

Abstract
Iron chelators, through their capacity to modulate the iron concentration in cells, are promising molecules for cancer chemotherapy. Chelators with high lipophilicity easily enter into cells and deplete the iron intracellular pool. Consequently, iron-dependent enzymes, such as ribonucleotide reductase, which is over-expressed in cancer cells, become nonfunctional. A series of calix[4]arene derivatives substituted at the lower rim by ICL670, a strong FeIII chelator, have been synthesized. Physicochemical properties and antiproliferative, angiogenesis, and tumorigenesis effects of two calix[4]arenes mono- (5a) or disubstituted (5b) with ICL670 have been studied. These compounds form metal complexes in a ratio of one to two ligands per FeIII atom as shown by combined analyses of the protometric titration curves and ESIMS spectra. The grafting of an ICL670 group on a calix[4]arene core does not significantly alter the acid–base properties, but improves the iron-chelating and lipophilicity properties. The best antiproliferative and anti-angiogenic results were obtained with calix[4]arene ligand 5a, which possesses the highest corresponding properties. Analyses of molecular dynamics simulations performed on the two calix[4]arenes provide three-dimensional structures of the complexes and proved 5a to be the most stable upon complexation.
Keywords: calixarenes, chelates, iron, lipophilicity, molecular dynamics
Introduction
Iron chelators such as deferoxamine, deferiprone, or deferasirox/ICL670, initially developed for iron-overload therapy, have shown antiproliferative effects.[1] The chemical structure of these iron chelators influences their capacity to modulate iron uptake in cells. Indeed, deficiency of labile iron in the cytoplasm results in inhibition of deoxyribonucleotide biosynthesis, which is the limiting step in DNA synthesis, and subsequently leads to the inhibition of cell proliferation.[2] Ribonucleotide reductase (RR) is over-expressed in rapidly dividing cancer cells, and is critical for deoxyribonucleotide biosynthesis and cell division. RR is the main target of iron chelators and during the last two decades, a new generation of iron chelators has been synthesized and studied in cancer chemotherapy. Triapine,[3] o-trensox,[4] tachpyridine,[5] di-2-pyridyl thiosemicarbazones like Dp44mT,[6] and the pyridoxal isonicotinoyl hydrazone (PIH) family[7] currently appear to be the most promising molecules. In the case of PIH analogues, Richardson and Lovejoy identified a significant chelator structure–activity relationship dependent on lipophilicity.[8] Within a series or family of ligands, the most lipophilic compounds have generally the best iron-clearing efficiency. Moreover, lipophilicity has a profound effect on organ distribution of the chelators.[9] Chelators with a high lipophilicity can easily enter cells and subsequently deplete the iron intracellular pool, which is necessary for the RR catalytic activity.
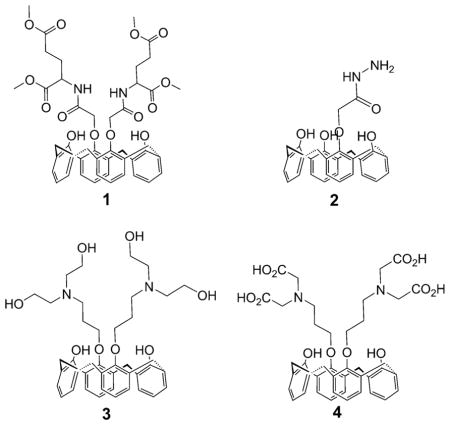
Owing to the development of supramolecular chemistry, a number of calixarene-based ligands have been extensively developed for their coordination properties,[10] in particular towards iron.[11] We have previously prepared new calix[4]arene compounds as selective cesium chelators.[12] In 2001, FeIII complexes of some amide-substituted calix[4]arenes were reported.[13] Later Rao et al. reported for the first time the synthesis and characterization of MnII, FeIII, CoII, NiII, CuII, and ZnII complexes of p-tert-butylcalix[4]arene-1,3-diacid derivatives.[14] More recently, Wang et al. described selective FeIII and CrIII recognition by fluorescent calix[4]arene sensors.[15] Many pharmacological properties have been described for calix[4]arenes including anticancer, antiviral, antibacterial, antifungal, and antithrombotic activities.[16] Sebti et al. have shown that a calix[4]-arene called GFA-116 is capable of selectively disrupting the binding of vascular endothelial growth factor (VEGF) to its receptor.[17] Consequently, the VEGF-dependent signaling is inhibited, which suppresses angiogenesis and tumorigenesis. We have shown recently that calix[4]arenes 1–4 exhibit interesting antiproliferative activities close to the ICL670 reference, and the correlation between the cytostatic effect and iron depletion was established clearly for tetra-acid 4.[18]
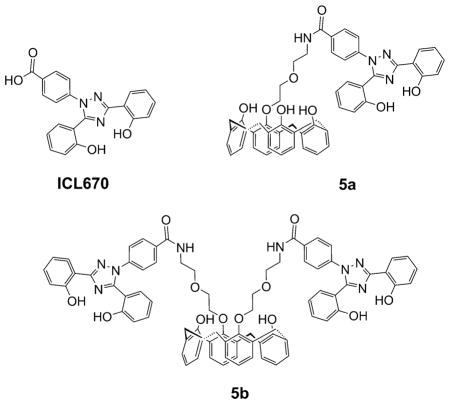
Herein, we report on the synthesis, physicochemical and molecular dynamics (MD) studies, as well as on the antiproliferative and antiangiogenic effects of new calix[4]arenes 5a,b substituted at the lower rim with ICL670, a strong FeIII complexant.
Results and Discussion
Ligand design and synthesis
The synthesis strategy for the new calix[4]arene derivatives 5 a,b starts from unsubstituted calix[4]arene 6 and iodo compound 7 to obtain the mono- and diaminocalix[4]arenes 9a,b (Scheme 1). The peptidic coupling of the 9a,b key intermediates with ICL670 leads to the corresponding calix[4]arenes 5 a,b in a cone conformation.
Scheme 1.

Synthesis of the cone calix[4]arene derivatives 5 a,b.
Calix[4]arenes 5a,b were synthesized in three steps starting from 6 and 7 (Scheme 1). Unsubstituted calix[4]arene 6 was first prepared according to a two-step synthesis developed by Gutsche et al.[19] from p-tert-butylphenol, whereas iodo compound 7 was synthesized as described previously.[20] Two main strategies are described to prepare monoalkylated calix[4]arene derivatives: either by a method of protection/deprotection[21] or by a selective approach.[22] In our case, the best result was obtained by using the CsF base (1.3 equiv.) in dimethylformamide (DMF) and monoalkylated calix[4]arene 8a in a cone conformation was isolated in a 62 % yield. The 1H NMR spectrum for 8a shows two singlet peaks at δ=9.05 (2H) and 9.68 ppm (1 H), which correspond to the phenolic hydroxyl protons and thereby confirm the monoalkylation. Calix[4]arene 8b was prepared in a 74 % yield by dialkylation of calix[4]arene 6 with 7 (4 equiv.) in the presence of K2CO3 in acetonitrile and heated at reflux for seven days (Scheme 1).
The alkylation selectively led to diametrically substituted calix[4]arene 8b in the cone conformation, as indicated by the 1H NMR spectrum. A typical “AB” pattern was observed for the methylene bridge Ar-CH2-Ar (Ar = aryl) protons (2J =12.9 Hz) at δ=3.06 ppm for the equatorial protons and δ=3.97 ppm for the axial protons. The treatment of phthalimidocalix[4]arenes 8 a,b with an excess amount of aqueous hydrazine in ethanol at reflux gave the corresponding aminocalix[4]arenes 9 a,b in the cone conformation in good yields (88–99 %; Scheme 1). Aminocalix[4]arenes 9 a,b were then coupled to the ICL670 moieties, subsequently synthesized in two steps according to Steinhauser et al.,[23] using 1-(3-dimethylaminopropyl)-3-ethyl-carboiimide/N-hydroxybenzotriazole (EDCI/HOBt) to afford amidocalix[4]arenes 5 a,b in moderate yields (37–51 %).
Analysis of the hydrophobicity of the ligands
The determination of the logarithm of the distribution coefficient (log D) was achieved by using HPLC for calix[4]arenes 5 a,b at pH 7.[24] In the case of ICL670, reported values for the logarithm of the partition coefficient P (log P) and log D are 3.8[25] and −0.1,[26] respectively. Compounds 5 a,b were found to be highly lipophilic with log D values of 4.59 (±0.06) and 3.49 (±0.05), respectively. The introduction of the calixarene backbone resulted in a significant increase of the lipophilicity for 5a and 5b relative to ICL670. The introduction of a second ICL670 substituent on 5b, in comparison with 5a, yielded a substantial reduction of the lipophilicity by about 12-fold. There is a clear relationship between the fact that chelators presenting a high lipophilicity can easily enter cells and deplete the intracellular iron that is necessary for the catalytic activity of RR. Compound 5a should present a better antiproliferative active profile than calix[4]arene 5b.
Metal-binding properties
The chelating abilities of ICL670, 5a, and 5b towards FeIII were estimated on the basis of the global formation constants of their complexes, which were determined by UV-visible spectrophotometric and potentiometric (Hyperquad program[27]) techniques. Because of the poor solubility of the chelators in an aqueous medium, all experiments were monitored in a water/DMSO media (v/v = 30:70; xDMSO = 0.5). The global stability constant βmlh of each ferric complex is defined by Equations (1) and (2):
| (1) |
| (2) |
in which m, l, and h are values in the general complex formula [MmLlHh]. M, L, and H correspond to the FeIII metal ion, the ligand (L1=ICL670, L2=5a, L3=5b), and the protons, respectively. For the sake of clarity, charges are omitted. The HYSS computer program[27] was used to obtain the species distribution curves.
Analysis of neutralization curves of the ligand solution shows that ICL670 and 5a have three sites able to be protonated in the 2.5–13 pH range, whereas 5b has five such sites. In the case of ICL670, pKa1 is ascribed to the deprotonation of the carboxylic functions, whereas pKa2 and pKa3 correspond to the deprotonation of the two phenolic groups. Although the two OH groups are structurally equivalent, a difference of two units is observed between pKa2 and pKa3. Such a difference has been observed already by Steinhauser et al.,[23] and can be explained by a hydrogen bond between the proton of one phenolic group and a nitrogen atom of the triazole moiety making the phenolic group more basic. Nevertheless, it appears highly probable that the two hydroxyl groups are deprotonated simultaneously. The observed pKa values thus represent averages of the individual (microscopic) acidity constants. On the other hand, pKa2 and pKa3 of ICL670 and 5a show that the grafting of the ICL670 group on a calix[4]arene core does not significantly alter the acid–base properties. In the case of 5b, deprotonation of the ICL670 phenolic groups corresponds to values in the range of pKa2 to pKa5. Finally, the pKa1 value for 5a and 5b is ascribed to the deprotonation of the phenolic groups of the calix[4]arene core. This assignation is corroborated by the study of the acid–base properties of calix[4]arene 6 in solution, which shows that the latter possesses a site able to be easily deprotonated with a pKa value of 4.6. The relatively low value of pKa1 relative to that of an isolated phenolic function is indicative of strong cooperation between all the phenolic groups of the calix[4]arene crown by means of hydrogen bonding, which stabilizes the negative charge of the phenolate moiety. The higher pKa1 values for 5a and 5b than calix[4]arene 6 are related to the decreasing number of free hydroxyl groups on the crown.[28]
The complexation study of L1 (ICL670), L2 (5a), and L3 (5b) with iron was then studied. In the case of Fe–L1 and Fe–L2 solutions, at the beginning of the titration (pH ≈ 2.5) FeIII is almost completely complexed as FeL1H−2 and FeL2H−3. For these species the formation constant was calculated on the basis of spectrophotometric titration monitored in the pH range 1 to 6.5 (see Figure SI1 in the Supporting Information). These data were then incorporated in the refinements of potentiometric curves to determine the formation constant of the whole species. In the pH range 1–3, complexation reactions induce a progressive change in the solution coloration from yellow to violet. In this pH range, a change in the solution absorbance induced by a pH increase reflects a single equilibrium between the free FeIII ion and the 1:1 species (corresponding to FeL1H−2, FeL2H−3). The electronic spectra of 1:1 species show similar features and are characterized by a single band centered at 510 nm and a molar extinction coefficient around 3200 mol−1L cm−1. This transition, corresponding mainly to a charge-transfer transition between the metal center and the deprotonated phenolic groups, is indicative of the binding of FeIII to ICL670 moieties. Above pH 4, the solutions turn orange, which results in the appearance of a new band at 400 nm. In this pH range, the spectral characteristics of the solution do not vary with the FeIII concentration, even when the amount of iron increases, thereby leading to a decrease of the [L]/[M] ratio below a value of 2. This indicates that the spectrum modifications of FeIII–L solutions cannot be ascribed to the formation of 1:2 species as postulated by Steinhauser et al.[23] This makes the spectrum more difficult to interpret and does not allow the calculation of stability constants for 1:2 species.
The protonation constants were determined from the refinement of neutralization curves. β11–2 for FeL1 and β11–3 for FeL2 were determined from spectrophotometric titrations and were used without further adjustment in the refinement of potentiometric titration. All the results are reported in Table 1. The corresponding species distribution curves are reported in Figure 1.
Table 1.
Logarithmic values of the overall protonation constants of the ligands (β01h) and of the stability constant of the FeIII complexes (βm1h).[a]
| Log βmlh | pKa | ICL670 [L1][b] | 5a [L2][b] | 5b [L3][b] |
|---|---|---|---|---|
| 0 1–1 | pKa1 | −5.56(5) 5.56 | −6.67(2) 6.67 | −9.88(3) 9.88 |
| 0 1–2 | pKa2 | −16.55(7) 10.99 | −17.12(2) 10.45 | −21.11(3) 11.23 |
| 0 1–3 | pKa3 | −29.53(7) 12.98 | −29.63(2) 12.51 | −33.21(3) 12.10 |
| 0 1–4 | pKa4 | – | – | −46.38(6) 13.17 |
| 0 1–5 | pKa5 | – | – | −59.85(4) 13.47 |
| 1 1–2 | – | −1.06(9)[c] | – | −2.35(8)[c] |
| 1 1–3 | – | −6.48(9) | −1.74(1)[c] | −5.08(6) |
| 1 2–5 | – | −13.72(9) | – | – |
| 1 2–6 | – | −20.35(8) | −19.23(8) | – |
The calculated uncertainties for log βm1h were determined on the basis of the standard deviation. Values in parentheses refer to estimated standard deviations for the last significant digit.
Italic numbers correspond to the pKa values.
Determined by potentiometric and spectrophotometric titrations; I =0.1 M (KCl); T =298 K, H2O/DMSO media (v/v 30:70).
Figure 1.

Distribution curves for the FeL systems (CL = 2.10−3 M; CM = 10−3 M): a) FeL1, b) FeL2, c) FeL3.
The stoichiometry of the complexes was confirmed by ESI-MS analysis of Fe–L solutions (spectra are reported in the Supporting Information). For the FeIII–L1 solution the spectrum recorded at pH 3 in the positive mode shows only 1:1 complex species detected as proton adducts at m/z 505 [Fe+L1+DMSO–2H]+, 523 [Fe+L1+DMSO+H2O–2H]+, and 583 [Fe+L1+2 DMSO–2H]+, which is compatible with the exclusive formation of FeL1H−2. The spectrum recorded at pH 9 in the negative mode corroborates the addition of a second ligand to the metal center and the formation of FeL12H−6 detected at m/z 798 [Fe+2L1–4H]−. For Fe–L2 solutions, the ESI-MS analysis shows a similar complexation scheme to the Fe–L1 system. The complex species detected at pH 4 at m/z 998.2 [Fe+L2+DMSO–2H]+ correspond to the formation of FeL2H−3, whereas those detected at pH 9 at m/z 1787.5 [Fe+2L2–2H]+ are ascribed to FeL22H−6 (Figure 1b). In the case of Fe–L3 solutions, only 1:1 species have been detected at m/z 1384 [Fe+L3+Na–3H]+ and 1406 [Fe+L3+2 Na–4H]+, which correspond to the formation of FeL3H−3.
In designing iron chelators for clinical applications, metal selectivity and ligand–metal complex stability are of paramount importance. The most suitable way to compare the FeIII chelating ability between ligands 5a, 5b, and ICL670 is to determine pFe, since, unlike stability constants, pFe takes into account the effects of ligand basicity, ligand protonation, and metal hydrolysis as well as differences in metal–ligand stoichiometries. pFe is defined as the negative logarithm of the free FeIII concentration in solution under defined conditions. The pFe was derived from ligand protonation and Fe-complex formation constants. For clinically relevant conditions, pFe values are typically calculated in pure water at pH 7.45 for total ligand and Fe concentrations equal to 10−5 and 10−6M, respectively. Under our conditions, taking into account the extended pH range due to DMSO, the free FeIII concentration was calculated at pH 9.16 (see the Experimental Section). The pFe values are 23.12, 26.44, and 23.23 for Fe–ICL670, Fe–5a, and Fe–5b, respectively. The ICL670 pFe value determined in this study is close to the one reported in the literature (pFe =23.5),[29] which validates our calculation procedure for water-insoluble systems. The pFe values determined in this study for ICL670-based calix[4]arene ligands (pFe >20) met the criteria for application as iron chelation therapy agents.[30] The stability of [FeL] at physiological pH decreases in the order ICL670 <5b <5a. The higher Fe-chelating ability at physiological pH of ligand 5a relative to 5b is probably due to the coordination of two ICL670 moieties, which leads to a more stable bis-tridentate structure similar to the one published in the case of ICL670.[23]
Force-field development for molecular dynamics simulations of the iron complexes
Efforts to obtain crystalline samples of the ferric complexes for X-ray diffraction experiments have been unsuccessful to date. Consequently, it was decided that MD studies would be performed to provide further insight into both of the ferric complex structures. In this study, the four phenolic ICL670 groups (corresponding to pKa2 to pKa5) of the FeIII–calix[4]arene 5a and 5b complexes were deprotonated, whereas the phenolic groups of the calix[4]arene crown (pKa1) were conserved since they are not involved in the iron complexation. The resulting total charge of each complex is negative (FeIII–4H =−1). Consequently, three molecular complexes have been studied by means of quantum chemistry (QC) and/or MD methods: the complex composed of two ICL670 molecules and FeIII, abbreviated as [Fe–ICL670]−; the complex composed of two 5a molecules and FeIII, abbreviated as [Fe–5a]−; and the complex composed of one 5b molecule and FeIII, abbreviated as [Fe–5b]−. The following approach was used:
The complexation sites in 5a and 5b were assumed to adopt a similar conformation to that observed in the octahedral [Fe–ICL670]− complex, therefore the [Fe–5a]− and [Fe–5b]− molecular systems were built accordingly.
[Fe–ICL670]− with a covalently bound FeIII was used for parameterization of iron-related force-field parameters.
MD simulations of complexes 5a and 5b with bound and nonbound iron were performed in explicit water.
The geometry of [Fe–ICL670]− was taken from the Cambridge Structural Database (CSD)[31] under the SAJFAL reference.[23] In this study, two empirical force fields for the FeIII-based complexes, namely, “q4md-FeF” and “q4md-FeB”, were developed in association with the Amber99SB force field,[32] which correspond to approaches in which the iron is nonbonded and bonded, respectively. Atomic charges were derived at the B3LYP/6-31G* level of theory using both the RESP and ESP methods using the latest version of the R.E.D. IV program available through “R.E.D. Server Development”[33] (see the Supporting Information for additional details about the charge derivation procedures). ESP and RESP charge values embedded in force-field libraries, and DFT-optimized geometries as well as the molecular fragments required to build the FeIII complexes in this study were submitted to the R.E.DD.B. database. The corresponding projects are available under the “F-88” (ESP charges) and “F-89” (RESP charges) R.E.DD.B. codes.[34] The method developed by Seminario[35] was used to extract missing bond and angle force constants.
Complex [Fe–ICL670]−
We performed 50 ns MD simulations using the q4md-FeB and q4md-FeF force fields. In both cases, the molecular system remained quite rigid with an RMSD mean value of (0.84 ±0.17) and (0.86 ±0.22) Å for q4md-FeB and q4md-FeF, respectively, using the heavy atoms only and with the crystallographic structure taken as the reference. These values indicate that both force fields are suitable to study the FeIII–5a and –5b complexes.
The [Fe–5a]− and [Fe–5b]− systems
Owing to the lack of any experimental three-dimensional structure of 5a and/or 5b complexed with FeIII, preliminary MD simulations were performed with the q4md-FeB force field to get more insight into a consistent structure. Hence, for each complex, we performed a series of MD simulations using different conditions accounting for 1 μs of simulation consisting of 30 independent trajectories. Analyses of the different trajectories for [Fe–5b]− indicated that, after a few picoseconds, the structure converges to a stable conformation. Superimposition of the different structures obtained after each independent MD run shows that they belong to the same family of conformation. A representative structure of this family in represented in Figure 2a. RMSD analyses performed for the [Fe–5a]− system with the q4md-FeB and q4md-FeF force fields indicate that, contrary to the [Fe–5b]− system, [Fe–5a]− does not converge towards a unique family of conformations (see Figure SI7 in the Supporting Information). Indeed, the [Fe–5a]− complex is composed of two calix[4]arene cavities, which, opposed to those in [Fe–5b]−, are not constrained and hence fluctuate around the Fe(ICL670)2 center. The superimposition of 30 structures randomly extracted from six 50 ns MD simulations is represented in Figure 2b. The Fe(ICL670)2 center fluctuates a bit more than that in the [Fe–5b]− system but remains nevertheless rigid relative to the calix[4]arene part of the complex. Many stable structures of the [Fe–5a]− system are observed because the two calix[4]arene cavities can adopt different stable positions and yet share a similar Fe(ICL670)2 geometry.
Figure 2.

Representative structure of the a) [Fe–5b]− and b) [Fe–5a]− complex obtained after MD simulations using the q4md-FeB force field. Similar geometries were obtained with the q4md-FeF force field after a B3LYP/6-31G* geometry optimization.
The nonbonded model demonstrated a robust performance, as no disruption of the coordination center and no significant conformational changes were observed throughout the simulation, neither for [Fe–5b]− nor for [Fe–5a]−. The obtained structures globally remained in the same conformation, which makes us confident in the reliability of the proposed [Fe–5b]− structure (its Cartesian coordinates are provided in the Supporting Information). The [Fe–5b]− and [Fe–5a] −− complexes optimize their structure by forming stabilizing intramolecular interactions between aromatic cycles of the ICL670 moiety and those of the calix[4]arene(s).[36] The calix[4]arene part of the complex remains in a cone conformation throughout the simulation, but is significantly distorted from its C4 symmetry. The cone conformation, however, adopts an approximate C2 symmetry. The rings bound to the ligands are more vertical with respect to the cone axis. Relative to its crystallographic state, the [Fe(ICL670)2]− part of the two calix[4]arene complexes is more distorted as demonstrated in Figure 3. Because of smaller constraints, the Fe(ICL670)2 center is able to adopt a conformation closer to its “native” state in the [Fe–5a]− complex.
Figure 3.

Superimposition of the Fe(ICL670)2 moiety of the [Fe–5a]− structure (blue) and of the [Fe–5b] − structure (green) with the crystallographic structure of [Fe–ICL670]− (pink).
In the [Fe–ICL670] − complex, aromatic intramolecular interactions are weak and two sandwich interactions between the two ICL670 fragments are observed. When grafted on the calix[4]arene scaffold, aromatic rings of the ICL670 moieties are able to interact with the newly added phenolic rings to form T-shaped aromatic interactions, thus stabilizing the entire system. Even though one of the ICL670 fragments moves around the Fe–N pseudoaxis, the octahedral conformation is kept as shown in Table SI2 (see the Supporting Information), which compares the distances and angles associated with the position of the FeIII ion. It has to be stressed that, independently of the system, the octahedral conformation is always kept during any MD simulation performed with the q4md-FeF force field.
Binding energies between FeIII and the ligands were evaluated using the MM-GBSA method[37] (see Table SI3 in the Supporting Information). The binding energy ΔH(GBtot : total energy from generalized Born implicit solvation) favors [Fe–5a]−, which is in agreement both with experimental results and with the better flexibility of this molecular system to rearrange around the ion binding site.
Antiproliferative effects in the human hepatocarcinoma HepaRG cell cultures
In the proliferating HepaRG cells, a dose-dependent decrease of the cell viability, measured by the succinate dehydrogenase (SDH) activity (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay), was observed after a 72 h cell treatment in the presence of increasing concentrations of the L1, L2, and L3 ligands (see Figure SI8 in the Supporting Information, 0 to 400 μM). As deduced from the dose–effects curves, compound 5a is cytostatic for concentrations lower than 10 μM, whereas it induces a cytotoxic effect, associated with membrane damages and lactate dehydrogenase (LDH) leakage for concentrations higher than 10 μM, which suggests a narrow therapeutic index. These two effects were partly reversed in the presence of exogenous iron. At 10 μM of compound 5a, SDH and LDH values were 6 and 105 % relative to the control (Table 2), respectively. The decrease in cell viability for chelator concentrations higher than 100 μM was associated with LDH leakage in the cell supernatants and was probably due to the cytotoxic effects of both the chelators and DMSO. In this range of concentrations, the addition of 20 μM of exogenous FeIII was ineffective in reversing the cytotoxic effect of the various compounds. Experiments of membrane diffusion were carried out on HepaRG cells that had been incubated with 5a and 5b. After cellular lysis, the two ligands were found in the intracellular medium by LC–MS analyses. These antiproliferative-effect results are consistent with the fact that 5a possesses a better antiproliferative active profile due to its higher iron-chelating abilities and lipophilicity than those of ICL670 and 5b.
Table 2.
Biological effect of a 72 h treatment of HepaRG cells with ICL670 and calix[4]arenes 5a and 5b. SDH activity (MTT assay) expressed by IC50 [μM] is given as a measurement of cell viability and LDH leakage in the cell supernatant for compound concentrations of 10 and 100 μM was used as an index of membrane damage (cytotoxicity). Data expressed as a percentage of the control (absence of compound) are averaged over three independent experiments.
| Compound | SDH, IC50 without FeIII | SDH, IC50 with FeIII 20 μM | % LDH/control without FeIII[a] | % LDH/control with FeIII 20 μM[a] |
|---|---|---|---|---|
| 5a | 6 ±1 | 37 ±3 | 105 ±11 200 ±15 |
67 ±6 187 ±5 |
| 5b | 90 ±7 | 125 ±5 | 76 ±4 103 ±2 |
91 ±11 69 ±6 |
| ICL670 | 11 ±4 | 61 ±7 | 87 ±7 101 ±13 |
80 ±5 88 ±1 |
Compound concentrations of 10 and 100 μM
Ligand 5a and 5b effects on angiogenesis and on HUVEC and MDA-MB-231 cell proliferation
ICL670, 5a, and 5b (10−8 to 10−5M) angiogenesis activity was evaluated by testing human umbilical vein endothelial cell (HUVEC) migration and tube capillary formation. ICL670, 5a, and 5b (10−8 to 10−5M) effects on HUVEC and MDA-MB-231 cell survival and proliferation were determined by using the BrdU assay as described in the Experimental Section. The relative cell proliferation and survival in the presence of ICL670, 5a, or 5b was normalized to the untreated controls after background subtraction. No statistical differences were found between the control and ICL670 for HUVEC or MDA-MB231 cell proliferation and survival. It was found that, after 48 h of treatment, 5a at 10−7 to 10−5M decreased HUVEC survival significantly by 28 % for 10−7 M (p: statistical significance from Student’s test <0.05), 56 % for 10−6 M (p <0.05), and 65 % for 10−5 M (p <0.001) versus the control (data shown in the Supporting Information). For MDA-MB-231 survival, 5a at 10−6 to 10−5 M decreased cell survival significantly by 51 % for 10−6M (p <0.001) and 58 % for 10−5M (p <0.001) versus the control. Ligand 5a at 10−7 to 10−5 M reduced the HUVEC proliferation significantly by 46 % for 10−7M (p <0.05), 51 % for 10−6M (p < 0.001), and 74 % for 10−5M (p <0.001) versus the control. For the MDA-MB-231 cell proliferation, 5a showed close results: from 10−7 to 10−5M cell proliferation decreased by 33 (p < 0.01), 62 (p <0.001), and 73 % (p <0.001), respectively, versus the control. ICL670 and 5b did not show any effect on HUVEC and MDA-MB-231 cell survival and proliferation. ICL670, 5a, and 5b effects on HUVEC migration have been evaluated using the wound-healing assay. As shown in Figure 4, the migration capacity of HUVECs was examined by counting cells that migrated through the wound after 16 h of treatment. Ligand 5a at 10−6 to 10−5M reduced HUVEC migration significantly by 51 (p <0.01) and 59 % (p <0.01) versus the control, whereas ICL670 and 5b did not show any effect on HUVEC migration versus the control.
Figure 4.

Effects of ICL670, 5a and 5b on HUVEC migration. 16 h after the wound was performed, cell migration was reduced by treatment with 5a at 10−5 and 10−6 M but not by treatment with ICL670 or 5b versus the control. Two controls were used: genistein (Gen) at 30 μg mL−1 as an inhibitor of HUVEC migration and EGF (50 ng mL−1) as an activator of HUVEC migration (**p <0.01, ***p <0.001 versus the control).
To examine whether ICL670, 5a, or 5b can induce morphogenetic changes resembling capillary-like structure formation, HUVECs were plated on Matrigel (Figure 5). After 16 h of treatment, 5a at 10−6 to 10−5M decreased the ability of HUVEC to form capillary tubes significantly by 42 (p <0.01) and 48 % (p < 0.01), respectively, versus the control. At the same concentrations, ICL670 and 5b did not show any effect on the HUVEC capillary tube formation versus the control. The most antiangiogenic efficient molecule tested in this study is 5a, which shows an inhibition of HUVEC and MDA-MB-231 survival and proliferation, as well as an inhibition of HUVEC migration and capillary-tube formation abilities.
Figure 5.

ICL670, 5a, and 5b effects on capillary-tube formation. The network length of the capillary-tube formation was quantified by using a CFM-S130T Digital Imaging System. 16 h after HUVECs were seeded directly on the Matrigel, they were treated with 10−6 or 10−5 M ICL670, 5a, or 5b. Ligand 5a but neither 5b nor ICL670 impaired endothelial cell differentiation. Untreated cells were used as a control (Ct). Two controls were used: genistein (30 μg mL−1) as an inhibitor of capillary-tube formation, and EGF (50 ng mL−1) as an activator (**p <0.01, ***p <0.001 versus the control).
Conclusion
Iron(III)-chelating ligands 5a and 5b have been synthesized by grafting one or two ICL670 moieties, respectively, onto a calix[4]arene core locked into a cone conformation. Ligand 5a is more lipophilic than ICL670 or 5b with a log D value of 4.59. The higher iron-chelating ability of the 5a ligand (pFe =26.4) relative to that of 5b (pFe =23.2) is likely due to the coordination of two ICL670 moieties, which lead to a more stable bis-tridentate structure similar to that published in the case of ICL670. These results are in agreement with the binding energies extracted from MD trajectories, the trend of which is as follows: [Fe–5a] −<[Fe–5b] −<[Fe–ICL670] −. The higher chelating ability of 5a along with its stability result from a better flexibility of the ICL670 moieties and from stabilizing intramolecular interactions with aromatic rings of the calix[4]arene. Furthermore, two force-field models for the [Fe–ICL670]−, [Fe–5a]−, and [Fe–5b]− molecular systems, namely, “q4md-FeB” and “q4md-FeF”, were developed for this study. Binding-energy calculations performed on the calix[4]arene complexes proved 5a to be the most stable by 7 kcal mol−1, likely because of the better flexibility and structural adaptation, hence demonstrating that the grafting of two ICL670 moieties onto a calix[4]arene does not afford any cooperativity effect upon iron binding.
The antiproliferative effects of these ligands on HepaRG cells show that 5a is more active than ICL670 and 5b is less active. The decrease in cell viability is reversed for 5a and ICL670 upon addition of 20 μM of exogenous FeIII. Only 5a shows interesting angiogenesis activity: at 10−6 to 10−5M it reduces HUVEC proliferation, migration, and capillary-tube formation abilities. In addition, MDA-MB-231 viability and proliferation was reduced by 5a at 10−6 to 10−5 M. These results indicate that 5a, relative to ICL670 and 5b, presents the best antitumoral profile due to its higher iron-chelating ability and lipophilicity.
Experimental Section
Chemicals and analysis
Column chromatography was performed on Kielselgel 60 (40–63 μm) ASTM (Merck). Reactions were analyzed on precoated silica gel 60 F254 plates (Merck) and the compounds were visualized with a UV lamp (254 nm) and phosphomolybdic acid in EtOH. Melting points were determined on a Stuart SMP3 apparatus and were reported uncorrected. Infrared measurements were performed on an FT/IR-4200 Jasco fitted with an ATR-golden gate allowing analysis of solids and liquids. 1H and 13C NMR spectra were recorded on a Bruker AC600, 500, or 300 spectrometer. Chemical shifts (δ) are reported in parts per million and the signals are quoted as s (singlet), br s (broad singlet), d (doublet), t (triplet), br t (broad triplet), q (quartet), or m (multiplet). Coupling constant (J) values are given in hertz. Signal assignment was made using HMBC, HSQC, COSY, and NOESY experiments when necessary. Mass spectra and high-resolution mass spectra (electrospray in positive mode, ESI +) were recorded on a Waters Q-TOF Ultima apparatus. Elemental analyses were performed on an elementary analyzer Flash EA 1112 series Thermo Finnigan. All commercially available products were used without further purification unless otherwise specified.
Synthesis
25-{1-[2-(2-Phthalimidoethoxy)ethoxy]}-26,27,28-trihydroxycalix[4]arene (8a)
A solution of iodide 7 (8.14 g, 23.6 mmol) in anhydrous DMF (25 mL) was added at RT under an argon atmosphere to a slurry of calix[4]arene 6 (5 g, 11.8 mmol) and CsF (2.32 g, 15.3 mmol) in anhydrous DMF (15 mL). The reaction was stirred at 50°C for 10 days. After cooling to RT, CH2Cl2 (300 mL) and water (300 mL) were added. The organic phase was collected and washed with water and brine. The organic phase was dried over Na2SO4, filtered, and then evaporated over silica (50 mL). The crude product was purified by chromatography (gradient elution CH2Cl2 to CH2Cl2/AcOEt 8:2). Some starting material was recovered (950 mg) and the desired product 8a was obtained as a white solid (4.7 g, 62 %). M.p. 230–232 °C; 1H NMR (500 MHz, CDCl3): δ = 3.27 (d, 2J(H,H) =13.7 Hz, 2 H; CqAr–CH2–CqAr), 3.36 (d, 2J(H,H) = 13.1 Hz, 2 H; CqAr–CH2–CqAr), 3.98–4.12 (m, 8 H; CH2–O–CH2–CH2–N, CqAr–CH2–CqAr), 4.26–4.30 (m, 2 H; CH2–O–CqAr), 4.36 (d, 2J(H,H) = 13.0 Hz, 2 H; CqAr–CH2–CqAr), 6.59 (t, 3J(H,H) =7.5 Hz, 2 H; CHAr calix), 6.65 (t, 3J(H,H) =7.5 Hz, 1H; CHAr calix), 6.83–6.88 (m, 3 H; CHAr calix), 6.93–6.98 (m, 4 H; CHAr calix), 7.05 (d, 3J(H,H) =7.5 Hz, 2 H; CHAr calix), 7.50–7.55 (m, 2 H; CH–CH–CqAr–C=O), 7.58–7.61 (m, 2 H; CH–CqAr– C=O), 9.05 (s, 2 H; OH), 9.68 ppm (s, 1H; OH); 13C NMR (125 MHz, CDCl3): δ=31.53 (2 C), 32.23 (2 C), 38.32 (1 C), 69.12 (1C), 70.31 (1 C), 75.57 (1 C), 121.39 (2 C), 122.01 (1C), 123.27 (2 C), 126.48 (1C), 128.71 (2 C), 128.86 (2 C), 129.03 (6 C), 129.13 (2 C), 129.64 (2C), 132.23 (2 C), 133.99 (2 C), 135.10 (2 C), 150.01 (1 C), 150.60 (2C), 151.66 (1 C), 168.77 ppm (2 C); IR (ATR): ν̃ =3317, 3171 (OH), 2920, 2873 (CH), 1764, 1701 cm−1 (C=O); MS: m/z: 664 [M+Na]+; HRMS: m/z calcd for [C40H35NO7Na] +: 664.2311; found: 664.2282.
25,27-Bis{1-[2-(2-phthalimidoethoxy)ethoxy]}-26,28-dihydroxycalix[4]arene (8b)
A slurry of calix[4]arene 6 (10 g, 23.6 mmol), anhydrous K2CO3 (6.5 g, 47.2 mmol), and iodide 7 (32.5 g, 94.4 mmol) in dry CH3CN (150 mL) was heated to reflux for 1 week. After removal of CH3CN, the residue was dissolved in CH2Cl2 (250 mL). The organic phase was washed successively with water (30 mL), of 1M aqueous HCl (2 × 20 mL), and brine (20 mL). After drying over Na2SO4, CH2Cl2 was removed by rotary evaporation and the crude product was purified by crystallization in CH2Cl2/MeOH (2:3). White crystals were obtained (14.9 g, 74 %). M.p. 199.5–200 °C; 1H NMR (500 MHz, CDCl3): δ=3.06 (d, 2J(H,H) =12.9 Hz, 4H; CqAr–CH2–CqAr), 3.97 (d, 2J(H,H) =12.8 Hz, 4H; CqAr–CH2–CqAr), 4.02–4.07 (m, 16 H; CH2–N, CH2–O), 6.47 (t, 3J(H,H) =7.4 Hz, 2 H; CHAr calix), 6.65 (t, J =7.6 Hz, 2 H; CHAr calix), 6.79 (d, 3J(H,H) =7.5 Hz, 4 H; CHAr calix), 6.82 (d, 3J(H,H) =7.6 Hz, 4H; CHAr calix), 7.38–7.42 (m, 4 H; CH–CH–CqAr–C=O), 7.47–7.51 (m, 4H; CH–CqAr–C=O), 7.88 ppm (s, 2H; OH); 13C NMR (125 MHz, CDCl3): δ = 31.43 (4 C), 38.53 (2 C), 68.98 (2 C), 70.44 (2C), 75.66 (2 C), 119.3 (2 C), 123.12 (4 C), 125.71 (2 C), 128.54 (4 C), 128.62 (4 C), 129.14 (4 C), 132.24 (4 C), 133.89 (4 C), 134.24 (4C), 151.88 (2 C), 153.06 (2 C), 168.89 ppm (4 C); IR (ATR): ν̃ =3320 (OH), 1773, 1709 cm−1 (C=O); MS: m/z: 881 [M+Na]+; HRMS: m/z calcd for [C52H46N2O10Na]+: 881.3050; found: 881.3008.
General procedure for the preparation of compounds 9a,b
A 35 % aqueous solution of hydrazine (10 mmol per mmol of phthalimidocalix[4]arene 8a,b) was added to a slurry of phthalimidocalix[4]arene 8a,b in absolute EtOH (50 mL per mmol−1 of phthalimidocalix[4]arene 8a,b). The reaction was heated to reflux for 7 h. Solvents were removed under reduced pressure. The residue was stirred in ethyl acetate (50 mL per mmol). The resulting precipitate was filtered and washed with small volumes of AcOEt. The combined organic phases were washed successively with a 2 M NaOH aqueous solution, water, and brine. The organic phase was dried over Na2SO4 and concentrated by rotary evaporation. After stirring in Et2O, a white gum 8a,d solidified after several days in air.
25-{1-[2-(2-Aminoethoxy)ethoxy]}-26,27,28-trihydroxycalix[4]arene (9a)
According to the general procedure, this compound was prepared from phthalimidocalix[4]arene 8a (2.7 g, 4.21 mmol). For solubility reasons, AcOEt was replaced by hot CHCl3. Compound 9a was obtained as a white solid (1.9 g, 88 %). M.p. 195–197°C; 1H NMR (500 MHz, CDCl3): δ = 2.83 (br t, 2 H; CH2–N), 3.37–3.41 (m, 4 H; CqAr–CH2–CqAr), 3.64 (br t, 2H; O–CH2–CH2–N), 3.95 (br t, 2H; CH2–O), 4.25 (br t, 2 H; CH2–O), 4.28 (d, 2J(H,H) =13.4 Hz, 2H; CqAr– CH2–CqAr), 4.42 (d, 2J(H,H) =12.8 Hz, 2H; CqAr–CH2–CqAr), 6.53 (brt, 1 H; CHAr), 6.65 (t, 3J(H,H) =7.4 Hz, 2 H; CHAr), 6.78 (t, 3J(H,H) = 7.2 Hz, 1 H; CHAr), 6.95 (d, 3J(H,H) =7.3 Hz, 4 H; CHAr), 7.01 (d, 3J(H,H) =7.2 Hz, 2 H; CHAr), 7.06 ppm (d, 3J(H,H) =7.2 Hz, 2H; CHAr); 13C NMR (125 MHz, CDCl3): δ = 31.61 (2C), 33.55 (2 C), 41.64 (1 C), 69.64 (1C), 72.76 (1C), 75.09 (1C), 120.16 (1 C), 125.78 (1 C), 128.56, 128.96, 129.20, 129.54, 129.86, 129.95, 134.08 (18 C), 152.83 ppm (4 C); IR (ATR): ν̃ =2917 cm−1 (CH); MS: m/z: 512 [M+H]+. HRMS: m/z calcd for [C32H33NO5Na] +: 534.2256; found: 534.2272.
25,27-Bis-{1-[2-(2-aminoethoxy)ethoxy]}-26,28-dihydroxycalix[4]-arene (9b)
According to the general procedure, this compound was prepared from phthalimidocalix[4]arene 8b (1.5 g, 1.75 mmol) to yield 9b as a white gum (1.03 g, 99 %). M.p. 104 °C; 1H NMR (500 MHz, CDCl3): δ = 2.07 (br s, 4 H; NH2), 2.98 (br t, 4 H; CH2–N), 3.38 (d, 2J(H,H) =13.0 Hz, 4H; CqAr–CH2–CqAr), 3.73 (brt, 4 H; O– CH2–CH2–N), 4.01 (br t, 4H; CH2–O), 4.22 (br t, 4H; CH2–O–CqAr), 4.43 (d, 2J(H,H) =13.0 Hz, 4H; CqAr–CH2–CqAr), 6.68 (t, 3J(H,H) = 7.4 Hz, 2 H; CHAr), 6.76 (t, 3J(H,H) =7.5 Hz, 2 H; CHAr), 6.92 (d, 3J(H,H) =7.5 Hz, 4 H; CHAr), 7.08 ppm (d, 3J(H,H) =7.4 Hz, 4H; CHAr); 13C NMR (125 MHz, CDCl3): δ = 31.58 (4 C), 42.39 (2 C), 70.11 (2 C), 73.92 (2 C), 75.86 (2 C), 119.51 (2 C), 125.81 (2 C), 128.54 (4 C), 128.88 (4 C), 129.38 (4 C), 133.84 (4 C), 152.20 (2 C), 153.45 ppm (2 C); IR (ATR): ν̃ =3359 (OH, NH), 2922, 2868 cm−1 (CH); MS: m/z: 599 [M+H]+; HRMS: m/z calcd for [C36H43N2O6] +: 599.3121; found: 599.3147.
General coupling procedure for preparation of compounds 5a,b
A solution of carboxylic acid (1.1 equiv. per equiv. of NH2 group), HOBt (1.1 equiv. per equiv. of NH2 group), and EDCI (equiv. per equiv. of NH2 group) in CH2Cl2 (10 mL per mmol of aminocalix[4]arene) was stirred at RT for 15 min. The aminocalix[4]arene 9a,b was added and the reaction was stirred at RT overnight. The reaction was diluted with CH2Cl2 (100 mL per mmol of aminocalix[4]arene 9 a,b) and this organic phase was washed successively with 1 M aqueous HCl, saturated aqueous NaHCO3, water, and brine. The organic phase was dried over Na2SO4 and then evaporated by rotary evaporation. The crude product was purified by chromatography.
25-{1-[2-(2-{4-[3,5-Bis-(2-hydroxyphenyl)-1,2,4-triazol-1-yl]phenyl}carbamoylethoxy)ethoxy]}-26,27,28-trihydroxycalix[4]arene (5a)
According to the general procedure this compound was prepared from aminocalix[4]arene 9 a (0.20 g, 0.391 mmol). The residue was purified by flash chromatography (CH2Cl2/AcOEt 9:1)), then by recrystallization in EtOH, to yield 5a as a beige solid (172 mg, 51 %). M.p. 195–200°C; 1H NMR (600 MHz, CDCl3): δ = 3.35 (d, 2J(H,H) =13.7 Hz, 2 H; CqAr–CH2–CqAr), 3.46 (d, 2J(H,H) =13.0 Hz, 2 H; CqAr–CH2–CqAr), 3.95–3.98 (m, 4H; O–CH2–CH2–N), 4.12 (d, 2J(H,H) =13.7 Hz, 2 H; CqAr–CH2–CqAr), 4.18–4.20 (m, 2H; CH2–O), 4.38–4.40 (m, 2 H; CH2–O–CqAr), 4.43 (d, 2J(H,H) =13.0 Hz, 2 H; CqAr– CH2–CqAr), 6.62–6.70 (m, 4 H; CHAr ICL+CHAr calix), 6.86 (d, 3J(H,H) = 6.9 Hz, 1 H; CHAr ICL), 6.93 (t, 3J(H,H) =7.6 Hz, 1 H; CHAr calix), 6.95 (d, 3J(H,H) =7.5 Hz, 2 H; CHAr calix), 6.99–7.02 (m, 4 H; CHAr calix), 7.08 (t, 3J(H,H) =7.7 Hz, 1H; CHAr ICL), 7.11–7.16 (m, 3 H; CHAr calyx+CHAr ICL), 7.18 (d, 3J(H,H) =8.2 Hz, 1 H; CHAr ICL), 7.21 (d, 3J(H,H) =8.4 Hz, 2H; CHAr ICL), 7.37 (dt, 3J(H,H) =7.8 Hz, 4J(H,H) =1.5 Hz, 1 H; CHAr ICL), 7.43 (dt, 3J(H,H) =7.8 Hz, 4J(H,H) =1.6 Hz, 1 H; CHAr ICL), 7.47 (br t, 1H; NH); 7.83 (d, 3J(H,H) =8.3 Hz, 2H; CHAr ICL), 8.17 (dd, 3J(H,H) =7.8 Hz, 4J(H,H) =1.5 Hz, 1H; CHAr ICL), 9.41 (s, 2H; OHcalix), 9.72 (s, 1 H; OHICL), 9.68 (s, 1 H; OHcalix), 11.41 ppm (s, 1 H; OHICL); 13C NMR (125 MHz, CDCl3): δ=31.32 (2 C), 31.77 (2 C), 40.52 (1 C), 69.81 (1C), 70.25 (1 C), 75.48 (1 C), 109.91 (1 C), 113.23 (1 C), 117.21 (1C), 118.40 (1 C), 119.06 (1C), 119.91 (1C), 121.70 (2C), 122.00 (1C), 126.01 (2 C), 126.47 (1 C), 127.56 (1 C), 127.66 (1 C), 128.48, 128.51, 128.64, 128.67, 128.80, 128.84, 128.98, 129.01, 129.45, 131.85 (1 C), 132.97 (1 C), 134.43 (2 C), 135.85 (1 C), 139.97 (1 C), 149.34 (1C), 149.91 (2 C), 151.19 (1 C), 152.07 (1 C), 156.57 (1 C), 158.08 (1C), 159.44 (1 C), 166.32 ppm (1 C); IR (ATR): ν̃ =3277 (OH), 1461 cm−1 (C=O); MS: m/z: 889 [M+Na]+; HRMS: m/z calcd for [C53H46N4O8Na]+: 889.3213; found: 889.3246; elemental analysis calcd (%) for C53H46N4O8 : C 73.43, H 5.35, N 6.46; found: C 73.32, H 5.37, N 6.49.
25,27-Bis-{1-[2-(2-{4-[3,5-bis(2-hydroxyphenyl)-1,2,4-triazol-1-yl]-phenyl}carbamoylethoxy)ethoxy]}-26,28-dihydroxycalix[4]arene (5b)
According to the general procedure, this compound was prepared from aminocalix[4]arene 9b (0.10 g, 0.167 mmol). The residue was purified by flash chromatography (CH2Cl2/MeOH 98:2), then by recrystallization in EtOH, to yield 5b (80 mg; 37 %) as a beige solid. M.p. 173 °C; 1H NMR (600 MHz, CDCl3): δ = 3.26 (d, 2J(H,H) =12.9 Hz, 4H; CqAr–CH2–CqAr), 3.85–3.93 (m, 8 H; O–CH2– CH2–N), 4.02–4.04 (m, 4H; CH2–O), 4.20–4.22 (m, 4H; CH2–O–CqAr), 4.27 (d, 2J(H,H) =12.9 Hz, 4H; CqAr–CH2–CqAr), 6.57 (t, 3J(H,H) = 7.5 Hz, 2H; CHAr calix), 6.64 (td, 3J(H,H) =7.6 Hz, 4J(H,H) =0.9 Hz, 2H; CHAr ICL), 6.78 (t, 3J(H,H) =7.6 Hz, 2 H; CHAr calix), 6.88 (dd, 3J(H,H) = 8.0 Hz, 4J(H,H) =1.4 Hz, 2 H; CHAr ICL), 6.93 (m, 8H; CHAr ICL+CHAr calix), 7.05 (t, 3J(H,H) =7.1 Hz, 2 H; CHAr ICL), 7.09 (d, 3J(H,H) =8.3 Hz, 2H; CHAr ICL), 7.14 (d, 3J(H,H) =8.5 Hz, 2H; CHAr ICL), 7.21 (d, 3J(H,H) = 8.5 Hz, 4 H; CHAr ICL), 7.35 (t, 3J(H,H) =7.1 Hz, 2H; CHAr ICL), 7.40 (t, 3J(H,H) =7.8 Hz, 2 H; CHAr ICL), 7.74 (br t, 2 H; NH), 7.81 (d, 3J(H,H) = 8.4 Hz, 4 H; CHAr ICL), 8.15 (dd, 3J(H,H) =7.8 Hz, 4J(H,H) =1.5 Hz, 2H; CHAr ICL), 8.24 (s, 2 H; OHcalix), 9.69 (s, 2 H; OHICL), 11.26 ppm (s, 2H; OHICL); 13C NMR (CDCl3, 125 MHz): δ = 31.10 (4 C), 40.53 (2 C), 69.95 (2 C), 70.20 (2 C), 75.47 (2 C), 110.10 (2 C), 113.26 (2 C), 117.22 (2 C), 118.40 (2 C), 119.07 (2 C), 119.95 (2 C), 120.66 (2 C), 125.86 (2 C), 125.99 (4 C), 127.67 (2 C), 127.70 (2 C), 128.66, 128.76, 128.98, 129.15, 131.87 (2 C), 132.98 (2 C), 133.64 (4C), 135.56 (2 C), 140.13 (2 C), 151.69 (2 C), 151.73 (2 C), 152.10 (2 C), 156.59 (2C), 158.04 (2 C), 159.54 (2 C), 165.58 ppm (2 C); IR (ATR): ν̃: 3296 (OH), 1459 cm−1 (C=O); MS: m/z: 1331 [M+Na]+; HRMS: m/z calcd for [C78H68N8O12Na]+: 1331.4854; found: 1331.4827; elemental analysis calcd (%) for C78H68N8O12 : C 71.55, H 5.23, N 8.56; found: C 71.68, H 5.20, N 8.59.
Reagents
Collagenase was purchased from Roche Diagnostic (Meylan, France); and M199 medium and phosphate-buffered saline (PBS) from Eurobio (Les Ulis, France). Endothelial cell grow supplement (ECGS) and endothelial growth factor (EGF) were from Promo Cell GmbH (Heidelberg, Germany); penicillin–streptomycin, gelatin 2 %, and genistein were from Sigma (Saint-Quentin Fallavier, France); fetal bovine serum (FBS) was purchased from AbCys (Paris, France), and Dulbecco’s modified Eagle’s medium (DMEM) low glucose was from Invitrogen (Cergy Pontoise, France). The BrdU kit was from Chemikon (Paris, France). Matrigel (Basement Membrane Matrix) was purchased from Becton Dickinson (Le pont de Claix, France).
Partition coefficients
In this study, the relative log D values were assessed at pH 7 by the micro-HPLC method. Determinations were performed with a chromatographic apparatus (Spectra Series, San Jose, USA) equipped with a model P1000XR pump and a model SCM 1000 vacuum membrane degasser, a model UV 150 ultraviolet detector (λ= 280 nm), and a ChromJet data module integrator (ThermoFinnigan, San Jose, USA). The reversed-phase column used was an Equisorb C8 (4.6 × 150 mm; 5 μm particle size, C.I.L. Cluzeau-France) with a mobile phase consisting of acetonitrile with trifluoroacetic acid (1 %, v/v)–water with trifluoroacetic acid (1%, v/v): 75:25, v/v (5b) and 65:35, v/v (5a). The compounds were partitioned between n-octanol (HPLC grade) and phosphate buffer (pH 7). Octanol was presaturated with the adequate phosphate buffer (1 %), and conversely. An amount of each compound (1 mg) was dissolved in an adequate volume of methanol to produce 1 mg mL−1 stock solutions. Then, an appropriate aliquot of these methanolic solutions was dissolved in buffer to obtain final concentrations of 50 μg mL−1. Under the above-described chromatographic conditions, the aqueous phase (50 μL) was injected into the chromatograph, leading to the determination of a peak area before partitioning (W0). In screw-capped tubes, the aqueous phase (Vaq; 1000 μL) was then added to n-octanol (Voct; 10 μL). The mixture was shaken by mechanical rotation for 20 min, followed by centrifugation at 3000 rpm for 10 min. An amount of the lower phase (20 μL) was injected into the chromatograph column. This led to the determination of a peak area after partitioning (W1). For each compound, the log D value was calculated by using the formula [Eq. (3)]:
| (3) |
Protonation and iron-binding constants in solution
All commercial reagents used were of the highest purity (>99 %) and were used without further purification. All stock solutions were used fresh. Because of the poor solubility of the chelators in aqueous media, all experiments were monitored in H2O/DMSO media (v/v =30:70; xDMSO = 0.5). Ligand solutions were prepared in pure DMSO. These solutions were then diluted to the 10−4 to 10−5 M range. FeCl3 solutions were prepared at a concentration of 10−2 M in H2O media in the presence of an excess amount of hydrochloric acid (0.5 M). Their concentrations were determined by ethylenediaminetetraacetic acid (EDTA) titration. The ionic strength was kept constant (I =0.1) by addition of potassium chloride (Pro-labo) of the highest purity (>99 %). The titrating solutions of carbonate-free base KOH and hydrochloric acid 0.1 M were prepared from standardized 1 M solutions (Prolabo). All solutions were prepared with glass-distilled, deionized water and were degassed by argon saturation to remove all dissolved CO2. Protometric titrations were carried out with an automatic titrator composed of a microprocessor burette (Metrohm Dosimat 665), a glass electrode (metrohm 6 204100) with an incorporated Ag/AgCl reference (filled with KCl 3 mol dm−3 in H2O/DMSO 30:70 (v/v)) and a pH meter (Metrohm 713) connected to a computer. The combined electrode has a very low alkaline error. The titration was fully automated. All measurements were performed within a thermoregulated cell at 25°C under an argon stream to avoid the dissolution of carbon dioxide. For a classical titration, a total of 120 to 150 points (volume of titrant, pH) was recorded. All equilibrium measurements were carried out in 5.00 mL sample volumes with magnetic stirring. The ionic strength was adjusted to 0.1 with potassium chloride solution. An HCl solution at exactly 10−2 M was used to calibrate the electrode. The electrode slope was checked by titration with an HCl solution, and no correction was necessary in the pH range of 2–15.3. The titrant, a carbonate-free KOH solution (Normadose), was standardized against a 0.05M potassium hydrogen phthalate solution by pH potentiometry. The ionic product of water was determined under these conditions (pKw = 17.16) and was used in the calculations. For the determination of FeIII formation constants, metal/ligand mixtures of 1:3, 1:2, and 1:1 molar ratios were studied. For spectrophotometric titrations, FeIII–ligand solutions were prepared with an excess amount of hydrochloric acid and were titrated with potassium hydroxide until pH 6.5. UV-visible spectra were recorded after each base addition at (25.0±0.1) °C on a Cary 500 spectrophotometer equipped with a thermostated cell carrier. Protonation and formation constants were calculated from potentiometric and spectrophotometric data by the HYPERQUAD[27] software. The computer program HYSS[27] was used to obtain the species distribution curves.
ESI-MS
All experiments (MS and MS/MS) were performed on a hybrid tandem quadrupole/time-of-flight (Q-TOF) instrument, equipped with a pneumatically assisted electrospray (Z-spray) ion source (Micromass, Manchester, UK) operated in positive mode. The electrospray potential was set to 3 kV in positive- and negative-ion mode and the extraction cone voltage was usually varied between 30 and 90 V to obtain optimized mass spectra. The FeL solutions were prepared in similar conditions to those set out for the potentiometric study and were introduced using a syringe pump with a flow rate of 5 μL min−1. The stoichiometries of the molecular associations were determined in accordance with the greater isotopic peak (corresponding to species with 56Fe) and further checked by simulating the different parts of the spectrum with the Isopro 3.0 software.[38] The differences between the experimental and calculated m/z values were less than or equal to 0.1 unit.
pFe determination
For ligands that have a low water solubility, the classical methodology to determine pFe consists of determining the overall formation constant (log β) values in a water/solvent mixture by varying the molar fraction of the used solvent (xsolvent). Then, the values are extrapolated in a pure H2O medium by taking into account a linear dependence approximation between log β and xsolvent.[23] The complex and ligand insolubility in a water/DMSO medium for xDMSO < 0.2 renders such a methodology irrelevant to the pFe determination in the case of Fe–5a and Fe–5b systems. This problem can be overcome by performing pFe calculations with data collected in a H2O/DMSO medium (xDMSO = 0.5) at a pH corresponding to the same [OH−]/[H3O+] ratio as that in pure water (pH 7.45). For xDMSO equal to 0.5, the pH scale is extended from 0 to 17.2, and a neutral solution has a pH value of 8.6. Consequently, a pH of 7.45 in water corresponds to 9.16 in a H2O/DMSO medium (xDMSO = 0.5). This value was experimentally confirmed by pH measurements of a phosphate buffer solution prepared with the same H2PO4−/HPO42− ratio in water and H2O/DMSO medium (xDMSO = 0.5). pFe calculations were monitored by using the HYSS software.[27] The protonation constants were determined from nonlinear least-squares refinements of potentiometric titration in H2O/DMSO (xDMSO = 0.5) media using the Hyperquad software.[27] Iron-complex formation was studied by spectrophotometric and potentiometric titrations in the 1–13 pH range. The stability constants were calculated using Hyperquad refinements. ESIMS analyses were also used to confirm the stoichiometry of the major complexes.
Molecular modeling
Models
The initial geometry for [Fe–ICL670] − was taken from the Cambridge structural database (CSD)[31] under the SAJFAL reference.[25] The LEaP program from the AmberTools 1.4 distribution was used to build the topologies of the FeIII–calix[4]arene systems.
QC calculations
Calculations were performed using the Gaussian 03 (revision E.01) program.[39] The [Fe–ICL670] −, [Fe–5a] −, and [Fe–5b] − structures were optimized with the hybrid B3LYP[40] DFT method using the 6-31G* basis set. According to Steinhauser et al.,[23] we assumed the iron complex to be in a high-spin state. This was confirmed by DFT calculations as well, as the sextet state was found to be the most stable relative to the doublet and quartet. Frequencies were calculated after geometry optimization at the same level of theory to confirm that the optimized structures were true energy minima and to provide the Hessian matrix necessary for developing force-field parameters for iron by means of the FUERZA procedure.[35]
Charge derivation and force-field topology database building
The geometries of the different building blocks constituting the ICL670- and calix[4]arenes–ICL670-based FeIII chelators were optimized by using the B3LYP/6-31G* theory level with the 2003 (version E.01) program.[39] For each building block the lowest minimum found after a conformational search was selected except for 2-methoxyethanol, for which two conformations were chosen. The MEP computation involved the Connolly surface algorithm, a radius of 1.8 Å for iron, and the B3LYP/6-31G* theory level implemented in the Gaussian 2003 (version E.01) program. As defined by Steinhauser et al. the high-spin state (S =5/2) of FeIII was considered in DFT calculations.[23] For each building block two molecular orientations based on the rigid-body reorientation algorithm implemented in the R.E.D. program were involved in the MEP computation, which ensured the reproducibility of the charge values.[33] The molecular fragments required for MD simulations were constructed by setting specific intra- and intermolecular charge constraints between fully characterized connecting groups during the charge fitting step (Figure SI6 in the Supporting Information). An additional intramolecular charge constraint of +3.0 e was used to define iron as an individual bound ion. ESP charge fitting was carried out by using a stand-alone version of the RESP program and by following a single-stage fitting procedure.[41]
Molecular dynamics simulations
The SANDER module of the AMBER10 program suite was used to perform MD simulations on the aforementioned complexes.[42] The systems were solvated in a truncated octahedral box with a buffer distance of 10.0 Å along with one sodium counterion to neutralize the complex net charge. The parameters used for water were taken from the TIP3P model.[43] After minimization, the systems were brought to the target temperature by increasing the temperature over periods of 25 ps followed by a run of 200 ps to relax and equilibrate the system. Classical MD simulations of 50 ns were then performed using the NPT ensemble at a pressure of 1 atm and a temperature of 300 K. The weak coupling algorithm[44] was used to regulate the temperature and pressure. The temperature was maintained close to the intended value by weak coupling to an external temperature bath with a relaxation time of 2 ps and the pressure to an external pressure bath of 1 atm with a coupling constant of 2 ps. The SHAKE algorithm[45] was used to constrain C–H bonds, and a time step of 2 fs was used to integrate the equations of motion. Periodic boundary conditions were imposed during the simulation. A distance cutoff of 9.0 Å was applied to nonbonded interactions and the PME method[46] was used to compute long-range interactions. Configurations of the systems were stored at intervals of 1 ps. Analyses of the trajectories were performed using the PTRAJ module available in AmberTools 1.4. Visualization of the trajectories was achieved by using the VMD package.[47] The Hornak et al.[32] all-atom model force field parameters (i.e., Amber99SB) were used to model the [Fe–ICL670] −, [Fe–5a] −, and [Fe–5b] − systems when available, and those of GAFF[48] otherwise. Additional parameters relevant to iron, not available within Amber99SB or GAFF, are required. The van der Waals parameters for iron were taken from Ref. [49].
Chelator solutions
Biological properties of the various calix[4]arenes were compared to tridentate hydroxyphenyltriazole ICL670 (Exjade from Novartis Pharma).[50] Stock solutions of each molecule (10 mM) were prepared in DMSO. The 100 μM chelator concentration corresponds to an addition in the culture medium of 1 % DMSO, which was preliminary verified to be inefficient on HepaRG cell viability. However, a low but significant cytotoxicity (20 %) was observed with 2 % DMSO. This percentage was reached for chelator concentrations of 200 μM. The solubility of each derivative in the culture medium in the concentration range of 0–200 μM was preliminary verified by turbidity measurements. Two controls have been performed for each experiment: one with the standard culture medium, and the other with a culture medium supplemented with DMSO at the concentration used for ICL670 and calix[4]arenes tests.
Solubility of the FeIII chelators
Solubility of the new calix[4]arenes were estimated in PBS solutions and in cell culture media containing 10 % FCS. Solutions for the various compounds (200, 100, 50, 25 μM) were prepared in 96-well microplates by diluting the 2.5 mM stock solutions in DMSO in PBS (200 μL) and a culture medium. The absorbance (turbidity) of the solutions was measured at 590 nm, out of the absorption range of the chromophores.
HepaRG cell cultures
HepaRG cells were isolated from a liver tumor of a female patient suffering from hepatocarcinoma and a hepatitis C infection.[51] HepaRG cells plated at 20 × 104 cells cm−2 in the presence of insulin and corticosteroids proliferated for 3 to 4 days with a doubling time of 24 h. During the proliferation phase, undifferentiated HepaRG cells appeared as a homogeneous epithelial phenotype cell population without regular structural organization. Progressive morphological changes, including the appearance of granular polygonal hepatocyte-like cells and flat, clear epithelial cells, occurred after reaching confluency two weeks after plating. In this study, chelator exposures were performed one day after cell seeding in proliferating HepaRG cells. HepaRG cells were cultured as previously described.[1h] They were maintained in William’s E medium supplemented with 10 % fetal bovine serum, penicillin (100 U mL−1), streptomycin (100 mgmL−1), hydrocortisone hemisuccinate (5 × 10−5 M), and insulin (5 mg mL−1). Cells were seeded at 2 × 104 cells cm−2 in 96-well microplates for viability measurements including LDH and cell-nuclei counting after Hoescht staining.
Cytostatic and cytotoxic effects measurements
The viability assay of the compounds on cells was as follows: 2×104 cells cm−2 were seeded in 96-multiwell plates and were left for 24 h for attachment, spreading, and growth. Then, they were exposed for 72 h to increasing concentrations of the compounds, ranging from 0.1 to 400 μM in a final volume of 100 μL culture medium. The cytotoxic and cytostatic effects of the various compounds were evaluated by measuring extracellular LDH activity (cytotoxicity detection kit–LDH, Roche, Penzberg, Germany) and mitochondrial SDH activity by the tetrazolium colorimetric assay (MTT, Sigma, St Louis, MO). Extracellular LDH activity was measured as described by the manufacturer on a 20 μL aliquot of cell-free medium obtained by centrifugation (2500 rpm min−1 for 5 min). LDH activities were detected by reading the absorbance at 485 nm. An LDH standard curve (0 to 4000 mU mL−1) was used to quantify the enzyme activity (L-lactate dehydrogenase, Sigma, St Louis, MO) and the values were corrected from the intracellular protein content measured according to the method of Bradford.[52] Extracellular LDH activity was reported as a percentage of the control value. After a 72 h treatment, cells supernatants were removed and the HepaRG cells were washed twice with PBS (50 mM, pH 7). SDH activity was detected after 3 h of incubation in a 100 μL serum-free medium containing MTT (0.5 mgL−1). Formazan salts were dissolved in DMSO and the absorbance was read at 535 nm in a microplate fluorescence reader (Packard, Fusion). Data are reported as a percentage of SDH activity with respect to the control value. Three independent replicates were performed for each experiment and each experiment was repeated three times. Data reported are the average results of three independent experiments. Parameters of the dose–response curves were deduced from a four-parameters curve fit according to Rodbar [Eq. (4)]:[53]
| (4) |
in which y represents the SDH activity with respect to the control; ACmin and ACmax are the minimal and maximal chelator concentrations, respectively; C is the chelator concentration (Cip at the inflection point); and Pip is the slope at the inflection point of the sigmoid curve. Due to their biphasic feature, the dose–response curves were fitted as the sum of two sigmoids (double four-parameter fit). Percentages of HepaRG cells involved in each viability response were deduced from the ACmin, ACmax, and IC50 values of each sigmoid, which were obtained from these fits. Statistical analysis was performed using the nonparametric Mann–Whitney test. The significant level was set to 0.01.
Chelator-uptake measurement
Chelator uptake was investigated in HepaRG cell cultures seeded in 25 cm2 culture flasks (20000 cells cm−2) according to a previously reported procedure.[1h] Cell treatments were performed in triplicate, in proliferating cells (D4) or after differentiation (D21), in the absence (control) or in the presence of the various compounds (50 μM). After 72 h of treatment, supernatants were removed and cells were then washed three times with ice-cold PBS solutions (3 mL). They were collected by scraping after adding water (1 mL) and sonication for 30 s at 0°C. The protein content in these cell extracts was measured by using the Bradford method.[52] Samples for calibration were prepared in triplicate by adding various compound concentrations (0.5, 1, and 5 μM) to cell extracts obtained from untreated control cells. Ultrafiltration of the cell lysates (200 μL) was performed by centrifugation for 20 min at 15 000g in a NANOSEP 3K centrifugal device (Pall Filtron Co). A volume of 1 μL of each intracellular medium was injected into a liquid chromatograph mass spectrometer (LCMS 2020 Shimadzu) with a flow rate of 0.5 mL min−1 for 10 min. This chromatograph was equipped with a diode-array detector (SPD-M20A) and the reversed column used was a Shim pack XR-ODSII (2.0 × 75 mm) with a mobile phase consisting of acetonitrile/water 70:30 (v/v) until 100:0 (v/v) (5a, tR = 3.4 min) or acetonitrile/water 80:20 (v/v) (5b, tR = 4.6 min).
HUVEC cultures
HUVECs were freshly isolated from umbilical veins of newborn babies by collagenase digestion.[54] The umbilical cords were obtained from the Centre de Gynécologie Obstétrique d’Amiens. HUVECs were seeded in a 25 cm2 plastic flask coated with 0.2 % gelatin in M199 medium supplemented with penicillin–streptomycin (100 units mL−1, 20 μg mL−1), 20 % FBS, and ECGS (10 ng mL−1). Plastic culture flasks were maintained at 37 °C in a humidified atmosphere of 5 % CO2. For cell-culture experiments, HUVECs were used between the second and the fifth passage.
BrdU assay
For cell-culture experiments, ICL, 5a and 5b were dissolved in methanol at a 10−3 M concentration, removed as aliquots, and then frozen at −20°C until further utilization. The 5a and 5b effects on HUVEC and MDA-MB-231 cell survival and proliferation were tested using “BrdU Cell Proliferation Assay Kit” as per the manufacturer description.[55] HUVECs were seeded, in triplicate, at 2 × 104 cells per well and MB-231 cells at 1.5 × 104 cells per well in a 96-well microplate, in the appropriate cell-culture medium. The day after, cells were synchronized in the G1 phase for 24 h with serum-free medium (100 μL) without red phenol. Cells were then incubated in the presence or absence of 5a or 5b (10−8–10−5 M) freshly prepared in growth medium (100 μL). We tested two experimental conditions: low serum conditions, 0% FBS (for MDA-MB-231) or 1 % FBS (for HUVEC) for cell survival determination, and 10 % FBS conditions for cell-proliferation determination. After 48 h of incubation, BrdU (20 μL) was added to each well at a final concentration of 0.01 mM; the cells were then re-incubated for 2–24 h. Optical densities were then measured at 450 and 595 nm using an Envision HTS microplate reader (Turku, Finland).
Matrigel assay and wound-healing assay
A Matrigel assay allows in vitro endothelial cell differentiation in capillary tubes. This technique gives a simple initial assessment of the potential pro- or anti-angiogenic properties of the molecules tested.[56] The capillary-tube formation was quantified by using image-analysis techniques and the network length was assessed.[54] Forty thousand HUVECs were seeded directly on top of the Matrigel, in a 24-well microplate, in M199 supplemented with 10 % FBS (500 μL) with different concentrations of ICL670, 5a and 5b (10−5 to 10−6 M) and incubated for 16 h.[57] To determine whether ICL670 or 5a or 5b modified the endothelial cell migration and proliferation, we performed a wound healing assay. Four hundred thousand HUVECs per well were seeded on 0.2 % gelatin-covered 24-well plates. After 24 h, the wound was performed on confluent cells, then cells were treated, for 16 h, with different concentrations of 5a and 5b (10−5 to 10−6 M) freshly prepared in M199 medium (250 mL) without red phenol supplemented with 10 % FBS.[57] In both cases, pictures were taken after 16 h of incubation by using a JVC video camera connected to the VisioL software and mounted on a Leica microscope. The results are given as an average of at least three separate (replicate) experiments for each treatment. Results were presented as means plus or minus the SEM. The statistical significance of the differences between the values in the treated and control cells was determined by ANOVA and the test of student. Statistical differences between controls and treated cells were considered significant when p is <0.05.
Supplementary Material
Acknowledgments
This study was supported by Région Picardie (TsFer project). P.R. was the recipient of a grant from the Région Picardie. We thank Dr. D. Harakat for mass spectra recording. We also thank P. Gans, A. Sabatini, and A. Vacca for the free distribution of the HYSS program. We thank “Fonds Européen de Développement Régional et la Région Picardie” and “La ligue nationale contre le cancer” for financial support. We also thank the “Service de gynécologie obstétrique et médecine de la reproduction du Centre de Gynécologie Obstétrique d’Amiens (CGO)” for providing the umbilical cords.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cplu.201200141.
References
- 1.a) Richardson DR. Crit Rev Oncol Hematol. 2002;42:267–281. doi: 10.1016/s1040-8428(01)00218-9. [DOI] [PubMed] [Google Scholar]; b) Donfrancesco A, Deb G, Dominici C, Pileggi D, Castello MA, Helson L. Cancer Res. 1990;50:4929–4930. [PubMed] [Google Scholar]; c) Chantrel-Groussard K, Gaboriau F, Pasdeloup N, Havouis R, Nick H, Pierre JL, Brissot P, Lescoat G. Eur J Pharmacol. 2006;541:129–137. doi: 10.1016/j.ejphar.2006.05.001. [DOI] [PubMed] [Google Scholar]; d) Ohyashiki JH, Kobayashi C, Hamamura R, Okabe S, Tauchi T, Ohyashiki K. Cancer Sci. 2009;100:970–977. doi: 10.1111/j.1349-7006.2009.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kalinowski DS, Richardson DR. Pharmacol Rev. 2005;57:547–583. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]; f) Wallace HM, Fraser AV, Hughes A. Biochem J. 2003;376:1–14. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Lescoat G, Chantrel-Groussard K, Pasdeloup N, Nick H, Brissot P, Gaboriau F. Cell Proliferation. 2007;40:755–767. doi: 10.1111/j.1365-2184.2007.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Gaboriau F, Leray AM, Ropert M, Gouffier L, Cannie I, Troadec MB, Loreal O, Brissot P, Lescoat G. Biometals. 2010;23:231–245. doi: 10.1007/s10534-009-9281-9. [DOI] [PubMed] [Google Scholar]
- 2.Hileti D, Panayiotidis P, Hoffbrand AV. Br J Haematol. 1995;89:181–187. doi: 10.1111/j.1365-2141.1995.tb08927.x. [DOI] [PubMed] [Google Scholar]
- 3.Finch RA, Liu M, Grill SP, Rose WC, Loomis R, Vasquez KM, Cheng Y, Sartorelli AC. Biochem Pharmacol. 2000;59:983–991. doi: 10.1016/s0006-2952(99)00419-0. [DOI] [PubMed] [Google Scholar]
- 4.Rakba N, Loyer P, Gilot D, Delcros JG, Glaise D, Baret P, Pierre JL, Brissot P, Lescoat G. Carcinogenesis. 2000;21:943–951. doi: 10.1093/carcin/21.5.943. [DOI] [PubMed] [Google Scholar]
- 5.a) Bowen T, Planalp RP, Brechbiel MW. Bioorg Med Chem Lett. 1996;6:807–810. [Google Scholar]; b) Turner J, Koumenis C, Kute TE, Planalp RP, Brechbiel MW, Beardsley D, Cody B, Brown KD, Torti FM, Torti SV. Blood. 2005;106:3191–3199. doi: 10.1182/blood-2005-03-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Whitnall M, Howard J, Ponka P, Richardson DR. Proc Natl Acad Sci USA. 2006;103:14901–14906. doi: 10.1073/pnas.0604979103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rao VA, Klein SR, Agama KK, Toyoda E, Adachi N, Pommier Y, Shacter EB. Cancer Res. 2009;69:948–957. doi: 10.1158/0008-5472.CAN-08-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Lovejoy DB, Richardson DR. Curr Med Chem. 2003;10:1035–1049. doi: 10.2174/0929867033457557. [DOI] [PubMed] [Google Scholar]; b) Richardson DR, Tran EH, Ponka P. Blood. 1995;86:4295–4306. [PubMed] [Google Scholar]
- 8.Lovejoy DB, Richardson DR. Blood. 2002;100:666–676. doi: 10.1182/blood.v100.2.666. [DOI] [PubMed] [Google Scholar]
- 9.Bergeron RJ, Wiegand J, McManis JS, Bharti N. J Med Chem. 2006;49:7032–7043. doi: 10.1021/jm0608816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Wieser C, Dieleman CB, Matt D. Coord Chem Rev. 1997;165:93–161. [Google Scholar]; b) Ikeda A, Shinkai S. Chem Rev. 1997;97:1713–1734. doi: 10.1021/cr960385x. [DOI] [PubMed] [Google Scholar]; c) Harvey PD. Coord Chem Rev. 2002;233–234:289–309. [Google Scholar]; d) Creaven BS, Donlon DF, McGinley J. Coord Chem Rev. 2009;253:893–962. [Google Scholar]
- 11.a) Arduini A, Casnati A, Fabbi M, Minari P, Pochini A, Sicuri AR, Ungaro R. Supramol Chem. 1993;1:235–246. [Google Scholar]; b) Dasaradhi L, Stark P, Huber VJ, Smith P, Jarvinen G, Gopalan AS. J Chem Soc Perkin Trans. 1997;2:1187–1192. [Google Scholar]; c) Canevet C, Libman J, Shanzer A. Angew Chem. 1996;108:2842–2845. [Google Scholar]; Angew Chem Int Ed Engl. 1996;35:2657–2660. [Google Scholar]; d) Haino T, Shio H, Takano R, Fukazawa Y. Chem Commun. 2009:2481–2483. doi: 10.1039/b900599d. [DOI] [PubMed] [Google Scholar]
- 12.a) Guillon J, Leger JM, Sonnet P, Jarry C, Robba M. J Org Chem. 2000;65:8283–8289. doi: 10.1021/jo001023z. [DOI] [PubMed] [Google Scholar]; b) Guillon J, Sonnet P, Malval JP, Massip S, Gosse I, Léger JM, Lapouyade R, Rochette J, Monti JP, Jarry C. Supramol Chem. 2002;14:437–451. [Google Scholar]; c) Guillon J, Léger J-M, Dapremont C, Apollonia Denis L, Sonnet P, Massip S, Jarry C. Supramol Chem. 2004;16:319–329. [Google Scholar]
- 13.Ogden MI, Skelton BW, White AH. J Chem Soc Dalton Trans. 2001:3073–3077. [Google Scholar]
- 14.Ali A, Salunke-Gawali S, Rao CP, Linares J. Inorg Chem Commun. 2004;7:1298–1301. [Google Scholar]
- 15.Wang HW, Feng YQ, Chen C, Xue JQ. Chin Chem Lett. 2009;20:1271–1274. [Google Scholar]
- 16.a) Sansone F, Segura M, Ungaro R. In: Calixarenes. Asfari M-Z, Böhmer V, Harrowfield J, Vicens J, editors. Kluwer Academic Publishers; Dordrecht: 2001. pp. 496–512. [Google Scholar]; b) Mutihac L. Curr Drug Discovery Technol. 2008;5:98–104. doi: 10.2174/157016308784746256. [DOI] [PubMed] [Google Scholar]; c) Rodik RV, Boyko VI, Kalchenko VI. Curr Med Chem. 2009;16:1630–1655. doi: 10.2174/092986709788186219. [DOI] [PubMed] [Google Scholar]; d) de Fatima A, Fernandes SA, Sabino AA. Curr Drug Discovery Technol. 2009;6:151–170. doi: 10.2174/157016309788488302. [DOI] [PubMed] [Google Scholar]; e) Coleman AW, Baggetto LG, Lazar AN, Michaud MH, Magnard S. 20060003406 20060418. F. Demande (France): FR. 2007; f) Sugiyama T, Zhou XP, Koizumi Y, Koyota S, Hamada F, Kondo Y. 2011111438 A 20110609. J. K. T. Koho (Japan): JP. 2011
- 17.Sun J, Blaskovich MA, Jain RK, Delarue F, Paris D, Brem S, Wotoczek-Obadia M, Lin Q, Coppola D, Choi K, Mullan M, Hamilton AD, Sebti SM. Cancer Res. 2004;64:3586–3592. doi: 10.1158/0008-5472.CAN-03-2673. [DOI] [PubMed] [Google Scholar]
- 18.a) Pires VS, Gaboriau F, Guillon J, Nascimento S, Dassonville A, Lescoat G, Desplat V, Rochette J, Jarry C, Sonnet P. J Enzyme Inhib Med Chem. 2006;21:261–270. doi: 10.1080/14756360600700384. [DOI] [PubMed] [Google Scholar]; b) Rouge P, Pires VS, Gaboriau F, Dassonville-Klimpt A, Guillon J, Nascimento SD, Léger JM, Lescoat G, Sonnet P. J Enzyme Inhib Med Chem. 2010;25:216–227. doi: 10.3109/14756360903169949. [DOI] [PubMed] [Google Scholar]; c) Latxague L, Gaboriau F, Chassande O, Léger JM, Pires V, Rouge P, Dassonville-Klimpt A, Fardeau S, Jarry C, Lescoat G, Guillon J, Sonnet P. J Enzyme Inhib Med Chem. 2011;26:204–215. doi: 10.3109/14756366.2010.489898. [DOI] [PubMed] [Google Scholar]
- 19.Gutsche CD, Dhawan B, Levine JA, Hyun No K, Bauer LJ. Tetrahedron. 1983;39:409–426. [Google Scholar]
- 20.Rouge P, Becuwe M, Dassonville-Klimpt A, Da Nascimento S, Raimbert JF, Cailleu D, Baudrin E, Sonnet P. Tetrahedron. 2011;67:2916–2924. [Google Scholar]
- 21.a) Gutsche CD, Lin LG. Tetrahedron. 1986;42:1633–1640. [Google Scholar]; b) Casnati A, Arduini A, Ghidini E, Pochini A, Ungaro R. Tetrahedron. 1991;47:2221–2228. [Google Scholar]; c) Matvieiev Y, Boyko VI, Podoprigorina A, Kalchenko V. J Inclusion Phenom Macrocyclic Chem. 2008;61:89–92. [Google Scholar]
- 22.a) Groenen LC, Ru+l BHM, Casnati A, Verboom W, Pochini A, Ungaro R, Reinhoudt DN. Tetrahedron. 1991;47:8379–8384. [Google Scholar]; b) Boyko VI, Podoprigorina AA, Yakovenko AV, Pirozhenko VV, Kalchenko VI. J Inclusion Phenom Macrocyclic Chem. 2004;50:193–197. [Google Scholar]; c) Cunningham ID, Woolfall M. J Org Chem. 2005;70:9248–9256. doi: 10.1021/jo051312t. [DOI] [PubMed] [Google Scholar]; d) Mbemba C, Sigaud K, Perret F, Suwinska K, Shkurenko O, Coleman A. J Inclusion Phenom Macrocyclic Chem. 2008;61:29–40. [Google Scholar]
- 23.Steinhauser S, Heinz U, Bartholomä M, Weyherm4ller T, Nick H, Hegetschweiler K. Eur J Inorg Chem. 2004;2004:4177–4192. [Google Scholar]
- 24.Bhal SK, Kassam K, Peirson IG, Pearl GM. Mol Pharm. 2007;4:556–560. doi: 10.1021/mp0700209. [DOI] [PubMed] [Google Scholar]
- 25.Nick HP, Acklin P, Lattmann R, Buehlmayer P, Hauffe S, Schupp J, Alberti D. Curr Med Chem. 2003;10:1065–1076. doi: 10.2174/0929867033457610. [DOI] [PubMed] [Google Scholar]
- 26.Huang XP, Spino M, Thiessen JJ. Pharm Res. 2006;23:280–290. doi: 10.1007/s11095-005-9258-5. [DOI] [PubMed] [Google Scholar]
- 27.a) Gans P, Sabatini A, Vacca A. Talanta. 1996;43:1739–1753. doi: 10.1016/0039-9140(96)01958-3. [DOI] [PubMed] [Google Scholar]; b) Alderighi L, Gans P, Ienco A, Peters D, Sabatini A, Vacca A. Coord Chem Rev. 1999;184:311–318. [Google Scholar]
- 28.Arena G, Cali R, Lombardo GG, Rizzarelli E, Sciotto D, Ungaro R, Casnati A. Supramol Chem. 1992;1:19–24. [Google Scholar]
- 29.Harrington JM, Chittamuru S, Dhungana S, Jacobs HK, Gopalan AS, Crumbliss AL. Inorg Chem. 2010;49:8208–8221. doi: 10.1021/ic902595c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albrecht-Gary AM, Crumbliss AL. Met Ions Biol Syst. 1998;35:239–327. [PubMed] [Google Scholar]
- 31.Allen FH. Acta Crystallogr Sect B. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 32.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.a) Dupradeau FY, Pigache A, Zaffran T, Savineau C, Lelong R, Grivel N, Lelong D, Rosanski D, Cieplak P. Phys Chem Chem Phys. 2010;12:7821–7839. doi: 10.1039/c0cp00111b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vanquelef E, Simon S, Marquant G, Garcia E, Klimerak G, Delepine JC, Cieplak P, Dupradeau FY. Nucleic Acids Res. 2011;39:W511–W517. doi: 10.1093/nar/gkr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dupradeau FY, Cézard C, Lelong R, Stanislawiak E, PÞcher J, Delepine JC, Cieplak P. Nuc Acids Res. 2008:D360–D367. doi: 10.1093/nar/gkm887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seminario JM. Int J Quantum Chem. 1996;60:1271–1277. [Google Scholar]
- 36.a) Hunter CA, Sanders JKM. J Am Chem Soc. 1990;112:5525–5534. [Google Scholar]; b) Hunter CA. Chem Soc Rev. 1994;23:101–109. [Google Scholar]
- 37.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE., III Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 38.Senso M. Isopro 3.0 MS/MS software, Isotopic Abundance Simulator Version 3.0. National High Magnetic Field Laboratory; 243 Buena Vista Avenue, #502, Sunnyvale CA 94086, USA: [Google Scholar]
- 39.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 40.a) Becke AD. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; b) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; c) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 41.Bayly CI, Cieplak P, Cornell WD, Kollman PA. J Phys Chem. 1993;97:10269–10280.b)http://q4mdforcefieldtools.org/RED/resp
- 42.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, Merz KM, Wang B, Hayik S, Roitberg A, Seabra G, Kolossvary I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Mathews DH, Seetin MG, Sagui C, Babin V, Kollman PA. AMBER 10. University of California; San Francisco: 2008. [Google Scholar]
- 43.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 44.Berendsen HJC, Postma JPM, van Gunsteren WF, Dinola A, Haak JR. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 45.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 46.Essmann U, Perera L, Berkowitz ML, Darden TA, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 47.Humphrey W, Dalke A, Schulten K. J Molec Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 48.Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 49.Giammona DA. PhD thesis. University of California; Davis: 1984. [Google Scholar]
- 50.Rouan MC, Marfil F, Mangoni P, Séchaud R, Humbert H, Maurer G. J Chromatogr B. 2001;755:203–213. doi: 10.1016/s0378-4347(01)00079-2. [DOI] [PubMed] [Google Scholar]
- 51.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. Proc Natl Acad Sci USA. 2002;99:15655–15660. doi: 10.1073/pnas.232137699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 53.Rodbard D, McClean SW. Clin Chem. 1977;23:112–115. [PubMed] [Google Scholar]
- 54.Taraboletti G, Giavazzi R. Eur J Cancer. 2004;40:881–889. doi: 10.1016/j.ejca.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 55.Rocha A, Azevedo I, Soares R. Angiogenesis. 2007;10:279–286. doi: 10.1007/s10456-007-9080-2. [DOI] [PubMed] [Google Scholar]
- 56.Benelli R, Albini A. Int J Biol Markers. 1999;14:243–246. doi: 10.1177/172460089901400408. [DOI] [PubMed] [Google Scholar]
- 57.Zilberberg L, Shinkaruk S, Lequin O, Rousseau B, Hagedorn M, Costa F, Caronzolo D, Balke M, Canron X, Convert O, Lain G, Gionnet K, Goncalves M, Bayle M, Bello L, Chassaing G, Deleris G, Bikfalvi A. J Biol Chem. 2003;278:35564–35573. doi: 10.1074/jbc.M304435200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.