

Abstract
Objective: Isolated growth hormone deficiency (IGHD) is defined as a medical condition associated with growth failure due to insufficient production of GH or lack of GH action. Mutations in the gene encoding for GH-releasing hormone receptor (GHRHR) have been detected in patients with IGHD type IB. However, genetic defects on GHRHR causing IGHD in the Turkish population have not yet been reported. To identify mutations on GHRHR gene in a population of Turkish children with IGHD.
Methods: Ninety-six Turkish children with IGHD were included in this study. Exon1-13 and exon/intron boundaries of GHRHR were amplified by suitable primers. The polymerase chain reaction products for GHRHR gene were sequenced with primers.
Results: We analyzed the GHRHR gene for mutations in ninety-six patients with IGHD based on sequence results. We identified novel p.K264E, p.S317T, p.S330L, p.G369V, p.T257A and C base insertion on position 380 (c.380inserC) mutations. In 5 of the patients, the mutation was homozygote and in 1-heterozygote (p.S317T).
Conclusion: Six new missense mutations and one first case of insertion mutations for the GHRHR gene are reported.
Keywords: IGHD, GHRHR gene, short stature
INTRODUCTION
Growth hormone (GH) is a 22 kDa protein involved mainly in skeletal and visceral growth but also in carbohydrate, protein and lipid metabolisms (1). GH is synthesized and secreted by somatotropes in the anterior pituitary gland. The expression and secretion of GH are regulated multifactorially, but predominantly by hypothalamic hormones, GH-releasing hormone (GHRH), GH secretagogue (GHS) and somatostatin (SS) (2).
GH deficiency (GHD) is defined as deficient or insufficient production/secretion of GH from the pituitary gland (3,4,5). The prevalence of short stature associated with GHD is between 1/4000 and 1/10 000 live births (6,7). Although most of the cases are sporadic and thought to be caused by environmental cerebral insults or developmental anomalies, 5-30% of cases are familial (8).
Based on their severity and mode of inheritance, there are three types of familial isolated GHD (IGHD) (9). While types 1 A and 1 B show a recessive autosomal transmission, type 2 shows an autosomal dominant transmission. Type 3 shows an X-linked chromosome pattern. Patients with type 1 A have severe short stature, they lack any detectable GH and generally produce GH antibodies. These cases are mainly caused by deletion of the entire GH (GH-1) gene (10). Patients with type 1 B are milder and they respond to GH treatment very well. IGHD type 1 B is caused by mutations in both GH-N gene which is one of the GH-gene clusters (hGH-N, hCS-L, hCS-A, hCS-B and hGH-V) encoding 22 kDa GH protein and GHRH receptor (GHRHR) gene (2,11,12,13).
GHRH, through GHRHR, plays an important role in GH expression and secretion (14,15). The human GHRHR gene is located on the short arm of chromosome 7, is mostly expressed in the anterior pituitary gland and belongs to a G protein-coupled receptor superfamily (16). The GHRHR gene consists of 13 exons and encodes a 423-amino acid protein with an N-terminal and a C-terminal domain linked by 7 alpha-helical transmembrane domains (16).
More than 20 mutations for GHRHR have been reported in patients with IGHD; homozygous and compound heterozygous mutations lead to a loss of GHRHR function. These are missense, splice, nonsense, microdeletion and promoter mutations (17,18,19,20,21).
In this study, we analyzed the GHRHR protein coding region and the exon/intron boundary of the GHRHR gene for mutations in 96 children with IGHD.
METHODS
A total of 96 patients with IGHD (59 boys, 37 girls) were included in this study; 6 of these patients had GHRHR mutations. At least two GH stimulation tests were performed in each patient. Height and weight standard deviation scores (SDS) were calculated according to standard reference values for age, sex and pubertal maturation. GH stimulation tests (ITT and L-DOPA) were performed (deficiency defined as a GH peak <10 ng/mL) and other pituitary hormone deficiencies were ruled out by measuring free thyroxine (fT4) and cortisol levels. Serum GH levels were measured by RIA or ELISA, insulin-like growth factor-1, insulin-like growth factor binding protein-3 levels were determined by immunoradiometric assays (22). FT4 and thyroid-stimulating hormone levels were assessed on the AxSYM system by microparticle enzyme immunoassay and cortisol was measured using a chemiluminescence immunoassay. Clinical and hormonal data of the 6 children identified to have GHRHR gene mutations are shown Table 1.
Table 1. Clinical and hormonal data of six patients with IGHD.
DNA Isolation and Specific Exon and Exon/Intron Boundary Polymerase Chain Reaction (PCR)
Genomic DNA was isolated from bloods of children with IGHD based on salting out method (23). Exon 2-3 and 8-9 together and other exons (1,4,5,6,7,10,11,12,13) alone and their flanking splice sites were amplified by PCR using the exon-specific primers shown in Figure 1. PCR products were visualized on agarose gels to rule out large deletion and insertions.
Figure 1. Primers for PCR. The primers for GHRHR were used for the amplifications of specific exons and exon/intron boundaries for exons 1-13. F shows forward primer and R refers reverse primer.

DNA Sequencing
The PCR products of GHRHR exons for each patient were purified and sequenced with direct sequencing of the DNA Cycle Sequencing System (ABI Prism kit) with the dideoxychain termination method and applied on an autosequencer (ABI Prism 377 DNA sequencer).
Silico Functional Analysis
PolyPhen-2 and PROVEAN programs were used in order to predict the function of mutated amino acids on the GHRHR protein structure.
Ethics Approval
This research was approved by the ethics committee of Marmara University. All human rights were protected and any necessary approval was secured from the ethics committee. Written informed consent was taken from the parents of each patient.
RESULTS
Ninety-six children were diagnosed as IGHD by at least two GH stimulation tests; they presently are under GH treatment. Six of the patients had GHRHR mutations and prevalence of mutation in this series was 6.25%. A summary of mutations on the GHRHR gene in these 6 children with IGHD is shown in Table 2.
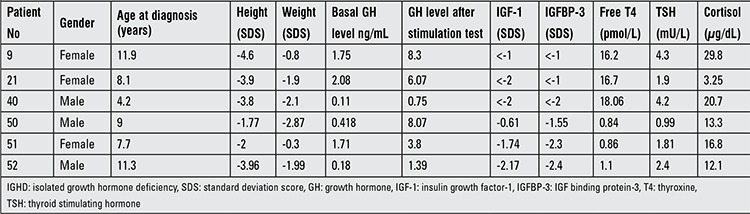
Five different missense mutations were determined in the exons 8, 10, 11 and 12 of the GHRHR gene. The p.T257A homozygous mutation was located at exon 8 and was seen in patient 9. This mutation was created by the substitution of the A residue of ACT codon encoding threonine (T) to G and GCT codon encodes alanine (A) (Figure 2A). p.K264E mutation seen in patient 50 was homozygous, located at exon 8 and was created by replacement of the first A residue of AAA encoding lysine (K) to G and GAA codon encoding glutamic acid (E). This mutation is novel (Figure 2B). Another missense mutation was found in patient 51 - p.S317T located in exon 10. This mutation occurred by changing of G residue of AGC codon to C (ACC) (Figure 2C) and is a heterozygous mutation. p.S330L mutation is located at exon 11 and was seen in patient 21. This mutation was created by changing the C residue of TCG to T residue (TTG) (Figure 2D). The last missense mutation was p.G369V mutation located at exon 12 and observed in patient 40. This mutation was created by substitution of the second G residue of GGC to T (GTC) (Figure 2E).
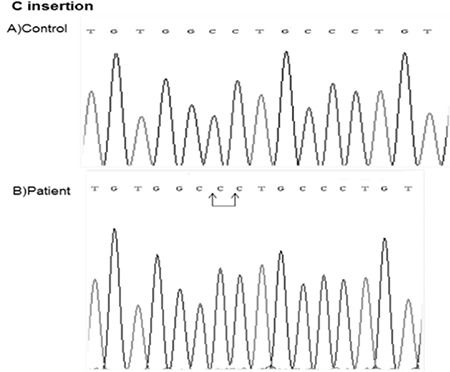
Another interesting and rare mutation found in this Turkish population was C insertion at position 380 residue (c.380inserC) - a homozygote mutation located in exon 4 which was found in patient 52 (Figure 3). This mutation changed Open Reading Frame of GHRHR and introduced stop codon (p.C112Lfs*9).
Figure 2. Determination of missense GHRHR gene mutations. Sequence analysis of A) T257A Mutation, Normal person carries Threonine (T) amino acid residues on position 257 in GHRHR gene encoded by ACT. The A residue of ACT was changed to G (GCT) encoding Alanine (A) in IGHD patient (sequence from reverse), B) K264E Mutation, Lysine (K) is encoded by AAA in the healthy person and the first A residue of AAA codon was converted to G (GAA) in the patient (reverse sequences), C) S317T Mutation, healthy person has AGC codon encoding Serine (S) and Threonine (T) occurred by changing of G residue of AGC codon to C (ACC) in IGHD patient, D) S330L Mutation, The healthy person has Serine (S) amino acid on position 330 encoded by TCG and Leucine (L) mutation was created by substitution of the C residue of TCG to T (TTG) in the patient, E) G369V Mutation, the control has GGC codon encoding Glycine (G), but this amino acid is changed to Valine (V) by the substitution of the second G residue of GGC to T (GTC) in the patient.
Furthermore, we found two sense mutations p.Y163Y and p.L247L (Table 2). Also, we found two polymorphisms, p.A57T and p.E121D, located in exons 3 and 4, respectively in Turkish population (Table 2). The homozygote p.A57T polymorphism was found only in patient 40 containing p.G369V mutation (Table 2); however, the homozygote pE121D polymorphism was seen in patient 21 containing p.S330L mutation and in patients 32 and 50 containing p.K264E mutation,78 (Table 2). p.A57T and p.E121D polymorphisms were reported previously. However, p.K264E, p.S317T, p.S330L, p.G369V, p.T257A and C insertion at exon 4 mutations are novel.
DISCUSSION
GHRH induces GH expression and secretion through GHRHR (24). Although GHS, also known as ghrelin, is involved in GH secretion via GHS receptor (GHSR), somatostatin suppresses GH secretion in the pituitary. Deficiency of GH secretion leads to growth failure and to changes in metabolic activities. Mutations in GH-1 cause mainly types 1 and 2 of IGHD; however, they have been determined only in less than 10% of total cases and in less than 2% of patients with type 1 B IGHD suggesting the involvement of other genes. GHRH is supposed to be involved in GH secretion; however, no mutation was detected in GHRH gene. It has been reported that mutations in GHRHR gene are being detected with increasing frequency. More than 20 GHRHR mutations were reported to date, including missense mutations (13,25,26,27,28,29), nonsense mutations (13,25,29,30,31), splice site mutations (12,29,32,33,34,35,36,37,38,39), promoter mutation (20), micro-deletion mutations (19,26,40) that lead to loss of GHRHR function. The missense mutations show defective ligand binding resulting in blocking GHRH-GHRHR signaling (33); promoter mutations affect promoter activity and thus change GHRHR expression (26); nonsense and splicing mutations produce truncated GHRHR receptor and can be involved in mRNA instability (29).
We identified five different missense (p.K264E, p.S317T, p.S330L, p.G369V and p.T257A) and one insertion mutation (C insertion) on the GHRHR gene from 96 IGHD patients. We also found p.A57T and p.E121D polymorphisms in GHRHR, reported previously. p.K264E, p.S317T, p.S330L, p.G369V, p.T257A and C insertion at exon 4 mutations are novel.
One of missense mutations in GHRHR is p.T257A homozygous mutation seen in patient 9 (Figure 2A, Table 2). This mutation occurred on the fourth transmembrane domain of GHRHR and probably causes neutral effect on the receptor based on functional silico analysis. We do not have functional analysis of this mutation for GHRHR signaling in the cell culture system, but this mutation is novel. It has been reported that CHO cells expressing p.F242C mutation on the same transmembrane domain of GHRHR did not alter the receptor surface expression, but the authors failed to show cAMP production after treatment of cells with GHRH (41). This result showed that mutation on the transmembrane domain affects GHRHR-GHRH binding affinity.
We observed homozygous p.K264E missense mutation, a finding not reported previously; thus, it is a novel mutation. This mutation occurred at the extracellular loop of the transmembrane domain 4 of GHRHR; it is probably detrimental for receptor based on silico functional analysis and may reduce the ligand binding to receptor, impairing the receptor ability to transmit intracellular signaling.
We also determined heterozygous p.S317T missense mutation that lied within the intracellular loop of the sixth transmembrane domain of GHRHR. This mutation has a damaging effect according to the PolyPhen-2 program. It is interesting that this is a heterozygote mutation, since most GHRHR mutations are inherited in an autosomal recessive way. Heterozygote mutations on GHRHR have been reported previously (12,18,26). The mother of the patient carrying S317T mutation was of short stature but not the father. However, we do not have any data on the genetic make-up of this patient’s parents. This mutation can change the structure of GHRHR and may lead to defectiveness of intracellular signaling. There is no reported mutation in this region of GHRHR receptor so far.
Another homozygous missense mutation is p.S330L mutation located on the sixth transmembrane domain of GHRHR. This mutation will have a deleterious effect on the receptor since polar amino acid (S) was converted to non-polar amino acid (L). This mutation probably has a reducing effect on GHRH binding to GHRHR, leading to a block in intracellular signaling. Another mutation called p.K329E and encoding an amino acid for GHRHR was also reported. Chinese hamster ovary cells transfected with cDNA encoding p.K329E GHRHR failed to induce cAMP production (20). This experiment showed that this mutation completely knocked out the function of GHRHR.
The last missense mutation is a p.G369V mutation located at the seventh transmembrane domain of GHRHR. This mutation is detrimental based on functional silico analysis. This mutation probably destroys GHRHR structure resulting in the inhibition of GHRH binding to GHRHR and thus disrupts GH signaling. A c.1120-1123delATCC mutation was reported in this transmembrane domain GHRHR (40) and four base pair deletion in exon 12 of the GHRH receptor caused a frameshift and the premature stop codon resulted in a C-terminally truncated GHRHR receptor that blocked GHRH-GHRHR signaling.
GH levels after GH-stimulation of patients 9 and 50 were 8.3 and 8.07 ng/mL, respectively. These levels are close to normal post-stimulation GH levels (10 ng/mL). Both mutations (p.T257A and p.K264E) are located at the GHRHR gene, but the biological activity of the receptor is different because of a different effect of the mutation on GHRHR or because of a possibly existing different domain of receptor. Height SDS of both patients (patients 9 and 50) was -4.6 and -1.77, respectively. The lower height SDS of patient 50 may be due to an additional effect of the polymorphism (p.V121D) noted in patient 50.
In patient 52, we detected a homozygote C insertion (c.380inserC) on position 380 which was located at the extracellular domain of GHRHR (Figure 3, Table 2). This insertion causes frameshift mutation (p.C112Lfs*9) that introduces stop codon (after changed 9 amino acid coding sequence) proceeding sequence of GHRHR gene. The mutated receptor is missing of the part of exon 4 and exons 5-13 and this mutation is also novel. It is interesting that there are no reports on insertion mutation on GHRHR gene to date. The function of novel mutations is under investigation.
Additionally, we identified two heterozygote sense mutations in this Turkish population which are p.Y163Y located at exon 6 and p.L247L found at exon 7 (Table 2). We do not know whether or not these mutations affect codon usage in the human.
Furthermore, we detected homozygote p.A57T and p.E121D polymorphisms reported previously (42,43). It has been reported that TSA cell lines carrying p.A57T and p.E121D mutations did not induce cAMP production in absence of GHRH but induced cAMP production in presence of GHRH (43). This result showed that these polymorphisms may not affect GHRHR function or the pathogenesis of IGHD. However, it has been reported that the tumors containing p.A57T polymorphism responded strongly to GHRH-based cAMP production in vitro (42).
In conclusion, we report five missense and one insertion mutations determined in a Turkish population consisting of 96 children with IGHD. These mutations are novel. These mutations are probably unique for Turkey since previously reported mutations were located in unique geographic locations.
Acknowledgement
This project was supported by a grant from the Scientific and Technical Research Council of the Turkish Republic.
Table 2. Growth hormone-releasing hormone receptor (GHRHR) mutations encountered in this series of Turkish children.
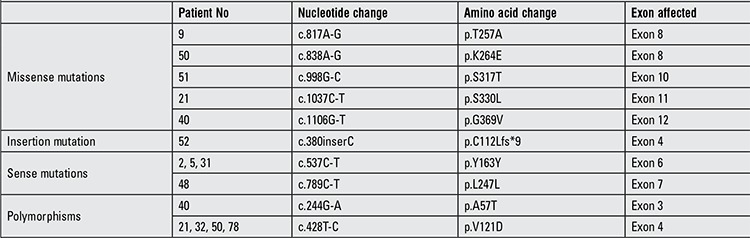
Figure 3. Determination of C insertion mutation at exon 4 of GHRHR gene. A shows the sequence from normal person, B shows C insertion at 380 residue located at exon 4 of GHRHR in the patient.
References
- 1.Davidson MB. Effect of growth hormone on carbohydrate and lipid metabolism. Endocr Rev. 1987;8:115–131. doi: 10.1210/edrv-8-2-115. [DOI] [PubMed] [Google Scholar]
- 2.Tannenbaum GS, Ling N. The interrelationship of growth hormone (GH)-releasing factor and somatostatin in generation of the ultradian rhythm of GH secretion. Endocrinology. 1984;115:1952–1967. doi: 10.1210/endo-115-5-1952. [DOI] [PubMed] [Google Scholar]
- 3.Hernández LM, Lee PDK, Camacho-Hübner C. Isolated growth hormone deficiency. Pituitary. 2007;10:351–357. doi: 10.1007/s11102-007-0073-3. [DOI] [PubMed] [Google Scholar]
- 4.Isaksson OG, Jansson JO, Gause IA. Growth hormone stimulates longitudinal bone growth directly. Science. 1982;216:1237–1239. doi: 10.1126/science.7079756. [DOI] [PubMed] [Google Scholar]
- 5.Savage MO, Burren CP, Rosenfeld RG. The continuum of growth hormone-IGF-I axis defects causing short stature: diagnostic and therapeutic challenges. Clin Endocrinol (Oxf) 2010;72:721–728. doi: 10.1111/j.1365-2265.2009.03775.x. [DOI] [PubMed] [Google Scholar]
- 6.Vimpani GV, Vimpani AF, Lidgard GP, Cameron EH, Farquhar JW. Prevalence of severe growth hormone deficiency. Br Med J. 1977;2:427–430. doi: 10.1136/bmj.2.6084.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas M, Massa G, Craen M, Zegher F, de, Bourguignon JP, Heinrichs C, De Schepper J, Du Caju M, Thiry-Counson G, Maes M. Prevalence and demographic features of childhood growth hormone deficiency in Belgium during the period 1986-2001. Eur J Endocrinol. 2004;151:67–72. doi: 10.1530/eje.0.1510067. [DOI] [PubMed] [Google Scholar]
- 8.Cogan JD, Phillips JA., 3rd Growth disorders caused by genetic defects in the growth hormone pathway. Adv Pediatr. 1998;45:337–361. [PubMed] [Google Scholar]
- 9.Phillips JA, 3rd, Cogan JD. Genetic basis of endocrine disease. 6. Molecular basis of familial human growth hormone deficiency. J Clin Endocrinol Metab. 1994;78:11–16. doi: 10.1210/jcem.78.1.8288694. [DOI] [PubMed] [Google Scholar]
- 10.Mullis PE, Akinci A, Kanaka C, Eblé A, Brook CG. Prevalence of human growth hormone-1 gene deletions among patients with isolated growth hormone deficiency from different populations. Pediatr Res. 1992;31:532–534. doi: 10.1203/00006450-199205000-00026. [DOI] [PubMed] [Google Scholar]
- 11.Baumann G. Genetic characterization of growth hormone deficiency and resistance: implications for treatment with recombinant growth hormone. Am J Pharmacogenomics. 2002;2:93–111. doi: 10.2165/00129785-200202020-00003. [DOI] [PubMed] [Google Scholar]
- 12.Salvatori R, Hayashida CY, Aguiar-Oliveira MH, Phillips JA, 3rd, Souza AH, Gondo RG, Toledo SP, Conceicão MM, Prince M, Maheshwari HG, Baumann G, Levine MA. Familial dwarfism due to a novel mutation of the growth hormone-releasing hormone receptor gene. J Clin Endocrinol Metab. 1999;84:917–923. doi: 10.1210/jcem.84.3.5599. [DOI] [PubMed] [Google Scholar]
- 13.Netchine I, Talon P, Dastot F, Vitaux F, Goossens M, Amselem S. Extensive phenotypic analysis of a family with growth hormone (GH) deficiency caused by a mutation in the GH-releasing hormone receptor gene. J Clin Endocrinol Metab. 1998;83:432–436. doi: 10.1210/jcem.83.2.4528. [DOI] [PubMed] [Google Scholar]
- 14.Iguchi G, Okimura Y, Takahashi T, Mizuno I, Fumoto M, Takahashi Y, Kaji H, Abe H, Chihara K. Cloning and characterization of the 5’-flanking region of the human growth hormone-releasing hormone receptor gene. J Biol Chem. 1999;274:12108–12114. doi: 10.1074/jbc.274.17.12108. [DOI] [PubMed] [Google Scholar]
- 15.Petersenn S, Rasch AC, Heyens M, Schulte HM. Structure and regulation of the human growth hormone-releasing hormone receptor gene. Mol Endocrinol. 1998;12:233–247. doi: 10.1210/mend.12.2.0057. [DOI] [PubMed] [Google Scholar]
- 16.Lin-Su K, Wajnrajch MP. Growth hormone releasing hormone (GHRH) and the GHRH receptor. Rev Endocr Metab Disord. 2002;3:313–323. doi: 10.1023/a:1020949507265. [DOI] [PubMed] [Google Scholar]
- 17.Corazzini V, Salvatori R. Molecular and clinical aspects of GHRH receptor mutations. Endocr Dev. 2013;24:106–117. doi: 10.1159/000342575. [DOI] [PubMed] [Google Scholar]
- 18.Soneda A, Adachi M, Muroya K, Asakura Y, Takagi M, Hasegawa T, Inoue H, Itakura M. Novel compound heterozygous mutations of the growth hormone-releasing hormone receptor gene in a case of isolated growth hormone deficiency. Growth Horm IGF Res. 2013;23:89–97. doi: 10.1016/j.ghir.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Shohreh R, Sherafat-Kazemzadeh R, Jee YH, Blitz A, Salvatori R. A novel frame shift mutation in the GHRH receptor gene in familial isolated GH deficiency: early occurrence of anterior pituitary hypoplasia. J Clin Endocrinol Metab. 2011;96:2982–2986. doi: 10.1210/jc.2011-1031. [DOI] [PubMed] [Google Scholar]
- 20.Salvatori R, Fan X, Mullis PE, Haile A, Levine MA. Decreased expression of the GHRH receptor gene due to a mutation in a Pit-1 binding site. Mol Endocrinol. 2002;16:450–458. doi: 10.1210/mend.16.3.0785. [DOI] [PubMed] [Google Scholar]
- 21.Inoue H, Kangawa N, Kinouchi A, Sakamoto Y, Kimura C, Horikawa R, Shigematsu Y, Itakura M, Ogata T, Fujieda K. Japan Growth Genome Consortium. Identification and functional analysis of novel human growth hormone-releasing hormone receptor (GHRHR) gene mutations in Japanese subjects with short stature. Clin Endocrinol (Oxf) 2011;74:223–233. doi: 10.1111/j.1365-2265.2010.03911.x. [DOI] [PubMed] [Google Scholar]
- 22.Blum WF, Horn N, Kratzsch J, Jørgensen JO, Juul A, Teale D, Mohnike K, Ranke MB. Clinical studies of IGFBP-2 by radioimmunoassay. Growth Regul. 1993;3:100–104. [PubMed] [Google Scholar]
- 23.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215–1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaylinn BD. Growth hormone releasing hormone receptor. Recept Channels. 2002;8:155–162. [PubMed] [Google Scholar]
- 25.Alatzoglou KS, Turton JP, Kelberman D, Clayton PE, Mehta A, Buchanan C, Aylwin S, Crowne EC, Christesen HT, Hertel NT, Trainer PJ, Savage MO, Raza J, Banerjee K, Sinha SK, Ten S, Mushtaq T, Brauner R, Cheetham TD, Hindmarsh PC, Mullis PE, Dattani MT. Expanding the spectrum of mutations in GH1 and GHRHR: genetic screening in a large cohort of patients with congenital isolated growth hormone deficiency. J Clin Endocrinol Metab. 2009;94:3191–3199. doi: 10.1210/jc.2008-2783. [DOI] [PubMed] [Google Scholar]
- 26.Salvatori R, Fan X, Phillips JA, 3rd, Prince M, Levine MA. Isolated growth hormone (GH) deficiency due to compound heterozygosity for two new mutations in the GH-releasing hormone receptor gene. Clin Endocrinol (Oxf) 2001;54:681–687. doi: 10.1046/j.1365-2265.2001.01273.x. [DOI] [PubMed] [Google Scholar]
- 27.Salvatori R, Aguiar-Oliveira MH, Monte LV, Hedges L, Santos NL, Pereira RM, Phillips JA. Detection of a recurring mutation in the human growth hormone-releasing hormone receptor gene. Clin Endocrinol (Oxf) 2002;57:77–80. doi: 10.1046/j.1365-2265.2002.01565.x. [DOI] [PubMed] [Google Scholar]
- 28.Carakushansky M, Whatmore AJ, Clayton PE, Shalet SM, Gleeson HK, Price DA, Levine MA, Salvatori R. A new missense mutation in the growth hormone-releasing hormone receptor gene in familial isolated GH deficiency. Eur J Endocrinol. 2003;148:25–30. doi: 10.1530/eje.0.1480025. [DOI] [PubMed] [Google Scholar]
- 29.Salvatori R, Fan X, Veldhuis JD, Couch R. Serum GH response to pharmacological stimuli and physical exercise in two siblings with two new inactivating mutations in the GH-releasing hormone receptor gene. Eur J Endocrinol. 2002;147:591–596. doi: 10.1530/eje.0.1470591. [DOI] [PubMed] [Google Scholar]
- 30.Maheshwari HG, Silverman BL, Dupuis J, Baumann G. Phenotype and genetic analysis of a syndrome caused by an inactivating mutation in the growth hormone-releasing hormone receptor: Dwarfism of Sindh. J Clin Endocrinol Metab. 1998;83:4065–4074. doi: 10.1210/jcem.83.11.5226. [DOI] [PubMed] [Google Scholar]
- 31.Kamijo T, Hayashi Y, Seo H, Yamamoto M, Ogawa M, Choski CS, Sawant NJ, Colaco MP, Desai MP. A nonsense mutation (E72X) in growth hormone releasing hormone receptor (GHRHR) gene is the major cause of familial isolated growth hormone deficiency in Western region of India: founder effect suggested by analysis of dinucleotide repeat polymorphism close to GHRHR gene. Growth Horm IGF Res. 2004;14:394–401. doi: 10.1016/j.ghir.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Sıklar Z, Berberoğlu M, Legendre M, Amselem S, Evliyaoğlu O, Hacıhamdioğlu B, Savaş Erdeve S, Oçal G. Two siblings with isolated GH deficiency due to loss-of-function mutation in the GHRHR gene: successful treatment with growth hormone despite late admission and severe growth retardation. J Clin Res Pediatr Endocrinol. 2010;2:164–167. doi: 10.4274/jcrpe.v2i4.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alba M, Hall CM, Whatmore AJ, Clayton PE, Price DA, Salvatori R. Variability in anterior pituitary size within members of a family with GH deficiency due to a new splice mutation in the GHRH receptor gene. Clin Endocrinol (Oxf) 2004;60:470–475. doi: 10.1111/j.1365-2265.2004.02003.x. [DOI] [PubMed] [Google Scholar]
- 34.Hilal L, Hajaji Y, Vie-Luton MP, Ajaltouni Z, Benazzouz B, Chana M, Chraïbi A, Kadiri A, Amselem S, Sobrier ML. Unusual phenotypic features in a patient with a novel splice mutation in the GHRHR gene. Mol Med. 2008;14:286–292. doi: 10.2119/2007-00128.Hilal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marui S, Trarbach EB, Boguszewski MC, França MM, Jorge AA, Inoue H, Nishi MY, Aguiar-Oliveira MH, Mendonca BB, Arnhold IJ. GH-releasing hormone receptor gene: a novel splice-disrupting mutation and study of founder effects. Horm Res Paediatr. 2012;78:165–172. doi: 10.1159/000342760. [DOI] [PubMed] [Google Scholar]
- 36.Osorio MG, Marui S, Jorge AA, Latronico AC, Lo LS, Leite CC, Estefan V, Mendonca BB, Arnhold IJ. Pituitary magnetic resonance imaging and function in patients with growth hormone deficiency with and without mutations in GHRH-R, GH-1, or PROP-1 genes. J Clin Endocrinol Metab. 2002;87:5076–5084. doi: 10.1210/jc.2001-011936. [DOI] [PubMed] [Google Scholar]
- 37.Roelfsema F, Biermasz NR, Veldman RG, Veldhuis JD, Frölich M, Stokvis-Brantsma WH, Wit JM. Growth hormone (GH) secretion in patients with an inactivating defect of the GH-releasing hormone (GHRH) receptor is pulsatile: evidence for a role for non-GHRH inputs into the generation of GH pulses. J Clin Endocrinol Metab. 2001;86:2459–24564. doi: 10.1210/jcem.86.6.7536. [DOI] [PubMed] [Google Scholar]
- 38.Walenkamp MJ, Pereira AM, Oostdijk W, Stokvis-Brantsma WH, Pfaeffle RW, Blankenstein O, Wit JM. Height gain with combined growth hormone and gonadotropin-releasing hormone analog therapy in two pubertal siblings with a growth hormone-releasing hormone receptor mutation. J Clin Endocrinol Metab. 2008;93:204–207. doi: 10.1210/jc.2007-1572. [DOI] [PubMed] [Google Scholar]
- 39.Wang Q, Diao Y, Xu Z, Li X, Luo XP, Xu H, Ouyang P, Liu M, Hu Z, Wang QK, Liu JY. Identification of a novel splicing mutation in the growth hormone (GH)-releasing hormone receptor gene in a Chinese family with pituitary dwarfism. Mol Cell Endocrinol. 2009;313:50–56. doi: 10.1016/j.mce.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 40.Horikawa R. Isolated GH deficiency due to inactivating mutation of GHRH receptor. Nihon Rinsho. 2002;60:297–305. [PubMed] [Google Scholar]
- 41.Salvatori R, Fan X, Phillips JA, 3rd, Espigares-Martin R, Freeman KL, Plotnick L, Al-Ashwal A, Levine MA. Three new mutations in the gene for the growth hormone (gh)-releasing hormone receptor in familial isolated gh deficiency type ib. J Clin Endocrinol Metab. 2001;86:273–279. doi: 10.1210/jcem.86.1.7156. [DOI] [PubMed] [Google Scholar]
- 42.Adams EF, Symowski H, Buchfelder M, Poyner DR. A polymorphism in the growth hormone (GH)-releasing hormone (GHRH) receptor gene is associated with elevated response to GHRH by human pituitary somatotrophinomas in vitro. Biochem Biophys Res Commun. 2000;275:33–36. doi: 10.1006/bbrc.2000.3247. [DOI] [PubMed] [Google Scholar]
- 43.Lee EJ, Kotlar TJ, Ciric I, Lee MK, Lim SK, Lee HC, Huh KB, Mayo KE, Jameson JL. Absence of constitutively activating mutations in the GHRH receptor in GH-producing pituitary tumors. J Clin Endocrinol Metab. 2001;86:3989–3995. doi: 10.1210/jcem.86.8.7732. [DOI] [PubMed] [Google Scholar]