

CHESS beamline G1 has been newly upgraded for biological X-ray solution scattering.
Keywords: SAXS, high throughput, MacCHESS
Abstract
X-ray scattering of biological macromolecules in solution is an increasingly popular tool for structural biology and benefits greatly from modern high-brightness synchrotron sources. The upgraded MacCHESS BioSAXS station is now located at the 49-pole wiggler beamline G1. The 20-fold improved flux over the previous beamline F2 provides higher sample throughput and autonomous X-ray scattering data collection using a unique SAXS/WAXS dual detectors configuration. This setup achieves a combined q-range from 0.007 to 0.7 Å−1, enabling better characterization of smaller molecules, while opening opportunities for emerging wide-angle scattering methods. In addition, a facility upgrade of the positron storage ring to continuous top-up mode has improved beam stability and eliminated beam drift over the course of typical BioSAXS experiments. Single exposure times have been reduced to 2 s for 3.560 mg ml−1 lysozyme with an average quality factor I/σ of 20 in the Guinier region. A novel disposable plastic sample cell design that incorporates lower background X-ray window material provides users with a more pristine sample environment than previously available. Systematic comparisons of common X-ray window materials bonded to the cell have also been extended to the wide-angle regime, offering new insight into best choices for various q-space ranges. In addition, a quantitative assessment of signal-to-noise levels has been performed on the station to allow users to estimate necessary exposure times for obtaining usable signals in the Guinier regime. Users also have access to a new BioSAXS sample preparation laboratory which houses essential wet-chemistry equipment and biophysical instrumentation. User experiments at the upgraded BioSAXS station have been on-going since commissioning of the beamline in Summer 2013. A planned upgrade of the G1 insertion device to an undulator for the Winter 2014 cycle is expected to further improve flux by an order of magnitude.
1. Introduction
Small-angle X-ray scattering (SAXS) is routinely used to obtain low-resolution structural information of a single species of macromolecules dispersed in a buffer solution. The rising popularity of SAXS in structural biology has been largely driven by an increasing demand for an easy-to-use technique to obtain structural information from such macromolecules in a solution, and by recent technological advances in both software and hardware that make such a technique readily available. Advanced structural modeling algorithms are commonly used for the interpretation of scattering data, and the worldwide availability of high-brightness synchrotron sources provides the necessary flux, while efficient and low-noise pixel-array detectors accurately record scattering events even from dilute systems. A number of synchrotron beamlines worldwide are now capable of this type of X-ray scattering experiment (Pernot et al., 2010 ▶; Kirby et al., 2013 ▶; David & Pérez, 2009 ▶; Smolsky et al., 2007 ▶; Inoue et al., 2013 ▶; Blanchet et al., 2012 ▶; Graceffa et al., 2013 ▶), including the upgraded MacCHESS BioSAXS facility now located at beamline G1 at the Cornell High Energy Synchrotron Source (CHESS).
With the advent of modern third-generation synchrotron sources, higher flux and smaller source sizes are achieved, paving the way for more elaborate solution scattering experiments. Advances in data analysis algorithms have made it possible to reconstruct low-resolution models from small-angle scattering data (Svergun, 2013 ▶; Schneidman-Duhovny et al., 2013 ▶) and to deduce higher-resolution folding patterns from wider-angle scattering data (Makowski, 2010 ▶). In addition, algorithms are now increasingly capable of extracting basic structural information from more challenging systems where traditional X-ray crystallography is not feasible; polydisperse samples (Williamson et al., 2008 ▶), protein–RNA complexes (Daugherty et al., 2010 ▶), as well as investigations of protein structural dynamics in time-resolved experiments (Cho et al., 2010 ▶) are notable examples. The trend towards more sophisticated problems is likely to continue as advances in source properties are made (Graewert & Svergun, 2013 ▶).
In order to more efficiently utilize available X-ray beam time, and to enable higher sample throughput, several beamlines now have automated sample-loading capabilities. Robotic sample loading greatly reduces the sample loading time, increases typical throughput and provides consistent control over the sample volume and environment used for each experiment. For example, the BioSAXS sample loaders at beamlines X33 at the EMBL Hamburg (Round et al., 2008 ▶) and at beamline BM29 at the ESRF (Pernot et al., 2010 ▶) can accommodate 96-well plates in a temperature-controlled environment. Further, several beamlines have demonstrated the capability to couple size-exclusion chromatography (SEC) with SAXS, thus providing a new way to generate a monodisperse sample stream from challenging samples (Watanabe & Inoko, 2013 ▶; Mathew et al., 2004 ▶; Gunn et al., 2011 ▶; David & Pérez, 2009 ▶). A considerable amount of effort has also been placed on reducing background scattering (Kirby et al., 2013 ▶) to further improve the signal-to-noise for weakly scattering samples. In practice, all these approaches are adopted to varying extents at modern solution scattering beamlines.
Here, we outline the upgraded X-ray scattering capabilities of biological macromolecules in solution at the BioSAXS beamline G1 for MacCHESS, previously located at beamline F2 (Nielsen et al., 2012 ▶). In the current setup, an upstream multilayer monochromator splits a 2 mrad fan of radiation from a 49-pole wiggler and provides half to beamline G1. The beam is collimated at the sample location with a FWHM of 250 µm × 250 µm with a flux of 2 × 1011 photons s−1 and a typical single exposure time of 2 s for lysozyme at 3.560 mg ml−1 to resolve basic information, such as R g and molecular weight to within 5%. We recommend repeated exposures (ca 10) to ensure sufficient data quality for structural modeling. A planned facility upgrade will replace the shared 49-pole wiggler with a novel dedicated 1.5 m CHESS Compact Undulator (CCU) (Temnykh et al., 2013 ▶) along with upgrades to upstream X-ray optics, and is expected to improve photon flux at the sample to 2 × 1012 photons s−1 before the start of the Winter 2014 cycle.
The upgrades and improvements in both the hardware and software aspects of the BioSAXS capabilities at MacCHESS pave the way for a new set of experiments, such as microfluidic-based experiments, use of simultaneous SAXS and WAXS to probe changes in domain structures at a higher resolution, as well as experiments that make use of shorter exposure times. We expect that a further improvement in flux due to the undulator upgrades will enable more elaborate in-line SEC–SAXS and SEC–MALS/DLS–SAXS experiments, as well as sub-second exposure times for some systems.
2. Beamline overview
The MacCHESS BioSAXS endstation was moved from the 24-pole wiggler beamline F2 to the 49-pole wiggler beamline G1 in Fall 2013 (Table 1 ▶). G1 receives radiation produced by the positron beam in the Cornell Energy Storage Ring, which operates at 5.3 GeV and 200 mA in continuous top-up mode. The insertion device at G1 delivers a photon flux upwards of 3 × 1012 photons s−1 mm−2 and delivers a 2 mrad fan of radiation (horizontal) with a critical energy of 14.9 keV and a peak magnetic field of 0.8 T. In the G-line optics hutch, an electroless-nickel-plated Glidcop white-beam collimating mirror aids in vertical focusing and reduces the heat load on subsequent optics. A pair of water-cooled W/B4C multilayer monochromators with an energy bandwidth of 1.5% ΔE/E operates in a fixed-exit geometry and splits off the upper half of the beam towards endstation G1. A downstream single-crystal silicon monochromatic mirror provides further vertical focusing (Fig. 1 ▶). A set of slits located directly upstream of the hutch can be set to reduce divergence before beam enters the G1 hutch.
Table 1. Experimental parameters for typical solution scattering experiments at the BioSAXS beamlines F2 and G1.
| Beamline | |||
|---|---|---|---|
| F2 | G1 (current) | G1 (Winter 2014)† | |
| Source | e+, 170200 mA | e+, 200mA | e+, 200mA |
| Insertion device | 24-pole 1.2T wiggler | 49-pole 0.8T wiggler | Cornell Compact Undulator |
| Energy (keV) | 9.881 | 10.5 | 8.0, 11.6 |
| Beam size (m) | 250 250 | 250 250 | 250 250, 100 100 |
| Collimated flux (photons s1) | 1 1010 | 2 1011 | 2 1012 (at 250 250) |
| Sample-to-detector distance (mm) | SAXS: 1500; WAXS: 430 | SAXS: 1500; WAXS: 430 | SAXS: 1500; WAXS: 430 |
| q-range (1) | 0.008 q 0.8 | 0.007 q 0.7 | 0.005 q 0.7 |
| Short exposure time (s) | 1030 | 15 | 1 |
Expected values based on preliminary 2012 undulator tests.
Figure 1.
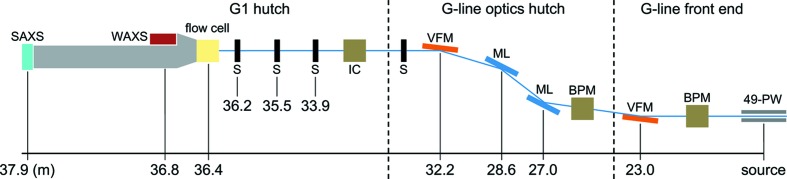
Schematic of the beamline setup at the MacCHESS BioSAXS beamline G1. A pair of vertical focusing mirrors and multilayer monochromators split a 2 mrad fan from the wiggler into the endstation G1 hutch. Abbreviations: 49-pole wiggler (49-PW), beam-position monitor (BPM); vertical-focusing mirror (VFM); multilayer monochromator (ML); slits (S); ion chamber (IC).
The 4.5 m-long experimental hutch at beamline G1 has a source-to-hutch distance of 33.5 m. Two sets of collimating slits are situated at 33.9 m and 35.5 m (0.6 mrad horizontal collimation). An additional set of guard slits is mounted at 36.2 m, directly in front of the sample location, to remove parasitic scatter (Advanced Design Consulting USA Inc., Lansing, NY, USA), all of which have been mounted in vacuo to reduce air scattering. In a standard configuration, the BioSAXS sample cell is located at a distance of 36.4 m where it receives a 250 µm × 250 µm (FWHM) collimated monochromatic beam with a typical photon flux of 2 × 1011 photons s−1 at 10.5 keV. At this energy, the optimum path length for X-ray exposures is 2 mm, as calculated by the X-ray mass attenuation coefficient of water.
2.1. SAXS and WAXS configuration
The configuration of identical Pilatus 100K-S detectors (Dectris, Baden, Switzerland) for SAXS and WAXS allows for simultaneous data collection with a combined q ranging from 0.007 to 0.7 Å−1 where q = 4πsin(θ)/λ, capturing both the overall shape as well as details of the internal structure of macromolecules. These detectors have a pixel size of 172 µm × 172 µm and an active area of 83.8 mm × 33.5 mm (487 × 195 pixels). To affix the SAXS (Fig. 1 ▶, cyan) and WAXS (Fig. 1 ▶, red) detectors at their respective exit ports on an evacuated flight path, a custom aluminium beam pipe was fabricated with a single entrance aperture facing the BioSAXS flow cell or sample area (Fig. 1 ▶, yellow) and exit apertures for both small- and wide-angle scattering events (Fig. 2 ▶). Aluminium (6061 alloy) was chosen over stainless steel to reduce possible signal from iron fluorescence. All detector apertures are sealed with 2 mil-thick Mylar film (99% transmission at 10.5 keV), and the beam pipe is evacuated to a pressure of <0.3 Pa prior to scattering measurements. The beam pipe is mounted on a track fastened to the optical table and can be moved upstream or downstream to increase or decrease the length of the sample area.
Figure 2.

(a) Illustration of the aluminium flight tube (grey), SAXS (cyan) and WAXS (red) detectors as well as the BioSAXS sample flow cell (yellow). (b) Full SAXS and WAXS scattering curves of AgBE, showing an overlap between 0.20 and 0.28 Å−1.
The SAXS detector is oriented vertically and located off-axis with the beam hitting the top-center of the detector. The sample-to-detector distance is 1500 mm and the detector is 2 mm removed from the SAXS exit port of the beam pipe. In this configuration the SAXS detector captures scattering events in the q-range 0.007–0.28 Å−1 with a 63° azimuthal range at a q of 0.11 Å−1 and a 30° azimuthal range at a q of 0.22 Å−1. A molybdenum beam stop with a VTS3085 photodiode (Excelitas Technologies, Waltham, MA, USA) is located inside the beam pipe directly at the SAXS exit port. The photodiode current is automatically integrated during exposure and stored in corresponding SAXS data files. The WAXS detector is oriented horizontally and located off-axis above the direct beam at a sample-to-detector distance of 430 mm, and is at a 2 mm distance to the WAXS exit port of the beam pipe. The WAXS detector captures scattering events in the q-range 0.20–0.7 Å−1 with a 104° azimuthal range at a q of 0.32 Å−1 and a 128° azimuthal range at a q of 0.54 Å−1. The overlap in q-range between 0.20 and 0.28 Å−1 aids in appending WAXS and SAXS scattering data. Calibration is typically performed using a silver behenate standard embedded in a plastic sample chip.
2.2. Hardware and software controls
Most beamline motors (e.g. for monochromator, shutters, slits, sample cell) are controlled by EPICS (Argonne National Laboratory, Lemon, IL, USA) and SPEC (Certified Scientific Software, Cambridge, MA, USA) from a control server located inside the hutch. Although all motor positions are optimized by beamline staff prior to the start of a user’s experiment and typically do not need further adjustment, users have access to several basic SPEC routines for troubleshooting, to realign optics or to further optimize slit positions if required. Further, SPEC macro commands can be used to initiate data collection on both SAXS and WAXS detectors for a pre-set amount of time.
The SAXS and WAXS detectors are controlled by separate dedicated data collection servers located inside the hutch. Data collection can be initiated from a workstation located outside the hutch either via SPEC, the command line interface or in-house-developed control software (Nielsen et al., 2012 ▶) to trigger both detectors to simultaneously start data collection for a single or multiple exposures, optionally with pauses of a predefined length between exposures. Data are automatically copied in real time from the data collection servers to a data processing computer.
2.3. BioSAXS sample preparation laboratory
A BioSAXS sample preparation laboratory space is available to users for last-minute sample preparation and characterization prior to BioSAXS measurements. This includes a MilliPore ultrapure water purification system and additional wet-chemistry laboratory equipment. An ÄKTA Purifier HPLC with several SEC columns and built-in UV absorptiometry as well as a fraction collector are available for size fractionation. Optionally, in-line SEC–MALS/DLS–dRI (Wyatt, Santa Barbara, CA, USA) may be used for molecular weight characterization and monodispersity validation prior to BioSAXS measurements, as well as an alternative method for determining concentration. A NanoVue (GE Life Sciences, Piscataway, NJ, USA) and Direct Detect IR spectrophotometer (Millipore, Billerica, MA, USA) are available as alternate faster methods for concentration determination. A cooled centrifuge may be used to spin down samples as an alternate means to remove aggregates and higher-order oligomers. Additional laboratory equipment is available at Cornell’s Department of Molecular Biology and Genetics located a short walk from the main CHESS facility with prior notice. To accommodate for shipping and short-term storage of samples, a 277 K refrigerator and a 188 K freezer are now available to scheduled users.
3. Sample handling
An in vacuo temperature-controlled sample area is highly desirable, if not required, for most X-ray scattering experiments of biological macromolecules in solution. As such, the upstream and downstream flight tubes with respect to the sample area have been designed to allow for custom sample compartments connected via KF-40 vacuum fittings to meet the specific needs of a variety of scattering experiments. The variable distance of the upstream flight tube with respect to the sample area allows for sample cell compartments ranging from 2 to 30 cm in width. The vacuum conditions (<0.3 Pa) in the upstream and downstream flight tubes can be extended into custom sample compartments, if desirable. Additional supporting custom equipment can be placed either in the direct path of the beam (such as sample cells and additional chambers) or directly onto the optical table adjacent to the sample cell (such as pumps, robotic sample changers and mixers). The most commonly used setup includes a position-adjustable temperature-controlled in-vacuum sample cell (Nielsen et al., 2012 ▶). Updated to hold a wide range of sample cells via face-sealing O-rings, this enclosure routinely accommodates flow cells of thickness 1–5 mm with X-ray transparent windows of up to 10 mm in diameter.
3.1. BioSAXS flow cell
The disposable BioSAXS flow cell features a multi-layered design with a 1 mm-tall by 2 mm-thick by 64 mm-long square channel (Gillilan et al., 2013 ▶). The outer walls can be made of 0.5–2 mm-thick PMMA with a 2 mm-tall and 5 mm-wide aperture. One of the outer walls also contains a 1 mm-diameter round drain port. A third, central, layer is composed of 2 mm-thick PMMA and forms the actual sample flow channel, thus creating a 2 mm path length for X-ray exposures. One end of this central channel layer forms a funnel to facilitate robotic sample loading while the other end connects to the drain port on the outer wall. Sandwiched between each wall is a 25 µm-thick window layer of polystyrene film that runs the length of the entire chip (Fig. 3a ▶). Although early designs utilized a layer adhesive film to bind PMMA and polystyrene pieces together, the current cell design incorporates a solvent vapour bonding technique (Ogilvie et al., 2010 ▶) to bond all components without the use of adhesives. Specifically, the outer PMMA walls are exposed on the central-wall-facing side to chloroform vapour at room temperature (RT) for 8 min. The polystyrene window material is then applied to the vapour-exposed PMMA walls and allowed to cure in air for at least 15 min. The central channel layer is subsequently exposed on both sides to chloroform vapour for 10 min at RT and pressed between the cured window pieces. To facilitate the pressing process, a custom alignment jig is used to ensure proper alignment of all components. Meanwhile, a constant stream of dry air is pumped through the channel to prevent damage to the X-ray windows due to outgassing of chloroform vapour from the assembled parts.
Figure 3.

(a) Schematic of the BioSAXS flow cell, showing the basic design elements. Two outer layers of PMMA (grey) provide structural support to the flow cell, while the central channel of PMMA contains the flow channel and effectively sets the X-ray path length. Sandwiched between the PMMA layers are X-ray transparent window layers of 25 µm-thick polystyrene (yellow). (b) Side view of the BioSAXS flow cell enclosed in the flow cell holder. A mirror on top of the flow cell holder allows a camera to capture video of the area of the flow cell where X-ray exposure occurs. (c) Configuration of the BioSAXS flow cell and flow cell holder at beamline G1. (d) Composition of detector image wedges of five candidate window materials in real space (equivalent q-range from 0 to 0.25 Å−1). High scattering intensities are indicated in white, low scattering intensities in black. Abbreviations: GL = quartz, KP = kapton, MI = mica, PS = polystyrene, SP = special glass. (e) Background-corrected SAXS and WAXS scattering curves of the five candidate window materials.
The simple design of this flow cell makes it cheap and fast to fabricate and easy to redesign. In addition, the dimensions of the flow cell were chosen such that the flow cell fits into our custom BioSAXS flow cell holder [Figs. 3(b) and 3(c) ▶]. In this manner, vacuum is maintained on the upstream side from the flow cell window layer to the upstream hutch entrance port, and on the downstream side from the flow cell window layer to the end of the flight tube. Further, sample volumes as small as 15 µL (7.5 mm-long plug) are routinely loaded using the robotic sample loader. In this setup, the robotic pipetter transfers a sample volume from a 96-well plate directly into the sample cell. Alternatively, a sample volume can be manually loaded into the sample cell using a manual pipetter. Once the sample is loaded into the sample cell, a pump attached to the exit port of the sample cell can be used to position the sample volume in the path of the X-ray beam. A mirror affixed to the top of the sample cell holder provides a top view of the sample cell channel and sufficient contrast to distinguish a sample plug, thereby aiding in positioning the sample plug directly in front of the X-ray beam. To facilitate oscillation of the sample plug, the recommended sample volume is 30 µL (15 mm-long plug). The scattering characteristics of several window materials were measured in the SAXS and WAXS ranges. Although quartz glass is a commonly used window material, several other candidate materials were found to outperform quartz, most notably in the SAXS regime [Figs. 3(d) and 3(e) ▶]. The transmission through 25 µm-thick polystyrene windows offered superior performance in the SAXS region up to ∼0.03 Å−1, and nearly matched 10 µm special glass up to ∼0.36 Å−1. The polystyrene window outperformed 25 µm mica over the entire measured range and was superior to 7 µm polyimide up to ∼0.12 Å−1. Although polyimide exhibited overall less scatter in the WAXS range, a strong scattering effect centered at 0.39 Å−1 makes it a less attractive window material for most applications. Since polystyrene is transparent to visible light, a camera can be used to directly observe the flow of sample plugs inside the flow cell to ensure proper operation of the device.
The lifetime of the SAXS flow cell is limited by sample aggregation onto the window resulting from radiation damage despite using aggressive cleaning protocols. The low cost and ease of manufacturing of this particular design makes it feasible to use a new cell for each experiment, if necessary. However, in our experience the performance of these flow cells is typically without deterioration for 24–48 h. Further, changing flow cells and readying the station for data collection is typically performed in 10 min, and can be performed by users.
3.2. Continuous flow-through and oscillation mode
At a flux density of 2 × 1011 photons s−1 on a 250 µm × 250 µm area with a path length of 2 mm, the absorbed dose is as high as 2300 Gy s−1 for a standard protein solution (Meisburger et al., 2013 ▶; Kuwamoto et al., 2004 ▶). Considering that structural changes are typically observable at doses over 400 Gy, it becomes necessary to reduce the absorbed dose per volume well below that limit for most protein solutions. The design of the flow cell and supporting apparatus (inlets, pump, etc.) offers two options to limit radiation damage to the sample. In oscillation mode, a fixed volume of sample is continuously oscillated through a narrow channel in the path of the beam at 2–4 µL s−1. Thus, the total dose is spread over a volume much larger (15 µL) than the instantaneously illuminated volume (0.125 µL), effectively reducing the absorbed dose by as much as two orders of magnitude (Nielsen et al., 2012 ▶). Another option is to continuously flow a sample volume past the X-ray beam and out of the flow cell. In either mode, we suggest collecting at least ten successive exposures. Upon confirmation that no radiation damage has occurred, the exposures are then averaged.
3.3. Robotic loading and temperature control of samples in a 96-well plate
A commercial pipetting robot (Hudson Robotics Inc., Springfield, NJ, USA) is used to load samples (15–60 µL) from a 96-well sample plate into the BioSAXS flow cell as previously described (Nielsen et al., 2012 ▶). The robot can be entirely controlled from outside the hutch using Robocon control software. Robocon also offers an option to clean the BioSAXS flow cell according to a pre-set cleaning protocol. The cleaning protocol, as outlined previously (Nielsen et al., 2012 ▶), is now optionally followed by a short purge (10 s) with compressed nitrogen, in order to blow-dry the flow cell. The entire cleaning procedure can be monitored via a camera mounted pointing at the flow cell assembly, as well as via the camera looking down and into the flow cell from the top. After the cleaning is completed, a background can be collected to confirm the cleanliness of the flow cell.
Temperature control of the 96-well sample plate from 263 K to RT (293 K) is accomplished using a vortex tube (Model 106, Vortec, Cincinnati, OH, USA). The vortex tube separates compressed air (∼80 p.s.i.) into cold and warm air streams, while a tunable muffler (Model 208MH, Vortec) attached to the warm air stream is used to manually control the temperature of the cold stream by regulating the cold fraction. When the cold stream is attached to the integrated tubing of the sample plate holder, the samples will reach a desired temperature after approximately 2 min with a stability of 1 K. Flow cell temperature is controlled in the 277–313 K range by circulating water through the copper block in the flow cell holder (Fig. 3b ▶).
3.4. Processing of SAXS and WAXS data
Analysis of scattering data can be performed on beamline data analysis computers equipped with an updated version of BioXTAS RAW (Nielsen et al., 2009 ▶). This software can be used to perform all data reduction steps in a graphical environment, including finding/setting the beam center, q-range calibration, masking, radial averaging, buffer subtraction, current normalization, water normalization and merging SAXS and WAXS data. The software can now be set to operate in online mode, in which new scattering patterns are automatically loaded, masked, radially integrated, buffer subtracted and plotted in the graphical interface during data collection. Further improvements include a revamped radial averaging algorithm, which now incorporates pixel supersampling of the scattering pattern as well as pixel splitting to assign fractions of individual pixel values to their respective q-bins. The resulting scattering plots can be exported in a variety of file formats. Further analysis and modeling of scattering data can be performed using the latest version of ATSAS installed on the data analysis computers (Petoukhov et al., 2012 ▶).
3.5. Sample data sets
30 µL volumes of lysozyme (LYS) and glucose isomerase (GI) were exposed to X-rays (λ = 1.2439 Å) in a continuous oscillating flow mode through a 2 mm-diameter BioSAXS flow cell for 20 successive exposures of 2 s at RT. Comparing individual exposures confirmed no radiation damage had occurred. Scattering curves were averaged followed by subtraction of buffer scattering. SAXS and WAXS curves were merged to arrive at a continuous q-range of 0.0073–0.7 Å−1. For 3.560 mg ml−1 LYS (Fig. 4a ▶), the scattering curve overlays well with a CRYSOL fit of a representative crystallographic dataset for LYS (PDB: 1lyz). In addition, a Guinier fit up to a qR g of 1.3 resolves an R g of 1.43 nm, matching the known R g of 1.43 nm for LYS (Mylonas & Svergun, 2007 ▶). For 0.390 mg ml−1 GI, the SAXS region up to 0.10 Å−1 overlays well with a CRYSOL fit (PDB: 1oad), but exhibits more noise up to 0.25 Å−1 (Fig. 4b ▶). The Guinier fit resolves an R g of 3.32 nm, closely matching the actual R g of 3.25 nm for GI (Mylonas & Svergun, 2007 ▶).
Figure 4.

(a) Scattering plot of 3.560 mg ml−1 lysozyme taken from an average of 20 successive 2 s exposures (red). A CRYSOL fit (black) using PDB 1lyz overlays well with the scattering data. Guinier analysis (inlay) resolves an R g of 1.43 nm and an average I/σ of 65. (b) Scattering plot of 0.390 mg ml−1 glucose isomerase taken from an average of 20 successive 2 s exposures (red). A CRYSOL fit (black) using PDB 1oad overlays well with the scattering data up to 0.10 Å−1. Guinier analysis (inlay) resolves an R g of 3.32 nm with an average I/σ of 210.
4. Signal quality and detection limits
An assessment of scattering data quality in the Guinier region as a function of data collection time was performed by measuring the average quality factor (QF), I/σ, of scattering data in the Guinier region as a function of data collection time from a dilution series of LYS. For a 3.560 ± 0.008 mg ml−1 sample and dilutions of 1.780, 0.890, 0.445 and 0.223 mg ml−1, the QF in their respective Guinier regions were computed and plotted as a function of data collection time. Total exposures of 2, 4, 10, 20 and 40 s were used (Fig. 5a ▶). In all cases scattering profiles were generated by averaging successive 2 s exposures with σ values scaled appropriately. For a second set of series dilutions to 0.223, 0.056 and 0.028 mg ml−1, QF values were calculated in a similar fashion but for longer data collection times, namely 20, 40, 100, 200, 400, 800 and 1600 s by averaging successive 20 s exposures. Data for 0.223 mg ml−1 samples were collected by both short (2 s) and long (20 s) exposures to ensure that these data sets are consistent.
Figure 5.

(a) Scattering data quality factor (QF), I/σ, as a function of data collection time for a range of dilutions of lysozyme. Data points for individual dilutions (indicated by markers) were best described using a power function (solid line). (b) Predicted correlation between sample concentration and estimated required data collection time for arbitrary QF for lysozyme.
For each dilution series, a power function of the form QF = at b could be fit to best describe the data, where parameter a described the scaling factor, while parameter b described the slope of the function and t the exposure time in seconds. It should be noted that the scaling factor is beamline specific as it indicates beamline performance in terms of signal quality (e.g. signal-to-noise). The exponent will generally approach 0.5, indicating an ideal increase in signal quality with measurement time, namely an increase proportional to the square root of the number or duration of measurements. Based on the dilution series, the exponent remained almost constant for all series at 0.4987 ± 0.0143. Further, the scaling factors of the fitted power functions were found to be dependent on sample concentration c (mg ml−1). Specifically, when plotted as a function of concentration, the scaling factors were best described by a second-degree polynomial of the form a = −0.5805c 2 + 6.1726c.
Subsequently, it was possible to estimate the required data collection time to achieve a desired QF in the Guinier region as a function of sample concentration for LYS. The power function exponent describing the gain in signal quality with collection time was held constant at 0.4987. Further, by using the second-degree polynomial fit describing beamline signal quantity, it was possible to calculate corresponding scaling factors for arbitrary sample concentrations. Given both these parameters, power functions were generated describing the relationship between the estimated required data collection time and sample concentration for arbitrary QF, namely 2, 5, 10, 20, 50 and 100 (Fig. 5b ▶). Although these plots are specific to beamline G1 at CHESS, this method of estimating required data collection time can be useful to BioSAXS users at other facilities. In addition, this analysis gives insight into reasonable detection limits for dilute samples. For example, to attain a QF of 10 for a 3.00 mg ml−1 LYS sample, the estimated data collection time is ∼0.6 s, while for a 0.30 mg ml−1 LYS sample the estimated data collection time to achieve a similar QF is ∼31 s. It should be noted that this analysis does not take into account potential sample aggregation at high concentrations and beam damage at longer exposure times. For proteins other that LYS, the required data collection time to yield a particular QF will scale inversely with the molecular weight squared, while the slope of the plots will remain unchanged. For example, GI has a molecular weight that is 12.1 times greater than that for LYS, and thus the required data collection time to achieve a QF of 10 for a 0.30 mg ml−1 sample will be ∼146 times shorter, or ∼0.2 s.
5. Facility access procedure
Interested users may apply for access to the BioSAXS beamline through the CHESS online express mode proposal form (http://express.chess.cornell.edu/EM_form.php) at any time. Proposals are peer-reviewed on a quarterly basis and scored based on technical difficulty and scientific impact. Particular emphasis is given to non-standard experiments that encourage the incorporation of new hardware and/or software methods or unique uses of the existing infrastructure. Successful requests are typically scheduled for beam time within three months of the initial request, although users are encouraged to contact scientific staff to assess feasibility and send in test samples prior to submitting beam time proposals. Users new to BioSAXS are encouraged to attend our annual three-day BioSAXS Essentials workshop, which includes lectures on the fundamentals and advanced topics in BioSAXS as well as actual hands-on experience at the beamline where attendees can test their own samples (http://www.macchess.cornell.edu/MacCHESS/biosaxs.html).
6. Conclusion
We have described the upgraded BioSAXS beamline for MacCHESS located at CHESS endstation G1. The BioSAXS beamline now receives a synchrotron X-ray beam from a more powerful 49-pole wiggler and improved flux to 2 × 1011 photons s−1. In addition, a storage ring upgrade to continuous top-up mode has improved beam current stability and reduced beam drift, thereby further improving signal stability at the sample location. Together, these factors have reduced single exposure time from ∼30 s at previous beamline F2 to 2 s for 3.560 mg ml−1 LYS with an average quality factor I/σ of 20 in the Guinier region, and increased sample throughput for batch experiments. We recommend repeated exposures (ca 10) to ensure sufficient data quality for structural modeling. In addition, a redesigned BioSAXS flow cell offers low background scattering and can be used in conjunction with a robotic sample loader for high-throughput automated BioSAXS experiments. To support BioSAXS user activities, a dedicated sample preparation laboratory has been outfitted with wet chemistry and protein characterization equipment.
An assessment of scattering data quality as a function of sample dilution and concentration at BioSAXS beamline G1 has made it possible to estimate the required data collection time to achieve a desired QF in the Guinier region for a particular sample concentration. This method of estimating required data collection time can assist in the planning of BioSAXS experiments, and gives insight into reasonable detection limits for unusually dilute samples. Although these estimates are specific for BioSAXS beamline G1 at CHESS, similar analysis can be performed elsewhere.
The upgraded BioSAXS station has been operational since Fall 2013. We anticipate that an upcoming upgrade to undulator insertion devices will further reduce exposure times to below a second.
Acknowledgments
We thank Jim Savino for designing and fabricating the 96-well plate holder. This work is based upon research conducted at the G1 Station of the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation (NSF) and the National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS) under NSF award DMR-0936384, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by NIH/NIGMS award GM-103485. This work was performed in part at the Cornell NanoScale Facility, a member of the National Nanotechnology Infrastructure Network, which is supported by the National Science Foundation (Grant ECCS-0335765).
References
- Blanchet, C. E., Zozulya, A. V., Kikhney, A. G., Franke, D., Konarev, P. V., Shang, W., Klaering, R., Robrahn, B., Hermes, C., Cipriani, F., Svergun, D. I. & Roessle, M. (2012). J. Appl. Cryst. 45, 489–495.
- Cho, H. S., Dashdorj, N., Schotte, F., Graber, T., Henning, R. & Anfinrud, P. (2010). Proc. Natl Acad. Sci. USA, 107, 7281–7286. [DOI] [PMC free article] [PubMed]
- Daugherty, M. D., Booth, D. S., Jayaraman, B., Cheng, Y. & Frankel, A. D. (2010). Proc. Natl Acad. Sci. USA, 107, 12481–12486. [DOI] [PMC free article] [PubMed]
- David, G. & Pérez, J. (2009). J. Appl. Cryst. 42, 892–900.
- Gillilan, R., Cook, M., Temnykh, G., Møller, M. & Nielsen, S. (2013). Trans. Am. Crystallogr. Assoc. Symp. 44, 40–50.
- Graceffa, R., Nobrega, R. P., Barrea, R. A., Kathuria, S. V., Chakravarthy, S., Bilsel, O. & Irving, T. C. (2013). J. Synchrotron Rad. 20, 820–825. [DOI] [PMC free article] [PubMed]
- Graewert, M. A. & Svergun, D. I. (2013). Curr. Opin. Struct. Biol. 23, 748–754. [DOI] [PubMed]
- Gunn, N. J., Gorman, M. A., Dobson, R. C. J., Parker, M. W. & Mulhern, T. D. (2011). Acta Cryst. F67, 336–339. [DOI] [PMC free article] [PubMed]
- Inoue, K., Doutch, J. & Terrill, N. (2013). Bunseki Kagaku, 62, 565–570.
- Kirby, N. M., Mudie, S. T., Hawley, A. M., Cookson, D. J., Mertens, H. D. T., Cowieson, N. & Samardzic-Boban, V. (2013). J. Appl. Cryst. 46, 1670–1680.
- Kuwamoto, S., Akiyama, S. & Fujisawa, T. (2004). J. Synchrotron Rad. 11, 462–468. [DOI] [PubMed]
- Makowski, L. (2010). J. Struct. Funct. Genom. 11, 9–19. [DOI] [PMC free article] [PubMed]
- Mathew, E., Mirza, A. & Menhart, N. (2004). J. Synchrotron Rad. 11, 314–318. [DOI] [PubMed]
- Meisburger, S. P., Warkentin, M., Chen, H. M., Hopkins, J. B., Gillilan, R. E., Pollack, L. & Thorne, R. E. (2013). Biophys. J. 104, 227–236. [DOI] [PMC free article] [PubMed]
- Mylonas, E. & Svergun, D. I. (2007). J. Appl. Cryst. 40, s245–s249.
- Nielsen, S. S., Møller, M. & Gillilan, R. E. (2012). J. Appl. Cryst. 45, 213–223. [DOI] [PMC free article] [PubMed]
- Nielsen, S. S., Toft, K. N., Snakenborg, D., Jeppesen, M. G., Jacobsen, J. K., Vestergaard, B., Kutter, J. P. & Arleth, L. (2009). J. Appl. Cryst. 42, 959–964.
- Ogilvie, I. R. G., Sieben, V. J., Floquet, C. F. A., Zmijan, R., Mowlem, M. C. & Morgan, H. (2010). J. Micromech. Microeng. 20, 065016.
- Pernot, P., Theveneau, P., Giraud, T., Fernandes, R. N., Nurizzo, D., Spruce, D., Surr, J., McSweeney, S., Round, A., Felisaz, F., Foedinger, L., Gobbo, A., Huet, J., Villard, C. & Cipriani, F. (2010). J. Phys. Conf. Ser. 247, 012009.
- Petoukhov, M. V., Franke, D., Shkumatov, A. V., Tria, G., Kikhney, A. G., Gajda, M., Gorba, C., Mertens, H. D. T., Konarev, P. V. & Svergun, D. I. (2012). J. Appl. Cryst. 45, 342–350. [DOI] [PMC free article] [PubMed]
- Round, A. R., Franke, D., Moritz, S., Huchler, R., Fritsche, M., Malthan, D., Klaering, R., Svergun, D. I. & Roessle, M. (2008). J. Appl. Cryst. 41, 913–917. [DOI] [PMC free article] [PubMed]
- Schneidman-Duhovny, D., Hammel, M., Tainer, J. A. & Sali, A. (2013). Biophys. J. 105, 962–974. [DOI] [PMC free article] [PubMed]
- Smolsky, I. L., Liu, P., Niebuhr, M., Ito, K., Weiss, T. M. & Tsuruta, H. (2007). J. Appl. Cryst. 40, s453–s458.
- Svergun, D. (2013). NATO Science for Peace and Security Series A: Chemistry and Biology, Advancing Methods for Biomolecular Crystallography, edited by R. Read, A. G. Urzhumtsev and V. Y. Lunin, pp. 343–351: Springer Netherlands.
- Temnykh, A., Dale, D., Fontes, E., Li, Y., Lyndaker, A., Revesz, P., Rice, D. & Woll, A. (2013). J. Phys. Conf. Ser. 425, 032004.
- Watanabe, Y. & Inoko, Y. (2013). J. Chromatogr. A, 1303, 100–104. [DOI] [PubMed]
- Williamson, T. E., Craig, B. A., Kondrashkina, E., Bailey-Kellogg, C. & Friedman, A. M. (2008). Biophys. J. 94, 4906–4923. [DOI] [PMC free article] [PubMed]