

Abstract
Background and Purpose
The Eph receptor tyrosine kinases and their ephrin ligands are key players in tumorigenesis and many reports have correlated changes in their expression with a poor clinical prognosis in many solid tumours. Agents targeting the Eph-ephrin system might emerge as new tools useful for the inhibition of different components of cancer progression. Even if different classes of small molecules targeting Eph-ephrin interactions have been reported, their use is hampered by poor chemical stability and low potency. Stable and potent ligands are crucial to achieve robust pharmacological performance.
Experimental Approach
UniPR129 (the L-homo-Trp conjugate of lithocholic acid) was designed by means of computational methods, synthetized and tested for its ability to inhibit the interaction between the EphA2 receptor and the ephrin-A1 ligand in an elisa binding study. The ability of UniPR129 to disrupt EphA2-ephrin-A1 interaction was functionally evaluated in a prostate adenocarcinoma cell line and its anti-angiogenic effect was tested in vitro using cultures of HUVECs.
Key Results
UniPR129 disrupted EphA2-ephrin-A1 interaction with Ki = 370 nM in an elisa binding assay and with low micromolar potency in cellular functional assays, including inhibition of EphA2 activation, inhibition of PC3 cell rounding and disruption of in vitro angiogenesis, without cytotoxic effects.
Conclusions and Implications
The discovery of UniPR129 represents not only a major advance in potency compared with the existing Eph-ephrin antagonists but also an improvement in terms of cytotoxicity, making this molecule a useful pharmacological tool and a promising lead compound.
Introduction
The Eph (erythropoietin-producing hepatocellular carcinoma) receptors represent the largest family of receptor tyrosine kinases in humans (Pasquale, 1997) and are activated by a class of membrane-associated ligands called ephrins. On the basis of the extracellular domain homology and ligand-binding properties, the Eph receptors are divided into two classes, EphA and EphB, both featuring an ephrin-binding globular domain and other extracellular domains, a relatively short transmembrane domain and an intracellular cytoplasmic signalling region containing a tyrosine kinase domain. The Eph receptor ligands are also divided in two groups, ephrin-A and ephrin-B, which differ in their linkage to the cell membrane. While ephrin-A ligands are tethered to the membrane by a glycosylphosphatidylinositol linkage, ephrin-B ligands possess a single transmembrane domain and a short cytoplasmic domain that contains a cytosolic PDZ-binding motif. In humans, the nine EphA receptors (EphA1–A8, EphA10) bind the five ephrin-A ligands (ephrin-A1–A5), while the five EphB receptors (EphB1–EphB4, EphB6) bind the three ephrin-B ligands (ephrin-B1–B3; nomenclature follows Alexander et al., 2013). Eph-ephrin binding within the same class is highly promiscuous and examples of inter-class binding have also been reported (Himanen et al., 2004; Pasquale, 2005).
A unique feature of the Eph-ephrin system is its ability to generate bidirectional signals after Eph-ephrin interaction at the cell-cell interface. The assembly of Eph-ephrin complex activates a signalling cascade both in the Eph receptor-expressing cells (‘forward signalling’) and in the ephrin-expressing cells (‘reverse signalling’) (Mellitzer et al., 1999; Xu et al., 1999). In several cellular environments, Eph bidirectional signalling has been found to suppress cancer cell adhesion, migration, invasion and growth. In these cases, Eph receptor signalling was low in cancer cells due to the inability of receptors and ligands to interact effectively or to an imbalance of Eph and ephrin expression. Conversely, Eph receptors and ephrins can also promote cancer progression through mechanisms that mainly depend on crosstalk with oncogenic signalling pathways (Pasquale, 2010). In particular, the Eph-ephrin system is overexpressed in the tumour vasculature where, along with VEGF and angiopoietin pathways, it promotes angiogenesis (Héroult et al., 2006; Tandon et al., 2011). While VEGF is known for regulating the early steps of angiogenesis by stimulating the formation of a primitive vascular network, angiopoietin and ephrins contribute to later steps of vascular development including vessel branching and remodelling (Pasquale, 2010).
The therapeutic strategies currently used to block tumour angiogenesis involve the use of VEGF receptor (VEGFR) pathway inhibitors, such as the monoclonal antibody bevacizumab and the kinase inhibitors sunitinib and sorafenib. Although this approach has shown early clinical efficacy (Ferrara et al., 2005; Sebolt-Leopold and English, 2006; Jubb and Harris, 2010), it was often followed by development of tumour resistance and rapid and lethal tumour progression (Pàez-Ribes et al., 2009).
Due to its key role in blood vessel formation, the Eph-ephrin system might be a target of anti-angiogenic therapies and possibly to overcome resistance to anti-VEGFR therapies. In the last decade, intense drug discovery efforts have produced several classes of small molecules able to inhibit the kinase domain of Eph receptors (Lafleur et al., 2009; van Linden et al., 2012). Among the large number of identified compounds, NVP-BHG712 has emerged as the most promising inhibitor, being able to inhibit angiogenesis by targeting the EphB4 receptor (Martiny-Baron et al., 2010; Noberini et al., 2012b).
Unfortunately, the use of kinase inhibitors is hampered by critical pharmacological issues, including: (i) a poor selectivity, because these ATP mimicking agents typically target several different kinase families; (ii) poor access to their intracellular targets, a process that is strongly influenced by their physicochemical properties; and (iii) a limited efficacy due to competition with intracellular ATP, which under physiological conditions is present at millimolar concentrations.
Alternatively, Eph signalling can be blocked by the use of protein-protein inhibitors (PPIs) able to prevent the interaction between the extracellular ligand-binding domain of Eph receptors and their ephrin ligands. PPIs offer some practical advantages over classical kinase inhibitors because: (i) they can block the activation of both the Eph receptor (forward signalling) and the ephrin ligand (reverse signalling); (ii) they do not need to enter the cells to exert their pharmacological actions; and (iii) they can be significantly more selective than kinase inhibitors.
Currently identified PPIs working on the Eph-ephrin system include chimeric proteins (i.e. soluble forms of EphA2 or ephrin-A1), antibodies and peptides, whose use is hampered by poor metabolic stability and pharmacokinetic parameters (Tognolini et al., 2013a). Few classes of non-peptidic PPIs of the Eph-ephrin system acting in the low micromolar range have been recently reported. However, their precise mechanism of action remains unclear, probably involving compound oxidation (Noberini et al., 2008; 2011).
In this situation, we believe that the availability of a new generation of low molecular weight PPIs, able to interfere with the Eph-ephrin system with a clear mechanism of action, could significantly help the development of pharmacological tools to investigate and exploit the therapeutic opportunities offered by the Eph-ephrin system. The ephrin-binding site of the Eph receptors (Himanen et al., 2009) has favourable features allowing high-affinity binding of small molecules, as shown by lithocholic acid (LCA) (Giorgio et al., 2011; Mohamed et al., 2011). LCA is a natural PPI of the Eph-ephrin system and protects human cardiomyocytes from apoptosis by decreasing EphA2 tyrosine phosphorylation (Jehle et al., 2012). LCA has been employed as a template structure to design other PPIs (Tognolini et al., 2012b) leading to the identification of PCM126 (now UniPR126) as a antagonist of the EphA2 receptor, effective in low micromolar concentrations. In the present work, we report the pharmacological properties of UniPR129, a novel PPI designed starting from the in silico model of the EphA2-UniPR126 complex (Incerti et al., 2013). We show that UniPR129 exhibits an unprecedented pharmacological profile among low MW compounds targeting the Eph-ephrin system because it disrupts EphA2-ephrin-A1 binding with a competitive mechanism of action and an inhibition constant in the high nanomolar range.
Methods
Docking simulation and MM-GBSA calculations
The crystal structure of the EphA2–ephrin-A1 complex (PDB accession 3HEI) (Himanen et al., 2009) was used for molecular modelling simulations. Chains A and B of the crystal structure were extracted and subsequently processed with the Protein Preparation Wizard tool implemented in Maestro (version 9.2, Schrodinger, LLC, New York, NY, USA), which was used to assign bond orders and to add missing hydrogen atoms. The overall hydrogen bonding network was optimized by sampling the side chain amide orientation of asparagines and glutamines, the hydroxyl and thiol groups of serine, threonine and cysteine residues and water molecules, and also by adjusting the tautomerization states of histidines. A final restrained minimization was conducted with the OPLS2005 force field (Jorgensen et al., 1996) to a root mean square deviation value of 0.3 Å calculated on protein heavy atoms. A subset of conserved water molecules delimiting the binding site region of the EphA2 receptor was retained, whereas all the other water molecules as well as the ephrin-A1 segment were deleted. UniPR126 and UniPR129 were built with Maestro 9.2 and energy minimized applying the OPSL2005 force field and the GBSA water solvation treatment to a gradient of 0.05 kj·mol−1·Å−1. The minimized ligand structures were then docked within the EphA2 receptor using Glide (version 5.7, Schrodinger) in standard precision mode and applying a hydrogen bond constraint between the carboxylic group of the compound and the guanidine group of Arg103. Docking grids were centred within the EphA2 receptor-binding site, in a region delimited by Arg103, Phe156, and Arg159, and the dimensions of enclosing and bounding boxes were set to 29.4 and 10.0 Å respectively. If Van der Waals (VdW) radii of protein atoms were not scaled, a scaling factor of 0.8 was applied to the VdW radii of ligand atoms having partial atomic charges lower than |0.15|. Twenty docking poses were retained for each ligand and ranked according to the Emodel score. The best-scored poses of compounds UniPR126 and UniPR129 (Figure 1) were then submitted to a rescoring procedure applying the MM-GBSA approach implemented in Prime (version 3.0, Schrodinger). Briefly, the side chains of residues lying in a sphere of 12 Å from the ligand pose, as well as the compound molecule, were energy minimized using the OPLS2005 force field and the GBSA continuum model. The free energy of binding was then estimated from the free energy difference between the complex, the free protein and the free ligand, similar to what was reported in Incerti et al. (2013).
Figure 1.
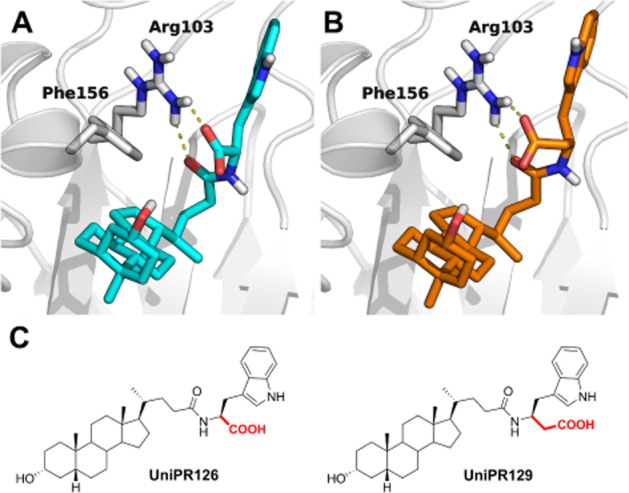
Theoretical binding mode of UniPR126 and UniPR129 to EphA2. UniPR126 (A, cyan carbon atoms) and UniPR129 (B, orange carbon atoms) docked to the ligand-binding domain of the EphA2 receptor. EphA2 carbon atoms and secondary structure elements are depicted in white. (C) Chemical structures of the two compounds are also shown.
Additional MM-GBSA simulations were performed using the whole ectodomain of EphA2 (PDB accession 3 MX0) (Himanen et al., 2010). Briefly, the protein was prepared according to the procedure reported above for the ligand-binding domain of EphA2. The docking grid was centred within the cysteine rich domain (CRD) of EphA2, in a region delimited by Pro221, Leu223 and Leu253 while the dimensions of enclosing and bounding boxes were set to 30 and 10.0 Å respectively. The minimized structure of UniPR129 was docked within the EphA2 receptor using Glide in standard precision mode. Twenty docking poses were retained for UniPR129 and ranked according to the Emodel score. The best-scored pose of UniPR129 was then submitted to the MM-GBSA rescoring procedure described above.
The structure of EphB4 receptor was taken from the PDB complex 2HLE (Chrencik et al., 2006) and then prepared for docking studies according to the procedure described above for the EphA2 receptor. The docking grid was centred within the ligand-binding domain of EphB4, with the dimensions of enclosing and bounding boxes set to 30 and 10.0 Å respectively. The minimized structure of UniPR129 was then docked within the EphA2 receptor using Glide in standard precision mode in the absence of bounding restraints. Twenty docking poses were retained and ranked according to the Emodel score.
Chemistry
UniPR129 synthesis is fully described in the Supporting Information.
Cell culture
PC3 human prostate adenocarcinoma cells (ECACC, Port Down, Salisbury, UK) were grown in Ham F12, supplemented with 5% FBS and 1% antibiotic solution. HUVEC (Life Technologies, Waltham, MA, USA) were maintained in MEM 200 supplemented with 1% penicillin-streptomycin solution, 1% fungizone solution, 2% low serum growth supplement and 10% FBS. PC3 and HUVEC were grown in a humidified atmosphere of 95% air, 5% CO2 at 37°C.
elisa assays and Ki/IC50 determination
elisa assays were performed as previously described (Giorgio et al., 2011). Briefly, 96-well elisa high-binding plates (Costar #2592) were incubated overnight at 4°C with 100 μL per well of 1 μg·mL−1 EphA2-Fc (R&D 639-A2) diluted in sterile PBS (0.2 g·L−1 KCl, 8.0 g·L−1 NaCl, 0.2 g·L−1 KH2PO4, 1.15 g·L−1 Na2HPO4, pH 7.4). On the next day, the wells were washed with washing buffer (PBS +0.05% Tween20, pH 7.5) and blocked with blocking solution (PBS +0.5% BSA) for 1 h at 37°C. Compounds were added to the wells at proper concentration in 1% DMSO and incubated at 37°C for 1 h. Biotinylated ephrin-A1-Fc (R&D Systems BT602, Minneapolis, MN, USA) was added at 37°C for 4 h at its KD in displacement assays or in a range from 1 to 2000 ng·mL−1 in saturation studies. Then wells were washed and incubated with 100 μL per well Streptavidin-HRP (Sigma S5512, Milano, Italy) for 20 min at room temperature, washed again and finally incubated at room temperature with 0.1 mg·mL−1 tetramethylbenzidine (Sigma T2885) reconstituted in stable peroxide buffer (11.3 g·L−1 citric acid, 9.7 g·L−1 sodium phosphate, pH 5.0) and 0.02% H2O2 (30% m/m in water), added immediately before use. The reaction was stopped with 3N HCl 100 μL/well and the absorbance was measured using an elisa plate reader (Sunrise, TECAN, Männedorf, Switzerland) at 450 nm. IC50 values were determined using one-site competition non-linear regression analysis with Prism software (GraphPad Software Inc., La Jolla, CA, USA). To assess the selectivity of compounds, all EphA (R&D Systems SMPK1) and EphB (R&D Systems SMPK2) receptors were incubated overnight similarly to EphA2 as previously described; biotinylated ephrin-A1-Fc or biotinylated ephrin-B1-Fc (R&D Systems BT473) at their KD were used towards EphAs or EphBs respectively.
Cell lysates
Cells were seeded in 12-well plates at concentration of 105 cells mL−1, 1 mL per well, in complete medium until they reached ∼70% confluence and serum starved overnight. On the next day, the cells were treated with the compounds under study, vehicle or standard drug, stimulated with the proper physiological agonist, rinsed with PBS and solubilized in lysis buffer. The lysates were resuspended and rocked at 4°C for 30 min and then centrifuged at 14 000× g for 5 min. The protein content of supernatant was measured with BCA protein assay kit (Thermo Scientific, Waltham, MA, USA) and standardized to 150 μg·mL−1.
Phosphorylation of EphA2, EphB4, VEGFR and EGF receptor (EGFR) in cells
EphA2, EphB4, VEGFR2 and EGFR phosphorylation was measured in cell lysates using DuoSet® IC Sandwich elisa (R&D Systems, #DYC4056, #DYC4057, #DYC1095 and #DYC1766, respectively) following manufacturer's protocol.
Briefly, 96-well elisa high-binding plates (Costar 2592) were incubated overnight with 100 μL per well of the specific capture antibody diluted in sterile PBS at the proper working concentrations. On the next day, the wells were washed and blocked for 1 h and 100 μL per well of lysates were added for 2 h. Then, wells were incubated with the specific detection antibody and the phosphorylation level was revealed utilizing a standard HRP format and tetra-methylbenzidine through a colorimetric reaction read at 450 nm. Each step was performed at room temperature and followed by the washing of each well.
LDH and dimethyl thiazolyl diphenyl tetrazolium (MTT) assays
Cytotoxicity of all compounds was evaluated with CytoTox 96® non-radioactive cytotoxicity assay, following the manufacturer's protocol (Promega, #1780, Madison, WI, USA). Cells were seeded in 96-well plates at a density of 105 cells mL−1 and the day after treated with compounds or lysis buffer for 2 or 15 h. After incubation, the released LDH in culture supernatants was measured using a 30 min coupled enzymatic assay, which results in conversion of a tetrazolium salt (INT) into a red formazan product. The amount of colour formed is proportional to the number of lysed cells and quantified by an elisa plate reader (Sunrise, TECAN) at 492 nm. The results were expressed as the ratio between absorbance of the cells treated with the compounds and the cells treated with lysis buffer.
Cell viability, instead, was evaluated using the MTT colorimetric assay. Cells were seeded in 96-well plates at a density of 105 cells mL−1 and the day after treated with compounds or 1% DMSO for 15 or 72 h. MTT was added at the final concentration of 1 mg·mL−1 and incubated for 2 h. The resulting formazan crystals were solubilized with DMSO 100 μL per well. The absorbance was measured at 550 nm using an elisa plate reader and the results were expressed as the ratio between absorbance of the cell treated with the compounds and untreated cells.
Cell cycle and apoptosis
For both cell cycle and apoptosis, PC3 cells were incubated for 48 h with UniPR129, 100 ng·mL−1 taxol or DMSO as a vehicle, harvested and washed in PBS.
Cells for apoptosis were immediately stained with Annexin-V and 7-AAD following the manufacturer's instructions (Millipore, 4500-0450, Billerica, MA, USA) whereas cells for cell cycle were fixed in cold 70% ethanol before staining with propidium iodide in the presence of RNase (Millipore, 4500-0220). Determination of apoptosis and cell cycle was performed by flow cytometry (Guava Easycyte 5; Millipore).
Retraction assay
PC3 cells (4 × 103 cells/well) were seeded in 96-well plates (Greiner Bio One, Frickenhausen, Germany) and grown for a day. The cells were then starved overnight in serum-free Ham F12, incubated for 20 min with the compounds or DMSO, and stimulated for 10 min with 0.5 μg·mL−1 ephrin-A1-Fc or Fc as a control. The cells were then fixed with 3.7% formaldehyde in PBS, permeabilized with 0.5% Triton X-100 in PBS, and stained with rhodamine-conjugated phalloidin (Invitrogen, Waltham, MA, USA). Nuclei were labelled with DAPI. Cells were photographed under a fluorescence microscope and the number of retracted cells was counted, without knowledge of the treatments.
In vitro angiogenesis
Twenty-four well tissue culture plates were coated with BD Matrigel (80 μL/well) and incubated for 30 min at 37°C in order to form a thin layer of gel on the bottom of the wells. HUVECs were treated with compounds (or DMSO as control) and 3.2 × 105 cells per well were seeded on Matrigel. After 15 h of incubation the cells were fixed with 3.7% formaldehyde for 15 min at room temperature. Photographs were taken through a digital camera mounted on a microscope and the number of polygons formed was counted. Data were normalized to control (100%).
Boyden chamber assay
The migration assay was performed using Neuro Probe 48-Well Micro Chemotaxis Chamber (#AP48#). Briefly, each bottom well of the Boyden chamber was loaded with the chemotactic solution or control (27 μL per well) and the membrane filter (previously moistened in PBS at 37°C for 30 min) was placed on the top. The chemotactic stimulus consisted of 2 μg·mL−1 ephrin-A1-Fc or cell culture medium containing 10% FBS, while the control was represented by 2 μg·mL−1 Fc or cell culture medium without serum. HUVEC were trypsinized and collected, resuspended in serum-free medium at the concentration of 6 × 105 cells mL−1 and treated with compounds or control (DMSO). The silicone gasket and the top chamber were placed over the membrane and 50 μL of the treated cell suspension was seeded in each well. After a 3 h incubation at 37°C, the membrane filter was removed and fixed with formaldehyde 3.7%, stained with DAPI solution after the scraping of non-migrated cells from the top and mounted on a glass slide with bottom facing up. The migrated cells were counted using Image J software (National Institute of Mental Health, Bethesda, MD, USA).
Scratch assay
HUVEC were seeded at a density of 106 cells per well in a 24-well plate and when they reached confluence, wells were scratched with a pipette tip and washed with PBS. Fresh medium with 0.5% FBS was added to the wells and photographs were taken at the base time (T0). Then, cells were incubated 15 h (T15) with different concentrations of compounds or control in the presence of VEGF (100 ng·mL−1) and new photographs were taken after fixing with formaldehyde 3.7%. The area delimited by the wound edges was measured using Image J software.
Results
Identification of UniPR129 as novel potential EphA2 small molecule ligand by MM-GBSA calculations
Based on the theoretical binding mode recently proposed for UniPR126 to EphA2 (Figure 1A) (Incerti et al., 2013), we hypothesized that it might be possible to improve its potency by reinforcing the interaction between the carboxylic acid function of the amino acid portion and the key EphA2 amino acid residue Arg103 (Figure 1, panel A). Specifically, the introduction of a methylene spacer between the carbon α atom and the carboxylic group of the L-Trp moiety of UniPR126 would yield a compound able to interact with Arg103 more strongly than UniPR126 (Figure 1B), hopefully endowed with higher potency.
We therefore evaluated the higher homologue of UniPR126, UniPR129 (the L-homo-Trp conjugate of LCA) for its ability to bind the EphA2 ligand-binding domain in silico using docking simulations in combination with MM-GBSA free energy evaluation (Guimarães and Cardozo, 2008). MM-GBSA is a computational method that employs a combination of molecular mechanics and continuum solvation to estimate binding free energy directly from structural information and was already successfully applied to the optimization of other EphA2 antagonists (Incerti et al., 2013).
In silico experiments showed that UniPR129 interacts with EphA2, using a binding mode comparable to that of UniPR126 (Figure 1, panel B) but much more efficiently in terms of estimated binding free energy. Indeed, the complex between UniPR129 and EphA2 was estimated to be energetically more stable (by 2.5 kcal·mol−1) than that formed by UniPR126 and EphA2. Encouraged by these computational results, UniPR129 was synthetized according to the procedure described in the Supporting Information and evaluated experimentally as an EphA2 antagonist.
While alternative binding modes (i.e. within the CRD of EphA2, see Supporting Information Fig. S1) can to some extent explain the binding properties of UniPR129 and of other amino acid derivatives of LCA, MM-GBSA calculations here performed (see Supporting Information Table S1) suggest that UniPR129 preferentially binds to the EphA2 receptor at its ligand-binding domain (Figure 1B).
UniPR129 competitively inhibits Eph receptor-ephrin binding
UniPR129 was tested for its ability to disrupt EphA2-ephrin-A1 interaction by using an elisa binding assay. We immobilized the EphA2-Fc ectodomain on elisa plates and detected the binding of biotinylated ephrin-A1-Fc using streptavidin-HRP and a colourimetric reaction with tetra-methylbenzidine according to a previously published protocol which also assessed selectivity and specificity of the employed protocol (Giorgio et al., 2011). UniPR129, added to the wells before the biotinylated ephrin-A1-Fc, dose-dependently reduced EphA2-ephrin-A1 interactions with an IC50 value of 945 nM (pIC50 = 6.04 ± 0.07, r2 = 0.90) (Figure 2A). Displacement curves of the parent compounds, UniPR126 and LCA, are also reported as comparison.
Figure 2.
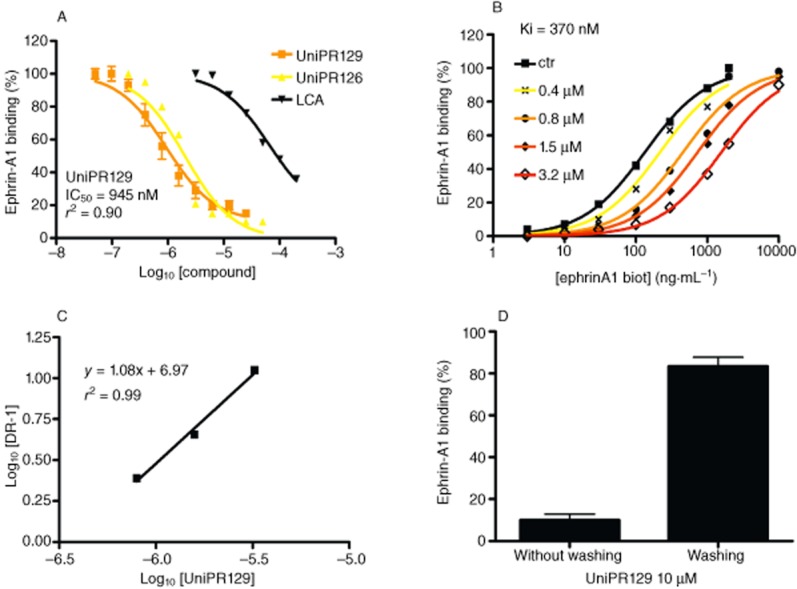
UniPR129 competitively displaces EphA2-ephrin-A1 binding. (A) UniPR129 dose-dependently displaced the binding of ephrin-A1-Fc from immobilized EphA2-Fc. (B) Binding of ephrin-A1-Fc to immobilized EphA2-Fc ectodomain in the presence of different concentrations of UniPR129. (C) The dissociation constants (KD) from the previous plot were used to calculate log (dose ratio –1) and to construct the Schild plot. pKi of UniPR129 was estimated by the intersection of the interpolated line with the X-axis. (D) EphA2-ephrin-A1 binding in the presence of 10 μM UniPR129 with or without washing three times with PBS before adding ephrin-A1-Fc. The histogram shows the means of at least three independent experiments ± SEM.
Next, we used saturation curves (Figure 2B) for EphA2-ephrin-A1 binding in the presence of increasing UniPR129 concentrations in order to calculate KD values and draw a Schild plot (Arunlakshana and Schild, 1959), where log[DR-1] is a function of the – log10[inhibitor] (Figure 2C). We obtained a well-interpolated regression line (r2 = 0.99) having a slope of 1.08, suggesting competitive binding. The pKi resulting from the intersection of the interpolated line with the X-axis was 6.43 ± 0.04 (corresponding to a Ki of 370 nM). In addition, we performed displacement experiments by incubating the wells with 10 μM UniPR129 for 1 h and washing away the compound in some wells before adding 100 ng·mL−1 ephrin-A1-Fc. Ephrin-A1-Fc displacement was detected only in unwashed wells, denoting the reversibility of UniPR129 binding to EphA2 (Figure 2D).
The interaction of UniPR129 with EphA and EphB receptors was studied using biotinylated ephrin-A1-Fc and biotinylated ephrin-B1-Fc, respectively, at their KD concentrations.
As shown in Figure 3, the compound inhibited the interaction of ephrin-A1 or ephrin-B1 with all Eph receptors, but the mechanism of inhibition is unknown for the B-class interaction.
Figure 3.
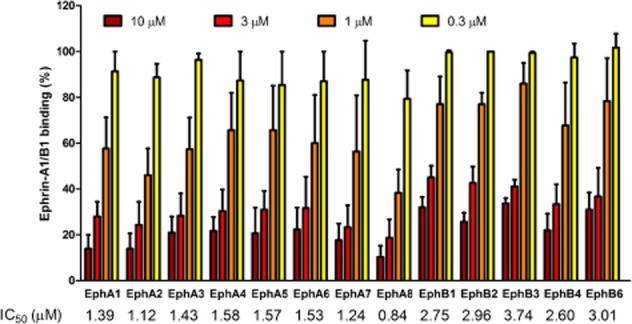
UniPR129 partially discriminates between A and B Eph receptor classes. UniPR129 dose dependently inhibited binding of biotinylated ephrin-A1-Fc or ephrin-B1-Fc to EphA or EphB receptor ectodomains respectively. Data are the means of at least three independent experiments ± SEM. IC50 values are indicated at the bottom.
The IC50 values for EphA receptors were in the 0.84–1.58 μM range and for EphB receptors in the 2.60–3.74 μM range, showing a slight preference of UniPR129 for EphA receptors.
UniPR129 dose-dependently inhibits EphA2 activation
We next tested UniPR129 for its effect in the PC3 prostate cancer cell line, which naturally expresses EphA2 and is a well-established pharmacological model to study this receptor (Miao et al., 2000). We stimulated EphA2 tyrosine phosphorylation with 0.25 μg·mL−1 ephrin-A1-Fc, with or without UniPR129. Under these conditions, UniPR129 dose-dependently inhibited EphA2 phosphorylation with an IC50 value of 5 μM. The compound did not act as an agonist because it did not stimulate EphA2 phosphorylation. The kinase inhibitor dasatinib, used as reference drug, completely abolished EphA2 phosphorylation at 1 μM, blocking kinase domain enzymatic activity (Figure 4).
Figure 4.
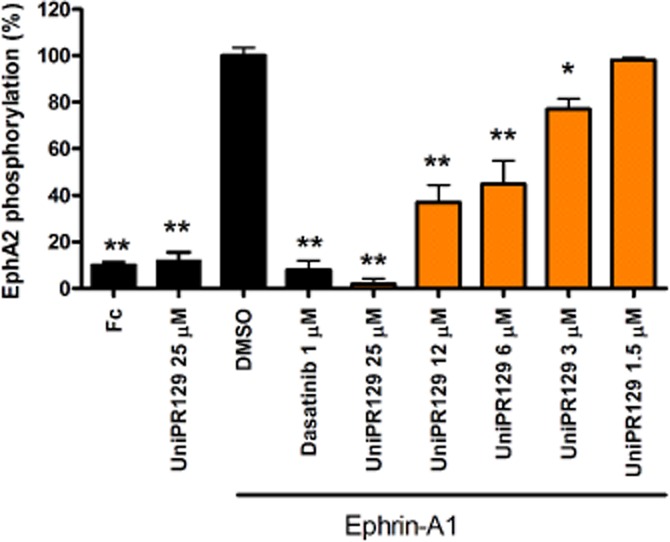
UniPR129 inhibits EphA2 activation induced by ephrin-A1 in PC3 cells. Cells were pretreated for 20 min with 1% DMSO, or the indicated concentrations (μM) of compounds, and then stimulated for 20 min with ephrin-A1-Fc. Phospho-EphA2 levels are relative to ephrin-A1-Fc + DMSO. Data are the means of at least three independent experiments ± SEM. One-way anova followed by Dunnett's post-test was performed to compare ephrin-A1-Fc + DMSO to all the other columns. *P < 0.05, **P < 0.01.
In order to make some assessment of the specificity of the interaction of UniPR129 with Eph kinases, we performed functional assays on other receptor tyrosine kinases including EGFR and VEGFR2 on PC3 and HUVECs respectively. In both cases, the compound was completely inactive (Supporting Information Fig. S2 for EGFR and Figure 8 for VEGFR2).
Figure 8.
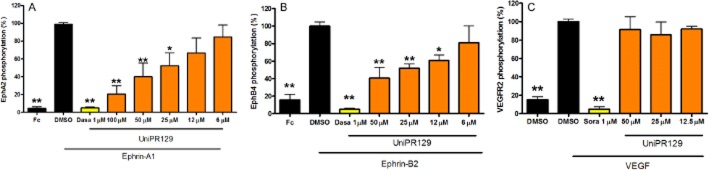
UniPR129 inhibits ephrin-induced EphA2 and EphB4 activation in HUVEC without affecting VEGFR2 phosphorylation induced by VEGF. Cells were pretreated for 20 min with the indicated concentrations of the compound, or 1% DMSO as a control, and then stimulated for 20 min with 0.25 μg·mL−1 ephrin-A1-Fc, 2 μg·mL−1 preclustered ephrin-B2-Fc or 20 ng·mL−1 VEGF (R&D Systems, 293-VE). The levels of phosphorylated EphA2 (A), EphB4 (B) and VEGFR2 (C) are normalized to the levels in cells treated with ephrin-A1-Fc, ephrin-B2-Fc or VEGF, respectively, in the absence of compound. Data are the means of at least three independent experiments ± SEM. One-way anova followed by Dunnett's post-test was performed to compare ephrin-A1-Fc + DMSO, ephrin-B2-Fc + DMSO or VEGF + DMSO to all other columns. *P < 0.05, **P < 0.01.
The potential interference of non-specific cytotoxicity in the functional assay conducted on PC3 cells was investigated by means of cell viability assays. UniPR129 did not show any toxic effect on PC3 cells even at high concentrations (50 μM) in the LDH assay but slightly decreased cell proliferation as indicated by MTT test, after 72 h (Figure 5A and B). Reduction of cell proliferation was not related to modifications in cell cycle nor to increased apoptosis (Figure 5C and D).
Figure 5.

UniPR129 slightly reduced cell proliferation without toxic effect and without affecting cell cycle or apoptosis. (A) Cells were incubated for 2 h with the indicated compounds and release of LDH was detected. (B) Cells were incubated for 72 h in the presence of the compound at the indicated concentrations and the metabolic activity of the cells was revealed with the MTT method; (C, D) Cells were incubated for 48 h with the indicated compounds and then cell cycle (C) or apoptosis (D) was determined by cytometry. Viable cells are negative towards both Annexin and 7-AAD, early apoptosis are Annexin positive cells and late apoptosis are 7-AAD positive cells. Data are the means of at least three independent experiments ± SEM. One-way anova followed by Dunnett's post-test (A and B) or two-way anova followed by Bonferroni's post-test (C and D) was performed to compare control to all other columns. *P < 0.05, **P < 0.01.
UniPR129 inhibits PC3 cell retraction
Retraction of the cell periphery and cell rounding is a typical morphological response of many cells, including PC3 cells, to ephrin-A1 stimulation as a result of integrin inhibition and RhoA activation (Figure 6) (Miao et al., 2000; Huang et al., 2008). Preincubation of PC3 cells with UniPR129 for 20 min dose dependently inhibited the rounding effect of ephrin-A1-Fc. A significant inhibition was detected for concentrations ≥ 6.25 μM and the resulting IC50 was estimated to be 6.2 μM (Figure 6). Furthermore, UniPR129 did not affect cell morphology when used alone (data not shown), showing that it did not have an agonist effect.
Figure 6.
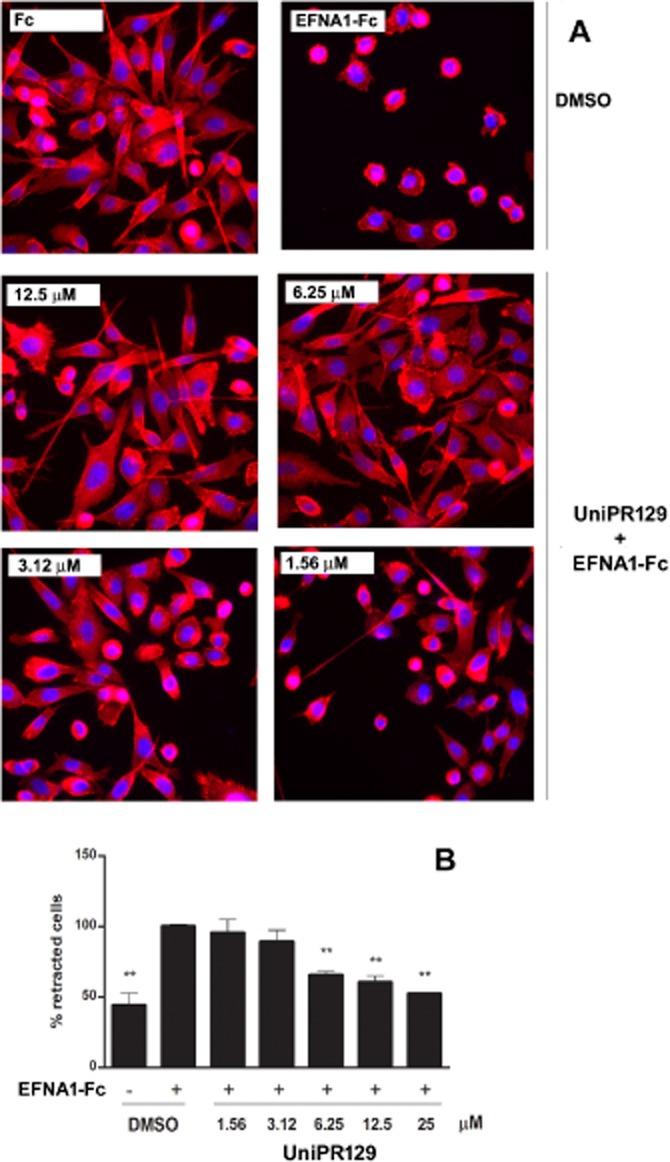
UniPR129 inhibits EphA2-dependent PC3 cell rounding. Serum-starved PC3 cells were preincubated with UniPR129 or DMSO for 20 min and then stimulated with 0.5 μg·mL−1 ephrin-A1-Fc or Fcfor 10 min. (A) Morphological changes induced by ephrin-A1-Fc or Fc in the presence of the indicated UniPR129 concentrations. Cells were stained with rhodamine–phalloidin to label actin filaments (red) and DAPI to label nuclei (blue). (B) Histogram showing the average percentage of retracted cells 10 min after the treatment with ephrin-A1-Fc or Fc in the presence of the indicated concentrations of UniPR129. Rounded cells having an area smaller than 20% of the initial value were scored as retracted. The number of retracted cells was normalized to the ephrin-A1-Fc-treated cells (corresponding to 100%). Data are the means of at least three independent experiments ± SEM. One-way anova followed by Dunnett's post-test was performed to compare control to all the other columns. *P < 0.05, **P < 0.01.
UniPR129 inhibits in vitro angiogenesis at low micromolar concentrations without directly interfering with VEGFR2 activation
We tested UniPR129 in an in vitro angiogenesis model with HUVEC in comparison with the parent compound UniPR126. Both compounds reduced polygon formation, but UniPR129 (with an IC50 value of 5.2 μM) was fourfold more potent than UniPR126 (IC50 value of 20.5 μM; Figure 7A). Moreover, UniPR126 was cytotoxic for HUVECs as it increased LDH release after 15 h and decreased MTT production in a dose-dependent manner at concentrations >25 μM. On the other hand, UniPR129 did not significantly increase LDH release, although it slightly reduced cell proliferation (Figure 7C and D), consistent with the previous findings on PC3 cells. Interference of UniPR129 with the Eph-ephrin system was further demonstrated by measuring EphA2 and EphB4 tyrosine phosphorylation in HUVECs stimulated by their physiological ligands, 0.25 μg·mL−1 ephrin-A1-Fc or 2 μg·mL−1 preclustered ephrin-B2-Fc respectively. UniPR129 dose dependently reduced phosphorylation of both receptors with IC50 values of 26.3 μM for EphA2 and 18.4 μM for EphB4. Due to toxicity, inhibition of EphA2 and EphB4 phosphorylation was not measured for UniPR126 (Figure 8).
Figure 7.

UniPR129 inhibits in vitro angiogenesis in HUVECs without cytotoxic effects. (A, B) HUVEC seeded on Matrigel were treated with UniPR126, UniPR129 or dasatinib, and images were taken 15 h later. The number of polygons is relative to 1% DMSO, which was used as the control. The histogram shows the means of at least three independent experiments ± SEM. One-way anova followed by Tukey's post-test was performed to compare the 1% DMSO control to all other conditions. *P < 0.05, **P < 0.01. (C, D) HUVEC were incubated for 15 h with UniPR126 or UniPR129 at the indicated concentrations in culture medium without FBS, but supplemented with 2% low serum growth supplement. Data are the means of at least three independent experiments ± SEM. One-way anova followed by Dunnett's post-test was performed to compare the 1% DMSO control (100%, not shown) to all other conditions for the MTT assay in (C) and the 1% DMSO control (0%, not shown) to all other conditions for the LDH assay in (D). *P < 0.05, **P < 0.01.
Anti-angiogenic activity of UniPR129 was not directly related to VEGFR2 inhibition, as this compound up to 50 μM did not reduce the phosphorylation of VEGFR2 when activated with 20 ng·mL−1 VEGF (Figure 8). The reference compound, sorafenib, at 1 μM, abolished VEGFR2 phosphorylation.
UniPR129 inhibits in vitro migration
In order to understand whether angiogenetic process was influenced by migration, we studied the effect of our compound in a transwell migration model and in the wound healing assay. In the transwell assay, ephrin-A1-Fc stimulated cell migration, consistent with Cheng's findings (Cheng et al., 2002), whereas 1 μM dasatinib, 30 and 10 μM UniPR129, significantly reduced the process. Similarly, as previously described (Cheng et al., 2002), VEGF stimulated wound healing but both 1 μM dasatinib and 30 μM UniPR129 reduced the healing (Figure 9).
Figure 9.
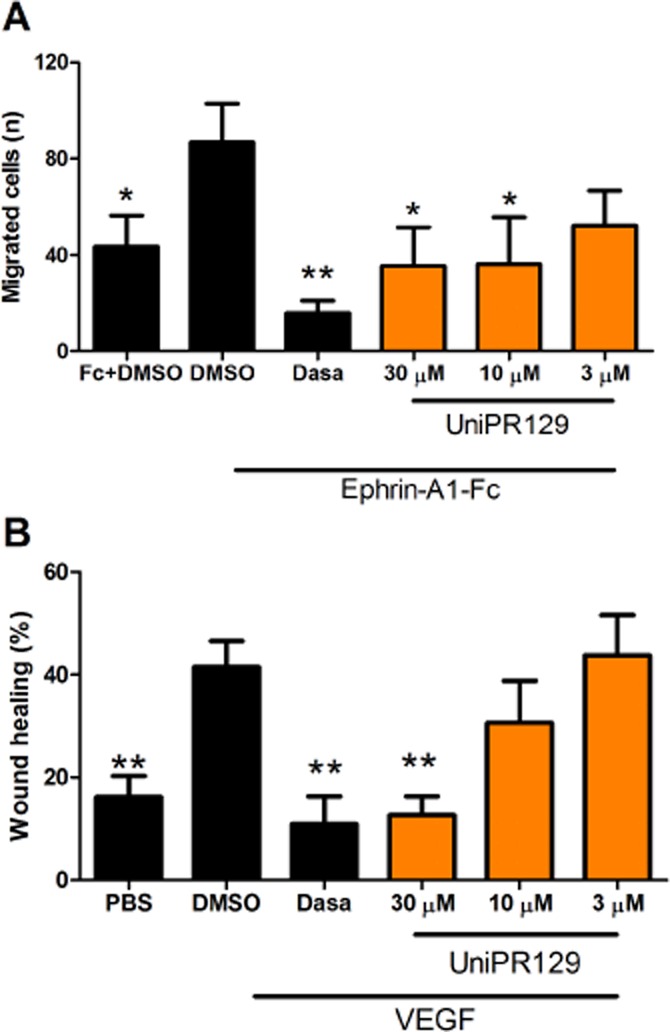
Effect of UniPR129 on HUVEC migration. (A) The bottom of the wells of the Boyden chamber was coated with ephrin-A1-Fc or with PBS for control. HUVEC treated with different concentrations of UniPR129 or DMSO were seeded on the top wells of the chamber. After 3 h at 37°C, non-migrated cells were gently removed from the membrane with a scraper, while migrated cells were fixed with formaldehyde and stained with DAPI. Photographs were taken in order to count the migrated cells in each well. The rate of migration for each group of chemotactic stimulus was normalized to the cells treated with DMSO (100% migration) in the absence of UniPR129. (B) Confluent HUVEC seeded on a 24-well plate were scratched with a pipette tip, washed with PBS and incubated for 15 h with different concentrations of UniPR129 or control (DMSO) in the presence or absence of 100 ng·mL−1 VEGF. Pictures were taken immediately after the scratch (T0) and after 15 h (T15). The wound area at T0 and T15 was measured with ImageJ software and wound healing percentage was calculated for each treatment. Dasatinib (Dasa;1 μM) was used as reference compound. Data are the means of at least three independent experiments ± SEM. *P < 0.05, **P < 0.01, comparing all the columns with DMSO + ephrin-A1-Fc in (A) and with DMSO + VEGF in (B).
Discussion and conclusions
Interest towards the Eph-ephrin system as a therapeutic target in various fields of pharmacology, including nervous system regeneration (Parrinello et al., 2010; Van Hoecke et al., 2012), haemostasis (Prevost et al., 2003), diabetes (Konstantinova et al., 2007) and targeted cancer chemotherapy (Tandon et al., 2011), is rapidly increasing. While intensive efforts have been made by several research groups to discover new PPI of the Eph-ephrin system, the compounds identified so far have less than ideal potency, stability and physicochemical properties (Noberini et al., 2011; Petty et al., 2012; Tognolini et al., 2012a,b2012a,2012b).
A structure-based drug design approach (Incerti et al., 2013) recently allowed us to identify UniPR126 (L-Trp conjugate of LCA, Figure 1C) as one of the most potent antagonists of the EphA2 receptor. Starting from the computational model of the EphA2-UniPR126 complex (Figure 1A), we designed UniPR129 (the L-homo-Trp conjugate of LCA, Figure 1C) to search for a more potent and effective PPI. Supported by free energy calculations, suggesting that UniPR129 would have higher affinity for the EphA2 receptor than its parent derivative, we synthesized UniPR129 and we investigated its pharmacological properties.
We found that UniPR129 was two- to threefold more potent than UniPR126 in elisa assays measuring inhibition of ephrin-A1 binding to EphA2, with a decrease in the IC50 from 2.04 to 0.95 μM. Moreover, the potency of UniPR129 was 60-fold higher than that of LCA (IC50 = 57 μM), confirming that it is possible to improve the potency of compounds with a cyclopenta[a]perhydrophenantrene scaffold. Moreover, the binding data in Figure 2B and C show surmountable saturation curves with a Hill coefficient close to 1, indicating both competitive binding and a 1:1 stoichiometry for the interaction of UniPR129 with EphA2 (Arunlakshana and Schild, 1959).
As found with the parent compound LCA and with UniPR126, UniPR129 is an promiscuous ligand for Eph receptors, with only a moderate preference for the EphA receptor class. The ability of UniPR129 to inhibit the binding of ephrin ligands to all members of the Eph receptor tyrosine kinase family at low micromolar concentrations suggests that it interacts with a highly conserved region (i.e. with a specific ‘hot-spot’ such as the basic chain of Arg103) within the ligand-binding pocket of both A and B classes of Eph receptors. The importance of this arginine residue in the Eph-ephrin recognition process has already been recognized for the EphA subfamily (Himanen et al., 2009), while its role seemed less clear for EphB receptors. A simple multiple sequence alignment shows that, with the only exception of EphB4, all the EphB receptors possess an arginine residue, corresponding to Arg103 of EphA2 (Supporting Information Fig. S3), which might be involved in a salt bridge with ephrin-B ligands and with small molecules. This hypothesis is supported by the X-ray structure of EphB2-ephrin-A5 complex (1SHW.pdb) (Himanen et al., 2004), where Arg103 of EphB2 interacts with Glu133 of ephrin-A5. It is thus likely that UniPR129 binds EphB1-B2-B3 and EphB6 receptors by targeting this conserved basic residue.
While the EphB4 receptor lacks a key arginine for the recognition of ephrin ligands, a visual inspection of the X-ray coordinates of the EphB4-ephrin-B2 complex (2HLE.pdb) (Chrencik et al., 2006) indicates that the association of these two proteins is reinforced by a salt bridge involving Lys149 of EphB4 and Glu128, lying on ephrin-B2. This suggests that Lys149 of EphB4 might act as an anchor point for UniPR129 binding (Supporting Information Fig. S4).
An improvement in the potency of UniPR129 compared with UniPR126 was also observed in cell functional studies, where the IC50 for inhibition of ephrin-A1-induced EphA2 tyrosine phosphorylation was 5 μM for UniPR129 and 12 μM for UniPR126. Furthermore, UniPR129 is approximately 20-fold more potent than LCA based on the potency of LCA reported in literature (Giorgio et al., 2011). The ability of UniPR129 to effectively antagonize the effects of EphA2 activation was further confirmed in the PC3 cell retraction assay, where cells stimulated with the physiological ligand ephrin-A1 undergo EphA2-mediated morphological changes (Miao et al., 2000). The IC50 for inhibition of PC3 cell retraction by UniPR129 was 6.2 μM, a value similar to that for its inhibition of EphA2 activation. When compared with the efficacy of previously studied LCA derivatives on PC3 cell retraction, UniPR129 was as potent as UniPR126 (Incerti et al., 2013), three- to fourfold more potent than cholanic acid (Tognolini et al., 2012b) and much more potent than LCA (Giorgio et al., 2011).
The compound slightly decreased PC3 proliferation without cytotoxic effect, modifications of cell cycle or increase of apoptosis. These findings are consistent with Binda's observations, in which the interference with Eph kinases affects proliferation of tumour-propagating cells (TPCs) with stem cell-like characteristics but does not affect non-TPCs (Binda et al., 2012). All together, these data suggest a good degree of specificity for Eph-ephrin system.
The most strikingly improved feature of UniPR129 was its in vitro anti-angiogenic activity at low micromolar concentrations (IC50 = 5.2 μM) together with the overcome of the toxicity limits encountered with UniPR126. Indeed, UniPR129 was not cytotoxic even at concentrations 10-fold higher than the ones active in the capillary-like tube formation assay. The anti-angiogenic activity of UniPR129 is mainly due to its ability to inhibit cell migration. Our findings demonstrate that UniPR129 can modify migration of endothelial cells interfering with Eph-ephrin system without directly affecting VEGFR2 activation. It is well known that VEGFR is a key player in angiogenesis, in cooperation with the Eph receptors (Cheng et al., 2002; Sawamiphak et al., 2010). At this stage, studies to clarify the mechanism of crosstalk between ephrins and VEGFR2 through the use of selective Eph-ephrin antagonists are needed.
When compared with other known low MW Eph receptor antagonists, UniPR129 is not the most potent PPI of the EphA2-ephrin-A1 interaction, being surpassed by gallic acid, which displays an IC50 of 260 nM (Tognolini et al., 2012a). However, gallic acid was completely inactive when tested in cellular functional assays. This confirms a possible pitfall of screening campaigns based on spectrophotometric assays, which might yield false-positive binders, thus rendering follow-up optimizations time-consuming, tedious and often unsuccessful (Baell and Holloway, 2010; Wu et al., 2013). On the other hand, UniPR129 is the most potent low MW compound currently available capable of inhibiting not only phosphorylation of endogenous EphA2 in cells, but also cell retraction and endothelial capillary-like tube formation. Furthermore, UniPR129 is active as an EphA2 antagonist in cellular functional assays at concentrations as low as 3–6 μM, whereas cholanic acid is active at 12–25 μM concentrations (Tognolini et al., 2012b), disalicylic derivatives at 25–50 μM (Noberini et al., 2011), the agonist doxazosin at 25–50 μM (Petty et al., 2012), green tea-derived epicathechin gallates at 25–100 μM (Noberini et al., 2012a), dimethylpyrrolyl derivatives at 100–200 μM (Noberini et al., 2008) and the recently discovered stilbene carboxylic acid derivatives (Tognolini et al., 2013b). Lastly, the potency of UniPR129 is comparable to that of the B11 antibody, which targets ephrin-B2 and has a threshold concentration in functional anti-angiogenesis assays of about 3 μM (Abéngozar et al., 2012).
Taken together, our data show that UniPR129 represents a significant advance in the development of effective Eph receptor-ephrin antagonists based on the LCA scaffold. Its improved potency, cytotoxicity profile and efficacy in functional in vitro assays suggest that UniPR129 could be a useful pharmacological tool and a promising lead compound.
Acknowledgments
This work was supported by Ministero dell'Università e della Ricerca, ‘Futuro in Ricerca’ program (project code: RBFR10FXCP) and NIH grant P01CA138390. I. H. M. thanks the Italian Society of Pharmacology (SIF) for supporting her permanence at the Sandford-Burnham Medical Research Institute. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Glossary
- BCA
bicinchoninic acid
- EGFR
EGF receptor
- LCA
lithocholic acid
- MTT
dimethyl thiazolyl diphenyl tetrazolium salt
- PPI
protein-protein inhibitor
- TPCs
tumour-propagating cells
- VEGFR
VEGF receptor
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12669
Figure S1 Docking of UniPR129 (cyan carbon atoms) in the CRD of EphA2 receptor (white ribbons with black carbon atoms). In evidence, Leu254 and Val255 of the leucine zipper region, as well as polar residues (i.e. Arg244 and Asp250).
Figure S2 UniPR129 does not affect EGF-induced EGFR phosphorylation on PC3 cells. Cells were pretreated for 20 min with the indicated concentrations of the compound, or 1% DMSO as a control and then stimulated for 20 min with 30 ng·mL−1 EGF (236-EG-200, R&D Systems). The levels of phosphorylated EGFR were detected with the Human Phospho-EGF-R sandwich elisa kit (DYC1095, R&D Systems) and normalized to the cells treated with DMSO + EGF in the absence of compound(s). Gefitinib (Gefi) 1 μM was used as a control. Data are the means of at least three independent experiments ± standard Error. One-way anova followed by Dunnet's post-test was performed to compare EGF + DMSO to all other columns. **P < 0.01.
Figure S3 Multiple sequence alignment of human Eph receptors. Secondary structure elements are shown above the sequences (h: helix; e: sheet) and are referred to the structure of EphA2 as it appears from the X-ray coordinates reported in the 3HEI.pdb complex.
Figure S4 Docking of UniPR129 (cyan carbon atoms) in the high-affinity ephrin-binding pocket of the EphB4 receptor (white ribbons with grey side chain carbon atoms). The G–H loop of ephrinB2 is also displayed (red ribbons). In evidence, Lys149 of EphB4 and Glu128 of ephrinB2.
Table S1 MM-GBSA calculations for UniPR129 in the LBD and in CRD domains of EphA2.
References
- Abéngozar MA, Frutos S, de Ferreiro S, Soriano J, Perez-Martinez M, Olmeda D, et al. Blocking ephrinB2 with highly specific antibodies inhibits angiogenesis, lymphangiogenesis, and tumor growth. Blood. 2012;119:4565–4576. doi: 10.1182/blood-2011-09-380006. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Binda E, Visioli A, Giani F, Lamorte G, Copetti M, Pitter KL, et al. The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell. 2012;22:765–780. doi: 10.1016/j.ccr.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng N, Brantley DM, Liu H, Lin Q, Enriquez M, Gale N, et al. Blockade of EphA receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol Cancer Res. 2002;1:2–11. [PubMed] [Google Scholar]
- Chrencik JE, Brooun A, Kraus ML, Recht MI, Kolatkar AR, Han GW, et al. Structural and biophysical characterization of the EphB4-ephrinB2 protein-protein interaction and receptor specificity. J Biol Chem. 2006;281:28185–28192. doi: 10.1074/jbc.M605766200. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333:328–335. doi: 10.1016/j.bbrc.2005.05.132. [DOI] [PubMed] [Google Scholar]
- Giorgio C, Hassan Mohamed I, Flammini L, Barocelli E, Incerti M, Lodola A, et al. Lithocholic acid is an Eph-ephrin ligand interfering with Eph-kinase activation. PLoS ONE. 2011;6:e18128. doi: 10.1371/journal.pone.0018128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimarães CRW, Cardozo M. MM-GB/SA rescoring of docking poses in structure-based lead optimization. J Chem Inf Model. 2008;48:958–970. doi: 10.1021/ci800004w. [DOI] [PubMed] [Google Scholar]
- Héroult M, Schaffner F, Augustin HG. Eph receptor and ephrin ligand-mediated interactions during angiogenesis and tumor progression. Exp Cell Res. 2006;312:642–650. doi: 10.1016/j.yexcr.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Himanen J-P, Chumley MJ, Lackmann M, Li C, Barton WA, Jeffrey PD, et al. Repelling class discrimination: ephrin-A5 binds to and activates EphB2 receptor signaling. Nat Neurosci. 2004;7:501–509. doi: 10.1038/nn1237. [DOI] [PubMed] [Google Scholar]
- Himanen JP, Goldgur Y, Miao H, Myshkin E, Guo H, Buck M, et al. Ligand recognition by A-class Eph receptors: crystal structures of the EphA2 ligand-binding domain and the EphA2/ephrin-A1 complex. EMBO Rep. 2009;10:722–728. doi: 10.1038/embor.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, et al. Architecture of Eph receptor clusters. Proc Natl Acad Sci U S A. 2010;107:10860–10865. doi: 10.1073/pnas.1004148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Wu D, Jin H, Stupack D, Wang JYJ. Induction of cell retraction by the combined actions of Abl–CrkII and Rho–ROCK1 signaling. J Cell Biol. 2008;183:711–723. doi: 10.1083/jcb.200801192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incerti M, Tognolini M, Russo S, Pala D, Giorgio C, Hassan-Mohamed I, et al. Amino acid conjugates of lithocholic acid as antagonists of the EphA2 receptor. J Med Chem. 2013;56:2936–2947. doi: 10.1021/jm301890k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehle J, Staudacher I, Wiedmann F, Schweizer P, Becker R, Katus H, et al. Regulation of apoptosis in HL-1 cardiomyocytes by phosphorylation of the receptor tyrosine kinase EphA2 and protection by lithocholic acid. Br J Pharmacol. 2012;167:1563–1572. doi: 10.1111/j.1476-5381.2012.02117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- Jubb AM, Harris AL. Biomarkers to predict the clinical efficacy of bevacizumab in cancer. Lancet Oncol. 2010;11:1172–1183. doi: 10.1016/S1470-2045(10)70232-1. [DOI] [PubMed] [Google Scholar]
- Konstantinova I, Nikolova G, Ohara-Imaizumi M, Meda P, Kucera T, Zarbalis K, et al. EphA-Ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell. 2007;129:359–370. doi: 10.1016/j.cell.2007.02.044. [DOI] [PubMed] [Google Scholar]
- Lafleur K, Huang D, Zhou T, Caflisch A, Nevado C. Structure-based optimization of potent and selective inhibitors of the tyrosine kinase erythropoietin producing human hepatocellular carcinoma receptor B4 (EphB4) J Med Chem. 2009;52:6433–6446. doi: 10.1021/jm9009444. [DOI] [PubMed] [Google Scholar]
- van Linden OPJ, Farenc C, Zoutman WH, Hameetman L, Wijtmans M, Leurs R, et al. Fragment based lead discovery of small molecule inhibitors for the EPHA4 receptor tyrosine kinase. Eur J Med Chem. 2012;47:493–500. doi: 10.1016/j.ejmech.2011.11.020. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Holzer P, Billy E, Schnell C, Brueggen J, Ferretti M, et al. The small molecule specific EphB4 kinase inhibitor NVP-BHG712 inhibits VEGF driven angiogenesis. Angiogenesis. 2010;13:259–267. doi: 10.1007/s10456-010-9183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellitzer G, Xu Q, Wilkinson DG. Eph receptors and ephrins restrict cell intermingling and communication. Nature. 1999;400:77–81. doi: 10.1038/21907. [DOI] [PubMed] [Google Scholar]
- Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62–69. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- Mohamed IH, Giorgio C, Bruni R, Flammini L, Barocelli E, Rossi D, et al. Polyphenol rich botanicals used as food supplements interfere with EphA2-ephrinA1 system. Pharmacol Res. 2011;64:464–470. doi: 10.1016/j.phrs.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Noberini R, Koolpe M, Peddibhotla S, Dahl R, Su Y, Cosford NDP, et al. Small molecules can selectively inhibit ephrin binding to the EphA4 and EphA2 receptors. J Biol Chem. 2008;283:29461–29472. doi: 10.1074/jbc.M804103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noberini R, De SK, Zhang Z, Wu B, Raveendra-Panickar D, Chen V, et al. A disalicylic acid-furanyl derivative inhibits ephrin binding to a subset of Eph receptors. Chem Biol Drug Des. 2011;78:667–678. doi: 10.1111/j.1747-0285.2011.01199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noberini R, Koolpe M, Lamberto I, Pasquale EB. Inhibition of Eph receptor-ephrin ligand interaction by tea polyphenols. Pharmacol Res. 2012a;66:363–373. doi: 10.1016/j.phrs.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noberini R, Lamberto I, Pasquale EB. Targeting Eph receptors with peptides and small molecules: progress and challenges. Semin Cell Dev Biol. 2012b;23:51–57. doi: 10.1016/j.semcdb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello S, Napoli I, Ribeiro S, Wingfield Digby P, Fedorova M, Parkinson DB, et al. EphB signaling directs peripheral nerve regeneration through Sox2-dependent Schwann cell sorting. Cell. 2010;143:145–155. doi: 10.1016/j.cell.2010.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquale EB. The Eph family of receptors. Curr Opin Cell Biol. 1997;9:608–615. doi: 10.1016/s0955-0674(97)80113-5. [DOI] [PubMed] [Google Scholar]
- Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–180. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty A, Myshkin E, Qin H, Guo H, Miao H, Tochtrop GP, et al. A small molecule agonist of EphA2 receptor tyrosine kinase inhibits tumor cell migration in vitro and prostate cancer metastasis in vivo. PLoS ONE. 2012;7:e42120. doi: 10.1371/journal.pone.0042120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevost N, Woulfe D, Tognolini M, Brass LF. Contact-dependent signaling during the late events of platelet activation. J Thromb Haemost. 2003;1:1613–1627. doi: 10.1046/j.1538-7836.2003.00327.x. [DOI] [PubMed] [Google Scholar]
- Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465:487–491. doi: 10.1038/nature08995. [DOI] [PubMed] [Google Scholar]
- Sebolt-Leopold JS, English JM. Mechanisms of drug inhibition of signalling molecules. Nature. 2006;441:457–462. doi: 10.1038/nature04874. [DOI] [PubMed] [Google Scholar]
- Tandon M, Vemula SV, Mittal SK. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin Ther Targets. 2011;15:31–51. doi: 10.1517/14728222.2011.538682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognolini M, Giorgio C, Hassan Mohamed I, Barocelli E, Calani L, Reynaud E, et al. Perturbation of the EphA2-EphrinA1 system in human prostate cancer cells by colonic (poly)phenol catabolites. J Agric Food Chem. 2012a;60:8877–8884. doi: 10.1021/jf205305m. [DOI] [PubMed] [Google Scholar]
- Tognolini M, Incerti M, Hassan-Mohamed I, Giorgio C, Russo S, Bruni R, et al. Structure-activity relationships and mechanism of action of Eph-ephrin antagonists: interaction of cholanic acid with the EphA2 receptor. ChemMedChem. 2012b;7:1071–1083. doi: 10.1002/cmdc.201200102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognolini M, Hassan-Mohamed I, Giorgio C, Zanotti I, Lodola A. Therapeutic perspectives of Eph-ephrin system modulation. Drug Discov Today. 2013a doi: 10.1016/j.drudis.2013.11.017. doi: 10.1016/j.drudis.2013.11.017. [DOI] [PubMed] [Google Scholar]
- Tognolini M, Incerti M, Pala D, Russo S, Castelli R, Hassan-Mohamed I, et al. Target hopping as a useful tool for the identification of novel EphA2 protein-protein antagonists. ChemMedChem. 2013b;9:67–72. doi: 10.1002/cmdc.201300305. [DOI] [PubMed] [Google Scholar]
- Van Hoecke A, Schoonaert L, Lemmens R, Timmers M, Staats KA, Laird AS, et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat Med. 2012;18:1418–1422. doi: 10.1038/nm.2901. [DOI] [PubMed] [Google Scholar]
- Wu B, Zhang Z, Noberini R, Barile E, Giulianotti M, Pinilla C, et al. HTS by NMR of combinatorial libraries: a fragment-based approach to ligand discovery. Chem Biol. 2013;20:19–33. doi: 10.1016/j.chembiol.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Mellitzer G, Robinson V, Wilkinson DG. In vivo cell sorting in complementary segmental domains mediated by Eph receptors and ephrins. Nature. 1999;399:267–271. doi: 10.1038/20452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Docking of UniPR129 (cyan carbon atoms) in the CRD of EphA2 receptor (white ribbons with black carbon atoms). In evidence, Leu254 and Val255 of the leucine zipper region, as well as polar residues (i.e. Arg244 and Asp250).
Figure S2 UniPR129 does not affect EGF-induced EGFR phosphorylation on PC3 cells. Cells were pretreated for 20 min with the indicated concentrations of the compound, or 1% DMSO as a control and then stimulated for 20 min with 30 ng·mL−1 EGF (236-EG-200, R&D Systems). The levels of phosphorylated EGFR were detected with the Human Phospho-EGF-R sandwich elisa kit (DYC1095, R&D Systems) and normalized to the cells treated with DMSO + EGF in the absence of compound(s). Gefitinib (Gefi) 1 μM was used as a control. Data are the means of at least three independent experiments ± standard Error. One-way anova followed by Dunnet's post-test was performed to compare EGF + DMSO to all other columns. **P < 0.01.
Figure S3 Multiple sequence alignment of human Eph receptors. Secondary structure elements are shown above the sequences (h: helix; e: sheet) and are referred to the structure of EphA2 as it appears from the X-ray coordinates reported in the 3HEI.pdb complex.
Figure S4 Docking of UniPR129 (cyan carbon atoms) in the high-affinity ephrin-binding pocket of the EphB4 receptor (white ribbons with grey side chain carbon atoms). The G–H loop of ephrinB2 is also displayed (red ribbons). In evidence, Lys149 of EphB4 and Glu128 of ephrinB2.
Table S1 MM-GBSA calculations for UniPR129 in the LBD and in CRD domains of EphA2.