

Abstract
Due to the substantial interspecies differences in drug metabolism and disposition, drug-induced liver injury (DILI) in humans is often not predicted by studies performed in animal species. For example, a drug (bosentan) used to treat pulmonary artery hypertension caused unexpected cholestatic liver toxicity in humans, which was not predicted by preclinical toxicology studies in multiple animal species. In this study, we demonstrate that NOG mice expressing a thymidine kinase transgene (TK-NOG) with humanized livers have a humanized profile of biliary excretion of a test (cefmetazole) drug, which was shown by an in situ perfusion study to result from interspecies differences in the rate of biliary transport and in liver retention of this drug. We also found that readily detectable cholestatic liver injury develops in TK-NOG mice with humanized livers after 1 week of treatment with bosentan (160, 32, or 6 mg/kg per day by mouth), whereas liver toxicity did not develop in control mice after 1 month of treatment. The laboratory and histologic features of bosentan-induced liver toxicity in humanized mice mirrored that of human subjects. Because DILI has become a significant public health problem, drug safety could be improved if preclinical toxicology studies were performed using humanized TK-NOG.
Introduction
Drug-induced liver injury (DILI) has become a leading cause of acute liver failure in several western countries, and is the most common reason for regulatory actions after drug approval (Ostapowicz et al., 2002; Watkins and Seeff, 2006). Interspecies differences in the drug metabolism and disposition pathways used by humans and animal species (reviewed in Williams et al., 2008, and Peltz, 2013) have limited the predictive utility of animal toxicology studies. The results obtained from in vitro systems and from in vivo animal testing have not always accurately predicted the drug metabolism (Anderson et al., 2009; Leclercq et al., 2009; Walker et al., 2009) or transporter-mediated drug clearance (Williams et al., 2008) pathways in humans. Because of this, drugs that produced minimal toxicity in animal studies have sometimes caused significant DILI in humans. The fatalities occurring in 7 of 15 human subjects that were treated with fialuridine provide a striking example of an unexpected DILI that was not predicted by toxicology studies in animal species (Manning and Swartz 1995; McKenzie et al., 1995). Although its toxicity was less severe, bosentan, which is an endothelin receptor antagonist used for the treatment of pulmonary arterial hypertension (Rubin et al., 2002), provides another example of unanticipated DILI in humans (Fattinger et al., 2001). Bosentan did not cause liver toxicity in preclinical animal models, but it caused dose-dependent and reversible liver damage in ∼10% of treated humans, which is manifested by elevated transaminase levels (Fattinger et al., 2001; Humbert et al., 2007). This has significantly limited its therapeutic utility, and patients taking bosentan must undergo monthly liver function monitoring. Bosentan-mediated inhibition of bile salt export pump (BSEP) activity interferes with bile acid secretion (Fattinger et al., 2001), which is thought to be responsible for its cholestatic toxicity. However, because bosentan inhibits both rodent and human BSEP (Fouassier et al., 2002), the species-specific difference in susceptibility to bosentan-induced liver toxicity cannot be explained by BSEP inhibition alone. More broadly, species-specific differences in drug transport make it difficult to accurately assess a drug’s potential for causing cholestatic hepatotoxicity in humans. Pharmaceutical companies are now producing drugs with high aqueous solubility, which further compounds the problem, because their elimination is more dependent on transporter-mediated biliary excretion pathways (Luo et al., 2010).
Chimeric mice with humanized livers were produced to generate a more predictive platform, which would improve drug safety. The humanized liver is produced by transplantation of human liver cells into mice with genetically engineered modifications that facilitate human liver cell engraftment (Peltz, 2013). For example, a NOG mouse expressing a thymidine kinase transgene (TK-NOG) expresses a thymidine kinase transgene within the liver of an immunodeficient mouse strain (Hasegawa et al., 2011), which enables a brief exposure to a nontoxic dose of ganciclovir to induce the rapid and temporally controlled ablation of mouse liver cells. This enables transplanted human liver cells to develop into a mature human organ with a three-dimensional architecture and a gene expression pattern characteristic of mature human liver, which could be stably maintained for >6 months without exogenous drug treatment (Hasegawa et al., 2011). Chimeric TK-NOG mice were shown to be a predictive model for the pattern of human drug metabolism and the occurrence of a human drug-drug interaction for a drug in development (Nishimura et al., 2013), and for identifying human genetic factors affecting drug metabolism (Hu et al., 2013). Because TK-NOG mice do not have ongoing liver toxicity, and do not require treatment with other drugs to suppress their immune system or to prevent liver damage, they could provide an optimal platform for toxicology studies (Peltz, 2013). Consistent with this possibility, we recently demonstrated that DILI caused by fialuridine was easily detected in chimeric TK-NOG mice (Xu et al., 2014). The humanized livers in TK-NOG mice express mRNAs encoding many human drug transporters (including BSEP) at levels that are equivalent to those in human hepatocytes (Hasegawa et al., 2011). However, we do not know whether their biliary tract is functionally humanized, or if they can predict whether a drug will have a human-specific pattern of hepatobiliary clearance. One test drug was previously shown to have a humanized profile of liver clearance in another type of chimeric mouse (Okumura et al., 2007), which suggests that this may be possible. In this study, we demonstrate that the human-specific cholestatic toxicity caused by bosentan could have been predicted using chimeric TK-NOG mice.
Materials and Methods
Preparation and Characterization of Chimeric TK-NOG Mice.
All animal experiments were performed according to protocols that were approved by the Stanford Institutional Animal Care and Use Committee, and the results are reported according to the ARRIVE guidelines (Kilkenny et al., 2010). TK-NOG mice were obtained from and housed at In Vivo Sciences International (Sunnyvale, CA). TK-NOG mice with humanized livers were prepared using the gancylcovir-conditioning and human hepatocyte transplantation protocol, as previously described (Hu et al., 2013). Cryopreserved human hepatocytes were obtained from Celsis In Vitro (Baltimore, MD). The chimeric mice, the hepatocyte donors, and the level of human serum albumin in the humanized mice, which was measured 8 weeks after transplantation, are shown in Supplemental Table 1. Only chimeric mice having a human plasma albumin level greater than 9 mg/ml was used in this study. Human liver cells were transplanted when the mice were 8 weeks old, and the toxicology studies were performed 8 weeks after transplantation. The plasma human albumin level, which was shown to correlate with the extent of liver humanization, was measured by enzyme immunoassay (Hasegawa et al., 2011).
Cefmetazole Disposition.
Cefmetazole was obtained from Sigma-Aldrich (Sigma-Aldrich, St. Louis MO). For the pharmacokinetic studies, control and humanized TK-NOG mice (n = 3) were dosed with 25 mg/kg i.p. cefmetazole (Sigma-Aldrich). Blood samples were collected from the tail vein 0.25, 0.5, 1, 2, 4, and 24 hours after dosing. Bile fluid was collected at 0.5, 4, and 8 hours after dosing using a Vevo 770 image-guided ultrasound system (Visual Sonic, Toronto, ON, Canada). For the drug excretion study, control and humanized mice were dosed with 25 mg/kg i.p. cefametazole and placed in individual metabolic cages (Hatteras Instruments, Cary, NC). At 8 and 24 hours after dosing, bile, feces, and urine were collected for analysis.
Cefmetazole Analysis.
To 5 μl mouse plasma, urine, or bile, 15 μl cold acetonitrile (containing an internal standard) was added to precipitate protein. The solution was then centrifuged at 14,000 rpm for 5 minutes. A total of 10 μl supernatant was transferred to an autosampler vial with 90 μl water; the solution was vortexed; and 5 μl was injected for analysis.
Fecal samples were weighed, and then 20 μl/mg acetonitrile (with internal standard) was added. The samples were homogenized with Precelly metal beads (2.8 mm) in a Precellys 24-dual homogenizer (Bertin Technologies, Montigny le Bretonneux, France).
A Bruker ultra high–performance liquid chromatography system (Bruker Chemical and Applied Markets, Fremont CA) and a YMC-Pack Pro C18 RS, 2.0 × 50-mm, 3-µm column (YMC, Kyoto, Japan) maintained at 40°C were used for the analysis. The mobile phase consisted of 0.1% formic acid, 2 mM ammonium formate in water (mobile phase A), and 0.1% formic acid, 2 mM ammonium formate in methanol (mobile phase B), and gradient elution starting at 20% mobile phase B, kept for 1 minute, and ramping with a constant slope to 95% mobile phase B at 2 minutes, and kept at 95% mobile phase B for 0.5 minutes at flow rate 0.4 ml/min was used with a run time of 7 minutes, including re-equilibration to initial conditions. The injection volume was 5 µl. Cefzolin (Sigma-Aldrich) was used as the internal standard. A Bruker EVOQ Elite (Bruker Chemical and Applied Markets) was used with an electrospray ionization source and positive polarity. The source parameters were spray voltage of 400 V, cone gas flow of 15 unit, cone temperature of 300°C, heated probe gas flow of 40 unit, heated probe temperature of 450°C, Nebulizer gas flow of 50 unit, and exhaust gas on. The mass spectrometer parameters listed in Table 1 were used for the analysis.
TABLE 1.
Paramaters used for mass spectrometric analysis of cefmetazole and cefzolin
The ions used for both quantification and qualification of cefmetazole are indicated. Cefzolin was used as the internal standard.
| Compound Name | Retention Time | Precursor Ion | Product Ion | Collision Energy | Dwell Time |
|---|---|---|---|---|---|
| m/z | V | ms | |||
| Cefmetazole (quantify) | 2.24 | 472 | 328 | 10 | 75 |
| Cefmetazole (qualify) | 2.24 | 472 | 356 | 8 | 75 |
| Cefzolin (internal standard) | 2.4 | 455 | 155.9 | 15 | 150 |
In Situ Perfusion.
These studies were performed using a modified version of previously described methods (Zamek-Gliszczynski et al., 2006). The mice were fully anesthetized by injection of 0.2 ml per 10 g body weight of a 1.2% Avertin (2,2,2-tribromoethanol; Sigma-Aldrich) solution (intraperitoneally). The abdominal cavity of the anesthetized mouse was opened to expose the intestines, liver, and gallbladder. The common bile duct was ligated above the duodenum to prevent bile from entering the intestine. A loose suture was then placed around the inferior vena cava below the liver, and the portal vein was cannulated with a 24-gauge catheter. The liver was perfused at a rate of 2–5 ml/min with drug-free Krebs-Henseleit buffer (Sigma-Aldrich) solution containing 5 μM taurocholate (catalogue 86339; Sigma-Aldrich) that was continuously oxygenated. The portion of the inferior vena cava that was below the liver was then immediately severed by an incision placed below the suture. Next, the gallbladder was cannulated with PE-10 tubing, and then the superior vena cava was cannulated with a 24-gauge catheter. Then, the loose suture around the inferior vena cava was tied off to direct all perfusate outflow through the cannula inside the inferior vena cava above the liver. After a 10-minute preperfusion period, which allows for equilibration of the liver temperature and bile flow, the liver was perfused with buffer containing 5 μM carboxydichlorofluorescein (CDCF; Life Technologies, Grand Island, NY), 5 μM CDCF, and 20 μM rifampicin (Sigma-Aldrich), or 5 μM CDCF and 20 μM probenecid (Sigma-Aldrich). In other in situ perfusion experiments, 56 μg/ml cefmetazole was added to the perfusion buffer. Bile and the outflow perfusate samples were collected at 10-minute intervals. At the end of the experiment, the livers were isolated and snap frozen for subsequent analysis. The in situ perfusion data were analyzed using previously described methods (Zamek-Gliszczynski et al., 2006) and the following formulas:
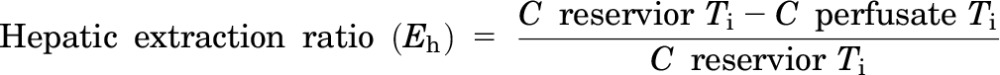 |
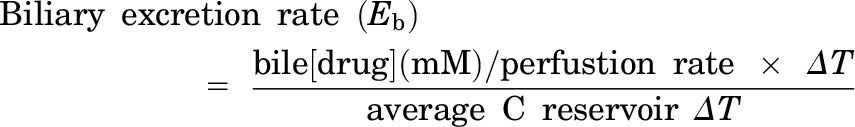 |
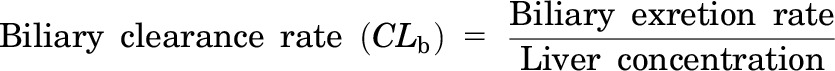 |
 |
where Ti is the time point for the measurement; C is the concentration in the reservoir or the perfusate; and ΔT is the time interval. The tissue concentration was determined by measurement of the bosentan or metabolite concentration in the perfused liver tissue, and the outlet concentration was the drug or metabolite concentration in the perfusate outflow at the last (40-minute) time point. The steady state start points were selected based upon when the drug concentration versus time curve reached its plateau level. All clearance values were normalized for liver mass.
Bosentan Toxicology Study.
Bosentan was synthesized by Bosche (New Brunswick, NJ); it was shown to be >99% pure by liquid chromatography and mass spectroscopy analysis, and its chemical structure was confirmed by two-dimensional NMR analysis. For drug formulation, a 20% Cavasol W7 HP Pharma (Wacker Chemical, München, Germany) was used as the solvent, which we refer to as CAVA. Bosentan powder was first dissolved in 20% CAVA to a concentration of 20 mg/ml, and the pH was adjusted to 7.7–7.9. Control or humanized TK-NOG mice were first treated with 40, 80, or 160 mg/kg per day bosentan by oral gavage. Then control and humanized TK-NOG mice were treated with 160 mg/kg per day bosentan by oral gavage for 28 days. Then, the liver tissue was harvested and placed in 10% formalin for H&E staining, 4% paraformaldehyde for immunohistochemistry, and OCT for immunofluorescence staining. The plasma levels of total cholesterol, total protein, and γ-glutamyltransferase (GGT) were measured using a Heska DryChem 7000 analyzer (HESKA, Loveland, CO), according to the manufacturer’s instructions. In a subsequent study, control and humanized TK-NOG mice were treated with 32 or 6 mg/kg per day bosentan by oral gavage for 14 days.
Statistical Analyses.
To facilitate comparisons between groups of mice, the data were first logarithm transformed. This enabled the group data being compared to be more homogeneous, and the data distribution was closer to a Gaussian distribution. Then, a two-sample, two-sided t test was applied to test the significance of the observed differences between drug-treated and the corresponding vehicle-treated humanized mice. Finally, when plasma alanine aminotransferase (ALT), alkaline phosphatase, GGT, total protein, and total cholesterol levels were investigated simultaneously, all P values were adjusted using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995) to control for the false-discovery rate.
Results
Chimeric TK-NOG Mice Have a Humanized Profile of Biliary Drug Clearance.
A cephalosporin antibiotic (cefmetazole) has been used to characterize species-specific pathways for drug excretion because it is eliminated in an unmetabolized form. Rodents clear this drug primarily via biliary excretion, whereas humans clear it by renal excretion (Okumura et al., 2007). Therefore, we compared the cefmetazole clearance pathways in control TK-NOG mice and TK-NOG mice with highly humanized livers (Supplemental Table 1). The plasma human albumin concentrations in the chimeric mice (12.4 ± 2.2 mg/ml), which correlate with the extent of liver humanization (Hasegawa et al., 2011), indicate that the livers were over 90% humanized. The amounts of cefmetazole in urine, feces, and bile obtained from control and humanized TK-NOG mice were measured for a 24-hour period after administration of cefmetazole (25 mg/kg i.p.). The amount of cefmetazole in feces was 8-fold below (P = 0.006) and in urine was 5-fold greater (P = 0.03) in humanized TK-NOG mice than in control TK-NOG mice (Supplemental Table 2). Cefmetazole had the same plasma pharmacokinetic profile in control and humanized mice, but their biliary profiles were substantially different. The maximal cefmetazole concentration in bile obtained from control TK-NOG mice was 55-fold greater than in humanized TK-NOG mice (P = 0.024) (Supplemental Fig. 1). In humanized TK-NOG mice, only 18% of this drug was cleared in an unchanged form through the feces (biliary excretion) and 81% was cleared by renal clearance. In contrast, 71% was cleared by biliary excretion and 26% by renal elimination in control TK-NOG mice (Fig. 1). These studies demonstrate that the livers in humanized TK-NOG mice have a humanized profile of biliary cefmetazole excretion.
Fig. 1.

To assess the pattern of cefmetazole clearance, control (n = 3) and humanized (n = 3) TK-NOG mice were dosed with cefmetazole (25 mg/kg i.p.), and the amount of cefmetazole in blood, urine, and bile was measured by liquid chromatography and mass spectroscopy analysis. The percentage of cefmetazole in urine, bile, or feces collected over a 24-hour period is shown for control and humanized TK-NOG mice. Although cefmetazole was primarily eliminated through renal excretion (81%) in humanized mice, it was mostly eliminated via the feces (71%) in control mice.
We wanted to determine whether interspecies differences in biliary cefmetazole clearance could be characterized using an in situ liver perfusion system (Fig. 2). As described in the supplement, in situ perfusion experiments using a fluorescent nonmetabolizable dye and transport inhibitors established that dye uptake into the liver and its transport into bile are saturable and carrier-dependent processes in the livers of both humanized and control mice (Supplemental Fig. 2). We next examined whether the in situ liver perfusion system could be used to characterize the basis for the different profiles of cefmetazole biliary transport in mice with control and humanized livers. To do this, cefmetazole (56 μg/ml) was added to the perfusion buffer, and the amount of cefmetazole in the liver and bile was measured as a function of the time of perfusion. Cefmetazole transport into bile reached its steady state rate after 20 minutes of perfusion, which persisted until the end of the perfusion period (Supplemental Fig. 3). The rate of hepatic cefmetazole uptake (i.e., the hepatic extraction ratio or Eh) was low in both control and humanized mice. However, the humanized liver had a 4-fold lower rate (P = 0.007) of biliary cefmetazole clearance (CLb = 0.02) relative to control mice (CLb = 0.08), and a 5-fold (P = 0.003) higher affinity for cefmetazole (KP = 2.7) than control mouse liver (KP = 0.5) (Supplemental Table 3). The perfusion data reveal that the humanized liver has a lower rate of cefmetazole biliary transport and higher cefmetazole retention, which explains why the biliary clearance of cefmetazole is reduced in humans.
Fig. 2.

Diagram of the in situ perfusion system. The liver is continuously perfused with a solution containing 100 μg/ml cefmetazole, which enters the liver via the portal vein. The inferior vena cava (IVC) is ligated, so the perfusate exits via the superior vena cava (SVC) after passing through the liver. The common bile duct is cannulated to direct all bile flow to the gallbladder, which is where the bile is collected.
Bosentan-Induced Liver Injury in Humanized TK-NOG Mice.
We next investigated whether the humanized TK-NOG model could predict whether bosentan would cause liver toxicity in humans. Chimeric and control TK-NOG mice (Supplemental Table 1) were first treated with 160 mg/kg per day by mouth bosentan for 28 days, which is a dose that is 23-fold above the total daily human dose (Rubin et al., 2002). This treatment was well tolerated by control and humanized TK-NOG mice; they did not lose weight over the 1-month period of treatment (P = 0.94) (Fig. 3A); there were no deaths; and none became overtly jaundiced. There was a temporary decrease in the weight of the vehicle-treated humanized mice (∼2 g) after 2 weeks, but their weight returned to the initial level by 4 weeks. Mice often experience a temporary decrease in weight during the early phase of a drug study. This is due to the fact that they are frequently handled and subject to daily weighing and daily injections, and their blood is frequently drawn. However, there was clear serologic evidence of DILI in the bosentan-treated humanized mice; their plasma ALT (875 ± 55.8) was increased 15-fold (P = 4.6 × 10−8) over bosentan-treated control (59.8 ± 6.6) TK-NOG mice. The ALT levels in bosentan-treated humanized mice were 6.3-fold increased (P = 7 × 10−6) relative to vehicle-treated humanized TK-NOG mice (Fig. 3B). The serum ALT levels were followed weekly in the bosentan-treated humanized mice; they were elevated after only 1 week of dosing, and remained elevated throughout the 4-week dosing period.
Fig. 3.

Control (nonhumanized) (n = 6) or chimeric (n = 6) TK-NOG mice were treated with 160 mg/kg per day bosentan by mouth or vehicle for 28 days, and their weights were measured on a daily basis (A). The plasma ALT (B), alkaline phosphatase, and GGT (C) levels were measured after 4 weeks of dosing. The plasma ALT levels were also measured after 1 and 2 weeks in (B). In (B and C), each bar is the average + S.D. of six measurements in control or humanized TK-NOG mice, and the dashed line indicates the upper limit of normal for the measured parameter. (D) Bosentan treatment induces an increase in plasma total bile acids in humanized TK-NOG mice. Plasma was collected from both control (n = 3) and humanized (n = 3) TK-NOG mice after 0 and 28 days of treatment with bosentan (160 mg/kg per day). After 28 days of bosentan treatment, the total plasma bile acid concentration in humanized mice was increased 5-fold (P = 0.046), whereas in control mice there was no change in the plasma total bile acid concentration (P = 0.38).
Analyses of other liver injury markers confirmed that bosentan caused liver toxicity in humanized, but not in control mice. After 28 days of dosing, plasma alkaline phosphatase levels in bosentan-treated humanized TK-NOG mice (508 ± 59) were increased by 7-fold (P = 3.1 × 10−7) relative to bosentan-treated control (73 ± 5.2) mice, and were 4-fold above (P = 4.0 × 10−5) that of vehicle-treated (123 ± 6.5) humanized TK-NOG mice (Fig. 3C). The plasma GGT levels in bosentan-treated humanized TK-NOG mice (21 ± 0.8) were also increased relative to bosentan-treated control (10 ± 0.8) (P = 8.0 × 10−5) or vehicle-treated humanized mice (11 ± 0.8) (P = 7.5 × 10−5) (Fig. 3C; see Supplemental Methods). Of importance, the serum ALT and alkaline phosphatase levels were not elevated above the upper limit of normal in the bosentan-treated control TK-NOG mice (Fig. 3, B and C). Because bosentan reportedly causes hepatic bile acid accumulation via inhibition of BSEP transporter activity, we measured the total bile acid concentration in the plasma of control (n = 3) and humanized mice (n = 3) after 0, 7, or 28 days of bosentan treatment. After 28 days of bosentan treatment, the total bile acid concentration in plasma obtained from humanized mice (45.1 ± 21.8 μM) was 5-fold higher than on day 0 (8.9 ± 1.6 μM) (P = 0.046). In contrast, in the plasma of control mice, there was no change in the total bile acid concentration on days 0 (1.9 ± 0.25 μM) and 28 (3.2 ± 1.1 μM) (P = 0.38) (Fig. 3D). Other liver function parameters, such as the total protein and cholesterol levels, were within normal limits in all groups of mice (Supplemental Fig. 3). Although their average values were slightly reduced in the bosentan-treated humanized mice, there was no significant difference in the total plasma cholesterol (P = 0.20) or protein (P = 0.37) between the bosentan and vehicle-treated groups of humanized mice. Examination of liver sections obtained from humanized TK-NOG mice revealed no histologic abnormalities except for occasional focal mild fatty change in areas with human hepatocytes, but these were noted in livers obtained from both vehicle- and bosentan-treated mice (Supplemental Fig. 4). Mouse hepatocytes in all groups of mice were unremarkable. Thus, bosentan-induced liver injury is clearly detected in chimeric (but not control) mice; the laboratory data and histology observed in chimeric mice mimic the cholestatic pattern seen in bosentan-treated human subjects (Fattinger et al., 2001).
To investigate whether this toxicity was dose dependent, highly humanized chimeric TK-NOG mice were treated with 32 or 6 mg/kg per day oral bosentan for 14 days, before drug dosing was electively terminated. There were no deaths, and the humanized mice treated with these doses did not lose body weight after 14 days of treatment with either the 32 (P = 0.5) or 6 mg/kg per day dose (P = 0.08) (Supplemental Fig. 5). However, there was clear evidence that bosentan caused liver toxicity in the humanized, but not in control mice. However, after treatment with the 6 or 32 mg/kg per day doses of bosentan, humanized TK-NOG mice had statistically significant elevations of plasma ALT (P = 0.006 and 0.003, respectively; Fig. 4A) and alkaline phosphatase (P = 0.002 and 0.003, respectively; Fig. 4B) levels relative to their pretreatment values. In contrast, neither bosentan dose caused the plasma ALT or alkaline phosphatase levels to increase in control mice, nor were the plasma ALT or alkaline phosphatase levels increased in vehicle-treated chimeric mice. These data establish that bosentan-induced liver toxicity was dose dependent, and could be detected in chimeric TK-NOG mice treated with a bosentan dose that is only 2-fold above the usual human dose.
Fig. 4.

Control (n = 6) or chimeric TK-NOG mice (n = 6 per group) were treated with 32 or 6 mg/kg per day bosentan by mouth or vehicle (n = 3) for 14 days, and their plasma ALT (A) and alkaline phosphatase (B) levels were measured on days 0, 7, and 14. Each bar is the average + S.D. of the measurements in control or humanized TK-NOG mice, and the dashed line indicates the upper limit of normal for the measured parameter.
Discussion
Our results demonstrate that the biliary tract of chimeric TK-NOG mice is functionally humanized, and that the TK-NOG model can be used to identify drugs that may cause cholestatic liver toxicity in humans. We also demonstrate that the basis for interspecies differences in biliary drug elimination can be characterized by coupling the TK-NOG model with an in situ liver perfusion system. The in situ perfusion results indicate that the interspecies differences in cefmetazole elimination pathways result from species-specific differences in the rate of biliary drug transport and in liver retention of the drug. Thus, the type of liver cell, irrespective of whether the cells in the biliary ducts are humanized, determines whether cefmetazole is predominantly eliminated by biliary (mouse) or renal pathways (human). Because of this, TK-NOG mice with humanized livers have a humanized profile of cefmetazole elimination, which is substantially different from that of control mice.
Given the significant public health problem caused by DILI, and the difficulties associated with regulatory actions occurring after drug approval (Ostapowicz et al., 2002; Watkins and Seeff, 2006), identification of candidate medications that are likely to cause human-specific DILI is of major importance. Bosentan-induced cholestatic toxicity could easily be detected in humanized TK-NOG mice after only 1 week of dosing, even when treatment of conventional mice for 1 month provided no evidence of its hepatotoxic potential in humans. At present, we do not understand the mechanism underlying the human-specific susceptibility to bosentan-induced liver toxicity. Because bosentan is an equipotent inhibitor of bile acid transport by murine and human BSEP (Mano et al., 2007), this activity alone cannot explain the human-specific hepatotoxicity. It is likely that interspecies differences in bile acid biosynthesis may underlie their differential susceptibility. Bosentan’s effect on bile transport resembles (in some ways) that of bile duct ligation, and humans and rodents exhibit very different responses to biliary obstruction. In mice (Zhang et al., 2012) and rats (Takita et al., 1988; Naito et al., 1996), bile duct ligation causes a rapid increase in the synthesis of mostly (95%) water-soluble muricholic acids that are excreted in the urine, which provides rodents with a route for elimination of hepatotoxic bile acids (Dueland et al., 1991; Naito et al., 1996). In contrast, human liver produces a very different set of hepatotoxic bile acids (chenodeoxycholic acid, cholic acid, and their glycine conjugates) (Russell, 2003), which could be more difficult to eliminate when BSEP is inhibited. Given their differential susceptibility to bosentan-induced liver toxicity, subsequent experimental analyses of bile acid production and transport in control and humanized mice could enable us to understand the increased susceptibility of the human liver to bosentan-induced toxicity.
Despite the bosentan-induced elevations in plasma liver enzymes and in total bile acid concentration, bosentan induced minimal histologic abnormalities in the livers of chimeric mice. Bosentan-induced liver toxicity in humans is reversible, and we do not have liver tissue from bosentan-treated humans for comparison. Hence, we do not know what type of histopathology to expect in the livers of the humanized mice. Nevertheless, if humanized TK-NOG mice had been used during the preclinical evaluation of bosentan, important information about its potential to cause cholestatic liver toxicity in humans would have been available to pharmaceutical companies and to government regulators at an early stage in its development. We have now demonstrated that the TK-NOG model could identify drugs causing human-specific DILI by two different mechanisms [mitochondrial-toxic (Xu et al., 2014) and cholestatic], which indicates that their use in preclinical drug assessment could improve drug safety.
Supplementary Material
Acknowledgments
The authors dedicate this work to Dr. Tatsuji Nomura, whose vision and leadership enabled this project to proceed.
Abbreviations
- ALT
alanine aminotransferase
- BSEP
bile salt export pump
- CDCF
carboxydichlorofluorescein
- DILI
drug-induced liver injury
- GGT
γ-glutamyltransferase
- TK-NOG
a NOG mouse expressing a thymidine kinase transgene
Authorship Contributions
Participated in research design: Guo, Peltz.
Conducted experiments: Xu, Wu, S. Nishimura, T. Nishimura, Takedai Takeda, Guan, Day, Hillgren.
Contributed new reagents or analytic tools: Yang, Yates.
Performed data analysis: Guo, Zheng, Michie.
Wrote or contributed to the writing of the manuscript: Xu, Peltz.
Footnotes
This work was supported by a transformative R01 award from the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant 1R01-DK0909921]. This work was also partially supported by Eli Lilly and Company through the Lilly Research Award Program.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Anderson S, Luffer-Atlas D, Knadler MP. (2009) Predicting circulating human metabolites: how good are we? Chem Res Toxicol 22:243–256. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc, B 57:289–300. [Google Scholar]
- Dueland S, Reichen J, Everson GT, Davis RA. (1991) Regulation of cholesterol and bile acid homoeostasis in bile-obstructed rats. Biochem J 280:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattinger K, Funk C, Pantze M, Weber C, Reichen J, Stieger B, Meier PJ. (2001) The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 69:223–231. [DOI] [PubMed] [Google Scholar]
- Fouassier L, Kinnman N, Lefèvre G, Lasnier E, Rey C, Poupon R, Elferink RP, Housset C. (2002) Contribution of mrp2 in alterations of canalicular bile formation by the endothelin antagonist bosentan. J Hepatol 37:184–191. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kawai K, Mitsui T, Taniguchi K, Monnai M, Wakui M, Ito M, Suematsu M, Peltz G, Nakamura M, et al. (2011) The reconstituted ‘humanized liver’ in TK-NOG mice is mature and functional. Biochem Biophys Res Commun 405:405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Wu M, Nishimura T, Zheng M, Peltz G. (2013) Human pharmacogenetic analysis in chimeric mice with ‘humanized livers.’ Pharmacogenet Genomics 23:78–83. [DOI] [PubMed] [Google Scholar]
- Humbert M, Segal ES, Kiely DG, Carlsen J, Schwierin B, Hoeper MM. (2007) Results of European post-marketing surveillance of bosentan in pulmonary hypertension. Eur Respir J 30:338–344. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. (2010) Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8:e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclercq L, Cuyckens F, Mannens GS, de Vries R, Timmerman P, Evans DC. (2009) Which human metabolites have we MIST? Retrospective analysis, practical aspects, and perspectives for metabolite identification and quantification in pharmaceutical development. Chem Res Toxicol 22:280–293. [DOI] [PubMed] [Google Scholar]
- Luo G, Johnson S, Hsueh MM, Zheng J, Cai H, Xin B, Chong S, He K, Harper TW. (2010) In silico prediction of biliary excretion of drugs in rats based on physicochemical properties. Drug Metab Dispos 38:422–430. [DOI] [PubMed] [Google Scholar]
- Manning FJ, Swartz MN. (1995) Review of the Fialuridine (FIAU) Clinical Trials, National Academy Press, Washington, D.C. [PubMed] [Google Scholar]
- Mano Y, Usui T, Kamimura H. (2007) Effects of bosentan, an endothelin receptor antagonist, on bile salt export pump and multidrug resistance-associated protein 2. Biopharm Drug Dispos 28:13–18. [DOI] [PubMed] [Google Scholar]
- McKenzie R, Fried MW, Sallie R, Conjeevaram H, Di Bisceglie AM, Park Y, Savarese B, Kleiner D, Tsokos M, Luciano C, et al. (1995) Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N Engl J Med 333:1099–1105. [DOI] [PubMed] [Google Scholar]
- Naito T, Kuroki S, Chijiiwa K, Tanaka M. (1996) Bile acid synthesis and biliary hydrophobicity during obstructive jaundice in rats. J Surg Res 65:70–76. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Hu Y, Wu M, Pham E, Suemizu H, Elazar M, Liu M, Idilman R, Yurdaydin C, Angus P, et al. (2013) Using chimeric mice with humanized livers to predict human drug metabolism and a drug-drug interaction. J Pharmacol Exp Ther 344:388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura H, Katoh M, Sawada T, Nakajima M, Soeno Y, Yabuuchi H, Ikeda T, Tateno C, Yoshizato K, Yokoi T. (2007) Humanization of excretory pathway in chimeric mice with humanized liver. Toxicol Sci 97:533–538. [DOI] [PubMed] [Google Scholar]
- Ostapowicz G, Fontana RJ, Schiødt FV, Larson A, Davern TJ, Han SH, McCashland TM, Shakil AO, Hay JE, Hynan L, et al. U.S. Acute Liver Failure Study Group (2002) Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 137:947–954. [DOI] [PubMed] [Google Scholar]
- Peltz G. (2013) Can ‘humanized’ mice improve drug development in the 21st century? Trends Pharmacol Sci 34:255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, et al. (2002) Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 346:896–903. [DOI] [PubMed] [Google Scholar]
- Russell DW. (2003) The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72:137–174. [DOI] [PubMed] [Google Scholar]
- Takita M, Ikawa S, Ogura Y. (1988) Effect of bile duct ligation on bile acid and cholesterol metabolism in rats. J Biochem 103:778–786. [DOI] [PubMed] [Google Scholar]
- Walker D, Brady J, Dalvie D, Davis J, Dowty M, Duncan JN, Nedderman A, Obach RS, Wright P. (2009) A holistic strategy for characterizing the safety of metabolites through drug discovery and development. Chem Res Toxicol 22:1653–1662. [DOI] [PubMed] [Google Scholar]
- Watkins PB, Seeff LB. (2006) Drug-induced liver injury: summary of a single topic clinical research conference. Hepatology 43:618–631. [DOI] [PubMed] [Google Scholar]
- Williams JA, Andersson T, Andersson TB, Blanchard R, Behm MO, Cohen N, Edeki T, Franc M, Hillgren KM, Johnson KJ, et al. (2008) PhRMA white paper on ADME pharmacogenomics. J Clin Pharmacol 48:849–889. [DOI] [PubMed] [Google Scholar]
- Xu D, Nishimura T, Nishimura S, Zhang H, Zheng M, Guo Y-Y, Masek M, Michie SA, Glenn J, Peltz G. (2014) Fialuridine induces acute liver failure in chimeric TK-NOG mice: a model for detecting hepatic drug toxicity prior to human testing. PLoS Med 11:e1001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamek-Gliszczynski MJ, Nezasa K, Tian X, Kalvass JC, Patel NJ, Raub TJ, Brouwer KL. (2006) The important role of Bcrp (Abcg2) in the biliary excretion of sulfate and glucuronide metabolites of acetaminophen, 4-methylumbelliferone, and harmol in mice. Mol Pharmacol 70:2127–2133. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Hong JY, Rockwell CE, Copple BL, Jaeschke H, and Klaassen CD (2012) Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver Int 32:58–69. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.