

Abstract
Background/Aims
Acute exacerbations in chronic obstructive pulmonary disease may be related to air pollution, of which ozone is an important constituent. In this study, we investigated the protein profiles associated with ozone-induced exacerbations in a smoking-induced emphysema model.
Methods
Mice were divided into the following groups: group I, no smoking and no ozone (NS + NO); group II, no smoking and ozone (NS + O); group III, smoking and no ozone (S + NO); and group IV, smoking and ozone (S + O). Bronchoalveolar lavage, the mean linear intercept (MLI) on hematoxylin and eosin staining, nano-liquid chromatography-tandem mass spectrometry (LC-MS/MS), and Western blotting analyses were performed.
Results
The MLIs of groups III (S + NO) and IV (S + O) (45 ± 2 and 44 ± 3 µm, respectively) were significantly higher than those of groups I (NS + NO) and II (NS + O) (26 ± 2 and 23 ± 2 µm, respectively; p < 0.05). Fourteen spots that showed significantly different intensities on image analyses of two-dimensional (2D) protein electrophoresis in group I (NS + NO) were identified by LC-MS/MS. The levels of six proteins were higher in group IV (S + O). The levels of vimentin, lactate dehydrogenase A, and triose phosphate isomerase were decreased by both smoking and ozone treatment in Western blotting and proteomic analyses. In contrast, TBC1 domain family 5 (TBC1D5) and lamin A were increased by both smoking and ozone treatment.
Conclusions
TBC1D5 could be a biomarker of ozone-induced lung injury in emphysema.
Keywords: Proteomics; Pulmonary disease, chronic obstructive; Ozone; Chromatography, liquid; Mass spectrometry
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is a common, preventable, and treatable disease, characterized by limited airflow that is persistent, typically progressive, and associated with chronic pulmonary inflammatory responses to noxious particles and gases [1]. The incidence of COPD has increased in recent decades and is estimated to become the third leading cause of death by 2020 [2]. Of the Korean population older than 45 years, 17.2% have some degree of airway obstruction; i.e., their forced expiratory volume in 1 seconds/forced vital capacity ratio is < 0.7, emphasizing the clinical importance of COPD [3]. A previous cross-sec tional study reported that the mortality rate increased to 2.5% in COPD patients with acute exacerbations of COPD (AECOPD) [4]. Additionally, acute exacerbations can reduce long-term survival in patients with COPD [5,6]. Two known common causes of AECOPD are respiratory infections and air pollution [1]. Among the air pollution causes of AECOPD, ozone is an important factor. One study found that the 2-day cumulative effect of a 5 ppb increase in ozone increased the hospital admissions of patients with COPD [7]. Other studies have also shown that an elevated level of ozone is associated with COPD-related hospital admissions [8,9].
Exogenous reactive oxygen species (ROS), from cigarette smoking, and endogenous ROS, from inflammatory cells, contribute to the pathogenesis of COPD [10,11]. However, no clear explanation of ozone-induced COPD exacerbations exists. We suggest that additive oxygen toxicity may play a pivotal role in the pathogenesis of COPD exacerbations, based on reports of elevated levels of hydrogen peroxide in exhaled air and interleukin 8 (IL-8) and soluble intercellular adhesion molecule-1 in the serum of patients with COPD exacerbations [12].
If biomarkers of COPD exacerbations due to ozone could be identified, the causative factors of COPD exacerbations and their early detection may be facilitated. To this end, biomarkers should be identified, as do the underlying mechanisms of this ailment. Several markers, including serum surfactant protein A, have been evaluated in patients with AECOPD, but, to date, no marker that can differentiate between COPD and AECOPD has been identified [13,14].
Identifying respiratory disease-specific proteins in the airway and alveolar lining fluids is important because such proteins will enable early detection, prognostic assessment, and treatment. Thus, large-scale, high-throughput, and whole-proteome studies of bronchoalveolar lavage (BAL) fluids using two-dimensional electrophoresis (2DE) and matrix-assisted laser desorption/ionization-time of flight mass spectrometry have been conducted to determine the proteomic contribution to asthma and idiopathic pulmonary fibrosis [15,16]. In the present study, we sought to identify proteins specific to ozone-induced AECOPD in lung tissues using differential-display proteomics.
To our knowledge, few reports have described experimental models of AECOPD. In particular, a study of ozone-induced lung damage in a smoking-induced emphysema model represented the first report related to AECOPD. In the present study, we identified AECOPD-specific proteins in a novel model of AECOPD, based on smoking and ozone.
METHODS
Animals
Male BALB/c mice, at 7 weeks of age, were purchased from Central Lab Animal Inc. (Seoul, Korea). The animals were housed in a temperature- and humidity-controlled room with free access to water and standard laboratory food. The animals were assigned randomly to four groups, for exposure to filtered air (group I, no smoking and no ozone, NS + NO), ozone (group II, no smoking and ozone, NS + O), smoking (group III, smoking and no ozone, S + NO), and smoking followed by ozone (group IV, smoking and ozone, S + O). Each group consisted of six animals.
Cigarette smoke exposure
Mice in the smoking groups were exposed to five cigarettes per day for 5 days per week for 6 months ('This' cigarettes, 6.5-mg tar and 0.65-mg nicotine; KT&G, Daejeon, Korea). On each day of exposure, animals were placed individually inside a Plexiglass cabinet (850 × 350 × 300 mm). Cigarette smoke was delivered into the cabinet through air inflow at a rate of 1.7 mL/sec with a burning cigarette using an automatic machine (Threeshine Co., Daejeon, Korea); the combustion time of the cigarette was less than 4 minutes. A fan inside the cabinet ensured rapid and equal distribution of the smoke. Fresh air was then delivered to the cabinet to remove the smoke. During exposure, animals, including control animals, did not receive food or water but were allowed free access to both after exposure.
Ozone exposure
Ozone exposure was performed 2 days after the last day of cigarette smoke inhalation. The ozone system delivered ozone concentrations of 0.1 to 3 ppm, generated by an electric discharge ozone generator (SW-OGMD-L001, Swater Inc., Bucheon, Korea). The concentrations of ozone were monitored continuously with an ozone monitor (IN2000-L2-LC, IN USA Inc., Needham, MA, USA), which can detect ozone concentrations between 0 and 3 ppm. On the day of the experiment, six mice were placed into the exposure chamber simultaneously and were exposed to 3 ppm for 2 hours. The mice were sacrificed 6 hours after ozone exposure.
Bronchoalveolar lavage
BAL was performed 3 hours after exposure to ozone as described previously [17]. Briefly, BAL fluid was obtained by instilling saline into the lungs three times through a tracheal tube using a total volume of 1.5 mL. The BAL fluid was centrifuged (150 ×g, 10 minutes, 4℃), and the cell pellet was resuspended in 1-mL phosphate-buffered saline. Then, 100 µL of cell suspension were mixed with the same volume of 4% trypan blue to determine cell counts and viability. To determine differential cell counts in BAL fluid, 5 × 103 cells were mounted on a slide by cytocentrifugation and stained with Diff-Quik (Baxter Healthcare, Miami, FL, USA).
Tissue preparation and fixation
Following BAL, the chest wall was opened. The right lungs were snap-frozen in liquid nitrogen and stored for protein analysis (Western blot and proteomic analysis). The left lungs were inflated with 10% phosphate-buffered formalin at a transpulmonary pressure of 25 cmH2O for 1 hour. The tissues were embedded in paraffin wax blocks and were cut for hematoxylin and eosin staining and immunohistochemistry for matrix metalloproteinase-12 (MMP-12).
Mean linear intercept
To calculate the change in alveolar destruction, we measured the air space size using the mean linear intercept (MLI) according to a modified version of a method described previously [18]. Briefly, after paraffin wax embedding, 5-µm sections were cut and stained with hematoxylin and eosin according to standard methods. Ten randomly selected fields at a magnification of ×100 from each lung were assessed. Fifteen lines were placed randomly on the lung sections, and the number of intercepts crossing the alveolar wall was determined. The MLI was calculated from the length of the lines multiplied by the number of the lines divided by the sum of all counted intercepts.
Sample preparation, two-dimensional electrophoresis, and image analysis
Protein (200 µg) from each group of lung tissue was used for the 2DE analysis. First, 1 mg of protein from each lung sample was precipitated with 10% trichloroacetic acid in acetone and resuspended in the sample solution. Immobiline Dry Strips (Amersham Biosciences, Little Chalfont, UK) were used for isoelectric focusing, which was carried out using 1 mg of the extracted protein in a MultiPhor II system (GE Healthcare, Little Chalfont, UK). After isoelectric focusing separation, the proteins were separated in the second dimension by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. For image analysis, the gels were visualized with Coomassie Brilliant Blue G-250 according to the manufacturer's instructions. The 2D gels were scanned with an ImageScanner (Bio-Rad Laboratories Inc., Hercules, CA, USA) in transmission mode. Spot detection and matching were performed using ImageMaster 2D version 5.0 (Amersham Biosciences). Digitized images were analyzed using the ImageMaster software to calculate the 2D spot intensity by integrating the optical density over the spot area (the spot "volume"); the images were then normalized [19].
Protein identification by nano liquid chromatography-tandem mass spectrometry and database searching
Differentially expressed protein spots were excised from the 2D gels, cut into smaller pieces, and digested with trypsin (Promega, Madison, WI, USA) as described previously [20]. All nano liquid chromatography-tandem mass spectrometry (LC-MS/MS) experiments were performed using an Agilent Nanoflow Proteomics Solution featuring an Agilent 1100 Series nano-LC system (Agilent Technologies, Santa Clara, CA, USA) for MS/MS coupled through an orthogonal nanospray ion source to an Agilent 1100 Series LC/mass selective detector (MSD) Trap XCT ion trap mass spectrometer.
The nano-LC system was operated in the sample enrichment/desalting mode with a Zorbax 300SB-C18 enrichment column (0.3 × 50 mm; 5 µm). Chromatography was performed using a Zorbax 300SB-C18 (75 µm × 150 mm) nanocolumn. The column was eluted with a gradient beginning with isocratic application of 3% solvent B (0.1% formic acid in acetonitrile) and 97% solvent A (0.1% formic acid in water) for 5 minutes. Then, the gradient mixture was changed to 10% B over 5 minutes (from 5 to 10), 45% B over 40 minutes (10 to 50), 90% B (isocratic) for 5 minutes (55 to 60), and 3% B over 1 minute (60 to 61); finally, the column was washed with 3% B for 10 minutes.
The LC/MSD Trap XCT was operated in the unique peptide scan Auto-MS/MS mode. The ionization mode was positive nanoelectrospray with an Agilent orthogonal source. The drying gas flowed at 5 L/min at a temperature of 300℃. Vcap was typically 1,800 to 1,900 V with skim 1 at 30 V, and the capillary exit was offset at 75 V. The trap drive was set at 85 V with averages of one or two. The ion charge control was on with a maximum accumulation time of 150 ms, the smart target was 125,000, and the MS scan range was 300 to 2,200. Automatic MS/MS was performed in the ultrascan mode with the number of parents at 2, averages of 2, a fragmentation amplitude of 1.15 V, SmartFrag on (30% to 200%), active exclusion on (after two spectra for 1 minute), prefer +2 on, an MS/MS scan range of 100 to 1,800, and ultrascan on. Each acquired MS/MS spectrum was searched against the non-redundant protein sequence database using the Spectrum Mill software [20].
Immunohistochemistry
A VectaStatin rabbit ABC Elite kit (Vector Laboratories, Burlingame, CA, USA) was used for immunostaining. Tissue sections (5 µm) were deparaffinized, endogenous peroxidase was blocked with 1.4% H2O2 in methyl alcohol for 30 minutes, and nonspecific binding was blocked with 1.5% normal rabbit serum for 30 minutes. The sections were incubated with rabbit polyclonal anti-mouse MMP-12 antibody (1:100; Abcam, Cambridge, UK) at 4℃ for 16 hours. After washing with Tris-buffered saline (TBS), the sections were sequentially incubated with biotinylated rabbit anti-rabbit immunoglobulin G (H + L; 1:200, Vector Laboratories) and avidin-biotin peroxidase complex (1:50, ABC kit, Vector Laboratories) for 30 minutes. The color reaction was developed with 3,3'-diamino-benzidine tetrachloride (Zymed Laboratories, San Francisco, CA, USA), and Harris hematoxylin was used as a counterstain.
Western blotting
First, 20-µg protein from each lung tissue sample were electrophoresed on 15% polyacrylamide gels using a discontinuous gel. The proteins were transferred to nitrocellulose membranes at 120 V for 40 minutes. After blocking in 5% skim milk and 0.1% NP40 in TBS for 2 hours at room temperature, the membranes were incubated with the following primary antibodies: rabbit polyclonal antibody to vimentin (Cell Signaling, Beverly, MA, USA), rabbit polyclonal antibody to lactate dehydrogenase A (LDHA; Novus Biologicals, Littleton, CO, USA), mouse monoclonal antibody to TBC1 domain family 5 (TBC1D5, Santa Cruz Biotechnology, CA, USA), goat polyclonal to triose phosphate isomerase (TPI; Abcam), rabbit polyclonal antibody to gelsolin (Abcam), and rabbit polyclonal antibody to lamin A (Abcam). The membrane was then incubated with horseradish peroxidase-conjugated anti-rabbit IgG (1:5,000 dilution) for 1 hour at room temperature. The target protein was detected using enhanced chemiluminescence solution (Amersham Pharmacia Biotech).
Statistics
Kruskal-Wallis nonparametric tests were used to compare the number of cells in the BAL fluid and MLIs across the four groups. The Mann-Whitney U test was used for comparisons between each group, and then Bonferroni correction was applied to counteract the problem of multiple comparisons if the overall p value comparing all four groups was less than 0.05. All analyses were performed using the SPSS version 21 (IBM Co., Armonk, NY, USA).
RESULTS
Cellular profiles in BAL fluid
In group IV (S + O), significant increases were noted in the total cell count and number of macrophages compared with the other groups. The total cell counts and number of macrophages were higher in group II (NS + O) and III (S + NO) than in group I (NS + NO) (Fig. 1A and 1B). The number of macrophages was lower in group III (S + NO) than in group II (NS + O) (Fig. 1B). Significantly higher numbers of neutrophils and lymphocytes were found in group IV (S + O) than in the other groups. There were also significantly more neutrophils and lymphocytes in groups II (NS + O) and III (S + NO) than in group I (NS + NO). No difference was observed in the number of eosinophils among the groups (Fig. 1C).
Figure 1.

Total cell count (A), and macrophage (B), and neutrophil, lymphocyte, and eosinophil counts (C) in bronchoalveolar lavage f luid. There were signif icant increases in the total cell count and number of macrophages in group IV smoking and ozone (S + O) versus the other groups. In groups II no smoking and ozone (NS + O) and III smoking and no ozone (S + NO), the total cell count and number of macrophages were higher compared to those in group I no smoking and no ozone (NS + NO) (A, B). The number of macrophages was lower in group III (S + NO) than in group II (NS + O) (B). There were significant increases in the number of neutrophils and lymphocytes in group IV (S + O) versus the other groups. The numbers of neutrophils and lymphocytes were significantly higher in groups II (NS + O) and III (S + NO) than in group I (NS + NO). No difference was found in the eosinophil count among the groups (C). ap < 0.05, bp < 0.01, cp < 0.001 vs. I, dp < 0.05, ep < 0.001, vs. II, fp < 0.01, gp < 0.001, vs. III.
Histology and morphometric analysis
In groups I and II, the architecture of the alveolar septa and spaces were well preserved; however, in group III (S + NO) and IV (S + O), the alveoli were confluent and enlarged (Fig. 2). In group II (NS + O), more inflammatory cells had infiltrated into the interstitium than in group I (NS + NO) (small rectangle). The number of infiltrated cells was not different between groups III (S + NO) and IV (S + O).
Figure 2.
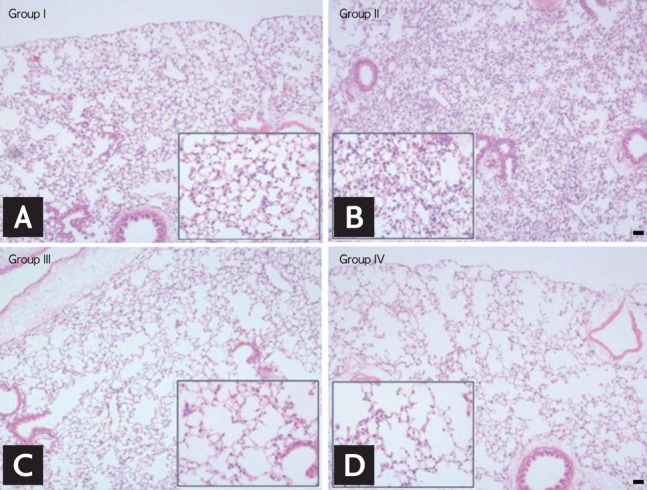
(A-D) Hematoxylin and eosin staining of the lung (×100) and magnified images (rectangle, ×400). In groups I (A) and II (B), the architecture of the alveolar septa and space was well preserved; however, in groups III (C) smoking and no ozone (S + NO) and (D) IV smoking and ozone (S + O), the alveoli were confluent and enlarged. In group II no smoking and ozone (NS + O), more inflammatory cells had infiltrated in the interstitium than in group I no smoking and no ozone (NS + NO) (rectangle). The numbers of infiltrated cells were not different between groups III (S + NO) and IV (S + O). Scale bar = 20 µm.
Immunohistochemical staining for MMP-12 showed no difference in the number of MMP-12-positive cells between groups I and II; additionally, no difference was noted in the number of MMP-12-positive cells between groups III (S + NO) and IV (S + O) (Fig. 3).
Figure 3.
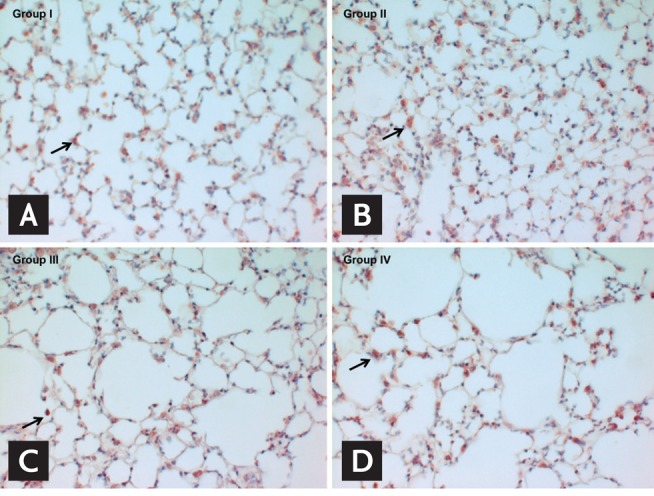
(A-D) Immunohistochemical staining for matrix metalloproteinase-12 (×400). MMP-12-positive cells are stained red (arrows). Group I, no smoking and no ozone; group II, no smoking and ozone; group III, smoking and no ozone; and group IV, smoking and ozone.
Alveolar destruction was quantified using the MLI, which was significantly higher in groups III (S + NO) and IV (S + O) (45 ± 2 and 44 ± 3 µm, respectively) compared with in groups I and II (26 ± 2 and 23 ± 2 µm, respectively) (Fig. 4).
Figure 4.

Mean linear intercept (MLI) in each group. MLI was signif icantly higher in groups III (smoking and no ozone, S + NO) and IV (smoking and ozone, S + O; 45 ± 2 and 44 ± 3 µm, respectively) compared with those in groups I (no smoking and no ozone, NS + NO) and II (no smoking and ozone, NS + O; 26 ± 2 and 23 ± 2 µm, respectively). Values are presented as means ± standard error of the mean. ap < 0.05 vs. groups I and II.
Two-dimensional electrophoresis and protein analysis
The differential expression of proteins in 1-mg total lung protein from each group was evaluated using 2DE (Fig. 5) and Coomassie blue staining. All of the identified spots were localized at a pH of 4 to 10 in the molecular mass range of 5 to 150 kDa. Fourteen spots that had significantly higher optical densities in group I (NS + NO) compared with the other groups, and six spots that had significantly lower optical densities in group I (NS + NO) compared with the other groups, were selected for analysis. The relative intensities of the spots are listed in Table 1, and the locations of the identified spots are shown in a master image (Fig. 5). The proteins of interest were analyzed by LC/MSD Trap XCT MS after tryptic digestion.
Figure 5.

Two-dimensional electrophoresis of lung proteins. Spots identified by liquid chromatography-tandem mass spectrometry are labeled with numbers. Group I, no smoking and no ozone; group II, no smoking and ozone; group III, smoking and no ozone; and group IV, smoking and ozone.
Table 1.
Proteins differentially expressed with and without ozone exposure in the smoking-induced emphysema lung model
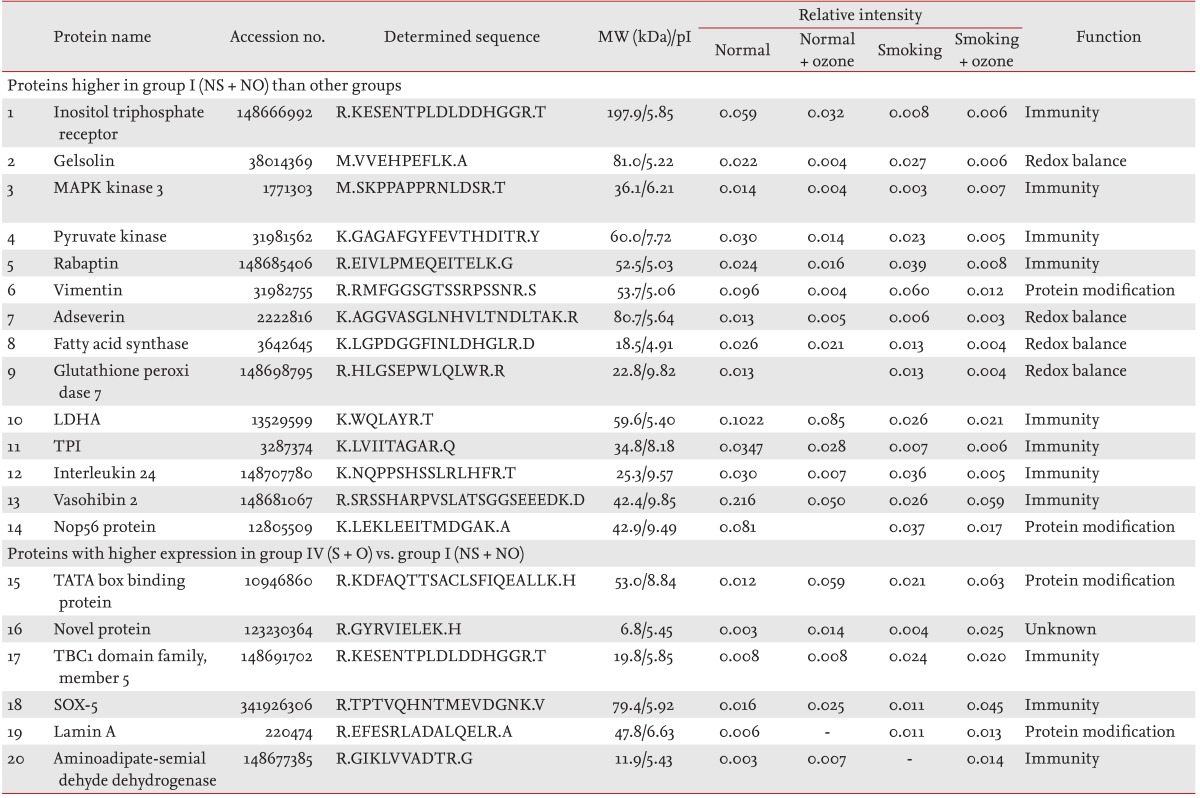
MW, molecular weight; NS + NO, no smoking and no ozone; MAPK, mitogen-activated protein kinase; LDHA, lactic dehydrogenase A; TPI, triose phosphate isomerase; S + O, smoking and ozone.
Spot 1 (inositol triphosphate receptor) showed a significantly higher relative intensity in group I (NS + NO) than in the other groups. Similar to spot 1, the 13 other proteins also showed higher relative intensities (Table 1). These proteins were involved in signaling, cytoskeleton, immunity, metabolism, and enzymatic activity.
We identified six proteins the levels of which were higher in groups II (NS + O), III (S + NO), and IV (S + O). These proteins were in spots 15 (TATA box-binding protein), 16 (novel protein), 17 (TBC1 domain family), 18 (SOX-5), 19 (Lamin A), and 20 (aminodipate semialdehyde dehydrogenase) and were involved in signaling, immunity, cytoskeleton, and enzymatic activity (Table 1).
Western blotting
The higher expression levels of the proteins inositol triphosphate receptor, gelsolin, vimentin, fatty acid synthase (FAS), LDHA, TPI, IL-24, and NOP56 in group I (NS + NO) versus the other groups (Table 1) were validated by Western blotting. Similar to the proteomics data, the densities of vimentin, LDHA, and TPI were higher in group I (NS + NO) (Fig. 6A). However, the densities of gelsolin were higher in groups I (NS + NO) and IV (S + O). No differences were found in the levels of inositol triphosphate receptor, FAS, IL-24, or NOP56 among the four groups by Western blotting (data not shown).
Figure 6.

Western blot analysis of proteins the expression of which was higher in group I no smoking and no ozone (NS + NO) (A) and group IV smoking and ozone (S + O) (B) by proteomic analysis. Expression levels of vimentin, lactic dehydrogenase A (LDHA), and triose phosphate isomerase (TPI) were higher in group I (NS + NO). Gelsolin expression was higher in groups I (NS + NO) and IV (S + O). Expression levels of TBC1 domain family 5 (TBC1D5) and lamin A were higher in groups III smoking and no ozone (S + NO) and IV (S + O). Expression of TBC1D5 increased in group IV (S + O) compared with group III (S + NO).
To determine whether TBC1D5 and lamin A were specific to group IV (S + O), Western blotting was performed for each group. The densities of TBC1D5 and lamin A were significantly higher in groups III (S + NO) and IV. In particular, the expression of TBC1D5 was increased in group IV (S + O) compared with group III (S + NO) (Fig. 6B).
DISCUSSION
We sought to identify proteins specific to ozone-induced exacerbations in an emphysema model. We found that the levels of several proteins in ozone-treated emphysema lungs were unusually high or low, and these results were validated by Western blotting. However, whether these proteins are pathogenic or end-products is unknown. We expect their roles to be involved in the pathogenesis of the ozone-induced exacerbation model and so they may be used biomarkers for diagnosis. To identify a biomarker of AECOPD, these proteins should be evaluated in human samples, such as sputa from patients with COPD and AECOPD.
We used ozone as an exacerbating factor in the emphysema model. Clinically, tracheal bronchitis due to bacterial infection is the most important cause of AECOPD; air pollution is also a causative factor of AECOPD [1]. A previously proposed animal model of COPD was not fully validated because it was established by only one study using Haemophilus influenzae as the exacerbating factor in elastin-induced emphysema [21]. In contrast, ozone has been used in many studies and is an established exacerbating factor. Furthermore, as described above, ozone has been reported to be related to AECOPD in clinical studies [7,8,9]. The amount of, and duration of exposure to, ozone depend on the objectives of the study. Multiple ozone exposures for 3 hours at 2.5 ppm for 6 weeks were used to generate chronic inflammation and pulmonary emphysema [22]. In contrast to multiple exposures, exposure for 3.0 ppm for 2 hours was used to induce lung inflammation related to pulmonary surfactant D [23], and 3.0 ppm for 3 hours to induce airway hyper-responsiveness [24]. In the present study, 3.0 ppm ozone exposure for 2 hours was used to produce lung and airway inflammation similar to AECOPD.
The function of TBC1D5, a member of the Rab GTPase-activating protein family, is unknown. Seaman et al. [25] reported that TBC1D5 released Rab7, resulting in the regulation of fusion of endosomes with lysosomes. However, the relevance of these properties in the ozone and smoking groups is unclear. In fact, the elevated TBC1D5 levels may represent end-products of the stimulation by ozone and smoking. Lamin A is a component of the fibrous layer on the nucleoplasmic side of the inner nuclear membrane, which is thought to provide a framework for the nuclear envelope and may also interact with chromatin [26]. Like TBC1D5, we suggest that the elevated levels of this protein may represent an end-product of stimulation by ozone and smoking. The densities of vimentin, LDHA, TPI, and gelsolin were lower following ozone and cigarette smoke exposure (group IV, S + O) (Fig. 5). Thus, TBC1D5 could be a biomarker in ozone-induced AECOPD, as illustrated by the finding of an elevated protein level. In contrast, lamin A is likely not a biomarker of ozone-induced AECOPD because the levels increased in both groups III (S + NO) and IV (S + O).
TPI is involved in glucose metabolism and energy production. LDHA catalyzes the conversion of L-lactate to pyruvate in anaerobic glycolysis. Unfortunately, we are unable to explain the decrease in the levels of these proteins in group IV (S + O). Gelsolin inhibits apoptosis by stabilizing mitochondria. The density of gelsolin by Western blot analysis differed from that determined in the proteomics analysis. We have at present no explanation for this.
We performed MMP-12 immunohistochemical staining to assess whether the infiltrating cells shown in Fig. 2 were MMP-12-secreting macrophages based on a previous report that these cells were increased in the lungs after 30 days of cigarette smoke exposure [27]. According to the immunohistochemical findings, the infiltrating cells were primarily macrophages.
The discrepancy between the BAL cellular profiles and lung histology and immunohistochemical staining may be due to conducting BAL prior to tissue fixation. To overcome this, BAL should not be performed in animals that are to be used to provide lungs for tissue fixation. However, in the present study, the change in cellular profiles was not a major focus, and it was challenging to maintain more than seven mice in each group for 6 months. Thus, we performed BAL before tissue fixation.
The main limitations of the current study were as follows. First, the COPD stage was different between patients with AECOPD and the animal model. Clinically, AECOPD is often observed in stage III or IV COPD; however, in the smoking-induced animal COPD model, the degree of COPD was equivalent to stage I or II of human COPD [28]. Second, we did not measure airway mechanics to confirm the smoking-induced COPD model, which has been performed in mouse and rat models by other researchers [29,30]. However, in the present study, the smoking-induced COPD model was appropriate because the MLIs were comparable to those in other studies [31,32]. Others have reported the MLI to be more than 30 µm; however, in the current study, the MLI increased to more than 40 µm in the smoking-induced COPD model in mice. Third, we used commercially available cigarettes instead of the "Kentucky cigarette" (Cigarette Laboratory, University of Kentucky), which has been used in much research on smoking-induced emphysema [33,34,35]. We described the contents of the cigarettes used in the present study, and the final emphysema models were appropriate, as confirmed by the MLI. Fourth, we sought to develop a mouse AECOPD model that resembled the human AECOPD situation. The definition of AECOPD in humans is acute aggravation of dyspnea, coughing, and/or sputum requiring a change of medication [1]. This definition cannot be applied fully to an animal model of AECOPD because clinical symptoms cannot be quantified in this way in an animal model. Thus, the present study model should be considered an ozone-induced toxicity model in an emphysema lung instead of true AECOPD. Fifth, lymphocytes, rather than neutrophils, increased in bronchoalveolar lavage fluid after ozone exposure in this study, which is similar to another study that used exposure to 2.0 ppm ozone in mice [36]. During AECOPD, there was a further increase in the neutrophil to lymphocyte ratio compared with the stable period [37]. Thus, strictly, ozone exposure to smoking-induced emphysema model is not an ideal model of AECOPD.
In conclusion, we developed an ozone-induced exacerbation model in a smoking-induced emphysema lung, and TBC1D5 was identified as a potential biomarker of ozone-induced lung injury in emphysema.
KEY MESSAGE
An ozone-induced exacerbation model in smoking-induced emphysema can be used in research related to acute exacerbations of chronic obstructive pulmonary disease.
TBC1 domain family 5 may be a potential biomarker of ozone-induced lung injury in emphysema.
Acknowledgments
This work was supported by grants from the Korea Health 21 R&D Project, Ministry of Health, Welfare, and Family Affairs, Republic of Korea (A010249, A090548, and A030003). This work was also supported by the Soonchunhyang University Research Fund.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Global Initiative for Chronic Obstructive Lung Disease (GOLD) Global Strategy for the Diagnosis, Management and Prevention of COPD 2011 [Internet] [place unknown]: Global Initiative for Chronic Obstructive Lung Disease; 2013. [cited 2013 Dec 15]. Available from: http://www.goldcopd.org. [Google Scholar]
- 2.Chapman KR, Mannino DM, Soriano JB, et al. Epidemiology and costs of chronic obstructive pulmonary disease. Eur Respir J. 2006;27:188–207. doi: 10.1183/09031936.06.00024505. [DOI] [PubMed] [Google Scholar]
- 3.Kim DS, Kim YS, Jung KS, et al. Prevalence of chronic obstructive pulmonary disease in Korea: a population-based spirometry survey. Am J Respir Crit Care Med. 2005;172:842–847. doi: 10.1164/rccm.200502-259OC. [DOI] [PubMed] [Google Scholar]
- 4.Patil SP, Krishnan JA, Lechtzin N, Diette GB. In-hospital mortality following acute exacerbations of chronic obstructive pulmonary disease. Arch Intern Med. 2003;163:1180–1186. doi: 10.1001/archinte.163.10.1180. [DOI] [PubMed] [Google Scholar]
- 5.Ai-Ping C, Lee KH, Lim TK. In-hospital and 5-year mortality of patients treated in the ICU for acute exacerbation of COPD: a retrospective study. Chest. 2005;128:518–524. doi: 10.1378/chest.128.2.518. [DOI] [PubMed] [Google Scholar]
- 6.Berkius J, Nolin T, Mardh C, Karlstrom G, Walther SM Swedish Intensive Care Registry. Characteristics and long-term outcome of acute exacerbations in chronic obstructive pulmonary disease: an analysis of cases in the Swedish Intensive Care Registry during 2002-2006. Acta Anaesthesiol Scand. 2008;52:759–765. doi: 10.1111/j.1399-6576.2008.01632.x. [DOI] [PubMed] [Google Scholar]
- 7.Medina-Ramon M, Zanobetti A, Schwartz J. The effect of ozone and PM10 on hospital admissions for pneumonia and chronic obstructive pulmonary disease: a national multicity study. Am J Epidemiol. 2006;163:579–588. doi: 10.1093/aje/kwj078. [DOI] [PubMed] [Google Scholar]
- 8.Ko FW, Tam W, Wong TW, et al. Temporal relationship between air pollutants and hospital admissions for chronic obstructive pulmonary disease in Hong Kong. Thorax. 2007;62:780–785. doi: 10.1136/thx.2006.076166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halonen JI, Lanki T, Tiittanen P, Niemi JV, Loh M, Pekkanen J. Ozone and cause-specific cardiorespiratory morbidity and mortality. J Epidemiol Community Health. 2010;64:814–820. doi: 10.1136/jech.2009.087106. [DOI] [PubMed] [Google Scholar]
- 10.Ciencewicki J, Trivedi S, Kleeberger SR. Oxidants and the pathogenesis of lung diseases. J Allergy Clin Immunol. 2008;122:456–468. doi: 10.1016/j.jaci.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD. 2004;1:59–70. doi: 10.1081/COPD-120028701. [DOI] [PubMed] [Google Scholar]
- 12.Gerritsen WB, Asin J, Zanen P, van den Bosch JM, Haas FJ. Markers of inflammation and oxidative stress in exacerbated chronic obstructive pulmonary disease patients. Respir Med. 2005;99:84–90. doi: 10.1016/j.rmed.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 13.Cazzola M, MacNee W, Martinez FJ, et al. Outcomes for COPD pharmacological trials: from lung function to biomarkers. Eur Respir J. 2008;31:416–469. doi: 10.1183/09031936.00099306. [DOI] [PubMed] [Google Scholar]
- 14.Lomas DA, Silverman EK, Edwards LD, et al. Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. Eur Respir J. 2009;34:95–102. doi: 10.1183/09031936.00156508. [DOI] [PubMed] [Google Scholar]
- 15.Magi B, Bini L, Perari MG, et al. Bronchoalveolar lavage fluid protein composition in patients with sarcoidosis and idiopathic pulmonary fibrosis: a two-dimensional electrophoretic study. Electrophoresis. 2002;23:3434–3444. doi: 10.1002/1522-2683(200210)23:19<3434::AID-ELPS3434>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 16.Larsen K, Malmstrom J, Wildt M, et al. Functional and phenotypical comparison of myofibroblasts derived from biopsies and bronchoalveolar lavage in mild asthma and scleroderma. Respir Res. 2006;7:11. doi: 10.1186/1465-9921-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haque R, Umstead TM, Freeman WM, Floros J, Phelps DS. The impact of surfactant protein-A on ozone-induced changes in the mouse bronchoalveolar lavage proteome. Proteome Sci. 2009;7:12. doi: 10.1186/1477-5956-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luthje L, Raupach T, Michels H, et al. Exercise intolerance and systemic manifestations of pulmonary emphysema in a mouse model. Respir Res. 2009;10:7. doi: 10.1186/1465-9921-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cha MH, Rhim T, Kim KH, Jang AS, Paik YK, Park CS. Proteomic identification of macrophage migration-inhibitory factor upon exposure to TiO2 particles. Mol Cell Proteomics. 2007;6:56–63. doi: 10.1074/mcp.M600234-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 21.Wang D, Wang Y, Liu YN. Experimental pulmonary infection and colonization of Haemophilus influenzae in emphysematous hamsters. Pulm Pharmacol Ther. 2010;23:292–299. doi: 10.1016/j.pupt.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 22.Triantaphyllopoulos K, Hussain F, Pinart M, et al. A model of chronic inflammation and pulmonary emphysema after multiple ozone exposures in mice. Am J Physiol Lung Cell Mol Physiol. 2011;300:L691–L700. doi: 10.1152/ajplung.00252.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kierstein S, Poulain FR, Cao Y, et al. Susceptibility to ozone-induced airway inflammation is associated with decreased levels of surfactant protein D. Respir Res. 2006;7:85. doi: 10.1186/1465-9921-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams AS, Nath P, Leung SY, et al. Modulation of ozone-induced airway hyperresponsiveness and inflammation by interleukin-13. Eur Respir J. 2008;32:571–578. doi: 10.1183/09031936.00121607. [DOI] [PubMed] [Google Scholar]
- 25.Seaman MN, Harbour ME, Tattersall D, Read E, Bright N. Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5. J Cell Sci. 2009;122(Pt 14):2371–2382. doi: 10.1242/jcs.048686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimi T, Pfleghaar K, Kojima S, et al. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 2008;22:3409–3421. doi: 10.1101/gad.1735208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valenca SS, de Souza da Fonseca A, da Hora K, Santos R, Porto LC. Lung morphometry and MMP-12 expression in rats treated with intraperitoneal nicotine. Exp Toxicol Pathol. 2004;55:393–400. doi: 10.1078/0940-2993-00322. [DOI] [PubMed] [Google Scholar]
- 28.Wright JL, Cosio M, Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2008;295:L1–L15. doi: 10.1152/ajplung.90200.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szabari MV, Parameswaran H, Sato S, Hantos Z, Bartolak-Suki E, Suki B. Acute mechanical forces cause deterioration in lung structure and function in elastase-induced emphysema. Am J Physiol Lung Cell Mol Physiol. 2012;303:L567–L574. doi: 10.1152/ajplung.00217.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu ZB, Song NN, Geng WY, et al. Orexin-A and respiration in a rat model of smoke-induced chronic obstructive pulmonary disease. Clin Exp Pharmacol Physiol. 2010;37:963–968. doi: 10.1111/j.1440-1681.2010.05411.x. [DOI] [PubMed] [Google Scholar]
- 31.Duan MC, Zhong XN, Huang Y, He ZY, Tang HJ. Mechanisms and dynamics of Th17 cells in mice with cigarette smoke-induced emphysema. Zhonghua Yi Xue Za Zhi. 2011;91:1996–2000. [PubMed] [Google Scholar]
- 32.Manoli SE, Smith LA, Vyhlidal CA, et al. Maternal smoking and the retinoid pathway in the developing lung. Respir Res. 2012;13:42. doi: 10.1186/1465-9921-13-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Churg A, Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2004;170:492–498. doi: 10.1164/rccm.200404-511OC. [DOI] [PubMed] [Google Scholar]
- 34.Churg A, Wang RD, Xie C, Wright JL. alpha-1-Antitrypsin ameliorates cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2003;168:199–207. doi: 10.1164/rccm.200302-203OC. [DOI] [PubMed] [Google Scholar]
- 35.Guerassimov A, Hoshino Y, Takubo Y, et al. The development of emphysema in cigarette smoke-exposed mice is strain dependent. Am J Respir Crit Care Med. 2004;170:974–980. doi: 10.1164/rccm.200309-1270OC. [DOI] [PubMed] [Google Scholar]
- 36.Cho HY, Gladwell W, Yamamoto M, Kleeberger SR. Exacerbated airway toxicity of environmental oxidant ozone in mice deficient in Nrf2. Oxid Med Cell Longev. 2013;2013:254069. doi: 10.1155/2013/254069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gunay E, Sarinc Ulasli S, Akar O, et al. Neutrophil-to-lymphocyte ratio in chronic obstructive pulmonary disease: a retrospective study. Inflammation. 2014;37:374–380. doi: 10.1007/s10753-013-9749-1. [DOI] [PubMed] [Google Scholar]