

Graphical abstract
Keywords: Amphimedine, Analogue, Ascididemin, Lead compound, Natural, Pyridoacridine
Abstract
Natural products are increasingly being considered “critical and important” in drug discovery paradigms as a number of them such as camptothecin, penicillin, and vincristine serve as “lead molecules” for the discovery of potent compounds of therapeutic interests namely irinotecan, penicillin G, vinblastine respectively. Derived compounds of pharmacological interests displayed a wide variety of activity viz. anticancer, anti-inflammatory, antimicrobial, anti-protozoal, etc.; when modifications or derivatizations are performed on a parent moiety representing the corresponding derivatives. Pyridoacridine is such a moiety which forms the basic structure of numerous medicinally important natural products such as, but not limited to, amphimedine, ascididemin, eilatin, and sampangine. Interestingly, synthetic analogues of natural pyridoacridine exhibit diverse pharmacological activities and in view of these, natural pyridoacridines can be considered as “lead compounds”. This review additionally provides a brief but critical account of inherent structure activity relationships among various subclasses of pyridoacridines. Furthermore, the current aspects and future prospects of natural pyridoacridines are detailed for further reference and consideration.
Introduction
The pivotal role of natural products in novel drug discovery programme can be ascertained from the fact that approximately 40% of Food and Drug Administration, USA (FDA) approved therapeutic drugs have natural origin [1]. The drugs derived via taking “lead” from nature, have shown immense potential in terms of their chemical diversity and biosynthetic molecular recognition usually absent in synthetically developed libraries. A “lead compound” can be defined as a compound responsible for synthesis of a series of compounds via chemical modifications in order to achieve optimal therapeutic activity [2]. A number of drugs derived from the natural sources are available in the market for different ailments (Table 1). In general, a lead compound is identified on the basis of its ability to bind to a therapeutic target. Once designated as a “tight-binder”, this lead compound can be chemically modified to improve the target specificity, bioavailability, and pharmacokinetics and finally tested for their therapeutic activity via pre-clinical and clinical studies [2].
Table 1.
Different medicinally important natural lead compounds and their derived drugs.
| S. no. | Lead compounds | Derived drugs | Medicinal Importance | References |
|---|---|---|---|---|
| 1 | Camptothecin | Irinotecan, Topotecan | Anticancer | [3,4] |
| 2 | Paclitaxel | Docetaxel | Anticancer | [5,6] |
| 3 | Vincristine | Vinblastine, Vindesine, Vinorelbine | Anticancer | [7,8] |
| 4 | Etoposide | Teniposide | Anticancer, Cytotoxic | [8,9] |
| 5 | Quinine | Quinidine | Antimalarial, Antiarrythmic | [10,11] |
| 6 | Digitoxin | Digoxigenin | In Cardiovascular diseases | [12,13] |
| 7 | Cephalosporin C | Cefixime, Cefuroxime | Antimicrobial | [14,15] |
| 8 | Morphine | Codeine, Pholcodeine, Ethylmorphine | Antitussives; Analgesic | [16,17] |
| 9 | Artemisinin | Artesunate, Artemether | Antimalarial | [18,19] |
| 10 | Penicillin G | Penicillin X | Antimicrobial | [20,21] |
| 11 | Tetracycline | Chlortetracyclins, Oxytetracyclins | Antimicrobial | [22,23] |
| 12 | Atropine | Hyoscine | Anticholinergic | [24,25] |
| 13 | Ergotamine | Ergotoxin, Ergometrine | α-adrenergic blockers, Uterine stimulants | [26,27] |
| 14 | Theophylline | Choline theophyllinate | Bronchodilators | [28,29] |
| 15 | Dopamine | Levodopa, Carbidopa | Parkinsonism | [30,31] |
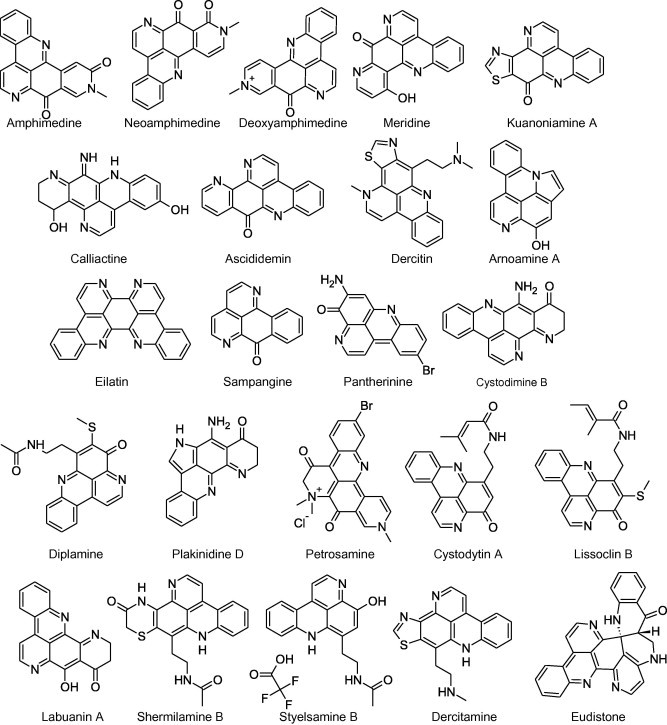
Pyridoacridines, a class of marine-derived alkaloids characterized by a 11H-pyrido[4,3,2-mn]acridine, fulfil all the requirements of being lead compounds in their respective therapeutic category [32]. With varied chemical compositions and conformations differing by (1) different side chains; (2) rings fused to ring C; (3) rings fused to the acridine nitrogen; (4) bromination at C2 in ring A; and (5) varied oxidation states, pyridoacridines present an array of biological activities with respect to, but not limited to, anticancer, anti-HIV, antimicrobial, anti-parasitic, anti-viral and insecticidal activities [33]. Furthermore, pyridoacridines are also associated with calcium ions release from sarcoplasmic reticulum, neuronal differentiation, metal chelation, and depict affinity towards GABA receptors [34]. Various subclasses of pyridoacridines such as amphimedines, ascididemins, petrosamines, dercitins, diplamines, and eilatins provide important chemical cues and clues to act as lead compounds (Fig. 1). For example, various pyridoacridines displayed their anticancer activities via different mechanism like binding with DNA, inhibition of DNA/RNA/protein synthesis, inhibition of topoisomerase, cleavage/catenation/damage of DNA, cell cycle arrest and hence can be employed as “hits” in further lead optimization [32–34].
Fig. 1.
Natural pyridoacridines.
Although some comprehensive reviews are available on pyridoacridines such as Molinski’s one [32] which describes the structure, synthesis and biological chemistry of pyridoacridines or the other by Marshall and Barrows [34], which reviewed the biological activities of pyridoacridines; the present review describes a correlation between various important structural aspects of pyridoacridines with respect to their biological activities demonstrating their potential as lead compounds for the future [32,34].
Different natural pyridoacridines are shown in Fig. 1. Inspired by these natural pyridoacridines, researchers around the world are synthesizing medicinally active derivatives. In view of the above mentioned facts, pyridoacridines can be considered as “Prospective lead compounds” of the future. The word “Prospective lead compounds” can be exemplified from the different representative examples of pyridoacridine derivatives to be covered in coming sections. In addition, structural activity relationship points may further prove their candidature as lead compounds for different ailments. Furthermore, for the interest of the readers, due care has been taken as the present review limit itself to discussion on different analogues of representative natural pyridoacridines with promising activities only and discussion has been provided with subheads namely: amphimedine analogues, ascididemin analogues, dercitin analogues, eilatin analogues, kuanoniamine analogues, sampangine analogues and current/future prospects.
Scope of the review
This review primarily focuses on the representative publications of last 20 years (i.e. from 1994 to 2014) related to chemical and medicinal aspects of pyridoacridines; each representing a unique study about pyridoacridine analogues.
Search terms like pyridoacridine, pyridoacridine derivative, lead, amphimedine, ascididemin, kuanoniamine, sampangine, dercitin, eilatin were used to find out different publications on natural as well as synthetic analogues of pyridoacridines by using various E-resources and databases like Google Scholar, American Chemical Society, Wiley-Blackwell Publishing, Elsevier Science, Nature, Royal Society of Chemistry, Springer Link, Taylor and Francis, Pubmed Scopus, Reaxys, Bielstein and Scifinder.
Amphimedine analogues
A small difference in the structure of a drug may impose noticeable effect on its pharmacological activity like three natural analogues of amphimedines namely: amphimedine, neoamphimedine, deoxyamphimedine bear only small differences in structures but neoamphimedine and deoxyamphimedine both possess antitumour activity. On the other hand, the study describes amphimedine as relatively inactive (Fig. 1). As per a study performed by Marshall and co-workers [35], neoamphimedine antitumour activity equals to well-known anticancer agent etoposide while in reference to work by Matsumoto et al. [36], authors speculate that mechanism behind anticancer activity of deoxyamphimedine could be redox cycling and reactive oxygen species (ROS) generation emanated from its iminoquinone moiety [35–37]. Authors further concluded that being a positively charged compound; deoxyamphimedine demonstrated significantly different biological activities as compared to neoamphimedine and deoxyamphimedine.
Ponder et al. [38], demonstrated the influence of carbonyl group position in biological activity with the help of molecular docking studies. Docking studies on ATPase site of TopoIIα enzyme revealed that carbonyl group of neoamphimedine interacts with Ser-148 residue at ATPase but in case of amphimedine, hydrogen bonding with Ser-148 was lost which resulted in unfavourable steric interaction with the active site Mg2+ and ultimately loss of biological activity [38].
Different analogues of amphimedine were synthesized via Diels–Alder reaction and their cytotoxic potential was assessed on human cancer cell lines with varying histopathological types (colon, lung, glioblastomas and bladder cancers) by using MTT assay. Interestingly, it was reported that most similar analogues of amphimedine i.e. compound 1 and compound 2 were found to exhibit highest cytotoxic potential with IC50 value less than 10−7 M [39].
Ascididemin analogues
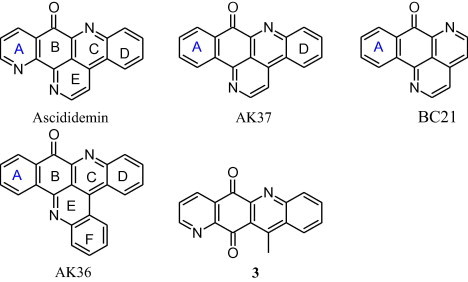
Marshall and co-workers [40] discussed structure activity relationship among different analogues of ascididemin i.e. AK37 and AK36 (Fig. 2). Structurally, the only difference between ascididemin and AK37 is the presence of an additional N-atom in ascididemin; while mechanistically AK37 act by inhibiting the catalytic activity of both topo I/II and stabilizes the DNA-Topo I cleavable complex. The DNA-Topo I cleavable complex stabilizing function of AK37, the first pyridoacridine to show this activity, as compared to the ROS-generating function of ascididemin, can be attributed to the absence of nitrogen in the ‘A’ ring of AK37. However, complete removal of ring ‘D’ from AK37 maintained the DNA-Topo I cleavable complex stabilization in BC21 which proves that ring ‘D’ does not exhibit any important role in AK37’s Topo I activity. In case of AK36, the presence of an additional ring ‘F’ prevented DNA intercalation by AK36 and rendered it lower cytotoxicity than the related compounds. Additionally, the presence of ring ‘F’ in AK36 diminished the DNA-Topo I cleavable complex stabilizing functions observed in AK37 [40].
Fig. 2.
The basic structure of ascididemin and their analogues.
In a study, Lindsay et al. [41], provided the structure activity relationship of various ascididemin analogues. In brief, the authors assessed the importance of ring A and ring E of ascididemin (Fig. 2) by synthesizing different analogues of ascididemin and carried out a range of assays for the evaluation of their biological activity. Presence of nitrogen in ring A is essential for the inhibition of Escherichia coli and Cladosporium resinae while its absence results in Trichophyton mentagrophytes inhibition. [41]. Alternatively, dramatic increase in antitumour/antifungal/antibacterial activity was evaluated by the presence or absence of ring E in ascididemin and compound 3 respectively (Fig. 2). Furthermore, it was concluded that ascididemin acts through multiple mechanisms towards mammalian cell systems [41]. Similar to these studies, Appleton and co-workers synthesized ring A analogues of ascididemin containing furan and thiophene rings which showed considerable antitubercular activity [42].
In their study, Guittat et al. [43], determined the affinity of ascididemin for DNA quadruplexes and findings of the study indicated that large flat aromatic surface of ascididemin might be responsible for their binding with DNA quadruplexes. Furthermore, authors asserted that presence of positive charge on nitrogen atom of ascididemin would enhance, as DNA quadruplex has high negative charge density [43].
Delfourne and co-workers [44], further suggested that the cytotoxic activity of the ascididemin isomer (compound 4) can be retained even after manipulating positions 5 and 7 on ring D [44].
In a study conducted by Delfourne and co-workers in [45], ascididemin analogues were synthesized and evaluated for their cytotoxic potential by MTT assay. Some ascididemin analogues (ring D-modified) appeared to be more cytotoxic than the reference compound ascididemin (Table 2). Furthermore, it was reported that substitution at R1 and R3 does not have much influence on cytotoxic activity [45].
Table 2.
Ascididemin derivatives along with mean IC50 of 12 different cell lines viz., two colon (HCT-15; LoVo); two breast (T-47D and MCF7); three glioblastomas (SW1088; U-373 MG and U-87 MG); one prostate (PC-3); two bladder (J82; T24) and two non small-cell lung (A549; A-427) cancer cell lines.
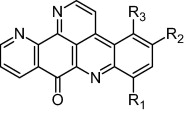 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Mean IC50 (nM) |
| 5 | NH2 | H | H | 53 |
| 6 | H | Br | H | 80 |
| 7 | H | NH2 | H | 21 |
| 8 | H | NHCH2CH2Cl | H | 7 |
| 9 | H | N(CH2CH2Cl)2 | H | 100 |
| 10 | H | NHCH2CH2N(CH3)2 | H | 60 |
| 11 | H | Cl | H | 270 |
| 12 | H | CH3 | H | 60 |
| 13 | H | OCH3 | H | 90 |
| 14 | H | N(CH3)2 | H | 37 |
| 15 | H | NHBn | H | 140 |
| 16 | H | NH2 | Br | 140 |
| Ascididemin | H | H | H | 100 |
Debnath and co-workers [46], carried out QSAR studies on the above mentioned ascididemin analogues reported by Delfourne et al. [45] and emphasized that the presence of an electron withdrawing substituent with higher molar refractivity value and the presence of NHR (R is hydrogen or alkyl group) at R1 and R3 positions, respectively, favoured the anti-tumour activity of ascididemin analogues [Fig. 3] [45,46].
Fig. 3.
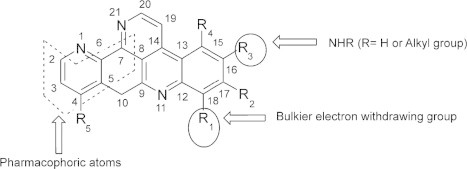
Pharmacophoric atoms and required physicochemical properties of substituents at R1 and R3 positions of ascididemin analogues for their anti-tumour activity ([46], Reproduced with permission from Elsevier B.V. Ltd.© 2003).
In this period of shortage of antitubercular drugs; Appleton and co-workers [42], synthesized and evaluated a series of ascididemin analogues and found thioethyl analogue i.e. compound 17 to exhibit potent antitubercular activity against Mycobacterium tuberculosis ((Mtb) H37Rv) strain (IC50 = 0.34 μM) as compared to reference compound rifampin (IC50 = 0.152 μM) [42]. Encouraged by these observations, authors made the following assertions:
-
1.
“Size-reduced analogues of ascididemin may provide a useful scaffold for future studies.”
-
2.
Iminoquinone moiety of ascididemin is essential for antitubercular activity.
Dercitin analogues
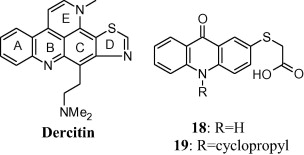
In 1992, Taraporewal and co-workers studied the anti-HIV and anti-tumour activities of dercitin analogues. Dercitin (Fig. 1), a pyridoacridine obtained from Dercitus sp. sponge namely Dercitus simplex, Dercitus lististinus [47], exerts its cytotoxic effect on mammalian cells through four basic nitrogen atoms capable of binding to the acidic amino acid residues. Furthermore, progressive removal of these basic nitrogen atoms results in lowering of cytotoxic potential of dercitin. In addition to nitrogen atoms, the presence of a fused thiazole ring contributed to the cytotoxic activity of dercitin while the presence of a nonlinearly fused pyridine ring E elicited no significant benefit. Compounds 18 and 19 possessed best anti-HIV activity. The presence of a sulphur atom seemed to be essential for antiviral activity of dercitin analogues while the acidic carboxylic group decreases viral affinity towards lymphocytes. Similarly, the occurrence of a mercaptoacetic acid group at the C-2 position in compounds 18 and 19 on the tricyclic ABC acridine nucleus demonstrated highest HIV-1 neutralization activity along with the partial inhibition of viral affinity towards lymphocytes [48].
Eilatin analogues
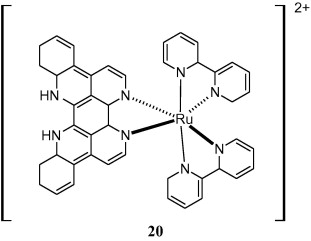
Eilatin (Fig. 1) is a highly symmetrical heptacyclic alkaloid isolated in 1988 from the red sea tunicate Eudistoma sp. A new family of eilatin-containing metal complexes is under investigation for their unusual nucleic acid binding specificity [49]. In a similar study, Zeglis and Barton [50], investigated the DNA-binding properties of Ru(bpy)2(eilatin)2+ (compound 20) to determine specificity of sterically expansive eilatin ligand for DNA [50]. From the results it was concluded that extended planar surface presented by eilatin is responsible for its specificity and anti-HIV activity [49,51].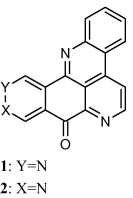
Kuanoniamine analogues
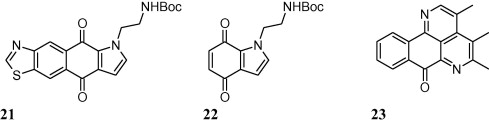
Different pentacyclic analogues of kuanoniamine A (Fig. 1) were synthesized for their antiprotozoal activity against virulent strains i.e. Leishmania donovani; Leishmania major and Toxoplasma gondii. As reported, most of the synthesized compounds were found to be more active than well-known drugs pentamidine, pyrimethamine, sulfadiazine and spiramycin against T. gondii. On the other hand, compound 21 and compound 22 displayed an IC50 of 6 nM against L. major as compared to reference compound pentamidine (IC50 = 1.8 nM). Authors neither discussed any structure activity relationship points nor they proposed any mechanism behind antiprotozoal activity. Although lipophilicity was mentioned as an important factor behind antiprotozoal activity while the presence of boc group and quinoneimine function might be responsible for antiprotozoal activity [52,53].
Sampangine analogues
A series of sampangine analogues were synthesized and screened for their antitubercular activity by Claes and co-workers [54]. As reported, most of the compounds showed promising activity against M. tuberculosis ((Mtb) H37Rv) strain of tuberculosis and minimal inhibitory concentrations (MIC) were measured for most potent sampangine derivative i.e. compound 23 as low as 0.39 μM [54].
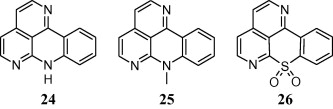
In another study, sampangine analogues were synthesized to evaluate their antimicrobial activity and to investigate the role of azaquinoid partial structure in the pharmacological activity. The microbial strains selected were namely Pseudomonas aeruginosa, E. coli, Staphylococcus aureus, Candida albicans and Aspergillus niger. In this study, authors concluded that presence of amino group in compound 24 and compound 25 while presence of sulphone moiety in compound 26 might be responsible for their significant antimicrobial activity [55].
In order to convert hit into lead, and lead into drug candidate it is very important to study their structural activity relationships; so that characteristic aspects like the nature of functional group, bulkiness of the molecule, electronegative effect, and inductive effect caused by the presence of a moiety/group, etc. should be considered. After discussing the representative pyridoacridine analogues and their important structure activity relationship (SAR) points, it can now be easily concluded that the “pyridoacridine moiety” will be helpful to design new medicinally active molecules and in view of these assertions, pyridoacridines can be designated as “lead compounds”.
Current status and future prospects
The literature discloses the biological potential of naturally available pyridoacridines, and the total synthesis of almost every natural pyridoacridine is available [32–34]. With the help of these available procedures, nowadays it is easy for medicinal chemists to prepare analogues of pyridoacridines in order to improve their biological activity and to lay down some interesting structure–activity relationship points for future research. Currently, search is in progress to identify novel biologically active pyridoacridine analogues on the basis of structure–activity relationship, such as, Marshall and co-workers [40], claimed AK37 as the first pyridoacridine reported to inhibit the catalytic activity of both topoisomerase I/II and stabilize the DNA-topo I cleavable complex [40]. Furthermore, in a similar study on pyridoacridine analogues, Delfourne and coworkers reported that some of the synthesized analogues are more cytotoxic than the reference compound ascididemin while Nukoolkarn et al. [56] opinionated that the quaternary ammonium group plays an important role in acetylcholinesterase inhibitory activity of ascididemin [56]. On the other hand, Guittat and co-workers identified quaternary ammonium group responsible for high affinity towards DNA quadruplex, due to high negative charge density present on DNA quadruplex [43].
Pyridoacridines were also found to possess biological activity equivalent to some well-known biologically active agents. For example, neoamphimedine showed anticancer activity equivalent to etoposide when tested in mice bearing human epidermoid-nasopharyngeal tumour cell line. Similarly neoamphimedine responds to HCT-116 tumours as effectively as in case of 9-aminocamptothecin, an anticancer drug in clinical development [35,57].
Because of continued interest in pyridoacridine analogues, a new class of “pyridoacridine-metal complexes” (like Ru(bpy)2(eilatin)2+) was discovered where due to planar surface presented by the eilatin, affinity towards nucleic acid binding improves [51].
Heterocyclic derivatives are well known for their biological potential [58,59] and similarly heterocyclic-pyridoacridine analogues were reported by some researchers in their studies. For example Appleton and coworkers, in their study published in 2010 reported furan and thiophene analogues of ascididemin with significant antitubercular activity [42] while in case of non-heterocyclic analogues like thioethyl analogue, which displayed considerable antitubercular activity and authors pointed out that this analogue could be considered as a useful scaffold for future studies [42]. The search is underway in some cases where the pyridoacridine analogues were synthesized but their biological activity and mechanism of action remain to be explored, for example synthesis of ascididemin analogues by Plodek and co-workers [60]; preparation of pyridoacridine analogues by Godard and co-workers [61]; and synthesis of sampangine derivatives by Vasilevsky and co-workers [62], etc. Looking at the promising results, the research in this area could provide some useful drugs of future.
Conflict of interest
The author has declared no conflict of interest.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Acknowledgements
Mr. Vikas Sharma is thankful to All India Council for Technical Education, India for providing National Doctoral Fellowship for PhD research work vide letter no. F.No. 14/AICTE/RIFD/NDF(Policy-I)/01/2012-13. Authors thankfully acknowledge Mr. Pradeep Kumar, Department of Pharmacy and Pharmacology, Faculty of Health Sciences, WITS Medical School, University of the Witwatersrand, Johannesburg, South Africa, for his valuable suggestions and contributions for the paper.
Biographies

Vikas Sharma is presently pursuing Ph.D from Institute of Pharmaceutical Sciences, Kurukshetra University, Kurukshetra, Haryana, India. He is the recipient of prestigious AICTE, National Doctoral Fellowship for his doctoral studies. He has around 10 national and international publications in different journals of repute. Earlier he completed his M. Pharmacy (Pharmaceutical Chemistry; 2008) from Rajiv Academy for Pharmacy, Mathura, Uttar Pradesh, India, before joining a CSIR project as Senior Research Fellow at Department of Biophysics, Post Graduate Institute of Medical Education & Research, Chandigarh, India. Later on, he worked as Assistant Professor at Institute of Pharmaceutical Sciences, Kurukshetra University, Kurukshetra, Haryana, India.

Dr. P.C. Sharma is Assistant Professor at Institute of Pharmaceutical Sciences, Kurukshetra University, Kurukshetra, Haryana, India. He obtained his Ph.D from Guru Jambheshwar University of Science and Technology, Hisar, India, after completing M. Pharmacy from J.S.S. College, Ooty, Tamilnadu, India. He is regular reviewer of various national and international journals. Besides that he worked as a Guest editor of a special issue for journal “Current Topics in Medicinal Chemistry”. He has authored around 60 publications in various national and international journals of repute. He has around 10 years of teaching and 2 years of industrial experience.

Dr. Vipin Kumar obtained his Ph.D. from Maharishi Dayanand University, Rohtak, Haryana, India and currently holds the position of Head at School of Pharmacy, Central University of Rajasthan, Ajmer, Rajasthan, India. He has approximately 10 years of teaching experience. His work on Graph theory and QSAR is noteworthy. He has co-authored 70 publications in various journals and is a regular reviewer of various national and international journals of repute.
Footnotes
Peer review under responsibility of Cairo University.

References
- 1.Pascolutti M., Quinn R.J. Natural products as lead structures: chemical transformations to create lead-like libraries. Drug Discov Today. 2014;19(3):215–221. doi: 10.1016/j.drudis.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 2.Lead compound. Scientific glossary (Pharmaceutical institute). <http://www.pharmainstitute.com/glossary/index.cfm?id=2128&search_term=lead%20 compound&select=TRUE> [accessed 20.08.14].
- 3.Ganesan A. Natural products and cancer drug discovery. Springer; New York: 2013. The impact of natural products upon cancer chemotherapy; pp. 3–15. [Google Scholar]
- 4.Singh P., Andola H.C., Rawat M.S.M., Joshi G., Haider S.Z. Himalayan plants as a source of anti-cancer agents: a review. Nat Prod J. 2013;3(4):296–308. [Google Scholar]
- 5.de Weger V.A., Beijnen J.H., Schellens J.H.M. Cellular and clinical pharmacology of the taxanes docetaxel and paclitaxel – a review. Anti-Cancer Drugs. 2014;25(5):488–494. doi: 10.1097/CAD.0000000000000093. [DOI] [PubMed] [Google Scholar]
- 6.Flora Z., Sergentanis T.N., Chrysikos D., Dimitrakakis C., Tsigginou A., Zografos C.G. Taxanes for breast cancer during pregnancy: a systematic review. Clin Breast Cancer. 2013;13(1):16–23. doi: 10.1016/j.clbc.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 7.Barker T.J., Duncan K.K., Otrubova K., Boger D.L. Potent vinblastine c20′ ureas displaying additionally improved activity against a vinblastine-resistant cancer cell line. ACS Med Chem Lett. 2013;4(10):985–988. doi: 10.1021/ml400281w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lv M., Xu H. Recent advances in semisynthesis, biosynthesis, biological activities, mode of action, and structure-activity relationship of podophyllotoxins: an update (2008–2010) Mini Rev Med Chem. 2011;11(10):901–909. doi: 10.2174/138955711796575461. [DOI] [PubMed] [Google Scholar]
- 9.Weiqiang L., Fu C., Zhao Y. Review on biosynthesis of podophyllotoxin. Zhongguo Zhong Yao Za Zhi. 2011;36(9):1109–1114. [PubMed] [Google Scholar]
- 10.de Villiers K.A., Gildenhuys J., le Roex T. Iron (III) protoporphyrin IX complexes of the antimalarial cinchona alkaloids quinine and quinidine. ACS Chem Biol. 2012;7(4):666–671. doi: 10.1021/cb200528z. [DOI] [PubMed] [Google Scholar]
- 11.Sullivan D.J. Treatment and prevention of malaria. Springer Basel; 2012. Cinchona alkaloids: quinine and quinidine; pp. 45–68. [Google Scholar]
- 12.Wurtele E.S., Chappell J., Jones A.D., Celiz M.D., Ransom N., Hur M. Medicinal plants: a public resource for metabolomics and hypothesis development. Metabolites. 2012;2(4):1031–1059. doi: 10.3390/metabo2041031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rao R.V., Vaidyanathan C.S. Chemistry and biochemical pharmacology of cardiac glycosides – a review. J Ind Inst Sci. 2013;71(4):329–364. [Google Scholar]
- 14.Erdem H., Kilic S., Pahsa A., Besirbellioglu B.A. Gram-negative bacterial resistance to cephalosporins in community-acquired infections in Turkey. J Chemother. 2005;17(1):61–65. doi: 10.1179/joc.2005.17.1.61. [DOI] [PubMed] [Google Scholar]
- 15.Weiming L., Neidert M.C., Groen R.J.M., Woernle C.M., Grundmann H. Third-generation cephalosporins as antibiotic prophylaxis in neurosurgery: what’s the evidence? Clin Neurol Neurosurg. 2014;116:13–19. doi: 10.1016/j.clineuro.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 16.Chen Z.R., Irvine R.J., Somogyi A.A., Bochner F. Mu receptor binding of some commonly used opioids and their metabolites. Life Sci. 1991;48(22):2165–2171. doi: 10.1016/0024-3205(91)90150-a. [DOI] [PubMed] [Google Scholar]
- 17.Mazák K., Hosztafi S., Rácz A., Noszál B. Structural and physicochemical profiling of morphine and related compounds of therapeutic interest. Mini Rev Med Chem. 2009;9(8):984–995. doi: 10.2174/138955709788681555. [DOI] [PubMed] [Google Scholar]
- 18.Das S. Artemisia annua (Qinghao): a pharmacological review. Int J Pharm Sci Res. 2012;3(12):4573–4577. [Google Scholar]
- 19.Dodoo A.N., Fogg C., Asiimwe A., Nartey E.T., Kodua A., Tenkorang O. Pattern of drug utilization for treatment of uncomplicated malaria in urban Ghana following national treatment policy change to artemisinin-combination therapy. Malaria J. 2009;8:2. doi: 10.1186/1475-2875-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merrikin D.J., Briant J., Rolinson G.N. Effect of protein binding on antibiotic activity in vivo. J Antimicrob Chemother. 1983;11(3):233–238. doi: 10.1093/jac/11.3.233. [DOI] [PubMed] [Google Scholar]
- 21.Craig W.A., Welling P.G. Protein binding of antimicrobials: clinical pharmacokinetic and therapeutic implications. Clin Pharmacokinet. 1977;2(4):252–268. doi: 10.2165/00003088-197702040-00002. [DOI] [PubMed] [Google Scholar]
- 22.Al-Omari A., Cameron D.W., Lee C., Corrales-Medina V.F. Oral antibiotic therapy for the treatment of infective endocarditis: a systematic review. BMC Infect Disease. 2014;14(1):140. doi: 10.1186/1471-2334-14-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodman J.J. The Tetracyclines. Springer; Berlin Heidelberg: 1985. Fermentation and mutational development of the tetracyclines; pp. 5–57. [Google Scholar]
- 24.Tramer M., Moore A., McQuay H. Prevention of vomiting after paediatric strabismus surgery: a systematic review using the numbers-needed-to-treat method. Brit J Anaesth. 1995;75(5):556–561. doi: 10.1093/bja/75.5.556. [DOI] [PubMed] [Google Scholar]
- 25.Maqbool F., Singh S., Kaloo Z.A., Jan M. Medicinal importance of genus Atropa (Royle) – a review. Int J Adv Res. 2014;2(2):48–54. [Google Scholar]
- 26.Mulac D., Lepski S., Ebert F., Schwerdtle T., Humpf H.U. Cytotoxicity and fluorescence visualization of ergot alkaloids in human cell lines. J Agric Food Chem. 2013;61(2):462–471. doi: 10.1021/jf304569q. [DOI] [PubMed] [Google Scholar]
- 27.Ayarragaray J.E.F. Ergotism: a change of perspective. Ann Vasc Surg. 2014;28(1):265–268. doi: 10.1016/j.avsg.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Parkes PC. Dose counter mechanisms for medicament delivery devices. U.S. Patent 8,511,302, Issued: August 20th; 2013.
- 29.Hendeles L., Weinberger M. Theophylline. A “State of the Art” review. Pharmacotherapy. 1983;3(1):2–44. doi: 10.1002/j.1875-9114.1983.tb04531.x. [DOI] [PubMed] [Google Scholar]
- 30.Volkmann J., Albanese A., Antonini A., Chaudhuri K.R., Clarke C.E., de Bie R.M.A. Selecting deep brain stimulation or infusion therapies in advanced Parkinson’s disease: an evidence-based review. J Neurol. 2013;260(11):2701–2714. doi: 10.1007/s00415-012-6798-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nyholm D. Duodopa® treatment for advanced Parkinson’s disease: a review of efficacy and safety. Parkinsonism Relat Disord. 2012;18(8):916–929. doi: 10.1016/j.parkreldis.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 32.Molinski T.F. Marine pyridoacridine alkaloids: structure, synthesis, and biological chemistry. Chem Rev. 1993;93:1825–1838. [Google Scholar]
- 33.Proksch P., Wray V., Steube K., Muller C.E., Eder C., Schupp P. Bioactive pyridoacridine alkaloids from the micronesian sponge oceanapia sp. J Nat Prod. 1998;61:301–305. doi: 10.1021/np9702704. [DOI] [PubMed] [Google Scholar]
- 34.Marshall K.M., Barrows L.R. Biological activities of pyridoacridines. Nat Prod Rep. 2004;21(6):731–751. doi: 10.1039/b401662a. [DOI] [PubMed] [Google Scholar]
- 35.Marshall K.M., Matsumoto S.S., Holden J.A., Concepcion G.P., Tasdemirc D., Ireland C.M. The anti-neoplastic and novel topoisomerase II-mediated cytotoxicity of neoamphimedine, a marine pyridoacridine. Biochem Pharmacol. 2003;66:447–458. doi: 10.1016/s0006-2952(03)00209-0. [DOI] [PubMed] [Google Scholar]
- 36.Matsumoto S.S., Biggs J., Copp B.R., Holden J.A., Barrows L.R. Mechanism of ascididemin-induced cytotoxicity. Chem Res Toxicol. 2003;16(2):113–122. doi: 10.1021/tx025618w. [DOI] [PubMed] [Google Scholar]
- 37.Marshall K.M., Andjelic C.D., Tasdemir D., Concepción G.P., Ireland C.M., Barrows L.R. Deoxyamphimedine, a pyridoacridine alkaloid, damages DNA via the production of reactive oxygen species. Mar Drugs. 2009;7:196–209. doi: 10.3390/md7020196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ponder J., Yoo B.H., Abraham A.D., Li Q., Ashley A.K., Amerin C.L. Neoamphimedine circumvents metnase-enhanced DNA topoisomerase IIα activity through ATP-competitive inhibition. Mar Drugs. 2011;9:2397–2408. doi: 10.3390/md9112397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brahic C., Darro F., Belloir M., Bastide J., Kiss R., Delfourne E. Synthesis and cytotoxic evaluation of analogues of the marine pyridoacridine amphimedine. Bioorg Med Chem. 2002;10:2845–2853. doi: 10.1016/s0968-0896(02)00148-7. [DOI] [PubMed] [Google Scholar]
- 40.Marshall K.M., Holden J.A., Koller A., Kashman Y., Copp B.R., Barrows L.R. AK37: the first pyridoacridine described capable of stabilizing the topoisomerase I cleavable complex. Anti-Cancer Drugs. 2004;15:907–913. doi: 10.1097/00001813-200410000-00012. [DOI] [PubMed] [Google Scholar]
- 41.Lindsay B.S., Barrows L.R., Copp B.R. Structural requirements for biological activity of the marine alkaloid ascididemin. Bioorg Med Chem Lett. 1995;5:739–742. [Google Scholar]
- 42.Appleton D.R., Pearce A.N., Copp B.R. Anti-tuberculosis natural products: synthesis and biological evaluation of pyridoacridine alkaloids related to ascididemin. Tetrahedron. 2010;66:4977–4986. [Google Scholar]
- 43.Guittat L., De Cian A., Rosu F., Gabelica V., Edwin De Pauw, Delfourne E. Ascididemin and meridine stabilise G-quadruplexes and inhibit telomerase in vitro. Biochim Biophys Acta. 2005;1724:375–384. doi: 10.1016/j.bbagen.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 44.Delfourne E., Kiss R., Le Corre L., Merza J., Bastide J., Frydmanc A. Synthesis and in vitro antitumor activity of an isomer of the marine pyridoacridine alkaloid ascididemin and related compounds. Bioorg Med Chem. 2003;11:4351–4356. doi: 10.1016/s0968-0896(03)00483-8. [DOI] [PubMed] [Google Scholar]
- 45.Delfourne E., Darro F., Portefaix P., Galaup C., Bayssade S., Bouteillé A. Synthesis and in vitro antitumor activity of novel ring d analogues of the marine pyridoacridine ascididemin: structure-activity relationship. J Med Chem. 2002;45:3765–3771. doi: 10.1021/jm0208774. [DOI] [PubMed] [Google Scholar]
- 46.Debnath B., Gayen S., Bhattacharya S., Samanta S., Jha T. QSAR study on some pyridoacridine ascididemin analogues as anti-tumor agents. Bioorg Med Chem. 2003;11:5493–5499. doi: 10.1016/j.bmc.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 47.Kobuk D.R., Van Soest R.W.M. Cavity-dwelling sponges in a southern caribbean coral reef and their paleontological implications. B Mar Sci. 1989;44:1207–1235. [Google Scholar]
- 48.Taraporewala I.B., Cessac J.W., Chanh T.C., Delgado A.V., Schinazi R.F. HIV-1 neutralization and tumor cell proliferation inhibition in vitro by simplified analogs of pyrido [4, 3, 2-mn] thiazolo [5, 4-b] acridine marine alkaloids. J Med Chem. 1992;35:2744–2752. doi: 10.1021/jm00093a005. [DOI] [PubMed] [Google Scholar]
- 49.Luedtke N.W., Hwang J.S., Nava E., Gut D., Kol M., Tor Y. The DNA and RNA specificity of eilatin Ru(II) complexes as compared to eilatin and ethidium Bromide. Nucleic Acid Res. 2003;31:5732–5740. doi: 10.1093/nar/gkg758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeglis B.M., Barton K.J. Binding of Ru(bpy)2(eilatin)2+ to matched and mismatched DNA. Inorg Chem. 2008;47:6452–6457. doi: 10.1021/ic8006537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luedtke N.W., Hwang J.S., Glazer E.C., Gut D., Kol M., Tor Y. Eilatin Ru(II) complexes display anti-HIV activity and enantiomeric diversity in the binding of RNA. ChemBioChem. 2002;3:766–771. doi: 10.1002/1439-7633(20020802)3:8<766::AID-CBIC766>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 52.Tapia R.A., Prieto Y., Pautet F., Walchshofer N., Fillion H., Fenetc B., Sarciron M.E. Synthesis and antiprotozoal evaluation of benzothiazolopyrroloquinoxalinones, analogues of kuanoniamine A. Bioorg Med Chem. 2003;11:3407–3412. doi: 10.1016/s0968-0896(03)00311-0. [DOI] [PubMed] [Google Scholar]
- 53.Kitahara Y., Nakahara S., Yonezawa T., Nagatsu M., Shibano Y., Kubo A. Synthetic studies on pentacyclic aromatic alkaloids, kuanoniamine A, ll-hydroxyascididemin, and neocalliactine acetate. Tetrahedron. 1997;53:17029–17038. [Google Scholar]
- 54.Claes P., Cappoen D., Mbala B.M., Jacobs J., Mertens B., Mathys V. Synthesis and antimycobacterial activity of analogues of the bioactive natural products sampangine and cleistopholine. Eur J Med Chem. 2013;67:98–110. doi: 10.1016/j.ejmech.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 55.Mink K., Bracher F. Hetero analogues of the antimicrobial alkaloids cleistopholine and sampangine. Arch Pharm Chem Life Sci. 2007;340:429–433. doi: 10.1002/ardp.200700064. [DOI] [PubMed] [Google Scholar]
- 56.Nukoolkarn V.S., Saen-oon S., Rungrotmongkol T., Hannongbua S., Ingkaninan K., Suwanborirux K. Petrosamine, a potent anticholinesterase pyridoacridine alkaloid from a Thai marine sponge Petrosia n. sp. Bioorg Med Chem. 2008;16:6560–6567. doi: 10.1016/j.bmc.2008.05.027. [DOI] [PubMed] [Google Scholar]
- 57.Khazir J., Mir B.A., Pilcher L., Riley D.L. Role of plants in anticancer drug discovery. Phytochem Lett. 2014;7:173–181. [Google Scholar]
- 58.Sharma V., Kumar V., Kumar P. Heterocyclic chalcone analogues as potential anticancer agents. Anti-Cancer Agent Med Chem. 2013;13(3):422–432. [PubMed] [Google Scholar]
- 59.Sharma V., Kumar P., Pathak D. Biological importance of the indole nucleus in recent years: a comprehensive review. J Het Chem. 2010;47(3):491–502. [Google Scholar]
- 60.Plodek A., Raeder S., Bracher F. A novel approach to ring A analogues of the marine pyridoacridine alkaloid ascididemin. Tetrahedron. 2013;69:9857–9864. [Google Scholar]
- 61.Godard A., Rocca P., Guillier F., Duvey G., Nivoliers F., Marsais F. Ortho-directed metallation of p-deficient heterocycles in connection with palladium catalysed biaryl cross-coupling: synthesis of marine alkaloids of the pyridoacridine series. Can J Chem. 2001;79:1754–1761. [Google Scholar]
- 62.Vasilevsky S.F., Baranov D.S., Mamatyuk V.I., Gatilov Y.V., Alabugin I.V. An unexpected rearrangement that disassembles alkyne moiety through formal nitrogen atom insertion between two acetylenic carbons and related cascade transformations: new approach to sampangine derivatives and polycyclic aromatic amides. J Org Chem. 2009;74:6143–6150. doi: 10.1021/jo9008904. [DOI] [PubMed] [Google Scholar]