

Abstract
Omega-3 fatty acids (n-3 FAs) are proposed to have many beneficial effects on human health. However, the mechanisms underlying their potential cancer preventative effects are unclear. G protein–coupled receptors (GPCRs) of the free fatty acid receptor (FFAR) family, FFA1/GPR40 and FFA4/GPR120, specifically bind n-3 FAs as agonist ligands. In this study, we examined the effects of n-3 FAs in human prostate cancer cell lines. Initial studies established that the long-chain n-3 FAs, eicosapentaenoic acid (EPA) and docosahexaenoic acid, inhibit proliferation of DU145 cells in response to lysophosphatidic acid (LPA), a mitogenic lipid mediator. When added alone to serum-starved DU145 cells, EPA transiently activates signaling events, including p70S6K phosphorylation. However, when added 15 minutes prior to LPA, EPA suppresses LPA-induced activating phosphorylations of ERK, FAK, and p70S6K, and expression of the matricellular protein CCN1. The rapid onset of the inhibitory action of EPA suggested involvement of a GPCR. Further studies showed that DU145 and PC-3 cells express mRNA and protein for both FFA4 and FFA1. TUG-891 (4-[(4-fluoro-4′-methyl[1,1′-biphenyl]-2-yl)methoxy]-benzenepropanoic acid), a selective agonist for FFA4, exerts inhibitory effects on LPA- and epidermal growth factor–induced proliferation and migration, similar to EPA, in DU145 and PC-3 cells. The effects of TUG-891 and EPA are readily reversible. The FFA1/FFA4 agonist GW9508 (4-[[(3-phenoxyphenyl)methyl]amino]-benzenepropranoic acid) likewise inhibits proliferation at doses that block FFA4. Knockdown of FFA4 expression prevents EPA- and TUG-891–induced inhibition of growth and migration. Together, these results indicate that activation of FFA4 initiates signaling events that can inhibit growth factor–induced signaling, providing a novel mechanism for suppression of cancer cell proliferation.
Introduction
Consumption of omega-3 fatty acids (n-3 FAs), as dietary constituents or supplements, has been linked to various positive health outcomes, particularly with respect to cardiovascular health (Calder and Yaqoob, 2009). With respect to cancer prevention or therapy, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) have been shown to suppress the growth of carcinoma cells in culture, including prostate cancer cells (Chung et al., 2001; Wang et al., 2012). Overexpression of FAT1, which converts n-6 to n-3 FAs, inhibits proliferation of DU145 and PC-3 prostate cancer cells (Lu et al., 2008). Inhibitory effects of n-3 FAs on prostate tumor growth in animal models have also been reported (Brown et al., 2006; Akinsete et al., 2012). Whether omega-3 fatty acid supplements are of benefit for prostate cancer prevention, based on human epidemiologic data, is quite controversial (Maclean et al., 2006; Gerber, 2012; Brasky et al., 2013). Nonetheless, the inhibitory effects of n-3 FAs on prostate cancer cells in culture suggest interference with growth factor signaling, which is worthy of further investigation.
The mechanisms underlying the potential cancer preventative effects of omega-3 fatty acids at the molecular level have not been firmly established (Gu et al., 2013a). Although numerous mechanisms have been considered, the prevailing hypothesis is that metabolites of n-3 FAs are responsible for the anticancer effects. It is known that uptake of omega-3 fatty acids modifies membrane phospholipid composition in a manner that decreases production of arachidonic acid (n-6)–derived eicosanoids that are proinflammatory (Norris and Dennis, 2012), and increases n-3 metabolites (e.g., “resolvins”) that are anti-inflammatory (Zhang and Spite, 2012). For example, prostaglandin E2 levels are decreased in prostate tumors following dietary supplementation with n-3 FAs in mice (Kobayashi et al., 2006). Thus, effects of n-3 fatty acids are usually tested after long-term exposure (e.g., overnight treatment in cell culture, or dietary interventions). More rapid effects have also been reported. For example, fatty acid derivatives can alter the organization of membrane microdomains (Ibarguren et al., 2013). One study suggests that modification of the FA content of phosphatidylinositol phosphates is responsible for some of the inhibitory effects on prostate cancer cells (Gu et al., 2013b). Other reported molecular targets for n-3 FAs include the nuclear receptor peroxisome proliferator–activated receptor-γ (Edwards and O’Flaherty, 2008).
Recently, omega-3 fatty acids were shown to act as agonist ligands for FFA1/GPR40 and FFA4/GPR120, G protein–coupled receptors (GPCRs) in the free fatty acid receptor (FFAR) family (Oh et al., 2010). This finding has been of particular interest with respect to the beneficial effects of omega-3 fatty acids on metabolism (Hara et al., 2011; Talukdar et al., 2011), but has not yet been fully explored with respect to cancer prevention or treatment. The roles of GPCRs in regulating growth and invasion of cancer cells are of increasing interest from a therapeutic standpoint (O’Hayre et al., 2014). GPCR ligands initiate cellular responses within minutes, in contrast to the longer-term effects of n-3 FAs studied previously with respect to lipid composition. However, responses to GPCR agonists can also be sustained over time. Both FFA1 and FFA4 are coupled to Gq/11 as well as arrestin-3, but many of their actions appear to be arrestin-mediated (Mancini and Poitout, 2013; Butcher et al., 2014). Selective agonists for FFA1 and FFA4 are rapidly being developed (Sun et al., 2010; Hudson et al., 2013b; Du et al., 2014), making it possible to test whether such agents elicit therapeutic effects of interest.
In the current study, we tested whether omega-3 fatty acids exert short-term inhibitory effects on growth factor signaling in human prostate cancer cell lines, and found that this was the case. We then used FFAR agonists to investigate whether GPCRs are responsible for the inhibitory effects observed. Lysophosphatidic acid (LPA) was used as the primary growth factor, based on the experience of our research group in studying LPA response (Xie et al., 2002; Gibbs et al., 2009) and on the importance of this lipid mediator in prostate cancer cells (Wang et al., 2004; Bektas et al., 2005; Guo et al., 2006; Choi et al., 2010). Our study also characterizes the effects of LPA and n-3 FAs on induction of CCN1, a matricellular protein that facilitates proliferation and migration (Jun and Lau, 2011). Together, the results raise new possibilities for mechanisms by which n-3 FAs can inhibit cancer cell growth and survival.
Materials and Methods
EPA and DHA (prepared in ethanol) were from Cayman Chemical (Ann Arbor, MI). The FFAR agonists TUG-891 (4-[(4-fluoro-4′-methyl[1,1′-biphenyl]-2-yl)methoxy]-benzenepropanoic acid; prepared in dimethylsulfoxide) and GW9508 (4-[[(3-phenoxyphenyl)methyl]amino]-benzenepropranoic acid; prepared in ethanol) were from Millipore (Billerica, MA) and Cayman Chemical, respectively. Vehicle controls for FFAR agonists had a final concentration of 0.03% (v/v) ethanol. LPA (18:1; oleoyl) was obtained from Avanti Polar Lipids (Birmingham, AL), and was delivered to cells as a 1000× stock solution that had been prepared in 4 mg/ml fatty acid–free bovine serum albumin (BSA). Vehicle controls for LPA had a final concentration of 4 µg/ml BSA. Epidermal growth factor (EGF) was from Sigma-Aldrich (St. Louis, MO). Anti-CCN1 (1:1000 dilution) was from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies recognizing the phosphorylated forms of p70S6 kinase (Thr389) (1:1000 dilution), extracellular signal-regulated kinase (ERK) (T202/Y204) (1:1000 dilution), and Akt (Ser473) (1:1000 dilution) were obtained from Cell Signaling Technologies (Beverly, MA). Antibody recognizing human focal adhesion kinase (FAK) phosphorylated at pY397 (1:1000 dilution) was obtained from BD Transduction Laboratories (Lexington, KY), as was anti-actin, which was used at a 1:5000 dilution. Anti-FFA4 was purchased from Abcam (Cambridge, MA) and used at a 1:1000 dilution. Goat anti-rabbit secondary antibody was purchased from Sigma-Aldrich and used at a 1:20,000 dilution, whereas goat anti-mouse secondary antibody was purchased from Invitrogen/Life Technologies (Grand Island, NY) and used at a 1:5000 dilution.
Cell Culture.
DU145, PC-3, LNCaP, and Caco-2 cells were obtained from the American Type Culture Collection (Manassas, VA). The prostate cancer cells were grown in RPMI 1640 medium with 10% fetal bovine serum (FBS; Hyclone/Thermo Fisher Scientific, Waltham, MA). Caco-2 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS (Invitrogen/Life Technologies). All cell lines were grown in an incubator at 37°C in 5% CO2 on tissue culture plastic.
Cell Proliferation Assays.
DU145 cells were seeded in six-well plates at 3 × 105 cells/well in serum-containing medium. After 1 day, the medium was generally changed to RPMI 1640 without serum. For some experiments testing serum response, cells were maintained continuously in serum. On the next day, the medium was changed to RPMI 1640 with 10% FBS, 10 µM LPA, or 10 nM EGF, in the absence or presence of 20 µM DHA, 20 µM EPA, or various concentrations of TUG-891 or GW9508. Control cells were incubated with the appropriate vehicle (0.03% ethanol, v/v). Duplicate wells were prepared for each experimental condition. Cell numbers were evaluated after 24, 48, and 72 hours by removing medium, incubating cells with trypsin/EDTA for 5 minutes, adding trypan blue, and counting the suspended live cells (excluding trypan blue) using a hemacytometer.
Cell Migration Assays.
Cell migration was assessed using a modified Boyden chamber method, as previously described (Zhang et al., 2011). Cells were serum starved for 24 hours and then seeded in serum-free medium at 2.5 × 104 cells per insert in the upper chambers of 8-μm transwell inserts (BD Biosciences, San Jose, CA). Cells were then treated with 10% FBS, 20 μM EPA, 1 μM TUG-891, 10 μM LPA, or 10 nM EGF, either alone or in combination. Serum-free medium was added to the lower wells. Following a 6-hour migration period, the insert membranes were fixed and stained using methanol and crystal violet. Cells that invaded the lower chambers were counted by microscopy.
Reverse-Transcription Polymerase Chain Reaction.
RNA from cells was isolated using an RNeasy Mini kit (Qiagen, Valencia, CA). First-strand cDNA was synthesized with SuperScript II Reverse Transcriptase (Invitrogen) following the manufacturer’s instructions using 20 μl of reaction mixture containing 2 μg of RNA. Polymerase chain reaction (PCR) was carried out using Platinum Pfx DNA Polymerase (Invitrogen) and Integrated DNA Technology (San Diego, CA) probes: FFA4 (Forward: 5′-CCTGGAGAGATCTCGTGGGA-3′; Reverse: 5′-AGGAGGTGTTCCGAGGTCTG-3′); FFA1 (Forward: 5′-CTCCTTCGGCCTCTATGTGG-3′; Reverse: 5′-AGACCAGGCTAGGGGTGAGA-3′); RPLP0 (Forward: 5′-CGCTATCCGCGGTTTCTGAT-3′; Reverse: 5′-AGACGATGTCACTTCCACGA-3′). Following the manufacturer’s instructions, 5 μg of cDNA template was used for each reaction. In brief, three-step cycling was used for 35 cycles, with denaturing at 94°C for 15 seconds, annealing at 55°C for 30 seconds, and extending at 68°C for 60 seconds. Products were separated by ethidium bromide agarose gel electrophoresis, and were then imaged using a ChemiDoc with Image Laboratory software (Bio-Rad, Hercules, CA).
Cell Incubations for Signal Transduction Assays.
Cells were grown in F12K or RPMI 1640 medium supplemented with 10% FBS until ∼80% confluent. Cells were serum starved for 24 hours in F12K or RPMI 1640 medium, then incubated with 10 µM LPA, 10 nM EGF, 20 µM DHA, and/or various concentrations of EPA, TUG-891, or GW9508 for various times. Cells were rinsed twice with ice-cold phosphate-buffered saline, harvested by scraping into 1 ml of ice-cold phosphate-buffered saline, collected by centrifugation at 10,000g for 10 minutes at 4°C, and resuspended in ice-cold lysis buffer [20 mM HEPES (pH = 7.4), 1% Triton X-100, 50 mM NaCl, 2 mM EGTA, 5 mM β-glycerophosphate, 30 mM sodium pyrophosphate, 100 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml aprotinin, 10 µg/ml leupeptin]. Insoluble debris was removed after centrifugation. Protein concentrations of these “whole-cell extracts” were determined using a Coomassie binding assay (Pierce Chemical, Rockford, IL).
Immunoblotting.
Whole-cell extracts containing equal amounts of protein (30 µg) were separated by SDS-PAGE on 10% Laemmli gels, transferred to nitrocellulose, and incubated with primary (overnight at 4°C) and then secondary (1–2 hours at room temperature) antibodies. Blots were developed using enhanced chemiluminescence (GE Healthcare, Piscataway, NJ) and imaged using a Gel Doc system (Bio-Rad). Protein expression was quantified by densitometry using Quantity One software (Bio-Rad). Results were normalized to the actin loading control, and then to the value obtained for untreated control cells.
RNA Interference.
Predesigned human Silencer Select small interfering RNA (siRNA) specific to FFA4 (s50346) was purchased from Life Technologies, along with Opti-MEM reduced serum medium and Lipofectamine 3000 reagent. The siRNA was resuspended in RNase-free water at a final concentration of 60 μM and stored at −20°C. AllStars Negative Control siRNA oligos were purchased from Qiagen and were used for all negative controls. Per the manufacturer’s directions for RNAi transfections, cells were plated in 1.7 ml of medium per well in RPMI medium plus 10% FBS without antibiotics in six-well plates 1 day before transfection. At the time of transfection, cells were approximately 50% confluent. For each transfection, two mixtures were prepared: 1) duplex siRNA added to 150 μl of Opti-MEM, and 2) 4 µl of Lipofectamine 3000 added to 150 µl of Opti-MEM. The two solutions were gently mixed and then incubated at room temperature for 5 minutes. Final concentrations of siRNA were 180 nM. The mixtures were added to each well and cells were cultured for an additional 24–72 hours. For LPA- and EGF-treated cells, the same procedure was followed, except cells were seeded in six-well plates 2 days before transfection and serum starved overnight the day before transfection.
Statistical Analysis.
Data were analyzed by two-way analysis of variance (ANOVA) followed by either Tukey's or Sidak’s multiple comparisons test. The only exceptions were assays in which there was only one time point (e.g., migration assays), which were analyzed by one-way ANOVA followed by Tukey’s multiple comparisons test. All analyses were performed using Prism software (GraphPad Software, San Diego, CA). The data presented are representative of at least two experiments, unless otherwise indicated in the figure legends.
Results
Effects of EPA and DHA on DU145 Cell Proliferation.
DU145 cells were serum starved for 24 hours to remove serum growth factors (including LPA), then treated with 10% serum, 10 µM LPA, and/or 20 µM DHA or 20 µM EPA for 24, 48, and 72 hours. The doses initially used for EPA and DHA were selected based on a literature survey; detailed dose-response data will be presented later. Notably, the doses used here are considerably lower than those used in previous studies with prostate cancer cells (Chung et al., 2001; Wang et al., 2012). Ethanol was used as a vehicle control for EPA/DHA, and BSA as a vehicle control for LPA; neither vehicle affected responses at the concentrations used (data not shown).
As shown in Fig. 1, LPA significantly stimulates proliferation of DU145 cells. Compared with serum-free control cells, the number of viable cells was decreased by treatment with EPA, but not by DHA, at 48 and 72 hours. However, cell number was not reduced below the number initially seeded in the dish, and the number of nonviable cells was not increased, indicating that EPA inhibits proliferation and does not cause cell death. Cells achieved confluence after 72 hours in the presence of serum; cells maintained in medium without serum remained viable for 72 hours (data not shown). Both EPA and DHA block LPA-induced proliferation in DU145 cells. However, since EPA was consistently more efficacious than DHA in this and other assays (data not shown), subsequent studies focused on the effects of EPA.
Fig. 1.

Effects of EPA and DHA on proliferation of DU145 cells. DU145 cells were serum starved overnight and then incubated in duplicate wells for the indicated times in the absence or presence of LPA (10 µM) or 10% fetal bovine serum in the absence or presence of 20 µM EPA or DHA. Live cells were counted. Each data point represents the mean ± S.E.M. for values from triplicate experiments. (A) The time course of effects of serum, DHA, and EPA alone. *P values for control versus serum; #P values for control versus EPA (control versus DHA had no significant difference). (B) The time course of the effects of LPA, LPA plus DHA, and LPA plus EPA. *P values for LPA versus LPA + EPA; #P value for LPA versus LPA + DHA. (C) Results for the 48-hour incubation. All panels show data derived from the same three experiments. Data for (A) and (B) were analyzed by two-way ANOVA followed by Tukey’s multiple comparisons test; data for (C) were analyzed by one-way ANOVA followed by Tukey’s multiple comparisons test. #P < 0.05; **,##P < 0.01; ****,####P < 0.0001.
Effects of EPA on LPA-Induced Signal Transduction.
We reasoned that inhibitory effects of EPA on proliferation would likely be reflected in the effects on growth factor–induced signal transduction. Since we had previously characterized signaling responses to LPA in DU145 cells (Gibbs et al., 2009), we tested whether EPA influences representative LPA responses. A 15-minute pretreatment with EPA was used to screen for effects that might be receptor-mediated.
The first response tested was activation of the Erk mitogen-activated protein kinase pathway, which plays an important role in LPA-induced proliferation in prostate cancer cells (Kue et al., 2002; Gibbs et al., 2009). A time course was conducted in which cells were preincubated with 20 µM EPA for 15 minutes, then incubated with LPA for various times. As shown in Fig. 2, A and B, EPA inhibits LPA-induced Erk phosphorylation in DU145 cells.
Fig. 2.

Effects of EPA on LPA-induced phosphorylation of Erk and FAK in DU145 cells. (A) Serum-starved DU145 cells were incubated with or without 20 µM EPA for 15 minutes. LPA (10 µM) was then added for the indicated times. Whole-cell extracts, equalized for protein, were immunoblotted for phospho-Erk (p-Erk) and for actin (loading control). (B) Immunoblotting results were quantified from two separate experiments. (C) Serum-starved DU145 cells were incubated with or without 20 µM EPA for 15 minutes. LPA (10 µM) was then added for the indicated times. Whole-cell extracts, equalized for protein, were immunoblotted for phospho-FAK (p-FAK) and for actin. The actin blot was the same as shown for (A), since (A) and (C) were from the same experiment. (D) Immunoblotting results were quantified from two separate experiments. For (B) and (D), results were normalized to actin, and then to the untreated control. Each value represents the mean ± S.E.M. from duplicate experiments. Data for (B) and (D) were analyzed by two-way ANOVA followed by Tukey’s multiple comparisons test. Asterisks denote values that are significantly different from untreated control (*P < 0.05; **P < 0.01; ***P < 0.001).
We next examined tyrosine phosphorylation of FAK, another response elicited by LPA in prostate cancer cells (Gibbs et al., 2009). EPA inhibits LPA-induced FAK phosphorylation over the 6-hour time course of the experiment (Fig. 2, C and D).
The effects of EPA on activation of p70S6K, which is activated via the phosphatidylinositol 3-kinase (PI3K)/Akt/mTORC1 pathway, were investigated. Signaling through this pathway is frequently upregulated in prostate cancer (Morgan et al., 2009). As shown in Fig. 3A, LPA stimulates the activating phosphorylation of p70S6K within 10 minutes, after which the response gradually declines over 6 hours. LPA also stimulates activating phosphorylation of phospho-Akt in these cells (data not shown). EPA suppresses LPA response throughout the time course. Figure 3B presents quantification, including the effects of EPA alone. EPA alone causes a transient partial activation of p70S6K (10 minutes). However, in marked contrast to LPA, EPA is not able to induce sustained activation of p70S6K. Because EPA alone has the ability to initiate transient signaling, significant inhibitory effects of EPA on LPA signaling are not always observed at early time points, when the stimulatory responses to EPA alone can obscure inhibitory effects on LPA signaling. In Fig. 3B, the difference between LPA alone and LPA plus EPA was only significant at 60 minutes, as analyzed by ANOVA.
Fig. 3.

Effects of EPA on LPA-induced phosphorylation of p70S6K in DU145 and Caco-2 cells. (A) Serum-starved DU145 cells were incubated with or without 20 µM EPA for 15 minutes. LPA (10 µM) was then added for the indicated times. Whole-cell extracts, equalized for protein, were immunoblotted for phospho-p70S6K (p-p70S6K) and for actin (loading control). (B) Immunoblotting results were quantified from experiments in which cells were incubated for the indicated times with EPA, LPA, or EPA plus LPA. Results were normalized to actin, and then to the untreated control. Each value represents the mean ± S.E.M. from duplicate experiments, except for EPA alone, which was from a single experiment. *Values that are significantly different from untreated control. (C) Serum-starved Caco-2 cells were incubated with or without 20 µM EPA or 10 µM LPA for the indicated times. Whole-cell extracts, equalized for protein, were immunoblotted for phospho-p70S6K and for actin (loading control). Immunoblotting results were quantified from three separate experiments. Results were normalized to actin, and then to the untreated control. Each value represents the mean ± S.E.M. from duplicate experiments. When there were two separate experiments for the same time point, the line on the graph was drawn through the mean of the data points. *P < 0.05.
We noted that the inhibitory effects of EPA on activation of the PI3K/Akt/p70S6K pathway in prostate cancer cells, as observed in our study, contrast with the role reported for FFAR agonists in some other cancer cell models (Wu et al., 2013). In the study by Wu and coworkers (2013), the FFA1/FFA4 agonist GW9508 was shown to rapidly stimulate Akt activation when applied alone to serum-starved colon cancer cells. We therefore tested the effects of LPA and EPA in Caco-2, a human colon cancer cell line that expresses FFA4 (Mobraten et al., 2013; Wu et al., 2013). LPA has been shown to induce proliferation of Caco-2 cells (Yun et al., 2005). Our data show that LPA increases activating phosphorylation of p70S6K in Caco-2 cells (Fig. 3C); similar results were seen for phospho-Akt (data not shown). However, the response to EPA alone was similar to or greater than the response to LPA (e.g., ∼4-fold increase in phospho-p70S6K at 30 minutes with EPA alone). Both magnitude and duration of the EPA response were in contrast with the weak effect of EPA alone in DU145 cells (Fig. 3B). The significant p70S6K response to EPA in Caco-2 interfered with efforts to determine whether EPA exerts an inhibitory influence in these cells, i.e., the response to a combination of EPA and LPA was approximately additive (data not shown). The stimulatory effect of EPA alone in Caco-2 cells is generally consistent with the GW9508-induced activation of Akt reported previously for other colon cancer cell lines (Wu et al., 2013).
The final signaling response assessed in prostate cancer cells was induction of CCN1. This response requires transcription and translation and is therefore a later response to growth factor action. Expression of CCN1 was first examined in the three most commonly used human prostate cancer cell lines: PC-3, LNCaP, and DU145. As shown in Fig. 4A, all three cell lines express CCN1 when grown in the presence of serum; levels are highest in DU145 cells, and lowest in the androgen-independent cell line LNCaP. Although CCN1 is eventually secreted, cellular levels were assessed in our experiments. LPA induces robust expression of CCN1 in PC-3 cells (Fig. 4B), a response that is blocked by the LPA receptor antagonist Ki16425 [3-(4-[4-([1-(2-chlorophenyl)ethoxy]carbonyl amino)-3-methyl-5-isoxazolyl] benzylsulfanyl) propanoic acid]. The time course of the effects of LPA on CCN1 levels is presented in Fig. 4C, where maximal induction was observed at 4 hours. These studies established that CCN1 is expressed in human prostate cancer cells, and that its expression can be induced by LPA. Figure 4, D and E, present data for DU145 cells, analogous to Figs. 2 and 3. LPA induces CCN1 expression in DU145 cells by 1 hour; this response is blocked by 20 µM EPA. EPA alone has no effect on CCN1 levels (Fig. 4F); this result was confirmed by a time course study in which DU145 cells were treated with EPA for up to 24 hours (data not shown).
Fig. 4.

Effects of LPA and EPA on CCN1 expression in prostate cancer cells. (A) Whole-cell extracts were prepared from PC-3, DU145, and LNCaP cells growing in serum. The extracts, equalized for protein, were immunoblotted for CCN1 and actin (loading control). (B) Serum-starved PC-3 cells were incubated for 4 hours with or without 10 µM LPA in the absence and presence of 10 µM Ki16425, a LPA receptor antagonist. Whole-cell extracts were immunoblotted for CCN1 and for total FAK (loading control). (C) Serum-starved PC-3 cells were incubated with 10 µM LPA for the indicated times. Whole-cell extracts were immunoblotted for CCN1 and actin (loading control). (D) Serum-starved DU145 cells were incubated with or without 20 µM EPA for 15 minutes. LPA (10 µM) was then added for the indicated times. Whole-cell extracts, equalized for protein, were immunoblotted for CCN1 and for actin (loading control). (E) Immunoblotting results, from experiments carried out as in (D), were quantified. Results were normalized to actin, and then to the untreated control. Each value represents the mean ± S.D. from duplicate experiments. *Values that are significantly different from the untreated control (P < 0.001). (F) The effects of EPA were quantified in experiments in which EPA alone was included as a control. Cells were incubated for 60 minutes with either LPA (10 µM) or EPA (20 µM), or with EPA for 15 minutes prior to incubation with LPA for an additional 60 minutes. Each data point represents the mean ± S.E.M. from duplicate experiments.
The effects of EPA on responses to another growth factor, EGF, were also tested. Previous work from our laboratory has shown that EGF and LPA have similar effects on signal transduction in human prostate cancer cells (Gibbs et al., 2009). Figure 5A illustrates the relative effects of serum, LPA, and EGF on proliferation of DU145 cells. Consistent with Fig. 1C, LPA is a weaker mitogen than fetal bovine serum, which contains a mixture of growth factors. EGF acts similarly to LPA. Figure 5B demonstrates that EPA inhibits activation of Erk and FAK in EGF-treated cells, whereas Fig. 5C shows that EPA also blocks induction of CCN1 in response to EGF. Our group has previously shown that EGF can increase production of LPA, resulting in transactivation of LPA receptors, in ovarian cancer cells (Snider et al., 2010). We therefore tested for receptor cross-talk in DU145 cells. As shown in Fig. 5D, the LPA receptor antagonist Ki16425 suppresses proliferation induced by either LPA or EGF, suggesting that LPA receptors play a role in EGF response in DU145. Similar results were obtained in PC-3, another human prostate cancer cell line (data not shown). Thus, the inhibitory effects of EPA on EGF signaling could conceivably be an indirect consequence of EPA-induced suppression of LPA signaling.
Fig. 5.

Effects of EPA on EGF responses in DU145 cells. (A) The effects of LPA (10 µM), EGF (10 nM), and fetal bovine serum (10%) on cell proliferation were assessed in a time course experiment, as described for Fig. 1; results of two separate experiments performed in duplicate are shown. (B) The effects of 10 nM EGF on activating phosphorylations of Erk and FAK (p-Erk and p-FAK) were examined by immunoblotting, as described for Figs. 2 and 3. (C) Serum-starved DU145 cells were incubated with and without 20 µM EPA for 15 minutes, followed by 10 nM EGF for 1 hour. Each data point represents the mean ± S.D. from duplicate experiments. (D) The effects of EGF and LPA on cell proliferation were assessed after 48 hours, in the presence and absence of 10 µM Ki16425, a LPA receptor antagonist. Each data point represents the mean ± S.D. of values from duplicate experiments, each performed with duplicate samples of cells.
Effects of FFAR Pharmacologic Agonists on Growth Factor Response.
The results presented earlier show that EPA can act rapidly (within 15 minutes) to inhibit mitogenic responses. This rapid action suggests a receptor-mediated response. The roles for FFARs were therefore investigated.
Two FFARs have been shown to bind long-chain n-3 FAs as agonists: FFA1/GPR40 and FFA4/GPR120. FFA4 is widely expressed, including in both normal and cancerous prostate tissue, according to a database search (www.oncomine.org), and thus seemed a likely candidate in prostate cancer cells. Reverse-transcription PCR was performed to determine whether this receptor is expressed in human prostate cancer cell lines. The results of reverse-transcription PCR experiments are shown in Fig. 6A. Caco-2 cells, which have previously been shown to express FFA4 (Mobraten et al., 2013), were used as a positive control. The data show that DU145 cells express mRNA for FFA4. PC-3, another androgen-independent human prostate cancer cell line, expresses lower levels of FFA4 mRNA. Interestingly, both cell lines also express FFA1 mRNA, with PC-3 expressing much higher levels than DU145. The expression of FFA4 protein was confirmed in both cell lines by immunoblotting, as shown in Fig. 6B.
Fig. 6.

Expression of FFA4 in prostate cancer cell lines. (A) Reverse-transcription PCR reactions were carried out using total mRNA prepared from DU145, PC-3, and Caco-2 cells to synthesize first-strand cDNA. The cDNA was then used for PCR using primers specific for human FFA4, FFA1, or Rplp0 (loading control). Products were separated by agarose gel electrophoresis and visualized under UV light. Results shown are representative of three separate experiments, each conducted in triplicate. (B) Immunoblotting was performed for FFA4 protein and actin (loading control) using whole-cell extracts, equalized for protein, prepared from DU145 and PC-3 cells. Results shown are representative of two separate experiments.
Based on these results, we tested whether TUG-891, a recently described pharmacologic agonist that is selective for human FFA4 (Shimpukade et al., 2012; Hudson et al., 2013a), exerts effects similar to those of EPA. TUG-891 has 1000-fold higher affinity for FFA4 than for FFA1. We also tested for effects of GW9508, an agonist that conversely is 1000-fold selective for FFA1 over FFA4 (Briscoe et al., 2006).
We first performed a concentration-effect (“dose-response”) study examining the effects of FFAR agonists on cell proliferation. Neither agonist affects DU145 cell proliferation when added alone to serum-starved cells (data not shown). Figure 7A shows that EPA inhibits LPA-induced proliferation of DU145 cells, with an IC50 of 5.7 µM. The FFA4-selective agonist TUG-891 also inhibits the ability of DU145 cells to proliferate in response to LPA. The IC50 for this effect is 73 nM, which is consistent with published EC50 values of ∼100 nM for TUG-891 for effects mediated by human FFA4 (Hudson et al., 2013b). The relative potencies of EPA, DHA, GW9508, and TUG-891 are also consistent with previously published values. TUG-891 was developed as a selective FFAR inhibitor, and has higher potency than the endogenous n-3 FAs (Hudson et al., 2013b). GW9508, the FFA1-selective agonist, inhibited proliferation with an IC50 of 950 nM. This value is much higher than the published EC50 of GW9508 at FFA1 (∼50 nM), but is consistent with its EC50 for FFA4 (∼2 µM) (Briscoe et al., 2006). In Fig. 7B, concentration-effect curves were obtained for inhibition of serum-induced proliferation by TUG-891, GW9508, EPA, and DHA; in these experiments, DU145 cells were maintained continuously in serum. It was noted that both agonists had approximately 10-fold higher IC50 values against serum as compared with LPA. In contrast, GW9508 had a lower IC50 value with serum than with LPA. There are at least two possible explanations for the differences in IC50 values between LPA and serum. First, serum contains proteins, lipids, and fatty acids that potentially interfere with the delivery and/or action of the FFAR agonists; this is especially likely to be the case for EPA and DHA, which are fatty acids that bind to plasma proteins. Second, serum contains multiple growth factors, some of which may act through mechanisms that are not inhibited by FFA4 agonists, or that are more sensitive to FFA1 agonists. In any case, the FFA4 agonist TUG-891 was the most potent inhibitor of proliferation induced by either LPA or serum. In summary, although the concentration-effect data do not completely exclude a role for FFA1, they are most consistent with a role for FFA4.
Fig. 7.

Dose response for the effects of EPA, TUG-891, and GW9508 on DU145 cell proliferation. (A) Serum-starved DU145 cells were incubated for 48 hours with and without 10 µM LPA in the absence and presence of the indicated concentrations of EPA, DHA, TUG-891, or GW9508. The number of cells achieved in response to LPA in the absence of other additions was defined as 100% response; the number of cells present in the absence of LPA was defined as 0% response. Each point represents the mean ± S.E.M. of values obtained from two to six experiments, each of which was performed using duplicate wells of cells for each experimental condition. (B) DU145 cells (not serum starved) were incubated for 48 hours with and without 10% fetal bovine serum in the absence and presence of the indicated concentrations of EPA (closed triangles), DHA, TUG-891, or GW9508. Each data point represents the mean ± S.E.M. of values obtained from two to three experiments, each of which was performed using duplicate wells of cells for each experimental condition. For both panels, data were fit by nonlinear regression, using GraphPad Prism software, with 0 and 100% response values defined by negative and positive controls within each experiment. (C and D) DU145 cells (C) and PC-3 cells (D) were serum starved, treated with or without 1 µM TUG-891 (C) or 20 µM EPA (D) for 15 minutes, and then treated with or without 10% serum for 24 hours. Cells were then washed, incubated with fresh medium without TUG-891 or EPA (but with serum as indicated), and counted (time “0”). The incubation continued, with live cells being counted every 24 hours from two to four experiments. Data analysis was performed using two-way ANOVA, followed by Tukey’s multiple comparisons test. (E) Serum-starved DU145 cells were incubated with or without 20 µM EPA or 1 µM TUG-891 for 15 minutes. LPA (10 µM) or vehicle was then added for 10 minutes. Whole-cell extracts, equalized for protein, were immunoblotted for phospho-p70S6K and actin. Immunoblots for p70S6K were quantified, normalized to actin, and then normalized to the untreated control. Each data point represents the mean ± S.D. of values from duplicate dishes of cells. The results shown are representative of three separate experiments.
Next, we asked whether the effects of TUG-891 on proliferation were reversible. TUG-891, a pharmacologic agent that is not a lipid, is not incorporated into phospholipid and acts reversibly (Hudson et al., 2013b). In Fig. 7, C and D, DU145 cells were incubated with TUG-891 and serum to inhibit proliferation, after which TUG-891 was removed and serum incubation was continued. Serum-induced proliferation resumes after TUG-891 is removed. These results demonstrate reversibility, confirm that TUG-891 is not toxic to DU145 cells, and indicate that the FFA4 agonist must be present continuously to inhibit proliferation. We next tested whether the effects of EPA are reversible. Since exogenous EPA has been shown to be readily incorporated into phospholipid in prostate cancer cells in culture (Gu et al., 2013a), effects of n-3 FAs on membrane fluidity, phospholipid composition, or generation of n-3 metabolites from FAs incorporated into phospholipid would be anticipated to be only slowly reversible. However, the reversibility of the inhibitory effects of EPA was observed to be similar to that of TUG-891 (Fig. 7, D and E). The rapid reversibility is more consistent with a receptor-mediated effect of EPA.
The effects of the FFA4 agonist TUG-891 on LPA-induced signal transduction were examined. Figure 7E shows that EPA and TUG-891 have significant inhibitory effects on p70S6K activation when LPA response is measured at 10 minutes. Thus, a selective FFA4 agonist can suppress responses to LPA.
In Fig. 8, we tested whether the effects of FFA4 agonists extend to another prostate cancer cell line. The effects of EPA and TUG-891 were tested on proliferation induced by serum, LPA, or EGF in both DU145 and PC-3 cells. The top panels show that both EPA and TUG-891 significantly inhibit serum-induced proliferation in both cell lines. In these experiments, the cells were serum starved prior to addition of the FFAR agonist and serum. The inhibitory effect of the agonists at the doses used is partial, which is consistent with the data shown in Fig. 7B. The remaining panels show that EPA and TUG-891 are both able to completely block proliferation in response to LPA (middle panels) and EGF (bottom panels) in both cell lines. Thus, TUG-891 and EPA inhibit proliferation in two different human prostate cancer cell lines.
Fig. 8.

Effects of TUG-891 on proliferation of DU145 and PC-3 cells. Proliferation assays were conducted using serum-starved PC-3 (left panels) or DU145 (right panels) cells as described in Fig. 1. Cells were incubated with or without fetal bovine serum (10%), LPA (10 µM), or EGF (10 nM) in the absence and presence of EPA (20 µM) or TUG-891 (1 µM) for the indicated times. Each data point represents the mean ± S.E.M. of values from duplicate experiments, each of which used duplicate wells of cells for each experimental condition. The control, EPA alone, and TUG-891 alone data are repeated in the three graphs for each cell line, since all data were from the same series of experiments. Asterisks represent the P values for LPA versus LPA + EPA and # represents the P values for LPA versus LPA + TUG-891; *,#P < 0.05; **,##P < 0.01; ***,###P < 0.001; ****,####P < 0.0001. There was no significant difference between control and LPA + EPA or LPA + TUG-891 at any time point. Data analysis was performed using two-way ANOVA, followed by Tukey’s multiple comparisons test.
FFA4 protein levels, as detected by immunoblotting, were not significantly altered by treatment of DU145 or PC-3 cells with either 20 µM EPA or 1 µM TUG-891 for up to 4 hours (data not shown). These results suggest that FFA4 is not subject to agonist-mediated proteolytic downregulation under the conditions tested.
Effects of FFAR Agonists on Cell Migration.
To evaluate the effects of FFAR agonists on another response relevant to cancer cell biology, we examined cell migration. A previous report showed that n-3 FAs inhibit migration of PC-3 cells (Brown et al., 2006). As shown in Fig. 9, LPA and EGF both enhance migration of DU145 and PC-3 cells, as assessed in a modified Boyden chamber assay. The response to either agonist was approximately 70% of the maximal response elicited by 10% serum in both cell lines (data not shown). EPA and TUG-891 significantly inhibit migration induced by either LPA or EGF, in both DU145 and PC-3 cells.
Fig. 9.

Effects of EPA and TUG-891 on migration of DU145 (A) and PC-3 (B) cells. Serum-starved DU145 and PC-3 cells were seeded in serum-free medium at 2.5 × 104 cells per insert in the upper chambers of 8-μm transwell inserts. Serum-free medium was added to the lower wells. Cells were then treated with 10% FBS, 20 μM EPA, 1 μM TUG-891, 10 μM LPA, or 10 nM EGF, either alone or in combination. Following a 6-hour migration period, the insert membranes were fixed and stained using methanol and crystal violet. Cells that invaded the lower chambers were counted by microscopy. Each bar represents the mean ± S.E.M. of values obtained from duplicate experiments, each of which used duplicate wells of cells for each experimental condition. Data analysis was performed using one-way ANOVA, followed by Tukey’s multiple comparisons test.
Effects of FFA4 Knockdown.
As another approach to test whether FFA4 mediates the inhibitory effects of FFAR agonists, siRNA was used to inhibit FFA4 expression in DU145 cells. siRNAs are normally administered to cells in the continuous presence of growth factors (i.e., not serum starved). Fortuitously, as shown previously (see Fig. 7B), FFAR agonists can inhibit proliferation of cycling DU145 cells. First, we optimized conditions for the FFA4 knockdown. As shown in Fig. 10A, the siRNA treatment was sufficient to achieve a maximal decrease in FFA4 protein levels at 24 hours. The extent of knockdown was 67.2 ± 7.0% (n = 5) at 24 hours, as measured in the absence of FFA4 agonist. Next, we tested whether FFAR agonists can inhibit serum- or LPA-induced proliferation in cells deficient in FFA4. In these experiments, serum or LPA was added at the same time as the FFA4 siRNA. FFA4 knockdown alone, in the absence of FFA4 agonists, had no effect on proliferation in response to either serum or LPA (data not shown). The results presented in Fig. 10, B and C, demonstrate that EPA and TUG-891 significantly inhibit serum-induced proliferation at 48 and 72 hours in DU145 and PC-3 cells treated with scrambled siRNA (negative control), but have no effect in cells treated with FFA4 siRNA. Similarly, in Fig. 10, D–G, we tested the effects of EPA and TUG-891 on LPA- and EGF-induced proliferation with FFA4 knockdown in both DU145 and PC-3 cells. Cells were serum starved for the LPA and EGF experiments, although the medium contained low levels of Opti-MEM, which contains proprietary growth factors. Time course studies for both LPA and EGF yielded similar results at 24, 48, and 72 hours; for clarity, only data for the 48-hour time point are presented. The results presented in Fig. 10 show that FFA4 knockdown eliminates the inhibitory effects of EPA and TUG-891 on proliferation.
Fig. 10.

Effects of FFA4 knockdown on inhibitory effects of EPA and TUG-891 on cell proliferation. (A) DU145 cells were incubated with scrambled siRNA (negative control) or FFA4 siRNA as described in Materials and Methods. Whole-cell extracts were immunoblotted for FFA4 and actin (loading control). The percent knockdown (KD) was calculated after quantification by densitometry, and normalization to actin. (B and C) DU145 cells (B) and PC-3 cells (C) were incubated with scrambled siRNA (Scr) or FFA4 siRNA (F4i), or without siRNA (control), as described in Materials and Methods. Simultaneously, cells were treated with and without 20 µM EPA or 1 µM TUG-891, in the continued presence of 10% serum. (D–G) DU145 cells (D and E) and PC-3 cells (F and G) were treated as in (B) and (C), except that serum was removed for 24 hours prior to the addition of scrambled or FFA4 siRNA, without or without 20 µM EPA, 1 µM TUG-891, 10 µM LPA, 10 nM EGF, or vehicle as indicated. For (B)–(G), data points represent the mean ± S.E.M. of values from duplicate experiments, each conducted in duplicate (n = 4). *Values that were significantly different (two-way ANOVA followed by Tukey’s multiple comparisons test; **P < 0.01; ****P < 0.0001) from the untreated control, as well as from the scrambled siRNA control, for the time point tested. UT, untransfected.
Similar studies were performed to test for effects of FFA4 knockdown on migration of DU145 and PC-3 cells. As shown in Fig. 11, inhibitory effects of TUG-891 and EPA on migration induced by LPA, EGF, and serum were absent when FFA4 was knocked down.
Fig. 11.
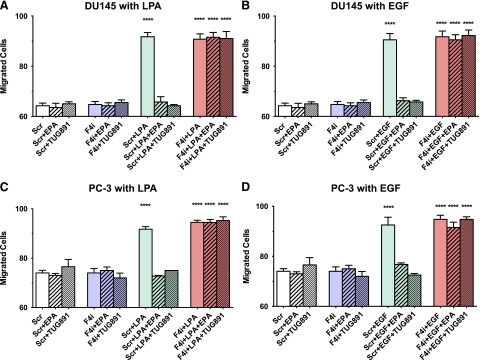
Effects of FFA4 knockdown on inhibitory effects of EPA and TUG-891 on cell migration. Serum-starved DU145 cells (A and B) and PC-3 cells (C and D) were incubated with scrambled siRNA (Scr) or FFA4 siRNA (F4i), or without siRNA (control) as described in Materials and Methods. After 24 hours, cells were seeded in a Boyden chamber and treated with and without 20 µM EPA or 1 µM TUG-891, with or without 10 µM LPA or 10 nM EGF. Migration experiments were carried out for 6 hours. Data points represent the mean ± S.E.M. of values from duplicate experiments, each conducted in duplicate (n = 4). *Values that were significantly different (two-way ANOVA followed by Tukey’s multiple comparisons test; ****P < 0.0001) from the untreated control, as well as from the scrambled siRNA control, for the time point tested.
It is interesting to note that FFA4 knockdown suppressed the effects of FFA4 agonists on proliferation in Fig. 10, even though FFA4 was expressed by the cells at the beginning of the incubation. In other words, the FFA4-mediated signaling that occurs initially, prior to FFA4 knockdown, is not sufficient to block proliferation occurring over a longer time course. These results, along with the reversibility results shown in Fig. 7, suggest that the inhibitory effects of FFA4 agonists require ongoing stimulation of FFA4, in the context of the continuous presence of growth factor receptor agonists.
Discussion
This study presents unexpected findings showing that omega-3 fatty acids and FFA4 agonists can inhibit growth factor–induced signaling. To our knowledge, ours is the first study to examine the roles of FFA4 in prostate cancer cells.
Most of the experiments described herein used LPA as the mitogenic agonist. The promitogenic and promigratory activities of LPA are mediated by GPCRs in prostate cancer cells (Hao et al., 2007; Gibbs et al., 2009). Several mechanisms could potentially be responsible for the inhibitory effect of FFA4 agonists on LPA response in prostate cancer cells.
First, activation of FFA4 could potentially activate signal transduction pathways that act downstream to “functionally antagonize” LPA action. For example, agents that increase cAMP levels have been shown to inhibit Erk activation in several model systems (Stork and Schmitt, 2002), and cAMP inhibits LPA-induced RhoA activation in PC-3 cells (Chen et al., 2005). However, the broad spectrum of the inhibitory action of EPA and FFA4 agonists argues against this type of mechanism. The signal transduction responses examined in our study, Erk, p70S6K, and FAK, are activated through signaling pathways that are largely separate, although cross-talk between pathways does occur. Erk is activated through the Ras/Raf/MEK pathway. The Akt/mTORC1 pathway initiates activation of p70S6K. FAK is subject to regulation by several pathways, including integrins, but the activating phosphorylation that was assessed in our study is performed by c-Src. Therefore, the demonstrated ability of FFA4 agonists to suppress all of these pathways simultaneously suggests a more general mechanism that lies upstream of the protein phosphorylation cascades.
A second type of inhibitory mechanism involves cross-talk between GPCRs. For example, activation of FFA4 could heterologously desensitize LPA receptors, so that subsequent responses to LPA are inhibited. Consistent with this idea, it has been noted that FFA4 agonists cause prominent homologous desensitization of FFA4 via β-arrestin binding (Watson et al., 2012; Dranse et al., 2013; Burns et al., 2014). The fact that EPA and TUG-891 also inhibit responses to EGF, in our studies, could argue against this mechanism. However, our data demonstrate that LPA antagonists can inhibit response to EGF, suggesting a significant role for EGF-induced transactivation of LPA receptors in EGF response. Thus, desensitization of LPA receptors could also inhibit responses to EGF. Another type of negative cross-talk can occur that involves heterodimerization of GPCRs (Milligan, 2004). This phenomenon has not been demonstrated to date for FFA4, but is conceivable. LPA receptors have been shown to heterodimerize with other GPCRs in prostate cancer cells (Ward et al., 2011). Cross-talk that involves changes in receptor localization is also conceivable, since FFA4 is efficiently internalized in response to agonists (Watson et al., 2012). Further work will be needed to explore these potential mechanisms of inhibition.
The experimental protocol used in our studies facilitated the acquisition of unique insights into the action of FFA agonists. Since we were interested in growth factor action, we routinely use serum-starved cells in which growth factor–induced stimulated signal transduction events, and inhibition of these signals, can be more readily assessed. However, subsequent studies showed that FFA4 agonists also inhibit proliferation of cells that are already actively growing in serum (Figs. 8 and 10). Also, since we examined the kinetics of signaling responses, which are best studied in serum-starved cells, we were able to discern that FFA4 agonists have transient stimulatory effects as well as inhibitory effects. Future studies can examine the extent to which these effects occur in the tumor microenvironment in vivo.
Three previously published studies have indicated that n-3 FAs can suppress signal transduction via activation of FFA4; all of these studies examined inflammatory responses. One group showed that n-3 FAs inhibit inflammasome activation in macrophages (Yan et al., 2013), whereas another showed that a 1-hour preincubation with n-3 FAs inhibits interleukin-1β response in Caco-2 colon cancer cells (Mobraten et al., 2013). A third group reported that DHA (24-hour preincubation) inhibits lipopolysaccharide responses mediated via Toll-like receptor 4, which include Akt/c-Jun N-terminal kinase phosphorylation (Li et al., 2013). Although these results are not directly analogous to the growth inhibitory responses examined in the current study, it is now clear that FFA4 can mediate inhibitory effects on signaling in multiple cellular models.
In contrast to the inhibitory effects discussed earlier, a recent report indicated that FFA4 plays an oncogenic role in human colon cancer, enhancing angiogenesis as well as migration (Wu et al., 2013). With regard to signal transduction, Wu and coworkers (2013) examined direct effects of GW9508 on two colon cancer cell lines, demonstrating activation of PI3K/Akt and nuclear factor κB. As was the case in our prostate cancer cell experiments, Wu and colleagues tested the effects of FFA4 agonists on the proliferation and migration of serum-starved cells. Our data (Fig. 3C) similarly show that the FFA4 agonist EPA activates p70S6K in Caco-2 colon cancer cells. The results with colon cancer cells stand in contrast to the results reported herein for prostate cancer cells. Specifically, the FFA1/FFA4 agonist GW9508 is mitogenic for colon cancer cells (Wu et al., 2013), but not for prostate cancer cells (Fig. 7). GW9508 (10 µM) stimulates migration of colon cancer cells treated for 72 hours prior to the migration assays (Wu et al., 2013), whereas TUG-891 inhibits migration of prostate cancer cells (Fig. 9). It will be important to further explore the mechanisms underlying the opposing effects of FFA4 agonists on proliferation in different types of cancer cells. These differences may reflect variations in the expression levels of FFA4, other GPCRs, G proteins, or GPCR effectors.
It is interesting to note that DU145 and PC-3 cells differ in many respects with regard to signal transduction. Their profiles of LPA receptor expression and response are different (Gibbs et al., 2009). PTEN, a tumor suppressor that attenuates activation of the Akt pathway, is expressed in DU145 but not in PC-3 cells (Fraser et al., 2012). Nonetheless, FFAR agonists have similar antiproliferative actions in both cell lines. Since FFA4 is expressed in normal as well as transformed prostate cancer cells, we speculate that FFA4 agonists can suppress growth of both precancerous and transformed prostate epithelial cells. This hypothesis requires further investigation.
In our study, CCN1 induction was used to assess a later response to LPA action. CCN1, which is induced in response to various stimuli including growth factors, plays roles in proliferation, apoptosis, and migration of prostate cancer cells (Sakamoto et al., 2004a; Franzen et al., 2009; Schmitz et al., 2013). CCN1 expression is induced by LPA in benign prostatic hyperplasia cells (Sakamoto et al., 2004b). Although several studies have implicated CCN1 in prostate tumor progression, including studies of DU145 cells (Sun et al., 2008), the roles of this interesting agonistic protein remain to be fully defined. It is possible that CCN1 induction is important for the second phase of the biphasic signaling response to LPA, which likely requires the production of additional promitogenic factors. Our results establish an experimental model in which CCN1 production can be inhibited as well as stimulated.
Our demonstration that FFA4 agonists can inhibit growth factor response in an acute and ongoing manner has implications for the administration of n-3 FA supplements or FFAR agonists with the intention of preventing cancer. If the major mechanism for the beneficial effects of n-3 FAs involves changes in phospholipid composition, as previously proposed, then the dosage regimen may be less critical. However, if the mechanism involves modulation of signal transduction pathways and requires the continuous presence of agonist, as our results indicate, then it may be important to administer the agonists more frequently. It will also be important to determine the underlying mechanism. For example, signaling events mediated by arrestins are less susceptible to desensitization, and therefore have a more sustained duration, than those mediated by G proteins (Luttrell and Gesty-Palmer, 2010). Our results show that continuous engagement of FFA4 is required for inhibitory effects, which persist as long as the agonist is present; these findings suggest arrestin involvement. Translational studies will be needed to determine whether FFA4 agonists suppress mitogenic signaling in tissues, and to define the dosage regimen required for such an effect.
The discovery that a pharmacologic FFA4 agonist can mimic the antiproliferative effects of an n-3 FA is particularly pertinent to proposed therapeutic approaches for the purposes of preventing or treating cancer. First, the fish oil preparations that are typically used for n-3 FA supplementation are composed of a mixture of FAs, primarily EPA and DHA. Although these compounds are efficacious as FFA4 agonists, a pharmacologic agent such as TUG-891 can be targeted much more specifically to FFA4, and its pharmacodynamic properties can be more easily defined. Second, the ability of n-3 FA metabolites (“resolvins”) to inhibit inflammation is of considerable interest (Zhang and Spite, 2012). Although inflammation promotes tumor formation, the direct effects of the numerous n-3 FA metabolites on cancer cells are understudied. Since small-molecule FFAR agonists are not metabolized to resolvins, utilization of these agonists will facilitate studies of the relative roles of FFARs in tumor progression. Finally, fish oil supplements are not well tolerated by all individuals. Dranse and colleagues (2013) have provided a thoughtful discussion of dietary versus pharmacologic approaches to targeting FFARs. FFA4 agonists are already of great interest with respect to the treatment of metabolic diseases (Holliday et al., 2011). The potential benefit of these agents with respect to cancer provides exciting new avenues for therapeutic intervention.
Acknowledgments
The authors thank Aimee Bell, Christian Legaspi, and Akriti Chaudhry for their supportive roles in various aspects of this project. Dr. Cliff Berkman (Washington State University) is acknowledged for his collegial assistance. The authors are grateful for advice provided by Dr. Harvey Motulsky (GraphPad Software, Inc.) regarding dose-response analyses.
Abbreviations
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- DHA
docosahexaenoic acid
- EGF
epidermal growth factor
- EPA
eicosapentaenoic acid
- ERK
extracellular signal-regulated kinase
- FA
fatty acid
- FAK
focal adhesion kinase
- FBS
fetal bovine serum
- FFAR
free fatty acid receptor
- GPCR
G protein–coupled receptor
- GW9508
4-[[(3-phenoxyphenyl)methyl]amino]-benzenepropranoic acid
- Ki16425
3-(4-[4-([1-(2-chlorophenyl)ethoxy]carbonyl amino)-3-methyl-5-isoxazolyl] benzylsulfanyl) propanoic acid
- LPA
lysophosphatidic acid
- n-3
omega-3
- PCR
polymerase chain reaction
- PI3K
phosphatidylinositol 3-kinase
- siRNA
small interfering RNA
- TUG-891
4-[(4-fluoro-4′-methyl[1,1′-biphenyl]-2-yl)methoxy]-benzenepropanoic acid
Authorship Contributions
Participated in research design: Meier, Hopkins, Liu, Zhang.
Conducted experiments: Liu, Hopkins, Zhang, Quisenberry, Fix, Galvan.
Performed data analysis: Hopkins, Liu, Meier, Zhang, Fix, Galvan.
Wrote or contributed to the writing of the manuscript: Meier, Liu, Hopkins, Zhang, Fix.
Footnotes
This work was supported by the College of Pharmacy at Washington State University (WSU), and by a Collaborative Translational Seed Grant [UL1-RR025014] to Z.Z. from the Institute of Translational Health Sciences at the University of Washington. C.R.Q. was the recipient of a Nutrition and Exercise Physiology Undergraduate Research Fellowship from the WSU College of Pharmacy. M.M.H., a member of the Graduate Program in Pharmaceutical Sciences in the College of Pharmacy, also received support from the National Institutes of Health National Institute of General Medical Sciences Protein Biotechnology Training Program at WSU [Grant T32-GM008336]. B.M.G. was the recipient of a Summer Undergraduate Research Fellowship funded by the American Society for Pharmacology and Experimental Therapeutics, and by the College of Pharmacy.
Liu Z (2013) Effects of Omega-3 Fatty Acids on Prostate Cancer Cells. M.Sc. thesis, Washington State University, Spokane, WA.
Aspects of this study have been previously discussed in the following abstracts: Zhang Z and Meier KE (2011) Effects of LPA antagonist and PLD inhibitor on proliferation of human prostate cancer cell lines. Experimental Biology 2011; 2011 Apr 9–13; Washington, DC; Zhang Z, Bailey CR, and Meier KE (2012) Effects of lysophosphatidic acid on CCN1 expression in human prostate cancer cell lines. Experimental Biology 2012; 2012 Apr 22–25; San Diego, CA; Hopkins MM, Liu Z, and Meier KE (2013) Effects of GPR120 agonists on lysophosphatidic acid signaling in prostate cancer cells. Southeast Regional Lipid Conference; 2013 Sept 13–15; Cashiers, NC; Hopkins MM, Liu Z, and Meier KE (2014) Effects of eicosapentaenoic acid and the GPR120 agonist TUG-891 on lysophosphatidic acid signaling in prostate cancer cells. Experimental Biology 2014; 2014 Apr 27–30; San Diego, CA; and Hopkins MM, Liu Z, Galvan BM, and Meier KE (1014) Omega-3 fatty acids inhibit LPA- and EGF-induced proliferation of human prostate cancer cells by acting as agonists for free fatty acid receptor 4 (FFA4). Southeast Regional Lipid Conference; 2014 Nov 5–7; Cashiers, NC.
References
- Akinsete JA, Ion G, Witte TR, Hardman WE. (2012) Consumption of high ω-3 fatty acid diet suppressed prostate tumorigenesis in C3(1) Tag mice. Carcinogenesis 33:140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bektas M, Payne SG, Liu H, Goparaju S, Milstien S, Spiegel S. (2005) A novel acylglycerol kinase that produces lysophosphatidic acid modulates cross talk with EGFR in prostate cancer cells. J Cell Biol 169:801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasky TM, Darke AK, Song X, Tangen CM, Goodman PJ, Thompson IM, Meyskens FL, Jr, Goodman GE, Minasian LM, Parnes HL, et al. (2013) Plasma phospholipid fatty acids and prostate cancer risk in the SELECT trial. J Natl Cancer Inst 105:1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe CP, Peat AJ, McKeown SC, Corbett DF, Goetz AS, Littleton TR, McCoy DC, Kenakin TP, Andrews JL, Ammala C, et al. (2006) Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol 178:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MD, Hart CA, Gazi E, Bagley S, Clarke NW. (2006) Promotion of prostatic metastatic migration towards human bone marrow stoma by Omega 6 and its inhibition by Omega 3 PUFAs. Br J Cancer 94:842–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns RN, Singh M, Senatorov IS, Moniri NH. (2014) Mechanisms of homologous and heterologous phosphorylation of FFA receptor 4 (GPR120): GRK6 and PKC mediate phosphorylation of Thr³⁴⁷, Ser³⁵⁰, and Ser³⁵⁷ in the C-terminal tail. Biochem Pharmacol 87:650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher AJ, Hudson BD, Shimpukade B, Alvarez-Curto E, Prihandoko R, Ulven T, Milligan G, Tobin AB. (2014) Concomitant action of structural elements and receptor phosphorylation determines arrestin-3 interaction with the free fatty acid receptor FFA4. J Biol Chem 289:18451–18465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calder PC, Yaqoob P. (2009) Omega-3 polyunsaturated fatty acids and human health outcomes. Biofactors 35:266–272. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang Y, Yu H, Wang F, Xu W. (2005) The cross talk between protein kinase A- and RhoA-mediated signaling in cancer cells. Exp Biol Med (Maywood) 230:731–741. [DOI] [PubMed] [Google Scholar]
- Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, Lin ME, Teo ST, Park KE, Mosley AN, et al. (2010) LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol 50:157–186. [DOI] [PubMed] [Google Scholar]
- Chung BH, Mitchell SH, Zhang J-S, Young CYF. (2001) Effects of docosahexaenoic acid and eicosapentaenoic acid on androgen-mediated cell growth and gene expression in LNCaP prostate cancer cells. Carcinogenesis 22:1201–1206. [DOI] [PubMed] [Google Scholar]
- Dranse HJ, Kelly ME, Hudson BD. (2013) Drugs or diet?—Developing novel therapeutic strategies targeting the free fatty acid family of GPCRs. Br J Pharmacol 170:696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Dransfield PJ, Lin DC-H, Wong S, Wang Y, Wang Z, Kohn T, Yu M, Brown SP, Vimolratana M, et al. (2014) Improving the pharmacokinetics of GPR40/FFA1 full agonists. ACS Med Chem Lett 5:384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards IJ, O’Flaherty JT. (2008) Omega-3 fatty acids and PPARgamma in cancer. PPAR Res 2008:358052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzen CA, Chen CC, Todorović V, Juric V, Monzon RI, Lau LF. (2009) Matrix protein CCN1 is critical for prostate carcinoma cell proliferation and TRAIL-induced apoptosis. Mol Cancer Res 7:1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser M, Zhao H, Luoto KR, Lundin C, Coackley C, Chan N, Joshua AM, Bismar TA, Evans A, Helleday T, et al. (2012) PTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: implications for radiotherapy and chemotherapy. Clin Cancer Res 18:1015–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber M. (2012) Omega-3 fatty acids and cancers: a systematic update review of epidemiological studies. Br J Nutr 107 (Suppl 2):S228–S239. [DOI] [PubMed] [Google Scholar]
- Gibbs TC, Rubio MV, Zhang Z, Xie Y, Kipp KR, Meier KE. (2009) Signal transduction responses to lysophosphatidic acid and sphingosine 1-phosphate in human prostate cancer cells. Prostate 69:1493–1506. [DOI] [PubMed] [Google Scholar]
- Gu Z, Suburu J, Chen H, Chen YQ. (2013b) Mechanisms of omega-3 polyunsaturated fatty acids in prostate cancer prevention. Biomed Res Int 2013:824563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Wu J, Wang S, Suburu J, Chen H, Thomas MJ, Shi L, Edwards IJ, Berquin IM, Chen YQ. (2013a) Polyunsaturated fatty acids affect the localization and signaling of PIP3/AKT in prostate cancer cells. Carcinogenesis 34:1968–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Kasbohm EA, Arora P, Sample CJ, Baban B, Sud N, Sivashanmugam P, Moniri NH, Daaka Y. (2006) Expression and function of lysophosphatidic acid LPA1 receptor in prostate cancer cells. Endocrinology 147:4883–4892. [DOI] [PubMed] [Google Scholar]
- Hao F, Tan M, Xu X, Han J, Miller DD, Tigyi G, Cui MZ. (2007) Lysophosphatidic acid induces prostate cancer PC3 cell migration via activation of LPA(1), p42 and p38alpha. Biochim Biophys Acta 1771:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Hirasawa A, Ichimura A, Kimura I, Tsujimoto G. (2011) Free fatty acid receptors FFAR1 and GPR120 as novel therapeutic targets for metabolic disorders. J Pharm Sci 100:3594–3601. [DOI] [PubMed] [Google Scholar]
- Holliday ND, Watson S-J, Brown AJH. (2011) Drug discovery opportunities and challenges at g protein coupled receptors for long chain free Fatty acids. Front Endocrinol (Lausanne) 2:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson BD, Murdoch H, Milligan G. (2013a) Minireview: The effects of species ortholog and SNP variation on receptors for free fatty acids. Mol Endocrinol 27:1177–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson BD, Shimpukade B, Mackenzie AE, Butcher AJ, Pediani JD, Christiansen E, Heathcote H, Tobin AB, Ulven T, Milligan G. (2013b) The pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol Pharmacol 84:710–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarguren M, López DJ, Encinar JA, González-Ros JM, Busquets X, Escribá PV. (2013) Partitioning of liquid-ordered/liquid-disordered membrane microdomains induced by the fluidifying effect of 2-hydroxylated fatty acid derivatives. Biochim Biophys Acta 1828:2553–2563. [DOI] [PubMed] [Google Scholar]
- Jun JI, Lau LF. (2011) Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov 10:945–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Barnard RJ, Henning SM, Elashoff D, Reddy ST, Cohen P, Leung P, Hong-Gonzalez J, Freedland SJ, Said J, et al. (2006) Effect of altering dietary ω-6/ω-3 fatty acid ratios on prostate cancer membrane composition, cyclooxygenase-2, and prostaglandin E2. Clin Cancer Res 12:4662–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kue PF, Taub JS, Harrington LB, Polakiewicz RD, Ullrich A, Daaka Y. (2002) Lysophosphatidic acid-regulated mitogenic ERK signaling in androgen-insensitive prostate cancer PC-3 cells. Int J Cancer 102:572–579. [DOI] [PubMed] [Google Scholar]
- Li X, Yu Y, Funk CD. (2013) Cyclooxygenase-2 induction in macrophages is modulated by docosahexaenoic acid via interactions with free fatty acid receptor 4 (FFA4). FASEB J 27:4987–4997. [DOI] [PubMed] [Google Scholar]
- Lu Y, Nie D, Witt WT, Chen Q, Shen M, Xie H, Lai L, Dai Y, Zhang J. (2008) Expression of the fat-1 gene diminishes prostate cancer growth in vivo through enhancing apoptosis and inhibiting GSK-3 beta phosphorylation. Mol Cancer Ther 7:3203–3211. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Gesty-Palmer D. (2010) Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev 62:305–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean CH, Newberry SJ, Mojica WA, Khanna P, Issa AM, Suttorp MJ, Lim YW, Traina SB, Hilton L, Garland R, et al. (2006) Effects of omega-3 fatty acids on cancer risk: a systematic review. JAMA 295:403–415. [DOI] [PubMed] [Google Scholar]
- Mancini AD, Poitout V. (2013) The fatty acid receptor FFA1/GPR40 a decade later: how much do we know? Trends Endocrinol Metab 24:398–407. [DOI] [PubMed] [Google Scholar]
- Milligan G. (2004) G protein-coupled receptor dimerization: function and ligand pharmacology. Mol Pharmacol 66:1–7. [DOI] [PubMed] [Google Scholar]
- Mobraten K, Haug TM, Kleiveland CR, Lea T. (2013) Omega-3 and omega-6 PUFAs induce the same GPR120-mediated signalling events, but with different kinetics and intensity in Caco-2 cells. Lipids Health Dis 12:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan TM, Koreckij TD, Corey E. (2009) Targeted therapy for advanced prostate cancer: inhibition of the PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets 9:237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris PC, Dennis EA. (2012) Omega-3 fatty acids cause dramatic changes in TLR4 and purinergic eicosanoid signaling. Proc Natl Acad Sci USA 109:8517–8522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan WQ, Li P, Wendell JL, Watkins SM, Olefsky JM. (2010) GRP120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142:687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hayre M, Degese MS, Gutkind JS. (2014) Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr Opin Cell Biol 27:126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto S, Yokoyama M, Aoki M, Suzuki K, Kakehi Y, Saito Y. (2004. a) Induction and function of CYR61 (CCN1) in prostatic stromal and epithelial cells: CYR61 is required for prostatic cell proliferation. Prostate 61:305–317. [DOI] [PubMed] [Google Scholar]
- Sakamoto S, Yokoyama M, Zhang X, Prakash K, Nagao K, Hatanaka T, Getzenberg RH, Kakehi Y. (2004. b) Increased expression of CYR61, an extracellular matrix signaling protein, in human benign prostatic hyperplasia and its regulation by lysophosphatidic acid. Endocrinology 145:2929–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz P, Gerber U, Jüngel E, Schütze N, Blaheta R, Bendas G. (2013) Cyr61/CCN1 affects the integrin-mediated migration of prostate cancer cells (PC-3) in vitro. Int J Clin Pharmacol Ther 51:47–50. [DOI] [PubMed] [Google Scholar]
- Shimpukade B, Hudson BD, Hovgaard CK, Milligan G, Ulven T. (2012) Discovery of a potent and selective GPR120 agonist. J Med Chem 55:4511–4515. [DOI] [PubMed] [Google Scholar]
- Snider AJ, Zhang Z, Xie Y, Meier KE. (2010) Epidermal growth factor increases lysophosphatidic acid production in human ovarian cancer cells: roles for phospholipase D2 and receptor transactivation. Am J Physiol Cell Physiol 298:C163–C170. [DOI] [PubMed] [Google Scholar]
- Stork PJS, Schmitt JM. (2002) Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 12:258–266. [DOI] [PubMed] [Google Scholar]
- Sun Q, Hirasawa A, Hara T, Kimura I, Adachi T, Awaji T, Ishiguro M, Suzuki T, Miyata N, Tsujimoto G. (2010) Structure-activity relationships of GPR120 agonists based on a docking simulation. Mol Pharmacol 78:804–810. [DOI] [PubMed] [Google Scholar]
- Sun ZJ, Wang Y, Cai Z, Chen PP, Tong XJ, Xie D. (2008) Involvement of Cyr61 in growth, migration, and metastasis of prostate cancer cells. Br J Cancer 99:1656–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S, Olefsky JM, Osborn O. (2011) Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol Sci 32:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liu M, Kozasa T, Rothstein JD, Sternweis PC, Neubig RR. (2004) Thrombin and lysophosphatidic acid receptors utilize distinct rhoGEFs in prostate cancer cells. J Biol Chem 279:28831–28834. [DOI] [PubMed] [Google Scholar]
- Wang S, Wu J, Suburu J, Gu Z, Cai J, Axanova LS, Cramer SD, Thomas MJ, Perry DL, Edwards IJ, et al. (2012) Effect of dietary polyunsaturated fatty acids on castration-resistant Pten-null prostate cancer. Carcinogenesis 33:404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward Y, Lake R, Yin JJ, Heger CD, Raffeld M, Goldsmith PK, Merino M, Kelly K. (2011) LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res 71:7301–7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson S-J, Brown AJH, Holliday ND. (2012) Differential signaling by splice variants of the human free fatty acid receptor GPR120. Mol Pharmacol 81:631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Wang H, Zhao X, Shi Y, Jin M, Wan B, Xu H, Cheng Y, Ge H, Zhang Y. (2013) Identification of G-protein-coupled receptor 120 as a tumor-promoting receptor that induces angiogenesis and migration in human colorectal carcinoma. Oncogene 32:5541–5550. [DOI] [PubMed] [Google Scholar]
- Xie Y, Gibbs TC, Mukhin YV, Meier KE. (2002) Role for 18:1 lysophosphatidic acid as an autocrine mediator in prostate cancer cells. J Biol Chem 277:32516–32526. [DOI] [PubMed] [Google Scholar]
- Yan Y, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C, Guarda G, Tian Z, Tschopp J, Zhou R. (2013) Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 38:1154–1163. [DOI] [PubMed] [Google Scholar]
- Yun CC, Sun H, Wang D, Rusovici R, Castleberry A, Hall RA, Shim H. (2005) LPA2 receptor mediates mitogenic signals in human colon cancer cells. Am J Physiol Cell Physiol 289:C2–C11. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Knoepp SM, Ku H, Sansbury HM, Xie Y, Chahal MS, Tomlinson S, Meier KE. (2011) Differential expression of FAK and Pyk2 in in metastatic and non-metastatic EL4 lymphoma cell lines. Clin Exp Metastasis 28:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MJ, Spite M. (2012) Resolvins: anti-inflammatory and proresolving mediators derived from omega-3 polyunsaturated fatty acids. Annu Rev Nutr 32:203–227. [DOI] [PubMed] [Google Scholar]