%; n = 180) and neurons (9.5 ± 0.6%; n = 141) compared with the controls (Fig. 2; p < 0.05; unpaired t test). Therefore, these data suggest that the majority of the neuronal response is due to direct astrocyte-to-neuron gliotransmission, but a small component of the neuronal response may be due to astrocyte-induced presynaptic glutamate release.
NST astrocytes release glutamate after PAR1 activation
Two of the most common excitatory gliotransmitters are ATP and glutamate (Parpura et al., 1994; Bezzi et al., 1998; Coco et al., 2003; Angulo et al., 2004; Fellin et al., 2004). Bath application of the purinergic P2 receptor antagonist suramin for 10 minutes before PAR1 stimulation (Evans et al., 1992) did not significantly affect the responses of either the astrocytes (25.1 ± 1.1%; n = 79) or neurons (13.6 ± 1.3%; n = 39) relative to their responses under control conditions (Fig. 3, Table 3; p > 0.05; one-way ANOVA with Tukey's post hoc test). These data suggest that astrocytes activated by PAR1 stimulation do not use ATP as a gliotransmitter to evoke neuronal calcium release.
Figure 3.

PAR1 stimulation induces astrocytic glutamate release. Hindbrain slices were perfused with the purinergic P2 receptor antagonist suramin for 10 min before SFLLRN-NH2 stimulation. Purinergic receptor block had no effect on either the neuronal or astrocytic responses to PAR1 stimulation. In contrast, other hindbrain slices that had been perfused with the excitatory amino acid transporter blocker TBOA for 10 min before SFLLRN-NH2 stimulation resulted in significantly higher peak calcium signals in both astrocytes and neurons compared with the control (one-way ANOVA with Tukey's post hoc test; *p < 0.05 compared with the control neuronal response; +p < 0.05 compared with the control glial response). These results suggest that glutamate, but not ATP, is used as a gliotransmitter in response to NST-astrocyte PAR activation.
Table 3.
NST astrocytes release glutamate after PAR1 activation
| Condition | (ΔF/F)% | n |
|---|---|---|
| Neurons | ||
| Control | 14.1 ± 1.3 | 39 |
| Suramin | 13.6 ± 1.3 | 39 |
| TBOA | 20.2 ± 1.1* | 48 |
| Astrocytes | ||
| Control | 25.7 ± 1.5 | 121 |
| Suramin | 25.1 ± 1.1 | 79 |
| TBOA | 31.5 ± 2.0** | 110 |
Slices were pretreated with either a purinergic receptor antagonist (suramin) or an excitatory amino acid reuptake blocker (TBOA) and were then stimulated with SFLLRN-NH2. Peak responses were determined for each cell type. Suramin had no effect on either neuronal or astrocytic responses to PAR1 activation. In contrast, pretreatment with TBOA enhanced the responses of both cell types to PAR1 activation (one-way ANOVA and Tukey's post hoc test;
* p < 0.05 compared with the control neuronal response;
** p < 0.05 compared with the control glial response.)
In contrast, bath application of the excitatory amino acid transporter blocker (i.e., reuptake blocker) TBOA for 10 min before PAR1 stimulation (Huda et al., 2013) significantly increased the evoked responses to SFLLRN-NH2 in both astrocytes (31.5 ± 2.0%; n = 110) and neurons (20.2 ± 1.1%; n = 48) compared with the control (Fig. 3, Table 3; p < 0.05; one-way ANOVA with Tukey's post hoc test). These data suggest that glutamate is the gliotransmitter released from astrocytes after PAR1 activation.
GluN2B-containing NMDA receptors mediate the NST neuronal response to PAR1 activation
We further explored which ionotropic or metabotropic glutamate receptors on the NST astrocytes and neurons were activated after PAR1 stimulation. Bath application of the general NMDA receptor antagonist dl-AP5 significantly reduced the responses to SFLLRN-NH2 in both astrocytes (9.8 ± 1.6%; n = 27) and neurons (4.4 ± 0.8%; n = 27) compared with the control conditions (Fig. 4, Table 4; p < 0.05; one-way ANOVA with Tukey's post hoc test).
Figure 4.

Astrocytic and neuronal responses to SFLLRN-NH2 are modulated by glutamatergic pharmacological tools. Hindbrain slices containing the NST were pretreated with one of the following conditions for 10–15 min before stimulation with PAR1 agonist: control, dl-AP5, DNQX, or dl-AP3. The peak responses to SFLLRN-NH2 are represented as the (ΔF/F)%. The nonspecific NMDA receptor antagonist dl-AP5 significantly reduced both neuronal and glial responses to SFLLRN-NH2. Application of the AMPAR antagonist DNQX significantly reduced glial but not neuronal responses to the PAR1 agonist; the broad spectrum metabotropic glutamate receptor antagonist dl-AP3 had no effect (one-way ANOVA with Tukey's post hoc test; *p < 0.05 compared with the control neuronal response; +p < 0.05 compared with the control glial response). These data suggest that glutamate gliotransmitter release after PAR1 stimulation activates NST-neuronal NMDA receptors as well as NST-astrocytic NMDA and AMPA receptors, which further enhances the astrocytic activation.
Table 4.
PAR1-dependent increases in calcium influx are diminished by glutamate receptor antagonists
| Condition | (ΔF/F)% | n |
|---|---|---|
| Neurons | ||
| Control | 14.1 ± 1.3 | 39 |
| dl-AP5 | 4.4 ± 0.8* | 27 |
| DNQX | 10.2 ± 0.9 | 45 |
| dl-AP3 | 11.4 ± 1.6 | 8 |
| Astrocytes | ||
| Control | 25.7 ± 1.5 | 121 |
| dl-AP5 | 9.8 ± 1.6** | 27 |
| DNQX | 17.5 ± 1.4** | 87 |
| dl-AP3 | 24.7 ± 2.7 | 40 |
Slices were pretreated with glutamate receptor antagonists dl-AP5, DNQX, or dl-AP3 before being stimulated with SFLLRN-NH2. Peak responses were determined for each cell type. The NMDAR antagonist dl-AP5 significantly inhibited both neuronal and glial responses to SFLLRN-NH2. However, the AMPAR antagonist DNQX significantly reduced glial but not neuronal responses to SFLLRN-NH2 and the broad spectrum metabotropic glutamate receptor antagonist dl-AP3 had no effect (one-way ANOVA with Tukey's post hoc test;
* p < 0.05 compared with the control neuronal response;
** p < 0.05 compared with the control glial response). These data suggest that glutamate gliotransmitter release after PAR1 stimulation activates NST-neuronal NMDA receptors as well as NST-astrocytic NMDA and AMPA receptors, which further enhances the astrocytic activation.
Bath application of the AMPAR antagonist DNQX also significantly reduced the peak response of the astrocytes (17.5 ± 1.4%; n = 87; p < 0.05; one-way ANOVA with Tukey's post hoc test; Fig. 4, Table 4), but did not influence the peak neuronal response (10.2 ± 0.9%; n = 45; p > 0.05; one-way ANOVA with Tukey's post hoc test; Table 4). These data are consistent with earlier studies showing that NST astrocytes express calcium-permeable AMPA receptors (McDougal et al., 2011). Finally, bath application of the mGluR antagonist dl-AP3 did not significantly affect the astrocytic (24.7 ± 2.7%; n = 40) or neuronal (11.4 ± 1.6%; n = 8) peak response (p > 0.05; one-way ANOVA with Tukey's post hoc test; Fig. 4, Table 4).
These data indicate that PAR1 stimulation with SFLLRN-NH2 induces astrocytic glutamate release. The glutamate gliotransmitter then binds to and activates astrocytic NMDA and AMPA receptors, which further enhances the astrocytic calcium signal. Furthermore, the glutamate released from the astrocytes after PAR1 stimulation primarily acts upon NMDA receptors to evoke a calcium response in neurons.
Because dl-AP5 inhibited the responses of both astrocytes and neurons after PAR1 activation, it is possible the decreased neuronal response was due to diminished astrocytic glutamate release and not the inhibition of neuronal NMDA receptors. Subunit-selective allosteric modulators for each of the four glutamate-binding GluN2 NMDA receptor subunits are available, allowing for selective targeting of the individual GluN2 subunits (Traynelis et al., 2010; Ogden and Traynelis, 2011). Previous studies indicate that the GluN2A and GluN2B subunits are expressed in NST neurons (Liu and Wong-Riley, 2010). Therefore, we used the GluN2A antagonist TCN 201 (Edman et al., 2012; Hansen et al., 2012) and the GluN2B antagonist ifenprodil (Williams, 1993) to attempt to selectively inhibit the neuronal NMDA receptors. Bath application of the GluN2B antagonist ifenprodil significantly inhibited the neuronal calcium response to PAR1 activation (8.7 ± 0.9%; n = 44) without inhibiting the astrocytic calcium response (25.7 ± 1.7%; n = 89; Fig. 5A,B, Table 5; p < 0.05; one-way ANOVA with Tukey's post hoc test). However, bath perfusion with TCN 201 did not significantly alter either the astrocytic (19.8 ± 1.2%; n = 30) or neuronal (10.9 ± 1.3%; n = 13) response to SFLLRN-NH2 stimulation (p > 0.05; one-way ANOVA with Tukey's post hoc test; Fig. 5A,C, Table 5). These data suggest that astrocytes stimulated by PAR1 activation release glutamate that in turn activates GluN2B-containing NMDA receptors on NST neurons.
Figure 5.

GluN2B-containing NMDA receptors mediate NST neuronal response to PAR1 activation. A, Subunit-specific NMDA receptor modulators were used to determine which GluN2 subunit contributed to the neuronal calcium response after PAR1 activation. Hindbrain slices were pretreated with either the GluN2A antagonist TCN 201 or the GluN2B antagonist ifenprodil for 10–15 min and then stimulated with SFLLRN-NH2. Ifenprodil selectively inhibited the neuronal response without altering the astrocytic response, suggesting that GluN2B receptors mediate the calcium signal in NST neurons. TCN 201 had no effect on either the astrocytic or the neuronal calcium response (one-way ANOVA with Tukey's post hoc test; *p < 0.05 compared with the control neuronal response). Responses from individual astrocytes (black) and neurons (gray) evoked by bath application of SFLLRN-NH2 after pretreatment with ifenprodil or TCN 201 are shown in B and C, respectively. These results suggest that most of the PAR1 astrocyte-to-neuron signal in the NST is carried by glial release of glutamate acting on NST-neuronal GluN2B-containing NMDA receptors.
Table 5.
GluN2B-containing NMDA receptors mediate neuronal calcium influx after PAR1 activation
| Condition | (ΔF/F)% | n |
|---|---|---|
| Neurons | ||
| Control | 14.1 ± 1.3 | 39 |
| Ifenprodil | 8.7 ± 0.9* | 44 |
| TCN 201 | 10.9 ± 1.3 | 13 |
| Astrocytes | ||
| Control | 25.7 ± 1.5 | 121 |
| Ifenprodil | 25.7 ± 1.7 | 89 |
| TCN 201 | 19.8 ± 1.2 | 30 |
Slices were pretreated with ifenprodil or TCN 201 before being stimulated with SFLLRN-NH2. Peak responses were determined for each cell type. Ifenprodil selectively inhibited the neuronal response without altering the astrocytic response, suggesting that GluN2B receptors mediate the calcium signal in NST neurons. TCN 201 had no effect on either the astrocytic or the neuronal calcium response (one-way ANOVA with Tukey's post hoc test;
* p < 0.05 compared with the control neuronal response). These results suggest that most of the PAR1 astrocyte-to-neuron signal in the NST is carried by glial release of glutamate acting on NST-neuronal GluN2B-containing NMDA receptors.
PAR1 activation increases mEPSC frequency in the NST
Our calcium-imaging data suggested that astrocytes may also modulate the presynaptic vagal afferent terminals that synapse in the NST. Therefore, we obtained whole-cell voltage-clamp recordings from NST neurons to determine whether astrocytic PAR1 increased the frequency of glutamate release from the vagal afferent synaptic terminals. mEPSC frequency, peak amplitude, and deactivation time course kinetics were evaluated. Action-potential-dependent glutamate release was inhibited by TTX, and bicuculline was included in the recording solution to block GABA receptors. Exposure to SFLLRN-NH2 increased mEPSC frequency because the inter-event interval significantly decreased compared with the TTX and bicuculline control (p < 0.05; Kolmogorov–Smirnov nonparametric analysis; Fig. 6A,B). This is reflected by an increase in frequency from 18.8 ± 7.8 Hz to 26.6 ± 9.5 Hz (n = 4). However, peak amplitude (13.1 ± 1.4 pA vs 14.0 ± 0.9 pA; Fig. 6C) and deactivation time course (4.3 ± 0.8 s vs 4.6 ± 0.8 s; Fig. 6D) were unchanged (paired t test, p > 0.05) compared with the control. The increase in mEPSC frequency was reversible, because the mEPSC frequency was reduced to 16.5 ± 6.9 Hz after the slices were perfused with the control recording solution. Bath application of DNQX fully inhibited the mEPSCs.
Figure 6.

SFLLRN-NH2 increases mEPSC frequency in the NST. A, mEPSCs (VHOLD = −60 mV) were recorded from NST neurons to evaluate whether SFLLRN-NH2 caused an increase in the frequency of glutamate release on to the postsynaptic NST neurons. A representative control recording (top) was obtained in the presence of bicuculline and TTX. SFLLRN-NH2 increased the frequency of events (bottom) in the same recording. B, The distribution of inter-event intervals was shifted significantly to the left in the presence of SFLLRN-NH2 (gray curve) compared with the control (black curve; Kolmogorov–Smirnov test, p < 0.05; see inset), indicating that the frequency of mEPSC events was increased after astrocytic activation by the PAR1 agonist. Neither peak amplitude (C) nor deactivation time course (D) was altered by SFLLRN-NH2 (paired t test, p > 0.05). These data reinforce the view that SFLLRN-NH2 has a significant, if small, effect to increase presynaptic excitatory inputs to NST neurons, as shown in Figure 2.
Previous behavioral studies have shown that GluN2B receptors on presynaptic vagal afferents control food intake (Guard et al., 2009a, 2009b). Therefore, we also evaluated whether the GluN2B subunit mediates a component of the increase in mEPSC frequency evoked by bath application of SFLLRN-NH2. Slices were exposed to the control TTX and bicuculline solution, then the control solution plus ifenprodil, and finally the control solution plus both ifenprodil and SFLLRN-NH2. Bath application of ifenprodil significantly increased inter-event interval compared with the control solution (p < 0.05; Kolmogorov–Smirnov nonparametric analysis; Fig. 7), which was reflected by a decrease in mEPSC frequency from 9.4 ± 2.9 Hz in the control solution to 5.6 ± 1.7 Hz in the presence of ifenprodil (n = 5). These data suggest that GluN2B-containing NMDA receptors may be tonically active under basal, resting conditions. Concurrent application of ifenprodil plus SFLLRN-NH2 increased mEPSC frequency to 6.5 ± 1.8 Hz (n = 5) and significantly reduced the inter-event interval (p < 0.05; Kolmogorov–Smirnov nonparametric analysis; Fig. 7). However, SFLLRN-NH2 plus ifenprodil increased mEPSC frequency by 119.3 ± 4.9% over ifenprodil alone, whereas SFLLRN-NH2 alone increased mEPSC frequency by 162.6 ± 23.2% over the control. These data suggest that GluN2B-containing NMDA receptors may mediate a component of the SFLLRN-NH2-induced increase in mEPSC frequency.
Figure 7.
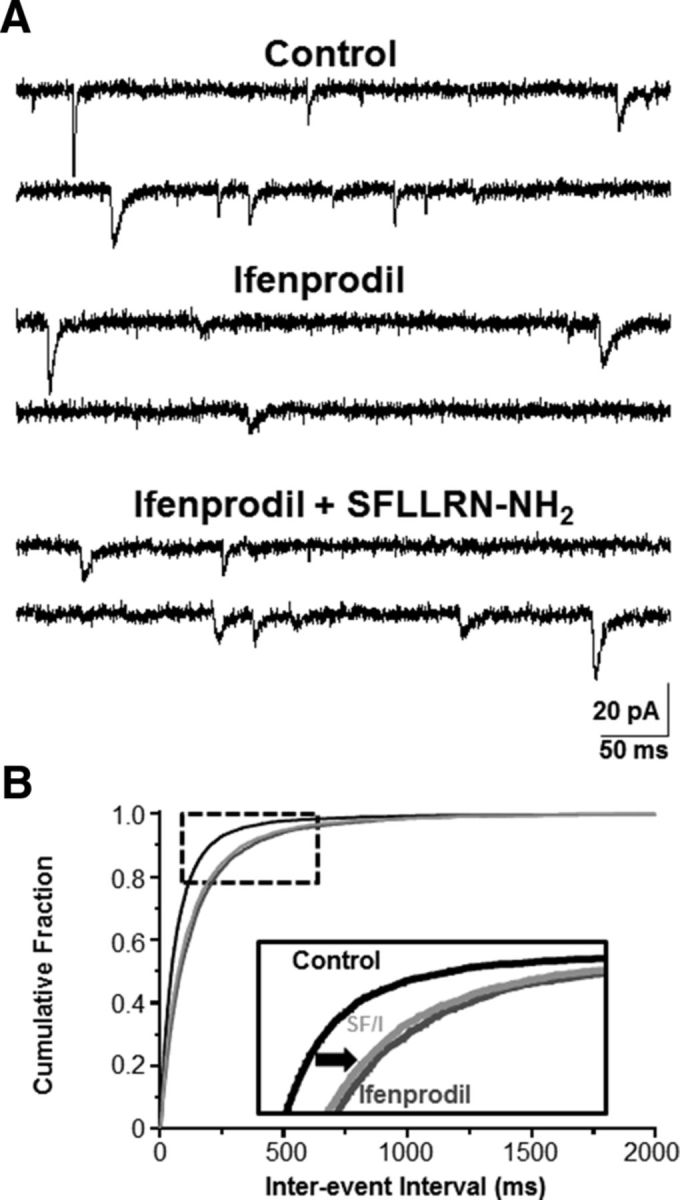
Presynaptic GluN2B-containing NMDA receptors mediate a component of the SFLLRN-NH2-induced response on mEPSC frequency. A, mEPSCs (VHOLD = −60 mV) were recorded from NST neurons under “control,” “ifenprodil,” and “ifenprodil + SFLLRN-NH2” conditions to evaluate effects on SFLLRN-NH2-induced increase in mEPSC frequency. B, The interevent interval was significantly increased by bath application of ifenprodil (dark gray curve) compared with the control (black curve; p < 0.05; Kolmogorov–Smirnov nonparametric analysis). Although bath application of ifenprodil + SFLLRN-NH2 (light gray curve) significantly decreased the inter-event interval compared with ifenprodil (p < 0.05; Kolmogorov–Smirnov nonparametric analysis; see inset), this was a very modest increase in mEPSC frequency relative to that we observed under control conditions (see Fig. 6). These data suggest that GluN2B-containing NMDA receptors on the presynaptic glutamatergic terminals (e.g., vagal afferents) may mediate a component of the mEPSC response evoked by SFLLRN-NH2-induced astrocytic glutamate release.
To confirm that this increase in mEPSC frequency was due to astrocytic excitation of the presynaptic terminals and not to PAR1 expressed on the terminals themselves, these experiments were repeated under astrocytic inhibition with fluorocitrate. Bath application of FC alone decreased mEPSC frequency, because the inter-event interval significantly increased compared with the TTX and bicuculline control (p < 0.05; Kolmogorov–Smirnov nonparametric analysis; Fig. 8A,B). This is reflected in a decrease in frequency from 8.6 ± 2.1 Hz under control conditions to 5.3 ± 1.4 Hz after astrocytic inhibition with FC (n = 4). These data suggest that astrocytes modulate the excitability of vagal afferent terminals even under basal conditions. Subsequent coapplication of SFLLRN-NH2 and FC did not significantly alter mEPSC interevent interval compared with FC alone (p > 0.05; Kolmogorov–Smirnov nonparametric analysis; Fig. 8A,B), as mEPSC frequency was 4.9 ± 1.4 Hz (n = 4). There was no significant difference in peak amplitude or deactivation time constant between the control, FC alone, or FC plus SFLLRN-NH2 groups (Fig. 8C,D; p > 0.05; one-way ANOVA with repeated measures and Tukey's post hoc test). Finally, bath application of DNQX fully inhibited the mEPSCs. These data show that astrocytic PAR1 activation induces the vagal afferent terminals to increase the frequency of neurotransmitter release from the presynaptic terminals onto the NST neurons.
Figure 8.

Astrocytes modulate the excitability of presynaptic vagal afferents. A, mEPSCs (VHOLD = −60 mV) were recorded from NST neurons to evaluate whether inhibiting astrocytes with fluorocitrate blocked the PAR1-mediated increase in mEPSC frequency. A representative recording is given under control conditions (top), after bath application of FC (middle), and after concurrent application of FC plus SFLLRN-NH2 (bottom). B, The distribution of inter-event intervals was shifted significantly to the right in the presence of FC (dark gray curve) compared with the control (black curve; Kolmogorov–Smirnov test, p < 0.05), whereas there was no significant difference in inter-event interval between the FC and SFLLRN-NH2 coapplication (light gray curve) compared with FC alone (Kolmogorov–Smirnov test, p > 0.05; see inset). Neither peak amplitude (C) nor deactivation time course (D) was altered with FC or FC plus SFLLRN-NH2 (one-way ANOVA with repeated measures and Tukey's post hoc test, p > 0.05).
Discussion
The data from this study support five main findings. First, our calcium-imaging and electrophysiological data corroborate studies suggesting that PAR1 receptors are functional in astrocytes and not neurons of the NST (Hermann et al., 2009). SFLLRN-NH2-induced increases in astrocytic and neuronal calcium signals are blocked when the cells are pretreated with the glial metabolic blocker, fluorocitrate. Second, the effects of fluorocitrate are not only reversible but also appear to be specific for metabotropic agonists in that astrocytes and neurons are still responsive to ligand-gated agonists even under FC conditions. Third, PAR1-stimulated astrocytes communicate directly with NST neurons. Blocking presynaptic vagal afferent glutamate release only partially inhibits the neuronal response after astrocytic activation. Therefore, a majority of the neuronal response is caused by direct astrocyte-to-neuron gliotransmission, but a minor component of the neuronal response may be due to astrocytic PAR1 stimulation of vagal afferent glutamate release. Fourth, astrocytes release glutamate, not ATP, after PAR1 activation, because TBOA potentiates the calcium responses in both astrocytes and neurons whereas dl-AP5 inhibits the calcium responses. Indeed, GluN2B-containing NMDA receptors mediate a significant component of the neuronal response after astrocytic PAR1 activation. Fifth, our electrophysiological data show that PAR1-activated astrocytes also release glutamate directly onto the presynaptic afferents that synapse upon the NST neurons. Bath application of SFLLRN-NH2 significantly increased mEPSC frequency compared with control. Furthermore, fluorocitrate significantly inhibited mEPSC frequency, suggesting that astrocytes also modulate afferent excitability under resting conditions. These data suggest that astrocytes modulate neurotransmission in the NST through two pathways: directly through astrocyte-to-neuron gliotransmission and indirectly through astrocyte-to-presynaptic terminal gliotransmission.
In summary, the NST neuronal population is under the influence of astrocytic PAR1 receptors. PAR1 activation induces astrocytic glutamate release and activation of neuronal GluN2B-containing NMDA receptors. Astrocytes also have significant effects on vagal afferent glutamate release both under resting conditions and in response to PAR1 stimulation. These data suggest that the autonomic dysfunction that occurs after traumatic brain injury and an increase in thrombin production (Young et al., 1992; Lu et al., 1997; Gingrich and Traynelis, 2000; Hamill et al., 2009) may be the result of the activation of NMDA receptors on NST neurons.
NST glial-neural interactions in physiological context
Astrocytes are the most abundant cells within the CNS. Long thought to only have a passive, supportive role in neurovascular coupling and maintaining neuronal networks, it now seems likely that astrocytes regulate neuronal excitability and synaptic plasticity directly. A single astrocyte may contact tens to hundreds of thousands of synapses and, along with presynaptic terminals and postsynaptic neurons, will form what has been termed the “tripartite synapse” (Araque et al., 1999; Bushong et al., 2002; Halassa et al., 2007; Perea et al., 2009; Halassa and Haydon, 2010). Astrocytes are activated by neurotransmitters released from neuronal presynaptic terminals, gliotransmitters released by other astrocytes, or other factors such as thrombin and all of these agents increase astrocytic calcium levels (Porter and McCarthy, 1996; Pasti et al., 1997; Bezzi et al., 1998; Kang et al., 1998; Araque et al., 2002; Scemes and Giaume, 2006; Lee et al., 2007; Hermann et al., 2009). This increase in astrocytic calcium is coupled to a release of gliotransmitters (Araque et al., 2001; Halassa et al., 2007; Perea et al., 2009; Halassa and Haydon, 2010) such as glutamate, ATP, and d-serine, among others (Parpura et al., 1994; Bezzi et al., 1998; Mothet et al., 2000; Coco et al., 2003; Angulo et al., 2004; Fellin et al., 2004).
Previous studies have demonstrated a role for astrocytic PAR1 in synaptic transmission in other regions of the brain, particularly in the hippocampus, where the receptor is expressed predominantly in astrocytes but is also expressed in neurons (Weinstein et al., 1995; Junge et al., 2004). Within the hippocampus, astrocytic PAR1 activation induces glutamate release and activates neuronal NMDA receptors, producing calcium influx and neuronal depolarization (Gingrich and Traynelis, 2000; Lee et al., 2007). Furthermore, the protease plasmin and the PAR1 agonist TFLLR both potentiate the NMDA receptor component of hippocampal mEPSCs (Lee et al., 2007; Mannaioni et al., 2008). Indeed, PAR1 activation and astrocytic glutamate release induces long-term potentiation via NMDA receptors and activated protein C (Maggio et al., 2008; Maggio et al., 2013). PAR1−/− mice have diminished NMDA-receptor-dependent long-term potentiation and learning and memory deficits (Almonte et al., 2007; Almonte et al., 2013). Recent studies show that PAR1-activated astrocytes in the CA1 release glutamate through the calcium-activated anion channel Bestrophin-1 (Park et al., 2009; Oh et al., 2012; Han et al., 2013; Park et al., 2013). It remains to be determined whether astrocytic PAR1s in the NST also release glutamate through this mechanism.
The studies we have presented here show that NST astrocytes activated by PAR1 stimulation communicate with both presynaptic terminals and with the NST neurons that process and relay visceral information. Astrocytes and neurons have robust calcium responses after PAR1 activation with SFLLRN-NH2 even when presynaptic glutamate release is pharmacologically inhibited, suggesting direct astrocyte-to-neuron gliotransmission. Our electrophysiological data suggest astrocyte-to-presynaptic terminal communication also occurs in the NST, because mEPSC frequency significantly increases after bath application of SFLLRN-NH2. Furthermore, inhibiting astrocytic activity with fluorocitrate significantly decreases the frequency of glutamate release from presynaptic vagal afferents under resting conditions, indicating that astrocytes may maintain regulatory control or “tone” over glutamatergic neurotransmission in the NST. Therefore, these studies are the first to determine the roles of astrocytes in the tripartite synapse in the NST after PAR1 activation.
In our calcium-imaging studies, dl-AP5 inhibits both the astrocytic and the neuronal calcium response to PAR1 stimulation. Although these data suggest that NMDA receptors may be expressed on NST astrocytes and are activated by astrocytic glutamate release, our previous work suggests that NMDA receptor expression in rat hindbrain astrocytes is not robust (McDougal et al., 2011). The diminished astrocytic response after NMDA receptor inhibition with dl-AP5 may be due to reduced neuron-to-astrocyte or vagal afferent-to-astrocyte signaling.
NST glial-neural communications in historical context
The foundation for the glial-neuron signaling controversy was laid >100 years ago by two of the most famous antagonists in neuroscience: Camillio Golgi and Santiago Ramon y Cajal. Golgi developed his silver chromate “black reaction” stains in 1872 specifically to investigate glial morphology. Golgi rightly concluded that astrocytes are connected in a syncytial fashion, but made the error of extending this argument to include neuronal interconnections. Cajal modified the Golgi procedure and used improved optical techniques to conclude correctly that neurons maintain synaptic relationships, but concluded incorrectly that glial cells could not communicate through syncytial connections. The fallout from Cajal winning the neuronal argument in Stockholm in 1906 probably set glial-neural physiology back 90 years. The relatively recent discovery of glial-neural interactions that could modify synaptic strength or initiate changes in neuronal excitability (Pasti et al., 1997; Perea et al., 2009; Halassa and Haydon, 2010) were revolutionary, requiring a modern reexamination of the role of glial cells in brain function (Agulhon et al., 2010; Smith, 2010).
However, the role of glia in regulating neuronal function is probably more conditional and circumstantial than has been conceived recently. Some of the earlier reports of glial-neural interactions involved strong activation of astrocytes in combined glial-neural culture systems, manipulations that could bias the preparation in favor of a demonstration of a glial effect on neurotransmission. A controversial follow-up study in the mouse strongly suggested that genetic manipulation of glial signaling pathways (and intrinsic glial “excitability”) had no significant effects on hippocampal neurotransmission (Agulhon et al., 2010). Although much of the controversy concerning the significance of glial-neural interactions is focused on hippocampus (Smith, 2010), a varied body of work has emerged showing that autonomic functions controlled by circuits in the medulla are powerfully modulated by inputs from chemosensing glial cells. Astrocytes, forming as they do the blood–brain barrier, are in the ideal position to provide autonomic control circuitry with essential chemosensor data. Glial detection of hypoglycemia, hypoxia, hypercapnia, markers of trauma (i.e., PAR), and immune activation may regulate the functions of adjacent autonomic control neurons. These effects, mediated by gliotransmission, can produce significant effects on gastric motility, respiration, and cardiovascular control (Hermann and Rogers, 2009; Funk, 2010; Gourine et al., 2010; Gourine and Kasparov, 2011; Lin et al., 2013; McDougal et al., 2013). Furthermore, autonomic glial-neural interactions are not unidirectional. There is now evidence to suggest that the responsiveness of “autonomic” astrocytes in the hindbrain can be regulated by visceral afferent neuronal inputs (McDougal et al., 2011; Accorsi-Mendonça et al., 2013). Astrocytes in the hindbrain are therefore emerging as full partners with neurons in exerting integrated control over critical autonomic functions (Agulhon et al., 2013).
Footnotes
The authors declare no competing financial interests.
This work was supported by the National Institutes of Health (Grant T32-AT004094 to K.M.V. and Grant NS60664 to R.C.R.).
References
- Accorsi-Mendonça D, Zoccal DB, Bonagamba LG, Machado BH. Glial cells modulate the synaptic transmission of NTS neurons sending projections to ventral medulla of Wistar rats. Physiol Rep. 2013;1:e00080. doi: 10.1002/phy2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science. 2010;327:1250–1254. doi: 10.1126/science.1184821. [DOI] [PubMed] [Google Scholar]
- Agulhon C, Boyt KM, Xie AX, Friocourt F, Roth BL, McCarthy KD. Modulation of the autonomic nervous system and behaviour by acute glial cell Gq protein-coupled receptor activation in vivo. J Physiol. 2013;591:5599–5609. doi: 10.1113/jphysiol.2013.261289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akindipe OA, Faul JL, Vierra MA, Triadafilopoulos G, Theodore J. The surgical management of severe gastroparesis in heart/lung transplant recipients. Chest. 2000;117:907–910. doi: 10.1378/chest.117.3.907. [DOI] [PubMed] [Google Scholar]
- Almonte AG, Hamill CE, Chhatwal JP, Wingo TS, Barber JA, Lyuboslavsky PN, David Sweatt J, Ressler KJ, White DA, Traynelis SF. Learning and memory deficits in mice lacking protease activated receptor-1. Neurobiol Learn Mem. 2007;88:295–304. doi: 10.1016/j.nlm.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almonte AG, Qadri LH, Sultan FA, Watson JA, Mount DJ, Rumbaugh G, Sweatt JD. Protease-activated receptor-1 modulates hippocampal memory formation and synaptic plasticity. J Neurochem. 2013;124:109–122. doi: 10.1111/jnc.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/S0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG. Dynamic signaling between astrocytes and neurons. Annu Rev Physiol. 2001;63:795–813. doi: 10.1146/annurev.physiol.63.1.795. [DOI] [PubMed] [Google Scholar]
- Araque A, Martín ED, Perea G, Arellano JI, Buño W. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J Neurosci. 2002;22:2443–2450. doi: 10.1523/JNEUROSCI.22-07-02443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz N, Schulman LL, McGregor C, Markowitz D. Gastroparesis after lung transplantation–potential role in postoperative respiratory complications. Chest. 1995;108:1602–1607. doi: 10.1378/chest.108.6.1602. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Bonansco C, Couve A, Perea G, Ferradas CÁ, Roncagliolo M, Fuenzalida M. Glutamate released spontaneously from astrocytes sets the threshold for synaptic plasticity. Eur J Neurosci. 2011;33:1483–1492. doi: 10.1111/j.1460-9568.2011.07631.x. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe KY, Olson JE, Bourque CW. Taurine release by astrocytes modulates osmosensitive glycine receptor tone and excitability in the adult supraoptic nucleus. J Neurosci. 2012;32:12518–12527. doi: 10.1523/JNEUROSCI.1380-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke DD. Fluoroacetate and fluorocitrate–mechanism of action. Neurochem Res. 1991;16:1055–1058. doi: 10.1007/BF00965850. [DOI] [PubMed] [Google Scholar]
- Coco S, Calegari F, Pravettoni E, Pozzi D, Taverna E, Rosa P, Matteoli M, Verderio C. Storage and release of ATP from Astrocytes in culture. J Biol Chem. 2003;278:1354–1362. doi: 10.1074/jbc.M209454200. [DOI] [PubMed] [Google Scholar]
- Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- Edman S, McKay S, Macdonald LJ, Samadi M, Livesey MR, Hardingham GE, Wyllie DJ. TCN 201 selectively blocks GluN2A-containing NMDARs in a GluN1 co-agonist dependent but non-competitive manner. Neuropharmacology. 2012;63:441–449. doi: 10.1016/j.neuropharm.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RJ, Derkach V, Surprenant A. ATP mediates fast synaptic transmission in mammalian neurons. Nature. 1992;357:503–505. doi: 10.1038/357503a0. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Funk GD. The ‘connexin’ between astrocytes, ATP and central respiratory chemoreception. J Physiol. 2010;588:4335–4337. doi: 10.1113/jphysiol.2010.200196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrick T, Mulvihill S, Buack S, Maeda-Hagiwara M, Tache Y. Intracerebroventricular pressure inhibits gastric antral and duodenal contractility but not acid-secretion in conscious rabbits. Gastroenterology. 1988;95:26–31. doi: 10.1016/0016-5085(88)90286-7. [DOI] [PubMed] [Google Scholar]
- Gingrich MB, Traynelis SF. Serine proteases and brain damage–is there a link? Trends Neurosci. 2000;23:399–407. doi: 10.1016/S0166-2236(00)01617-9. [DOI] [PubMed] [Google Scholar]
- Gourine AV, Kasparov S. Astrocytes as brain interoceptors. Exp Physiol. 2011;96:411–416. doi: 10.1113/expphysiol.2010.053165. [DOI] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K, Kasparov S. Astrocytes control breathing through pH-dependent release of ATP. Science. 2010;329:571–575. doi: 10.1126/science.1190721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guard DB, Swartz TD, Ritter RC, Burns GA, Covasa M. NMDA NR2 receptors participate in CCK-induced reduction of food intake and hindbrain neuronal activation. Brain Res. 2009a;1266:37–44. doi: 10.1016/j.brainres.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Guard DB, Swartz TD, Ritter RC, Burns GA, Covasa M. Blockade of hindbrain NMDA receptors containing NR2 subunits increases sucrose intake. Am J Physiol Regul Integr Comp Physiol. 2009b;296:R921–R928. doi: 10.1152/ajpregu.90456.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sánchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Hamill CE, Mannaioni G, Lyuboslavsky P, Sastre AA, Traynelis SF. Protease-activated receptor 1-dependent neuronal damage involves NMDA receptor function. Exp Neurol. 2009;217:136–146. doi: 10.1016/j.expneurol.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han KS, Woo J, Park H, Yoon BJ, Choi S, Lee CJ. Channel-mediated astrocytic glutamate release via Bestrophin-1 targets synaptic NMDARs. Mol Brain. 2013;6:4. doi: 10.1186/1756-6606-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Traynelis SF. Subunit-selective allosteric inhibition of glycine binding to NMDA receptors. J Neurosci. 2012;32:6197–6208. doi: 10.1523/JNEUROSCI.5757-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Harris RA. The anesthetic mechanism of urethane: the effects on rotransmitter-gated ion channels. Anesth Analg. 2002;94:313–318. doi: 10.1097/00000539-200202000-00015. [DOI] [PubMed] [Google Scholar]
- Hassel B, Paulsen RE, Johnsen A, Fonnum F. Selective inhibition of glial cell metabolism in vivo by fluorocitrate. Brain Res. 1992;576:120–124. doi: 10.1016/0006-8993(92)90616-H. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Rogers RC. TNF activates astrocytes and catecholaminergic neurons in the solitary nucleus: implications for autonomic control. Brain Res. 2009;1273:72–82. doi: 10.1016/j.brainres.2009.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Van Meter MJ, Rood JC, Rogers RC. Proteinase-activated receptors in the nucleus of the solitary tract: evidence for glial-neural interactions in autonomic control of the stomach. J Neurosci. 2009;29:9292–9300. doi: 10.1523/JNEUROSCI.6063-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Viard E, Rogers RC. Hindbrain glucoprivation effects on gastric vagal reflex circuits and gastric motility in the rat are suppressed by the astrocyte inhibitor fluorocitrate. J Neurosci. 2014;34:10488–10496. doi: 10.1523/JNEUROSCI.1406-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huda R, McCrimmon DR, Martina M. pH modulation of glial glutamate transporters regulates synaptic transmission in the nucleus of the solitary tract. J Neurophysiol. 2013;110:368–377. doi: 10.1152/jn.01074.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge CE, Sugawara T, Mannaioni G, Alagarsamy S, Conn PJ, Brat DJ, Chan PH, Traynelis SF. The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. Proc Natl Acad Sci U S A. 2003;100:13019–13024. doi: 10.1073/pnas.2235594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge CE, Lee CJ, Hubbard KB, Zhang Z, Olson JJ, Hepler JR, Brat DJ, Traynelis SF. Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp Neurol. 2004;188:94–103. doi: 10.1016/j.expneurol.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Katz E, Protti DA, Ferro PA, Rosato Siri MD, Uchitel OD. Effects of Ca2+ channel blocker neurotoxins on transmitter release and presynaptic currents at the mouse neuromuscular junction. Br J Pharmacol. 1997;121:1531–1540. doi: 10.1038/sj.bjp.0701290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson GM, Koch S, O'Dorisio TM, Osadchey B, McGraw P, Richardson JD. Gastric response to severe head injury. Am J Surg. 1984;147:97–105. doi: 10.1016/0002-9610(84)90041-2. [DOI] [PubMed] [Google Scholar]
- Lavrentieva A, Kontakiotis T, Bitzani M, Papaioannou-Gaki G, Parlapani A, Thomareis O, Tsotsolis N, Giala MA. Early coagulation disorders after severe burn injury: impact on mortality. Intensive Care Med. 2008;34:700–706. doi: 10.1007/s00134-007-0976-5. [DOI] [PubMed] [Google Scholar]
- Lee CJ, Mannaioni G, Yuan H, Woo DH, Gingrich MB, Traynelis SF. Astrocytic control of synaptic NMDA receptors. J Physiol London. 2007;581:1057–1081. doi: 10.1113/jphysiol.2007.130377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin LH, Moore SA, Jones SY, McGlashon J, Talman WT. Astrocytes in the rat nucleus tractus solitarii are critical for cardiovascular reflex control. J Neurosci. 2013;33:18608–18617. doi: 10.1523/JNEUROSCI.3257-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wong-Riley MT. Postnatal development of N-methyl-d-aspartate receptor subunits 2A, 2B, 2C, 2D, and 3B immunoreactivity in brain stem respiratory nuclei of the rat. Neuroscience. 2010;171:637–654. doi: 10.1016/j.neuroscience.2010.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WY, Rhoney DH, Boling WB, Johnson JD, Johnson D, Smith TC. A review of stress ulcer prophylaxis in the neurosurgical intensive care unit. Neurosurgery. 1997;41:416–425. doi: 10.1097/00006123-199708000-00017. discussion 425–426. [DOI] [PubMed] [Google Scholar]
- Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- Maggio N, Shavit E, Chapman J, Segal M. Thrombin induces long-term potentiation of reactivity to afferent stimulation and facilitates epileptic seizures in rat hippocampal slices: toward understanding the functional consequences of cerebrovascular insults. J Neurosci. 2008;28:732–736. doi: 10.1523/JNEUROSCI.3665-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio N, Itsekson Z, Dominissini D, Blatt I, Amariglio N, Rechavi G, Tanne D, Chapman J. Thrombin regulation of synaptic plasticity: Implications for physiology and pathology. Exp Neurol. 2013;247:595–604. doi: 10.1016/j.expneurol.2013.02.011. [DOI] [PubMed] [Google Scholar]
- Mannaioni G, Orr AG, Hamill CE, Yuan H, Pedone KH, McCoy KL, Berlinguer Palmini R, Junge CE, Lee CJ, Yepes M, Hepler JR, Traynelis SF. Plasmin potentiates synaptic NMDA receptor function in hippocampal neurons through activation of PAR1. J Biol Chem. 2008;283:20600–20611. doi: 10.1074/jbc.M803015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín ED, Fernández M, Perea G, Pascual O, Haydon PG, Araque A, Ceña V. Adenosine released by astrocytes contributes to hypoxia-induced modulation of synaptic transmission. Glia. 2007;55:36–45. doi: 10.1002/glia.20431. [DOI] [PubMed] [Google Scholar]
- McDougal DH, Hermann GE, Rogers RC. Vagal afferent stimulation activates astrocytes in the nucleus of the solitary tract via AMPA receptors: evidence of an atypical neural-glial interaction in the brainstem. J Neurosci. 2011;31:14037–14045. doi: 10.1523/JNEUROSCI.2855-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougal DH, Hermann GE, Rogers RC. Astrocytes in the nucleus of the solitary tract are activated by low glucose or glucoprivation: evidence for glial involvement in glucose homeostasis. Front Neurosci. 2013;7:249. doi: 10.3389/fnins.2013.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Parent AT, Wolosker H, Brady RO, Jr, Linden DJ, Ferris CD, Rogawski MA, Snyder SH. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 2000;97:4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niclou SP, Suidan HS, Pavlik A, Vejsada R, Monard D. Changes in the expression of protease-activated receptor 1 and protease nexin-1 mRNA during rat nervous system development and after nerve lesion. Eur J Neurosci. 1998;10:1590–1607. doi: 10.1046/j.1460-9568.1998.00183.x. [DOI] [PubMed] [Google Scholar]
- Nicole O, Goldshmidt A, Hamill CE, Sorensen SD, Sastre A, Lyuboslavsky P, Hepler JR, McKeon RJ, Traynelis SF. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J Neurosci. 2005;25:4319–4329. doi: 10.1523/JNEUROSCI.5200-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KK, Traynelis SF. New advances in NMDA receptor pharmacology. Trends Pharmacol Sci. 2011;32:726–733. doi: 10.1016/j.tips.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SJ, Han KS, Park H, Woo DH, Kim HY, Traynelis SF, Lee CJ. Protease activated receptor 1-induced glutamate release in cultured astrocytes is mediated by Bestrophin-1 channel but not by vesicular exocytosis. Mol Brain. 2012;5:38. doi: 10.1186/1756-6606-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EE, Lyuboslavsky P, Traynelis SF, McKeon RJ. PAR-1 deficiency protects against neuronal damage and neurologic deficits after unilateral cerebral hypoxia/ischemia. J Cereb Blood Flow Metab. 2004;24:964–971. doi: 10.1097/01.WCB.0000128266.87474.BF. [DOI] [PubMed] [Google Scholar]
- Park H, Oh SJ, Han KS, Woo DH, Park H, Mannaioni G, Traynelis SF, Lee CJ. Bestrophin-1 encodes for the Ca2+-Activated anion channel in hippocampal astrocytes. J Neurosci. 2009;29:13063–13073. doi: 10.1523/JNEUROSCI.3193-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Han KS, Oh SJ, Jo S, Woo J, Yoon BE, Lee CJ. High glutamate permeability and distal localization of Best1 channel in CA1 hippocampal astrocyte. Mol Brain. 2013;6:54. doi: 10.1186/1756-6606-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte neuron signaling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: A highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32:421–431. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scemes E, Giaume C. Astrocyte calcium waves: what they are and what they do. Glia. 2006;54:716–725. doi: 10.1002/glia.20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K. Neuroscience: settling the great glia debate. Nature. 2010;468:160–162. doi: 10.1038/468160a. [DOI] [PubMed] [Google Scholar]
- Sodhi SS, Guo JP, Maurer AH, O'Brien G, Srinivasan R, Parkman HP. Gastroparesis after combined heart and lung transplantation. J Clin Gastroenterol. 2002;34:34–39. doi: 10.1097/00004836-200201000-00007. [DOI] [PubMed] [Google Scholar]
- Striggow F, Riek-Burchardt M, Kiesel A, Schmidt W, Henrich-Noack P, Breder J, Krug M, Reymann KG, Reiser G. Four different types of protease-activated receptors are widely expressed in the brain and are up-regulated in hippocampus by severe ischemia. Eur J Neurosci. 2001;14:595–608. doi: 10.1046/j.0953-816x.2001.01676.x. [DOI] [PubMed] [Google Scholar]
- Tanahashi S, Yamamura S, Nakagawa M, Motomura E, Okada M. Clozapine, but not haloperidol, enhances glial d-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br J Pharmacol. 2012;165:1543–1555. doi: 10.1111/j.1476-5381.2011.01638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Trejo J. Protease-activated receptor signaling: new roles and regulatory mechanisms. Curr Opin Hematol. 2007;14:230–235. doi: 10.1097/MOH.0b013e3280dce568. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ubl JJ, Reiser G. Four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signaling. Glia. 2002;37:53–63. doi: 10.1002/glia.10012. [DOI] [PubMed] [Google Scholar]
- Weinstein JR, Gold SJ, Cunningham DD, Gall CM. Cellular-localization of thrombin receptor messenger RNA in rat brain–expression by mesencephalic dopaminergic neurons and codistribution with prothrombin messenger RNA. J Neurosci. 1995;15:2906–2919. doi: 10.1523/JNEUROSCI.15-04-02906.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- Young B, Ott L, Yingling B, McClain C. Nutrition and brain injury. J Neurotrauma. 1992;9:S375–S383. [PubMed] [Google Scholar]
- Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B, Augustine GJ, Deisseroth K, Luo M, Graybiel AM, Feng G. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods. 2011;8:745–752. doi: 10.1038/nmeth.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]