

Abstract
An expansive set of N-aryl-N’-(3-(substituted)phenyl)-N’-methylguanidines was prepared in a search for new leads to prospective PET ligands for imaging of the open channel of the N-methyl-D-aspartate (NMDA) receptor in vivo. The N-aryl rings and their substituents were varied, whereas the N-methyl group was maintained as a site for potential labeling with the positron-emitter, carbon-11 (t1/2 = 20.4 min). At micromolar concentration, over half of the prepared compounds strongly inhibited the binding of [3H]TCP to its binding site in the open NMDA receptor in vitro. Four ligands displayed affinities that are similar or superior to those of the promising SPECT radioligand ([123I]CNS1261). The 3’-dimethylamino (19; Ki 36.7 nM), 3’-trifluoromethyl (20; Ki 18.3 nM) and 3’-methylthio (2; Ki 39.8 nM) derivatives of N-1-naphthyl-N’-(phenyl)-N’-methylguanidine were identified as especially attractive leads for PET radioligand development.
Keywords: N-Methyl-d-aspartate receptor, Phencyclidine, Positron emission tomography
Dysfunction in N-methyl-D-aspartate (NMDA) receptors has been linked to several debilitating neurological and psychiatric illnesses, such as epilepsy, stroke, Parkinson's disease, Alzheimer's disease, and schizophrenia.1–4 The NMDA receptor is a voltage and ligand gated ion channel regulated by at least five binding sites for endogenous ligands, namely glutamate, glycine, Mg2+, Zn2+, and polyamines. NMDA receptors are involved in long-term potentiation, memory, neuroplasticity, neurodevelopment, and neuroprotection.3,5,6 Dysfunction in these receptors is typically characterized by over-activation, allowing for a high concentration of cation influx causing neuronal death.7–9 Several experimental drugs have been synthesized for the purpose of attenuating NMDA receptor activation. Many of these have been targeted at the phencyclidine (PCP) binding site located within the open ion channel.4 The development of a radioligand for imaging the PCP binding site in vivo would allow for the quantification of activated NMDA receptors in both diseased and healthy brain, thereby providing a useful tool for neuropsychiatric research. In addition, such a radioligand would be useful for the further development of drugs intended to act on the PCP binding site, for example, by allowing acquisition of data on target engagement.
Candidate radioligands from various structural classes have been considered for imaging the PCP binding site of the NMDA receptor in vivo.7,10–16 As recently reviewed,17 all of these radioligands display high affinity for the PCP binding site in vitro, as would be required for a prospective in vivo imaging agent. Among these radioligands, the single photon emission computed tomography (SPECT) radioligand [123I]CNS1261 ([123I]1) probably has the strongest evidence for receptor-specific signal in human brain in vivo (Chart 1), although the putative signal is small.9,15 We considered that this structure might be modified to produce a more effective radioligand for imaging the PCP binding site in living human brain with positron emission tomography (PET). With a view to developing such a PET radioligand, we synthesized a series of N-aryl-N’-phenyl-N’-methylguanidines that are all amenable to labeling with the short-lived positron-emitter carbon-11 (t1/2 = 20.4 min) or, potentially in some cases, with longer-lived fluorine-18 (t1/2 = 109.8 min).18 We focused on preparing N’-naphthyl and bicyclic analogs of CNS1261 (1). From this set of compounds, nearly half exhibited high binding to the PCP binding site at micromolar concentration, and a few exhibited binding affinities in the nanomolar range on par with that of 1.
Chart 1.
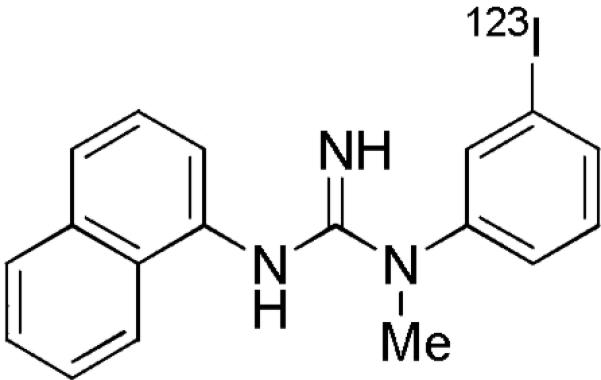
Structure of [123I]CNS1261 ([123I]1).
All the new N-aryl-N’-phenyl-N’-methylguanidines were prepared by reactions between arylamine hydrochlorides and aryl cyanamides by one of two methods (Scheme 1).19–21 In method 1, an N-methyl-N-aryl cyanamide was heated to 130 °C with the requisite arylamine hydrochloride salt for 21 h in toluene. In method 2 an N-methyl arylamine was used instead of an N-methyl-N-aryl cyanamide in order to reduce steric bulk at the cyanamide. This change led to increased yield, as demonstrated by the synthesis of 20, where Methods 1 and 2 gave 14 and 35% isolated yields, respectively. When the N’-methylguanidines could not be made by Method 1, we used Method 2 (see supplementary data).
Scheme 1.
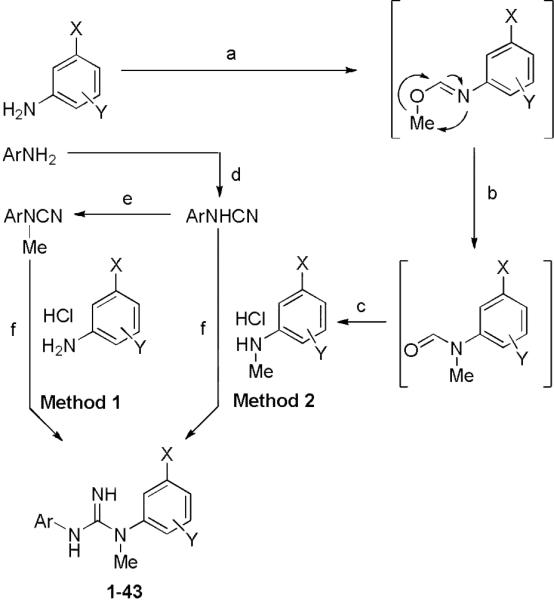
Syntheses of N-aryl-N’-(3-substituted)-N’-methylphenylguanidines. Reagents and conditions: (a) 1.5 eq. trimethylorthoformate, cat. H2SO4, 120 °C, 2 h; (b) 170 °C, 30 min; (c) 1. 10% aq. HCl, reflux, 3 h; 2. Et2O/HCl; (d) 0.67 eq. CNBr, Et2O, reflux, 24 h; (e) 1. 1.5 eq. NaH, THF, reflux, 3 h; 2. 1.5 eq. MeI, r.t., overnight; (f) toluene, 130 °C, 21 h.
The requisite N-aryl cyanamides were synthesized by refluxing cyanogen bromide (5 M in acetonitrile) with the appropriate arylamine in diethyl ether. The purified N-aryl cyanamides were used either directly or converted into their respective N-methyl-N-aryl cyanamides by treatment with sodium hydride and then iodomethane in tetrahydrofuran.21
Ligands were screened for affinity at the PCP site of the NMDA receptor in rat brain membrane suspensions with an assay using [3H]TCP as radioligand (Caliper Life Sciences). Initially ligands were tested at 1.0 μM concentration. Usually, if the inhibition of [3H]TCP binding exceeded 80%, a Ki value was determined, also with [3H]TCP as radioligand. In this assay, 1 inhibited [3H]TCP binding fully at 1.0 μM concentration with a Ki of 31.9 nM (Table 1). This value is appreciably higher than that reported when using [3H]MK801 as radioligand (4.65 nM).14,15
Table 1.
Inhibitions of [3H]TCP binding (%) at the PCP site of the NMDA receptor, binding affinities (Ki), and clogD values for 1 and 2.
Inhibition at 1.0 μM concentration (mean from two experiments).
Mean from two experiments.
Calculated using ACD Labs program.
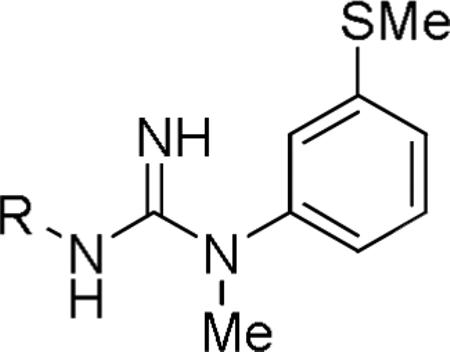
Initially, we focused on preparing non-iodinated derivatives of 1 where the metabolically labile 3-iodo substituent9,15 was replaced with another. We noted that a few such compounds have been reported previously, where the substituent at 3 position is Me, Et, NO2, NH2, or azido.20 However, only the 3-ethyl analog had shown a quite high IC50 value (36 nM) against [3H]MK801 binding to NMDA receptors. We first considered methylthio as an alternative to an iodo substituent because of its similar size and polarizability. The direct methylthio analog (2) was synthesized and was found to inhibit 93.1% of [3H]TCP binding at micromolar concentration and to exhibit a Ki value of 39.8 nM, which is very similar to that of 1 (31.9 nM) in the same assay (Table 1). Thus, a methylthio substituent was well tolerated in place of iodine.
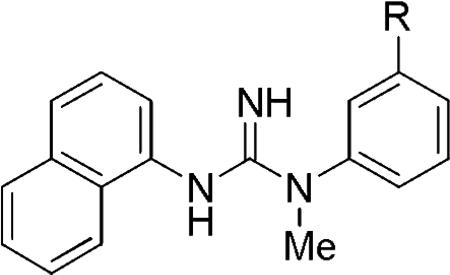
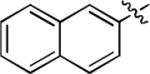
Subsequently, the methylthio group was retained while changes were made to the N’-(1-naphthyl) group of 2. First, the 1-naphthyl group was changed to a 2-naphthyl group, as in 3, but this change was highly detrimental to binding affinity (Table 2).
Table 2.
Inhibitions of [3H]TCP binding at the PCP site of the NMDA receptor (%), binding affinities (Ki), and clogD values for N-aryl-N’-(3-methylthiophenyl)-N’-methylguanidines.
| ||||
|---|---|---|---|---|
| Ligand | R | Inhibition (%)a | [3H]TCP Ki (nM)b | cLogDc |
| 3 |
|
3.4 | 1.86 | |
| 4 |
|
25.3 | 2.00 | |
| 5 |
|
80.7 | >1000 | 1.92 |
| 6 |
|
91.2 | 110 | 1.82 |
| 7 |
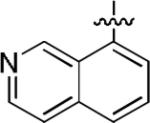
|
73.8 | 2.04 | |
| 8 |
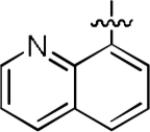
|
72.2 | 2.40 | |
| 9 |
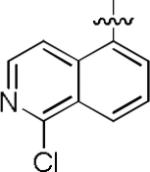
|
91.7 | 540 | 2.51 |
| 10 |
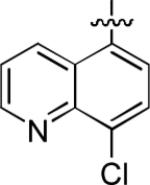
|
8.6 | 2.31 | |
| 11 |
|
14.4 | 2.69 | |
| 12 |
|
69.7 | 2.92 | |
| 13 |
|
23.7 | 1.87 | |
At 1.0 μM concentration. Mean from two experiments.
Binding affinities were measured for compounds whose inhibitions were ≥ 80%. Mean from two experiments.
Calculated with ACD Labs program.
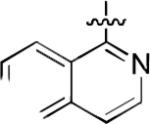
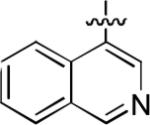
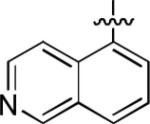
We next explored whether other bicyclic aryl groups, namely an isoquinolinyl or a quinolinyl group, would be advantageous over the 1-naphthyl group in 2. Five compounds that were formally isosteric with 2 were prepared in which the position of the benzopyridinyl nitrogen atom was varied (4–8) (Table 2). Compound 4 showed weaker inhibition than 2. Compounds 5–8, and especially 5 and 6, showed high inhibition of [3H]TCP binding at micromolar concentrations. The Ki values of 5 and 6 were however found to be much higher than that of 2 (Table 2).
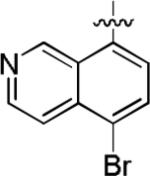
We noted that the computed lipophilicities of compounds 3−8 (Table 2) were lower than those of 1 and 2 (Table 1). Therefore, we attempted to compensate for this lower lipophilicity by adding a halogen to the heterobicyclic rings, as in compounds 9– 11. Insertion of chlorine in ortho position to the isoquinolinyl nitrogen of 6, as in compound 9, retained some affinity, but insertion of halogen (Cl or Br) into the benzenoid ring, as in 10 and 11, was highly detrimental to affinity (Table 2).
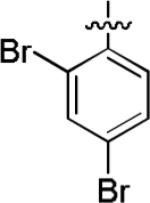
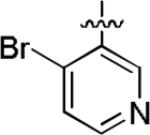
Among compounds 1−8 no relationship between affinity and lipophilicity emerged. Therefore, we next explored replacing the naphthyl substituent in 2 with monocyclic aryl substituents. The compound 12 having a 2,4-dibromophenyl substituent, intended to mimic the bulkiness of the naphthyl group in 2, displayed 69.7% inhibition of [3H]TCP binding at micromolar concentration (Table 2), but clearly had lower affinity than 2. When the naphthyl substituent was replaced with a 4-bromopyridin-3-yl substituent, as in 13, binding affinity was even lower (23.7% inhibition; Table 2). Therefore no improvement over the 1-naphthyl ring of 2 was found.
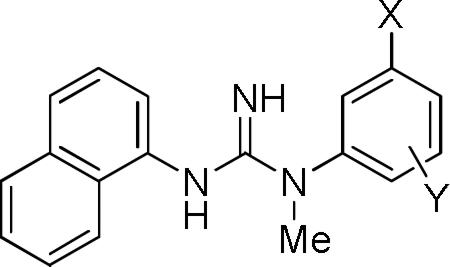
Our attention returned to varying substitution on the N’-phenyl ring of 1. Following on from the methylthio group replacement described earlier, we explored the effect of introducing various other substituents at the 3-position.20 Commercially available 3-substituted anilines were used to prepare compounds 14–33 (Table 3). From the assay data, it is apparent that many other substitutions are well tolerated at the 3-position. At micromolar concentration, over 60% of the monosubstituted compounds displayed appreciable (> 50%) inhibition of [3H]TCP binding at the PCP site of the NMDA receptor (15–17, 19–26, and 28–29). Lower affinity was observed in compounds with bulky aromatic (30–33) or small alcoholic substituents (18, 27). Also small and highly electronegative substituents were not well tolerated as shown in the halo derivatives (14–16 and 1); % inhibition/affinity increased across this series with halogen size
Table 3.
Inhibitions of [3H]TCP binding at the PCP site of the NMDA receptor (%), binding affinities (Ki), and clogD values for N-1-naphthyl-N’-(3-substituted-phenyl)-N’-methylguanidines and compound 30.
| |||||
|---|---|---|---|---|---|
| Ligand | X | Y | Inhibition (%)a | [3H]TCP Ki (nM)b | cLogDc |
| 14 | F | H | 36.9 | 1.80 | |
| 15 | Cl | H | 69.2 | 2.23 | |
| 16 | Br | H | 104 | 53.7 | 2.40 |
| 17 | CN | H | 62.9 | 1.13 | |
| 18 | OH | H | 11.9 | 0.83 | |
| 19 | N(Me)2 | H | 94.6 | 36.7 | 2.07 |
| 20 | CF3 | H | 107 | 18.3 | 2.23 |
| 21 | OCF3 | H | 79.2 | 2.49 | |
| 22 | SCF3 | H | 80.0 | 447 | 2.98 |
| 23 | OCF2H | H | 66.4 | 1.80 | |
| 24 | Ac | H | 82.3 | 152 | 1.07 |
| 25 | CH=CH2 | H | 73.9 | 2.38 | |
| 26 | CH≡CH | H | 74.3 | 1.61 | |
| 27 | CH2OH | H | 20.0 | 0.81 | |
| 28 | SCH2CH2F | H | 76.7 | 2.40 | |
| 29 | SCH=CH2 | H | 88.4 | 2.04 | |
| 30 | d | 55.0 | 4.15 | ||
| 31 | OPh | H | 30.0 | 3.33 | |
| 32 | Bn | H | 16.2 | 3.52 | |
| 33 | OBn | H | –2.5 | 3.52 | |
| 34 | Cl | 2-F | 84.0 | 701 | 2.20 |
| 35 | Cl | 4-F | 65.8 | 2.35 | |
| 36 | Cl | 5-F | 86.8 | 78.4 | 2.06 |
| 37 | Cl | 2,6-F | 37.6 | 2.33 | |
| 38 | Br | 4-F | 56.5 | 2.29 | |
| 39 | Br | 6-F | 13.8 | 2.13 | |
| 40 | CN | 4-F | 62.1 | 1.02 | |
| 41 | Me | 4-F | 46.5 | 1.77 | |
| 42 | OMe | 5-F | 32.9 | 1.48 | |
| 43 | NO2 | 4-F | 0.7 | 1.35 | |
At 1.0 μM concentration. Mean from two experiments.
Binding affinities were measured for compounds having inhibition ≥ 80%, except for 29. Mean from two experiments.
Calculated with ACD Labs program.
This compound has 2-naphthyl as the N’-aryl group.
From the Ki data, the preferred substituent at the 3-position is a CF3 group (20, 18.3 nM), which gave a nearly two-fold increase in binding affinity over 1. The Ki for the compound with an N(Me)2 substituent (19, 36.7 nM) is on par with that of both 1 and its methylthio analogue, 2. Compounds with other substituents, such as Br (16, 53.7 nM), Ac (24, 152 nM), and SCF3 (22, 447 nM), were less well tolerated (Table 3).
We were further curious to see the effect of introducing fluorine as an extra substituent on the phenyl ring, primarily because this might be a future site for labeling with fluorine-18 to give a PET radioligand. Moreover, fluorine-18 attached to an aryl carbon would be expected to be resistant to undesirable defluorination in vivo.22 As already noted, direct replacement of the 3-iodo substituent of 1 with a much smaller and highly electronegative 3-fluoro substituent, as in 14, decreased affinity substantially (Table 3). Nonetheless, because replacement of an aryl H with F causes little steric perturbation, we expected that some aryl fluoro derivatives might display binding affinities similar to those of their non-fluorinated counterparts.
A single fluoro substituent was added to the 3-chlorophenyl ring of 15, as in compounds 34–36 (Table 3). No appreciable effect on inhibition of binding was seen in 35, but beneficial effects were seen in the isomers, 34 and 36, which were then found to display Ki values of 701 and 78.4 nM, respectively. When fluoro substituents were added to 15 in both the 2- and 6-positions, as in 37, binding inhibition was greatly lowered. Addition of a single 4- or 6-fluoro substituent to the 3-bromo compound 16, as in 38 and 39, respectively, lowered affinity, whereas addition of a single 4-fluoro substituent to the 3-cyano compound 17, as in 40, left affinity unchanged. Finally, the prepared 3-methyl-4-fluoro and 3-methoxy-5-fluoro compounds, 41 and 42, respectively, displayed moderate binding inhibition. The 4-fluoro-3-nitro compound, 43, displayed negligible binding affinity.
Lipophilicity is an important parameter influencing many properties of PET radioligands, including plasma protein binding,23 and brain entry after intravenous administration.24 High plasma free fraction and moderate lipophilicity tend to promote good radioligand brain entry and brain uptake.24 Compound 1 is already known to readily enter animal and human brain after intravenous administration.9,15 Our computed logD value for 1 is 2.76 and quite close to the measured14 value of 2.19. Most of the prepared compounds, because of their close structural similarities to 1, have quite similar computed lipophilicity (Tables 1−3), and therefore they would be expected to be brain-penetrant after intravenous administration.
In conclusion, changes in the naphthyl group of 1 did not improve affinity towards the PCP site of the NMDA receptor. However, substitutions at the 3-position of the N’-aryl group of 1 led to new high-affinity ligands. Thus, two ligands, 2 (Ki, 39.8 nM) and 19 (Ki, 36.7 nM) were found to have affinities to the PCP site of the open NMDA receptor that are similar to that of 1 (Ki = 31.9 nM), and one was found with higher affinity (20, Ki = 18.3 nM). Moreover, the clogD values of 2, 19 and 20 are 2.49, 2.07 and 2.23, respectively, and notionally within the preferred range for PET radioligands for brain imaging. We have previously demonstrated that the labeling of 1 in its N-methyl group with carbon-11 is readily accomplished by treatment of desmethyl-1 with [11C]iodomethane,25 a technique that would be applicable to labeling any of the series of compounds reported here. Compounds 2, 19 and 20 therefore seem particularly attractive candidates for future labeling with carbon-11 for evaluation as PET radioligands for the open NMDA receptor.
Supplementary Material
Acknowledgements
This study was supported by the Intramural Research Program of the National Institutes of Health (NIH), specifically the National Institute of Mental Health (NIMH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Experimental procedures and spectroscopic characterization of all new compounds are available free of charge in the online version.
References and notes
- 1.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Pharmacol. Rev. 2010;62:405. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lau CG, Zukin RS. Nat. Rev. Neurosci. 2007;8:413. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 3.Paoletti P, Bellone C, Zhou Q. Nat. Rev. Neurosci. 2013;14:383. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- 4.Stone JM. Curr. Pharm. Design. 2009;15:2594. doi: 10.2174/138161209788957438. [DOI] [PubMed] [Google Scholar]
- 5.Limapichat W, Yu WY, Branigan E, Lester HA, Dougherty DA. ACS Chem. Neurosci. 2013;4:255. doi: 10.1021/cn300180a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamakura T, Shimoji K. Progr. Neurobiol. 1999;59:279. doi: 10.1016/s0301-0082(99)00007-6. [DOI] [PubMed] [Google Scholar]
- 7.Dumont F, Sultana A, Waterhouse RN. Bioorg. Med. Chem. Lett. 2002;12:1583. doi: 10.1016/s0960-894x(02)00235-4. [DOI] [PubMed] [Google Scholar]
- 8.Sattler R, Xiang ZG, Lu WY, Hafner M, MacDonald JF, Tymianski M. Science. 1999;284:1845. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- 9.Knol RJJ, de Bruin K, van Eck-Smit BLF, Pimlott S, Wyper DJ, Booij J. Synapse. 2009;67:557. doi: 10.1002/syn.20629. [DOI] [PubMed] [Google Scholar]
- 10.Kiesewetter DO, Finn RD, Rice KC, Monn JA. Int. J. Radiat. Appl. Instrum. [A] 1990;41:139. doi: 10.1016/0883-2889(90)90098-2. [DOI] [PubMed] [Google Scholar]
- 11.Samnick S, Ametamey S, Leenders KL, Vontobel P, Quack G, Parsons CG, Neu H, Schubiger PA. Nucl. Med. Biol. 1998;25:323. doi: 10.1016/s0969-8051(98)00003-1. [DOI] [PubMed] [Google Scholar]
- 12.Shiue CY, Vallabhahosula S, Wolf AP, Dewey SL, Fowler JS, Schlyer DJ, Arnett CD, Zhuo YG. Nucl. Med. Biol. 1997;24:145. doi: 10.1016/s0969-8051(96)00186-2. [DOI] [PubMed] [Google Scholar]
- 13.Haradahira T, Sasaki S, Maeda M, Kobayashi K, Inoue O, Tomita U, Nishikawa T, Suzuki K. J. Labelled Compd. Radiopharm. 1998;41:843. [Google Scholar]
- 14.Robins EG, Zhao Y, Khan I, Wilson A, Luthra SK, Årstad E. Bioorg. Med. Chem. Lett. 2010;20:1749. doi: 10.1016/j.bmcl.2010.01.052. [DOI] [PubMed] [Google Scholar]
- 15.Owens J, Tebbutt AA, McGregor AL, Kodama K, Magar SS, Perlman ME, Robins DJ, Durant GJ, McCulloch Nucl. Med. Biol. 2000;27:557. doi: 10.1016/s0969-8051(00)00102-5. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, Robins E, Turton D, Brady F, Luthra SK, Årstad E. J. Labelled Compd. Radiopharm. 2006;49:163. [Google Scholar]
- 17.Sorbio F, Gilbert G, Perrio C, Barré L, Debruyne D. Mini-Rev. Med. Chem. 2010;10:870. doi: 10.2174/138955710791608299. [DOI] [PubMed] [Google Scholar]
- 18.Cai L, Lu S, Pike VW. Eur. J. Org. Chem. 2008:2853. [Google Scholar]
- 19.Padmanabhan S, Reddy LN, Durant GJ. Synth. Commun. 1997;27:691. [Google Scholar]
- 20.Reddy NL, Hu LY, Cotter RE, Fischer JB, Wong WJ, McBurney RN, Weber E, Holmes DL, Wong ST, Prasad R, Keana JFW. J. Med. Chem. 1994;37:260. doi: 10.1021/jm00028a009. [DOI] [PubMed] [Google Scholar]
- 21.Padmanabhan S, Perlman ME, Zhang L, Moore D, Zhou D, Fischer JB, Durant GJ, McBurney RN. Bioorg. Med. Chem. Lett. 2001;11:501. doi: 10.1016/s0960-894x(00)00695-8. [DOI] [PubMed] [Google Scholar]
- 22.Pike VW. Trends Pharmacol. Sci. 2009;30:431. doi: 10.1016/j.tips.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zoghbi SS, Anderson KB, Jenko KJ, Luckenbaugh DA, Innis RB, Pike VW. J. Pharm. Sci. 2012;101:1028. doi: 10.1002/jps.22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laruelle M, Slifstein M, Huang Y. Mol. Imaging Biol. 2003;5:363. doi: 10.1016/j.mibio.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Naumiec G, Cai L, Morse CL, Pike VW. Abs. 244th Nat. Meeting Am. Chem. Soc. MEDI-165. 2012 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.