

Abstract
Primary malignant B-cell-type dural lymphoma is a rare subtype of primary central nervous system lymphoma (PCNSL). We herein report an unusual case of diffuse B-cell lymphoma that presents as a chronic subdural haematoma without extracranial involvement. The notable aspects of this case include the patient’s immunocompetence, a short clinical history of symptom onset, rapid neurological deterioration and a final diagnosis of high-grade PCNSL. This case highlights the challenges neurosurgeons face, especially in the emergency setting, when the disease manifests in varied presentations.
Keywords: chronic subdural haematoma, primary central nervous system lymphoma, primary dural lymphoma
INTRODUCTION
Primary central nervous system lymphomas (PCNSLs) are uncommon tumours of the central nervous system (CNS). They are a group of extranodal non-Hodgkin’s lymphomas (NHLs), primarily of B cell origin.(1) Primary malignant dural lymphoma, a subtype of PCNSL, is even rarer, comprising 0.6%–3% of all brain tumours.(2,3) We herein report the case of an immunocompetent man who presented with what was initially diagnosed to be a chronic subdural haematoma, but which turned out to be a primary B-cell lymphomatous lesion. In view of this unexpected diagnosis, we also reviewed the present case in the context of the relevant literature.
CASE REPORT
A previously well 62-year-old man presented with complaints of mild right upper limb and facial weakness for four days. He had no other significant past medical history (e.g. recent flu-like symptoms, family history, or autoimmune diseases). Physical examination demonstrated that the patient was well nourished and cooperative, with a Glasgow Coma Scale score of 15. However, there was notable right facial droop and grade 4+/5 power in the right upper limb. The rest of the clinical assessment and initial blood workup were unremarkable. Computed tomography (CT) of the brain reported a left, frontoparietal, acute-on-chronic subdural haematoma with rim calcification, associated with mass effect and oedema, as well as a midline shift of 0.8 cm to the right (Fig. 1).
Fig. 1.

Pre-contrast axial CT image shows the patient’s brain.
On the same day, multiplanar magnetic resonance (MR) imaging of the brain showed a large, left, parietofrontal, extra-axial mass with a crescentic configuration in its cranial part. The mass was well defined, with linear peripheral rim enhancement that merged with the adjoining normal dural enhancement. The mass showed signal heterogeneity on all sequences. Correlating both CT and MR imaging findings, the overall impression was still a chronic subdural haematoma. In the MR images, it was also noted that there was a small, extra-axial, polypoidal, enhancing nipple along the medial border of the haematoma. This lesion was hypointense on T1-weighted imaging, and mildly hyperintense on fluid-attenuated inversion recovery sequence and T2-weighted imaging with post-contrast enhancement and restricted diffusion. The differential diagnosis from findings was focal proliferation of granulation tissue (Fig. 2).
Fig. 2.
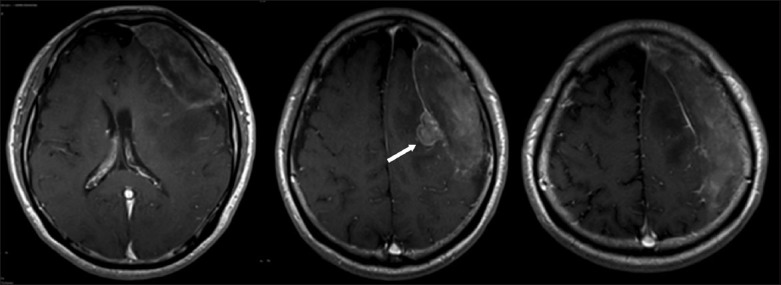
T1-weighted post-gadolinium MR image shows the extra-axial polypoidal nipple, which was thought to be a proliferation of granulation tissue (arrow).
The following day, the patient’s right-sided hemiparesis worsened, and this was associated with new-onset aphasia. Immediate surgery was decided on in view of the patient’s neurological deterioration. He underwent left frontoparietal craniotomy and removal of the left subdural collection. Intraoperatively, it was noted that the dura overlying the subdural area of interest had thickened into a hardened shell. There was no calvarial involvement. The contents of the subdural collection were greenish-yellow and paste-like in consistency. There was no evidence of haematoma or fluid in the lesion. A small, fleshy, granulating lesion was seen on the undersurface of the collection. The lesional dura, subdural collection, and attached granulating tissue were removed in gross total fashion, and sent for the relevant investigations.
Microbiological analyses of the specimens showed no evidence of intracranial infection. However, histological examination of the main bulk of resected specimen revealed an extensively necrotic lesion, with areas of fibrosis adherent to the underside of the dura fragments. Within these necrotic and fibrotic areas were islands of medium-to-large atypical lymphoid cells arranged in sheets. These cells contained enlarged pleomorphic vesicular nuclei with conspicuous nucleoli. On immunostaining, the neoplastic cells showed B-cell phenotype, being strongly positive for CD20. There was strong expression of BCL2 and aberrant expression of CD43 in the neoplastic cells. A small proportion of the cells was also positive for CycD1. CD45 staining is negligible in this case, and there was no staining for CD10, S100 and AE1/3. Small numbers of reactive CD3- and CD5-positive T cells, and some CD68-positive macrophages were also present within the lesion. The MIB-1 labelling index was markedly raised (Figs. 3 & 4). Kappa and lambda light chain restriction studies were considered unnecessary in this case, as the morphological features were fairly conclusive. Epstein-Barr virus (EBV) detection via in situ hybridisation was also not undertaken. The morphological features seen were consistent with diffuse large B-cell lymphoma.
Fig. 3.

Photomicrographs of the lesional tissue show (a) the dura at the bottom right, under low magnification (Haematoxylin & eosin); and (b) sheets of neoplastic cells with brisk mitotic activity, under high magnification (Haematoxylin & eosin). The neoplastic cells contain enlarged vesicular nuclei with small but conspicuous nucleoli, and show (c) strong and diffuse immunostaining for CD20; and (d) marked MIB-1 labelling index.
Fig. 4.

Photomicrographs of the immunostains of the neoplastic cells show positive tests for (a) BCL2 and (b) CD43. Photomicrographs of the scattered cells show (c) positive staining for CycD1; and (d) a small population of reactive CD5-positive T lymphocytes.
The patient underwent further workup to exclude primary lymphomatous lesion elsewhere in his body. Comprehensive CT studies of his body were negative for any suspicious tumour or lymphadenopathy. Serological tests for human immunodeficiency virus, EBV and autoimmune markers were negative. The patient was referred to the medical oncologist and radiation therapist for adjuvant treatment.
DISCUSSION
PCNSL is an uncommon form of extranodal NHL that occurs in the brain, leptomeninges, spinal cord or eyes without evidence of systemic disease.(2,3) PCNSL is histopathologically indistinguishable from NHLs that occur at other body sites,(4) and accounts for up to 1% of NHLs, 3%–6.6% of all primary cerebral neoplasms,(1,2,5) and less than 5% of all lymphomas.(1) More than 95% of PCNSLs are diffuse large B-cell lymphomas;(2,4) congenital or acquired immunodeficiency syndromes are well-recognised risk factors.(5) Other factors contributing to its pathogenesis include latent infection by EBV in immunosuppressed patients, the presence of B cell-attracting chemokine BCA-1, and B-cell growth factors such as interleukin 4.(7) However, our patient had neither a history of immunosuppression nor EBV infection, based on his blood tests.
The brain is usually involved as either a localised tumour mass resembling certain gliomas, or diffusely invasive neoplasms resembling exudative cellular inflammatory processes.(6) However, there is a variant of PCNSL known as primary dural lymphoma (PDL). PDL is an even more uncommon form that originates from the dura mater and differs biologically from other CNS lymphomas.(5,7,8) The question remains as to how a primary intracranial lymphoma can develop in a structure lacking a lymphatic system,(11) such as in the dura, as was the case in our patient. It is speculated that malignant transformation of B cells in the periphery will be recognised and destroyed by the immune system, whereas in the immune-privileged site of the CNS, any antitumour immune response is likely to be insufficient, leading to the development of PCNSL.(9,10) Furthermore, Tun et al’s work on identifying a ‘CNS signature’ via comparing genomics between PCNSL and non-CNS diffuse large B-cell lymphoma suggests that interactions between lymphoma cells and the CNS microenvironment are particularly important for PCNSL.(11) Recent research into the disease has demonstrated that aberrancy of genes such as SPP1 and PRDM1 may also help explain the exclusive manifestation of such tumours in the CNS.(12) Other postulations include the disease epigenetics involving the CpG island promoter hypermethylation, vascular endothelial growth factor-mediated angiogenesis, and dysfunctional folate and homocysteine metabolism. Altogether, the pathogenesis of PCNSL and its subtypes remain fragmented, and more studies are required to obtain a better understanding of the underlying mechanisms.(12)
Comparatively, PDLs are distinct from most primary intraparenchymal CNS lymphomas due to their predominance of low-grade forms. They are usually marginal zone lymphomas of mucosa-associated lymphoid tissue-type lymphomas. An even rarer subtype of PDLs are primary malignant dural lymphomas, which make up only 0.6%–3% of all brain tumours.(1,6) In Yamada et al’s review of primary malignant dural lymphomas, the epidemiology showed female predominance, immunocompetency and longer survival rate among patients when compared to those with primary malignant intracerebral lymphomas.(13) However, there is a paucity of overall knowledge of its epidemiology, risk factors and behaviour.
From our review of the literature, the top differential diagnosis for PDL is a meningioma. Neuroimaging often demonstrates extra-axial lesions with a ‘dural tail’ sign that is diffusely enhanced with the administration of gadolinium.(14) However, in our present case, MR imaging did not show convincing evidence of overall lesion contrast enhancement, overlying scalp mass, or cranial vault involvement. Putting it all together, this eventually led to the working diagnosis of a chronic subdural haematoma instead.
For the majority of CNS lymphoma patients, overall prognosis remains poor.(1) PCNSL is an aggressive disease with an average survival time of 1.5–3.3 months in untreated patients.(3,6) As opposed to most other types of PCNSLs, the course of primary malignant dural B-cell lymphomas is comparatively benign. As illustrated by Yamada et al(13) and Sanjeevi et al,(15) this particular subtype may be curable by surgical resection, with or without postoperative focal irradiation. However, it should be noted that, owing to the general lack of reported cases of primary malignant dural B-cell lymphoma, knowledge of this disease still remains limited.
In summary, we report an unusual presentation of a primary malignant B-cell dural lymphoma, a rare subtype of PCNSL, and the first in the Singapore context. This is also the first case of diffuse B-cell lymphoma that presents as a chronic subdural haematoma without extracranial involvement. Notable aspects of the present case include the short clinical history of the onset of the patient’s symptoms, rapid neurological deterioration, and the final diagnosis of a high-grade PCNSL. As the search continues for an improved understanding of this disease’s aetiology, clinicians have to be mindful to include primary malignant dural lymphoma as a differential diagnosis in the acute setting, even for an immunocompetent patient. The variances in presentation and neuroimaging make this evolving disease a mimicry entity, as illustrated by our case report.
REFERENCES
- 1.Chen TC, Abrey LE. Primary central nervous system lymphomas. J Neurosurg. 2006;21:1. doi: 10.3171/foc.2006.21.5.13. [DOI] [PubMed] [Google Scholar]
- 2.Lim T, Kim SJ, Kim K, et al. Primary CNS lymphoma other than DLBCL: a descriptive analysis of clinical features and treatment outcomes. Ann Hematol. 2011;90:1391–8. doi: 10.1007/s00277-011-1225-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schabet M. Epidemiology of primary CNS lymphoma. J Neurooncol. 1999;43:199–201. doi: 10.1023/a:1006290032052. [DOI] [PubMed] [Google Scholar]
- 4.Sacho RH, Kogels M, du Plessis D, Jowitt S, Josan VA. Primary diffuse large B-cell nervous system lymphoma presenting as an acute space-occupying subdural mass. J Neurosurg. 2010;113:384–7. doi: 10.3171/2010.2.JNS091554. [DOI] [PubMed] [Google Scholar]
- 5.Beriwal S, Hou JS, Miyamoto C, Garcia-Young JA. Primary dural low grade BCL-2 negative follicular lymphoma: a case report. J Neurooncol. 2003;61:23–5. doi: 10.1023/a:1021244228163. [DOI] [PubMed] [Google Scholar]
- 6.Zimmerman HM. Malignant lymphomas of the nervous system. Acta Neuropathol Suppl. 1975;6(Suppl):69–74. doi: 10.1007/978-3-662-08456-4_11. [DOI] [PubMed] [Google Scholar]
- 7.Kumar S, Kumar D, Kaldjian EP, et al. Primary low-grade B-cell lymphoma of the dura: a mucosa associated lymphoid tissue-type lymphoma. Am J Surg Pathol. 1997;21:81–7. doi: 10.1097/00000478-199701000-00009. [DOI] [PubMed] [Google Scholar]
- 8.Galarza M, Gazzeri R, Elfeky HA, Johnson RR., 2nd Primary diffuse large B-cell lymphoma of the dura mater and cranial vault. Case report and literature review. Neurosurg Focus. 2006;21:E10. doi: 10.3171/foc.2006.21.5.11. [DOI] [PubMed] [Google Scholar]
- 9.Riemersma SA, Jordanova ES, Schop RF, et al. Extensive genetic alterations of the HLA region, including homozygous deletions of HLA class II genes in B-cell lymphomas arising in immune-privileged sites. Blood. 2000;96:3569–77. [PubMed] [Google Scholar]
- 10.McCann KJ, Ashton-Key M, Smith K, Stevenson FK, Ottensmeier CH. Primary central nervous system lymphoma: tumor-related clones exist in the blood and bone marrow with evidence for separate development. Blood. 2009;113:4677–80. doi: 10.1182/blood-2008-09-179366. [DOI] [PubMed] [Google Scholar]
- 11.Tun HW, Personett D, Baskerville KA, et al. Pathway analysis of primary central nervous system lymphoma. Blood. 2008;111:3200–10. doi: 10.1182/blood-2007-10-119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roth P, Korfel A, Martus P, Weller M. Pathogenesis and management of primary CNS lymphoma. Expert Rev Anticancer Ther. 2012;12:623–33. doi: 10.1586/era.12.36. [DOI] [PubMed] [Google Scholar]
- 13.Yamada SM, Ikawa N, Toyonaga S, et al. Primary malignant B-cell-type dural lymphoma: Case report. Surg Neurol. 2006;66:539–43. doi: 10.1016/j.surneu.2006.02.033. [DOI] [PubMed] [Google Scholar]
- 14.Tu PH, Giannini C, Judkins AR, et al. Clinicopathologic and genetic profile of intracranial marginal zone lymphoma: a primary low-grade CNS lymphoma that mimics meningioma. J Clin Oncol. 2005;23:5718–27. doi: 10.1200/JCO.2005.17.624. [DOI] [PubMed] [Google Scholar]
- 15.Sanjeevi A, Krishnan J, Bailey PR, Catlett J. Extranodal marginal zone B-cell lymphoma of malt type involving the cavernous sinus. Leuk Lymphoma. 2001;42:1133–7. doi: 10.3109/10428190109097736. [DOI] [PubMed] [Google Scholar]