

Abstract
Background and Purpose
Opioid drugs are potent analgesics. However, their chronic use leads to the rapid development of tolerance to their analgesic effects and subsequent increase of significant side effects, including drug dependence and addiction. Here, we investigated the role of PPARγ in the development of analgesic tolerance to morphine in mice.
Experimental Approach
We monitored analgesia on alternate days using the tail immersion test.
Key Results
Daily administration of morphine (30 mg·kg−1, bid) resulted in the rapid development of tolerance to thermal analgesia. Co-administration of pioglitazone (10 and 30 mg·kg−1, bid) significantly attenuated the development and expression of tolerance. However, pretreatment with GW-9662 (5 mg·kg−1, bid), a selective PPARγ antagonist, completely abolished this effect. Injection of GW-9662 and a lower dose of morphine (15 mg·kg−1, bid) accelerated the development of tolerance to its antinociceptive effect. Subsequently, we found that conditional neuronal PPARγ knockout (KO) mice develop a more rapid and pronounced tolerance to morphine antinociception compared with wild-type (WT) controls. Moreover, in PPARγ KO mice, pioglitazone was no longer able to prevent the development of morphine tolerance.
Conclusions and Implications
Overall, our results demonstrate that PPARγ plays a tonic role in the modulation of morphine tolerance, and its pharmacological activation may help to reduce its development. These findings provide new information about the role of neuronal PPARγ and suggest that combining PPARγ agonists with opioid analgesics may reduce the development of tolerance and possibly attenuate the potential for opioid abuse.
Table of Links
| TARGETS | LIGANDS |
|---|---|
| μ opioid (MOP) receptor | IL-1β |
| PPARα | IL-6 |
| PPARδ | GW-9662 |
| PPARγ | Morphine |
| TNFα | Pioglitazone |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a,b,).
Introduction
Opioids are essential medications in the management of pain and are widely used to treat acute severe pain following trauma, extensive burns or surgery, as well as chronic pain. One of the major problems associated with chronic opioid use is the significant risk of developing drug dependence and possibly addiction (Kreek, 2001; Inturrisi, 2002). The rapid development of tolerance to the analgesic effect of opioids, which requires dose escalation to maintain adequate analgesia, clearly contributes to this risk. Moreover, increased opioid dosing is associated with side effects, such as severe constipation, nausea and urinary retention, which are highly disliked by patients. Identification of innovative strategies to attenuate opioid tolerance development could help reduce the risks of opiate abuse associated with their chronic use, thus offering important benefits in the management of chronic pain.
Opioid tolerance is a complex phenomenon and several mechanisms may be responsible for it. Chronic exposure to opioid agonists can cause opioid receptor down-regulation, internalization, uncoupling from G-proteins (Bailey and Connor, 2005; Martini and Whistler, 2007; Koch and Hollt, 2008) or desensitization mediated by PKC-dependent mechanism (Bailey et al., 2009). For instance, repeated morphine injections produce a marked decrease in brain μ-opioid receptor (MOP receptor) density (Davis et al., 1979; Tao et al., 1987; Diaz et al., 2000), down-regulation of the high-affinity MOP receptor site in rats and reduction of MOP receptor signalling in sensory neurons and brainstem nuclei (Sim et al., 1996; Johnson et al., 2006). Changes in transcription factor activation following chronic opioid treatment have also been proposed to play a relevant role in opioid tolerance and addiction (Carlezon et al., 2005; Zachariou et al., 2006). Finally, emerging evidence suggests that opioid hyperalgesia and tolerance are influenced by the activation of the central immune system. For instance, it has been shown that the activation of glial cells, induction of the NF-κB signalling pathway and up-regulation of the transcription, translation and release of pro-inflammatory cytokines, such as IL-1 β, IL-6 and TNF-α, may all contribute to the attenuation of opioid analgesia and tolerance (Hutchinson et al., 2011). In contrast, blockade of these pro-inflammatory mechanisms via inhibition of the cytokines IL-1β, IL-6 and TNF-α (Raghavendra et al., 2002; Shavit et al., 2005; Hutchinson et al., 2007; 2008a,) and inhibition of microglia activation following anti-inflammatory treatments have been shown to be effective in reducing the development of morphine tolerance (Hutchinson et al., 2007; 2008b; 2011,,). Of particular interest, inhibitors of glial activation, such as ibudilast and minocycline, have also been shown to reduce the expression of opioid withdrawal and reward, indicating the anti-abuse potential of these agents (Bland et al., 2009; Hutchinson et al., 2009).
PPARs are a group of nuclear receptor proteins that primarily regulate gene expression through their role as ligand-activated transcription factors (Michalik et al., 2006). PPARγ is one of the three distinct isoforms identified (the other two are PPARα and PPARδ) and is predominantly expressed in adipose tissue and macrophages. PPARγ regulates adipocyte differentiation and is involved in sugar and lipid homeostasis and in the control of inflammatory responses (Landreth and Heneka, 2001; Berger and Wagner, 2002; Kapadia et al., 2008). Recent studies, however, have shown that PPARγ is also expressed in the CNS, where it has been found not only in neurons but also in astrocytes and microglia (Moreno et al., 2004; Gofflot et al., 2007; Sarruf et al., 2009). Importantly, the activation of PPARγ results in a marked neuroprotective and anti-inflammatory response in the CNS (Landreth and Heneka, 2001; Berger and Wagner, 2002; Kapadia et al., 2008; Tontonoz and Spiegelman, 2008). These effects have been attributed to the ability of PPARγ agonists to act as inhibitors of glial activation and to their subsequent ability to reduce NF-κB and pro-inflammatory cytokine expression (Tureyen et al., 2007; Xing et al., 2007). This background led us to hypothesize that inhibition of pro-inflammatory mechanisms following activation of PPARγ might result in the reduction of opioid tolerance and possibly an increase in their analgesic effect. To test this hypothesis, we studied the effect of pioglitazone, a brain-penetrating PPARγ agonist with modest effects on PPARα (Gillies and Dunn, 2000; Smith, 2001), on the development of analgesic tolerance following chronic morphine administration. Using GW-9662, a selective PPARγ antagonist, we also examined the effect of receptor blockade on the development of morphine tolerance. Finally, using conditional neuronal PPARγ knockout (KO) mice, we explored the effect of cell-specific deletion of PPARγ on morphine tolerance and the effects of pioglitazone.
Methods
Animals
Experiments were performed on male C57 mice (Harlan, Correzzana, Italy) and on conditional neuronal PPARγ KO mice and their wild-type (WT) counterparts (bred at the School of Pharmacy, University of Camerino, Italy). Mice weighed 28–30 g at the beginning of the experiment. KO mice had neuron-specific PPARγ deletion generated using nestin Cre-LoxP technology. Neural deletion of PPARγ was achieved by crossing mice with a floxed PPARγ allele [TgH(PPARγ lox)1Mgn, TgH(PPARγ del)2Mgn] with Nestin-Cre mice [B6.Cg-Tg(Nes-cre)1Kln/J]. All mice were fully backcrossed to a C57/BL6 genetic background (Jones et al., 2002; Sarruf et al., 2009). Animals were housed in ventilated plastic common cages (five animals per cage) in rooms with constant temperature (20–22°C) and humidity (45–55%) under a normal day/night cycle. During the experiments, animals were offered free access to tap water and food pellets (4RF18, Mucedola, Settimo Milanese, Milan, Italy). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). A total of 269 C57 male mice and 60 male PPARγ KO mice were used in the experiments. Experiments were approved by the ethical committee (Comitato Etico di Ateneo per la Protezione Animale) of the University of Camerino and are in adherence with the European Community Council Directive for Care and Use of Laboratory Animals.
Drugs
Pioglitazone was prepared from Actos® 30 mg tablets (Takeda Pharmaceuticals, Tokyo, Japan). It was dissolved in sterile water and given p.o. via gavage in a volume of 10 mL·kg−1 at doses of 10 or 30 mg·kg−1 (Stopponi et al., 2011).
GW-9662 was purchased from Sigma-Aldrich (Milan, Italy). It was dissolved in 5% DMSO, 5% cremophor and 90% distilled water, and injected i.p. in a volume of 10 mL·kg−1 at the doses of 2.5 or 5 mg·kg−1 (Ciccocioppo et al., 2012).
Morphine hydrochloride was purchased from Salars (Milan, Italy). It was dissolved in 0.9% NaCl and injected i.p. in a volume of 10 mL·kg−1.
Experimental procedure
To induce tolerance to morphine-induced antinociception, we injected morphine twice daily (between 0900 h and 1000 h and 1700 h and 1800 h) for 9 days (Contet et al., 2008). On the first and last day of the experiment, we reduced the dose of morphine injected (15 mg·kg−1) to better show the development of tolerance. For the other 7 days of the experiment, the morphine dose was 30 mg·kg−1 (in Exp. 1, 2 and 4) or 15 mg·kg−1 (in Exp. 3 and 5). In certain experiments, mice received pioglitazone (0, 10 or 30 mg·kg−1) and/or GW-9662 (0, 2.5 or 5 mg·kg−1) before the morphine injections. For the evaluation of the effects of pioglitazone on reversal of morphine tolerance, animals received pioglitazone only in the evening of day 8 and on day 9, before the morphine injections. Analgesia was monitored on alternate days using the tail immersion test 45 min after the evening injection. The tail immersion test is based on thermal (heat) noxious stimulus. This test was chosen because it involves a spinally mediated reflex response and can be repeated several times on the same animal (Mogil et al., 2000; Le Bars et al., 2001). Briefly, each mouse was restrained in a soft tissue pocket, and the distal half of the tail was dipped into a water bath set at 52°C. The latency to withdrawal of the tail from the water bath was measured. Two tail-withdrawal measures (separated by 30 s) were recorded and averaged. A 10 s cut-off time was applied to avoid tail burns. The tail immersion test was performed on alternate days 45 min after the evening morphine injection.
Locomotor activity test and assessment of body temperature
Tolerance to morphine antinociception was induced in 32 male C57 mice with the same procedure described earlier. Body temperature was measured with a digital thermometer and a thermistor probe manufactured by Physitemp Instruments, Inc. (Clifton, NJ, USA). The body temperature was measured 45 min after the last injection of morphine by inserting the lubricated probe 2 cm into the rectum for 30 s.
Automated locomotor activity boxes (Med Associate, St. Albans, VT, USA) were used to quantify behavioural activity. Each animal was placed in the activity box, a square plastic box measuring 43 × 43 × 30 cm, and spontaneous locomotor activity parameters were monitored. Activity was recorded for 15 min, starting 5 min after placing the animal in the test cage. Locomotor activity of each mouse was automatically recorded by interruption of two orthogonal light beams, which were connected to an automatic software. The behavioural parameter observed was locomotion (as reflected by the number of beam breaks). Between each test session, the apparatus was cleaned with alcohol (70%) and dried with a clean cloth. Locomotor activity test was performed on day 9, 10 min after the assessment of body temperature.
Statistical analysis
The effect of pioglitazone, of GW-9662, or their combination on the development of morphine tolerance or in the reversal of its expression was analysed by mixed-factorial anova with treatments (pioglitazone, morphine or their vehicles) as the between-subject factor and time (days) as the within-subject factor. Differences in the development of morphine tolerance in PPARγ KO mice and their WT counterparts were assessed by mixed-factorial anova with strains (KO and WT) and treatment (morphine or saline) as the between-subject factors and time (days) as the within-subject factor. Where appropriate, the Newman–Keuls post hoc test was performed. Statistical significance was set at P < 0.05.
Results
Tolerance to the antinociceptive effect of morphine is attenuated by co-administration of the PPARγ agonist pioglitazone
C57 mice (n = 59) were divided into six groups. Group 1 (n = 10) received drug vehicles (veh/veh); group 2 (n = 10) received pioglitazone vehicle plus morphine (veh/mor) at 15 (day 1 and 9) or 30 mg·kg−1 (day 2–8); group 3 (n = 10) and group 4 (n = 10) received pioglitazone at 10 (pio10/veh) or 30 mg·kg−1 plus morphine vehicle (pio30/veh); group 5 (n = 9) and group 6 (n = 10) received pioglitazone at 10 (pio10/mor) or 30 mg·kg−1 (pio30/mor) plus morphine at 15 (day 1 and 9) or 30 mg·kg−1 (day 2–8).
The anova demonstrated a significant effect of treatment [F(5.53) = 25.443: P < 0.0001], time [F(4.53) = 9.78: P < 0.0001] and the treatment × time interaction [F(20.212) = 9.056: P < 0.0001]. As shown in Figure 1, the Newman–Keuls test revealed that, compared with the vehicle, morphine retained a significant antinociceptive effect on day 1, 3 (P < 0.01) and 7 (P < 0.05). Its antinociceptive efficacy progressively decreased, and on test day 9 no difference from controls was found. Treatment with pioglitazone attenuated the development of morphine tolerance, and, as shown in Figure 1, in the mice that received morphine and pioglitazone (10 and 30 mg·kg−1), opioid analgesia was maintained for the entire treatment period (P < 0.01). Treatment with pioglitazone did not result in analgesic effects.
Figure 1.
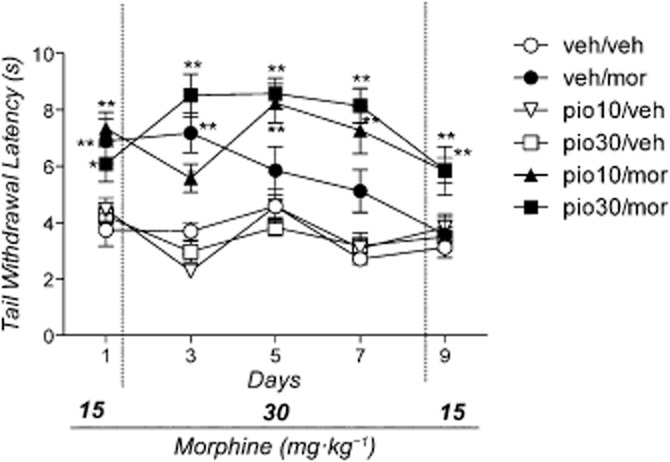
Effect of morphine, pioglitazone or their combination on tail immersion test. Mice (n = 59) were divided into six groups: group 1 (n = 10) received drug vehicles (veh/veh); group 2 (n = 10) received pioglitazone vehicle plus morphine (veh/mor); group 3 (n = 10) and group 4 (n = 10) received 10 or 30 mg·kg−1 of pioglitazone followed by morphine vehicle (pio10/veh and pio30/veh); group 5 (n = 9) and group 6 (n = 10) received 10 or 30 mg·kg−1 of pioglitazone followed by morphine (pio10/mor and pio30/mor). Significantly different from controls (veh/veh): **P < 0.01 and *P < 0.05.
Treatment with the PPARγ antagonist GW-9662 blocked the effect of pioglitazone in the development of tolerance to the antinociceptive effect of morphine
Mice (n = 59) were divided into six groups. Group 1 (n = 10) received drug vehicles (veh/veh/veh); group 2 (n = 10) received GW-9662 and pioglitazone vehicles followed by morphine (veh/veh/mor); group 3 (n = 10) received pioglitazone (10 mg·kg−1) plus morphine and GW-9662 vehicles (pio/veh/veh); group 4 (n = 10) received GW-9662 (5 mg·kg−1) followed by pioglitazone and morphine vehicles (veh/GW5/veh); group 5 (n = 10) received pioglitazone 10 mg·kg−1), morphine and GW-9662 vehicle (pio/veh/mor); group 6 (n = 9) received pioglitazone, morphine and GW-9662 (5 mg·kg−1) (pio/GW5/mor).
The anova revealed a significant effect of treatment [F(5.41) = 62.79: P < 0.001], time [F(4.41) = 2.41: P < 0.05] and the treatment × time interaction [F(20.164) = 3.54: P < 0.001]. As shown in Figure 2, the Newman–Keuls test revealed that morphine significantly increased the latency of tail withdrawal (P < 0.01). However, its effect progressively decayed, and on day 9 it was no longer evident. This result confirmed the previous results, in that the co-administration of pioglitazone (10 mg·kg−1) attenuated the development of morphine tolerance, and the analgesic effect was maintained throughout the treatment period (P < 0.01). Of note, pretreatment with GW-9662 (5 mg·kg−1) completely antagonized the effect of pioglitazone (10 mg·kg−1), and the analgesic curve with this treatment was indistinguishable from that generated by morphine alone.
Figure 2.
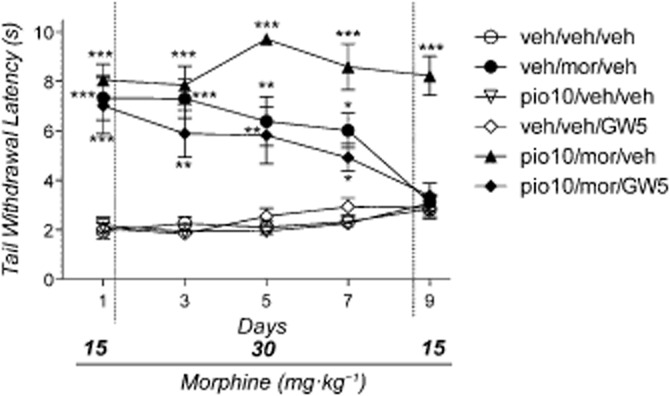
Effect of morphine, pioglitazone, GW-9662 or their combination on tail immersion test. Mice (n = 59) were divided into six groups: group 1 (n = 10) received drug vehicles (veh/veh/veh); group 2 (n = 10) received pioglitazone and GW-9662 vehicles and morphine (veh/veh/mor); group 3 (n = 10) received pioglitazone (10 mg·kg−1) and morphine and GW-9662 vehicles (pio10/veh/veh); group 4 (n = 10) received pioglitazone and morphine vehicles and GW-9662 (5 mg·kg−1) (veh/veh/GW5); group 5 (n = 10) received pioglitazone (10 mg·kg−1), morphine and GW-9662 vehicle (pio10/veh/mor); group 6 (n = 9) received pioglitazone, morphine and GW-9662 (5 mg·kg−1) (pio10/mor/GW5). Significantly different from controls (veh/veh/veh): ***P < 0.001, **P < 0.01 and *P < 0.05.
Treatment with pioglitazone or GW-9662 did not provide analgesic effects.
Blockade of PPARγ accelerates the development of tolerance to the antinociceptive effect of morphine
In this experiment, a lower dose (15 mg·kg−1) of morphine was used for the duration of treatment to elicit mild tolerance. Mice (n = 39) were divided into four groups. Group 1 (n = 10) received drug vehicles (veh/veh); group 2 (n = 10) received GW-9662 vehicle and morphine (veh/mor); group 3 (n = 10) received GW-9662 (5 mg·kg−1) and morphine vehicle (GW5/veh); group 4 (n = 9) received GW-9662 (5 mg·kg−1) and morphine (GW5/mor).
The anova revealed a significant effect of treatment [F(3,35) = 28.84: P < 0.0001], time [F(3,35) = 17.16: P < 0.001] and the treatment × time interaction [F(9,105) = 3.01: P < 0.01]. As shown in Figure 3, in morphine-treated animals, no overt signs of tolerance were observed and the Newman–Keuls test revealed a significant antinociceptive effect of morphine throughout the treatment period (P < 0.01). However, in the group treated with 5.0 mg·kg−1 GW-9662 plus morphine, the antinociceptive effect of the opioid progressively decreased and the difference from vehicle-treated mice was no longer significant at days 7–9.
Figure 3.
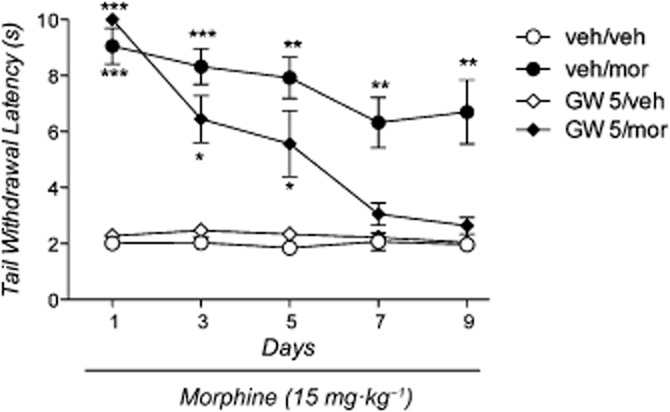
Effect of morphine, GW-9662 or their combination on tail immersion test. Mice (n = 39) were divided into four groups: group 1 (n = 10) received drug vehicles (veh/veh); group 2 (n = 10) received GW-9662 vehicle and morphine (veh/mor); group 3 (n = 10) received GW-9662 (5 mg·kg−1) and morphine vehicle (GW5/veh); group 4 (n = 9) received GW-9662 (5 mg·kg−1) and morphine (GW5/mor). Significantly different from controls (veh/veh): ***P < 0.001, **P < 0.01 and *P < 0.05.
Development of tolerance to the antinociceptive effect of morphine is more pronounced in conditional neuronal PPARγ KO mice than in WT controls
As in the previous experiment, 15 mg·kg−1 of morphine was used for the duration of treatment to elicit mild tolerance. PPARγ KO mice (n = 20) and WT controls (n = 20) were divided into two groups each (n = 10/group) and were treated with 15 mg·kg−1 of morphine (morph) or with its vehicle (veh).
The anova revealed a significant effect of treatment [F(3,36) = 251.41: P < 0.0001], time [F(4,36) = 20.37: P < 0.0001] and the treatment × time interaction [F(12,144) = 20.37: P < 0.0001]. Significant effects of strain [F(1,36) = 11.11: P < 0.01], strain × treatment interaction [F(1,36) = 28.5: P < 0.0001], time × strain interaction [F(4,44) = 6.09: P < 0.001] and time × strain × treatment interaction [F(4,144) = 2.23: P < 0.05] were also observed. As shown in Figure 4, the Newman–Keuls post hoc analysis demonstrated that morphine elicited a significant antinociceptive effect in both PPARγ KO mice and WT controls. However, over the course of the treatment, the antinociceptive effect of morphine decreased faster in KO mice than in WT controls. This phenomenon was confirmed by the statistical analysis, which showed a significant effect of morphine throughout the treatment period in WT mice (P < 0.001), whereas in KO mice the effect remained significant until day 5 and disappeared on day 7.
Figure 4.
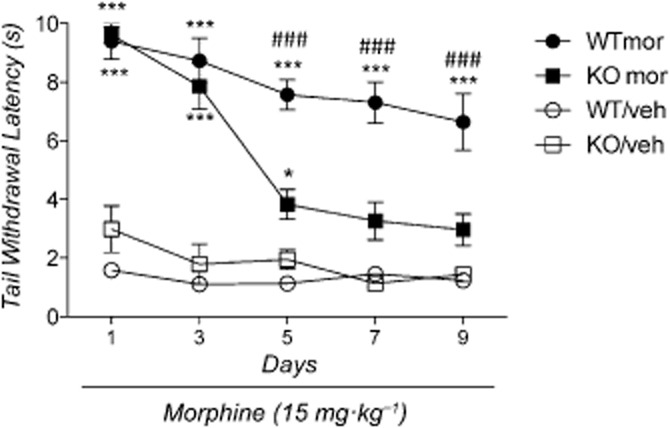
Effect of morphine on tail immersion test in PPARγ knockout (KO) and wild-type (WT) mice. Each line was divided into two groups containing (n = 10/group): groups 1 (WT/veh) and 2 (KO/veh) received morphine vehicle; groups 3 (WT mor) and 4 (KO mor) received morphine at the dose of 15 mg·kg−1. Significantly different from respective control vehicle-treated mice: *P < 0.05 and ***P < 0.0001. Significant difference between KO (mor) and WT (mor) mice: ###P < 0.0001.
Treatment with pioglitazone attenuated the development of tolerance to the antinociceptive effect of morphine in WT but not in PPARγ KO mice
PPARγ KO (n = 40) and WT (n = 40) mice were both divided into four groups of 10 animals each. Group 1 received drug vehicles (veh/veh); group 2 received morphine (15 mg·kg−1 on day 1 and 9; 30 mg·kg−1 on days 2–8) plus pioglitazone vehicle (veh/mor); group 3 received 10 mg·kg−1 of pioglitazone and morphine vehicle (pio10/veh); group 4 received morphine (15 mg·kg−1 on day 1 and 9; 30 mg·kg−1 on days 2–8) plus 10 mg·kg−1 of pioglitazone (pio10/mor).
As shown in Figure 5A, the anova revealed a significant effect of the treatment [F(3,36) = 251.41: P < 0.0001], time [F(4,36) = 20.37: P < 0.0001] and treatment × time interaction [F(12,144) = 20.37: P < 0.0001] in WT mice. Post hoc Newman–Keuls analysis showed that morphine significantly increased the latency of tail withdrawal on days 1, 3 and 5 (P < 0.001). The antinociceptive effect of morphine progressively decreased over the course of the treatment, and on day 9 it was no longer significant compared with vehicle-treated animals. Pretreatment with pioglitazone markedly attenuated the development of tolerance. Compared with the vehicle, the antinociceptive effect of morphine remained significant for the entire treatment period (P < 0.001). Pioglitazone alone had no effect on the tail immersion test. As shown in Figure 5B, the anova revealed a significant effect of treatment [F(3,36) = 58.73: P < 0.0001], time [F(4,36) = 56.95: P < 0.0001] and treatment × time interaction [F(12,144) = 14.46: P < 0.0001] in PPARγ KO mice. The Newman–Keuls test showed that morphine significantly increased the latency of tail withdrawal on days 1–5 (P < 0.001) and on day 7 (P < 0.05). However, the antinociceptive effect of morphine progressively decreased over the treatment period, and on day 9 it was no longer different from vehicle-treated animals. Pretreatment with pioglitazone did not attenuate the development of tolerance to the antinociceptive effect of morphine, indicating its ineffectiveness in mice lacking neuronal PPARγ receptors.
Figure 5.
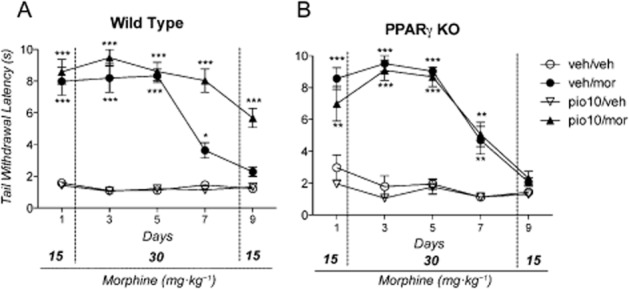
Effect of morphine, pioglitazone or their combination on tail immersion test in: (A) wild-type (WT) mice that were divided into four groups (n = 10/group): group 1 (veh/veh) received drug vehicles; group 2 (veh/mor) received pioglitazone vehicle and morphine; group 3 (pio10/veh) received 10 mg·kg−1 of pioglitazone plus morphine vehicle; group 4 (pio10/mor) received 10 mg·kg−1 of pioglitazone and morphine; (B) PPARγ knockout (KO) mice that were divided into four groups + (n = 10/group): group 1 (veh/veh) received drug vehicles; group 2 (veh/mor) received pioglitazone vehicle and morphine; group 3 (pio10/veh) received 10 mg·kg−1 of pioglitazone plus morphine vehicle; group 4 (pio10/morph) received 10 mg·kg−1 of pioglitazone and morphine. Significantly different from controls (veh/veh): ***P < 0.0001, **P < 0.01 and *P < 0.05.
Reversal of tolerance to the antinociceptive effect of morphine, by administration of the PPARγ agonist pioglitazone
C57 mice (n = 40) were divided into four groups. Group 1 (n = 10) received drug vehicles (veh/veh); group 2 (n = 10) received pioglitazone vehicle (evening day 8 and day 9) plus morphine (veh/mor) at 15 (day 1 and 9) or 30 mg·kg−1 (day 2–8); group 3 (n = 10) and group 4 (n = 10) received 10 (pio10/mor) or 30 mg·kg−1 (pio30/mor) of pioglitazone (evening day 8 and day 9) plus morphine at 15 (day 1 and 9) or 30 mg·kg−1 (day 2–8).
The anova demonstrated a significant effect of treatment [F(3.36) = 31.995: P < 0.0001], time [F(4.36) = 5.7775: P < 0.001] and the treatment × time interaction [F(12.144) = 2.412: P < 0.01]. As shown in Figure 6, the Newman–Keuls test revealed that, compared with the vehicle, morphine retained a significant antinociceptive effect on day 1, 3, 5 and 7 (P < 0.01). Its antinociceptive efficacy progressively decreased, and on test day 9 no difference from controls was found. Treatment with pioglitazone (on day 8 and 9) completely reversed the expression of morphine tolerance, and, as shown in Figure 6, in the mice that received morphine and pioglitazone (10 and 30 mg·kg−1), opioid analgesia was restored on day 9 (P < 0.01).
Figure 6.
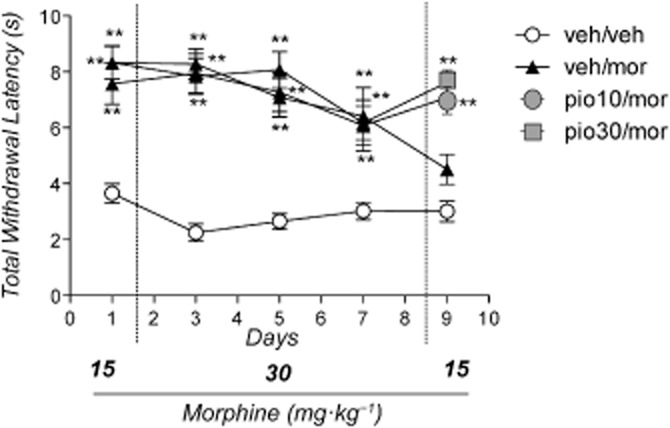
Effect of pioglitazone on reversal of morphine tolerance on tail immersion test. Mice (n = 40) were divided into four groups. Group 1 (n = 10) received drug vehicles (veh/veh); group 2 (n = 10) received pioglitazone vehicle (evening day 8 and day 9) plus morphine (veh/mor) at 15 (day 1 and 9) or 30 mg·kg−1 (day 2–8); group 3 (n = 10) and group 4 (n = 10) received 10 (pio10/mor) or 30 mg·kg−1 (pio30/mor) of pioglitazone (evening day 8 and day 9) plus morphine at 15 (day 1 and 9) or 30 mg·kg−1 (day 2–8). Significantly different from controls (veh/veh): **P < 0.01.
Effect of pioglitazone on locomotor activity and body temperature
C57 mice (n = 32) were divided into four groups. Group 1 (n = 8) received drug vehicles (veh/veh); group 2 (n = 8) received pioglitazone vehicle plus morphine (veh/mor) at 15 (day 1 and 9) or 30 mg·kg−1 (day 2–8); group 3 (n = 8) received pioglitazone 30 mg·kg−1 plus morphine vehicle (pio30/veh). Group 4 (n = 8) received 30 mg·kg−1 (pio30/mor) of pioglitazone plus morphine at 15 (day 1 and 9) or 30 mg·kg−1 (day 2–8). Results showed no significant differences between groups in terms of body temperature [F(3,28) = 2.316; P not significant] (data not shown). In the locomotor activity test, anova demonstrated a significant effect of the treatment [F(3,28) = 26.27; P < 0.001]. The total distance travelled was 2106.7 ± 175.9 cm for the group veh/veh, 2362.8 ± 177.7 cm for the group pio30/veh, 3712.0 ± 579.7 cm for the group veh/mor and 6066.8 ± 660.3 cm for the group pio30/mor. Newman–Keuls post hoc test showed that locomotor activity was increased in animals treated with morphine (P < 0.05). Treatment with pioglitazone was able to potentiate the morphine-induced increase of locomotor activity in mice (P < 0.001).
Discussion and conclusions
The results showed that repeated daily administration of morphine lead to a rapid development of tolerance to its antinociceptive effect. Concomitant administration of pioglitazone markedly reduced the development of tolerance. In addition, we demonstrated that acute treatment with pioglitazone was able to reverse an established morphine tolerance. Pioglitazone alone did not show any effect in response to noxious thermal stimuli, suggesting that this compound lacks antinociceptive properties in our experimental model. Hence, the observed effect on morphine tolerance cannot be secondary to changes in the pain threshold following pioglitazone administration. This finding is particularly relevant because previous studies have shown that activation of PPARγ elicits a pronounced attenuation of neuropathic pain following partial nerve ligation in rats (Churi et al., 2008; Iwai et al., 2008; Fehrenbacher et al., 2009; Takahashi et al., 2011; Morgenweck et al., 2013). Importantly, pioglitazone did not alter body temperature neither reduced locomotor activity, ruling out unspecific or sedative effects that might have influenced animals’ reactivity in the tail withdrawal test. To confirm that the effect of pioglitazone was mediated by PPARγ, in a subsequent set of experiments, we administered the selective receptor antagonist GW-9662 prior to pioglitazone. As expected, co-administration of the PPARγ blocker completely abolished the effect of pioglitazone on morphine tolerance. Confirming the lack of a role of PPARγ in thermal analgesia, GW-9662 alone did not show effects in the tail immersion test. Intriguingly, when animals were treated with GW-9662, we observed a tendency towards the facilitation of analgesic tolerance to morphine. We explored this possibility by treating animals with a lower dose of morphine (15 mg·kg−1), which, when given for the entire treatment period (9 days), led to a modest non-significant analgesic tolerance. Confirming our initial observations, co-administration of GW-9662 significantly accelerated the development of tolerance, and the analgesic effect of morphine started to decrease from treatment day 3. This is a major finding that indicates a role of PPARγ in the regulation of the endogenous mechanisms controlling the development of tolerance to the antinociceptive effect of morphine. An intriguing hypothesis, therefore, is that conditions associated with reduced tonic activity or down-regulation of PPARγ may facilitate opioid tolerance. Some evidence may support this view; for example, it has been shown that injections of the bacterial endotoxin lipopolysaccharide accelerate the development of analgesic tolerance to morphine, but they also reduce the expression of the PPARγ transcript (Johnston and Westbrook, 2005; Necela et al., 2008). Conversely, physical stress that has been associated with increased PPARγ expression (Garcia-Bueno et al., 2005), among other effects on analgesia, also reduces the development of morphine tolerance (MacRae and Siegel, 1987; Takahashi et al., 1988; Yamashiro et al., 1990). A growing body of evidence indicates that opioids activate glial cells and modulate the immune response and that these effects may have a major impact in opioid tolerance and possibly also addiction (Narita et al., 2006; Hutchinson et al., 2007; 2008a,). Hence, one potential mechanism through which PPARγ agonists may reduce tolerance to the antinociceptive effects of morphine is through their ability to act as a glial inhibitors and to reduce the expression of pro-inflammatory factors such as IL-1ß, IL-6, and TNF-α and NF-κB (Bernardo and Minghetti, 2006). However, we found that the effect of pioglitazone is not present in conditional KO mice lacking the PPARγ receptor in neurons. We also observed facilitation of the development of analgesic tolerance to morphine in these mice, which further confirms the role of neuronal PPARγ in the regulation of this phenomenon. Based on these findings, it seems, therefore, that neuron-mediated mechanisms are responsible for the effects of pioglitazone observed here. However, an indirect role of glia-mediated mechanisms that could be under the control of neuronal PPARγ cannot be excluded. Further studies are needed in order to clarify this point.
Like for opioids, chronic administration of cannabinoid agonists results in the development of tolerance to most of their acute pharmacological effects (Maldonado, 2002). Importantly, the interaction between cannabinoids and opioids on the development of tolerance is supported by cross-modulation of several pharmacological responses induced by these compounds, such as antinociception, hypolocomotion, catalepsy and hypothermia (Pertwee et al., 1993; Fan et al., 1994; Vigano et al., 2005; Maguma and Taylor, 2011). Based on these functional similarities between the opioid and the cannabinoid systems and given that PPARγ mediates some of the cannabinoid effects on neuroinflammation (O'Sullivan et al., 2006; Sun et al., 2006), we speculate that treatment with pioglitazone could also affect the development of cannabinoid tolerance. However, this aspect needs to be clarified with further studies.
Recently, important effects mediated by brain PPARγ in relation to drugs of abuse have been described. For instance, our laboratory has shown that administration of pioglitazone attenuates alcohol self-administration and relapse to alcohol seeking in rats (Stopponi et al., 2011; 2013,). More importantly, we also found that it reduced intravenous heroin self-administration and morphine-induced reward, possibly by inhibiting the opioid stimulation of mesolimbic dopamine transmission (Ciccocioppo et al., 2012). Altogether, these results suggest that the combination of PPARγ agonists with opioid agents may reduce the development of analgesic tolerance and possibly reduce the potential for abuse of these compounds. It is noteworthy that patients who have become physically dependent on opioids following chronic pain treatment may persist with drug use once the pain has dissipated to avoid withdrawal symptoms (Savage et al., 2008). Delayed development of tolerance should reduce the requirement for escalating its dosage to maintain adequate chronic analgesia. This, in turn, may diminish the development of physical dependence and therefore withdrawal when the treatment is discontinued. Moreover, reduced escalation of the opioid dose may also favourably impact the side effects associated with the use of high doses of these drugs such as sedation, respiratory depression, constipation, nausea and urinary retention. PPARγ agonists are also known to have intrinsic anti-inflammatory properties and to reduce neuropathic pain (Churi et al., 2008; Iwai et al., 2008; Napimoga et al., 2008; Fehrenbacher et al., 2009; Takahashi et al., 2011; Morgenweck et al., 2013). This effect, combined with the ability of PPARγ agonists to attenuate opioid tolerance and addiction, suggests the attractive possibility of combining them with opioids to achieve an enhanced analgesic effect and reduced potential for abuse. This approach could have a relevant clinical impact because management of chronic pain (including neuropathic pain) remains an important medical need that is limited by all the problems linked to protracted opioid use. Another major clinical problem is the use of opioid analgesics in patients at a high risk of developing dependence, such as individuals with a positive history of substance misuse (i.e., alcohol, benzodiazepines etc.). Currently, these patients are less likely to receive effective pain treatment due to concerns of opioid misuse (Rupp and Delaney, 2004). The combination of PPARγ agonists and opioids may help to address the ethical issue related to the appropriateness of the use of opioid analgesics in those patients. In summary, our results indicate that PPARγ plays an important role in the modulation of tolerance to morphine, and pioglitazone may potentially be used as an add-on medication to attenuate the development of opioid tolerance when these drugs are used for chronic pain. Clinical trials to test this possibility should be relatively easy to conduct because pioglitazone is already used clinically and its pharmacological and toxicological profile has been well documented.
Acknowledgments
This study was supported by the University of Camerino funds (to R. C.). Authors wish to thank Professor K.D. Neiswender at Vanderbilt University for providing the PPARγ KO mice employed in the study.
Glossary
- KO
knockout
- MOP receptor
μ-opioid receptor
- WT
wild type
Author contributions
G. d. G. and R. C. were responsible for the study concept and design. G. d. G. and G. S. contributed to the acquisition of animal data and drafted the manuscript. M. K. and S. S. assisted with interpretation of findings and contributed with data collection. G. D. and G. G. provided critical revision of the manuscript for important intellectual content. All authors critically reviewed the content and approved the final version for publication.
Conflict of interest
G. D. is Chairman and CEO and G. G. is Chief Scientific Officer of Omeros Corporation. Omeros exclusively controls the intellectual property rights from the University of Camerino. R. C. is the inventor on a number of patent applications, which have been assigned to Omeros, relating to the therapeutic use of PPARγ agonists in addiction. He is entitled to receive payments and royalties from Omeros under such licensing arrangement. The other authors have no conflict of interest.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear hormone receptors. Br J Pharmacol. 2013b;170:1652–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Connor M. Opioids: cellular mechanisms of tolerance and physical dependence. Curr Opin Pharmacol. 2005;5:60–68. doi: 10.1016/j.coph.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Llorente J, Gabra BH, Smith FL, Dewey WL, Kelly E, et al. Role of protein kinase C and mu-opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. Eur J Neurosci. 2009;29:307–318. doi: 10.1111/j.1460-9568.2008.06573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger J, Wagner JA. Physiological and therapeutic roles of peroxisome proliferator-activated receptors. Diabetes Technol Ther. 2002;4:163–174. doi: 10.1089/15209150260007381. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Minghetti L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr Pharm Des. 2006;12:93–109. doi: 10.2174/138161206780574579. [DOI] [PubMed] [Google Scholar]
- Bland ST, Hutchinson MR, Maier SF, Watkins LR, Johnson KW. The glial activation inhibitor AV411 reduces morphine-induced nucleus accumbens dopamine release. Brain Behav Immun. 2009;23:492–497. doi: 10.1016/j.bbi.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Churi SB, Abdel-Aleem OS, Tumber KK, Scuderi-Porter H, Taylor BK. Intrathecal rosiglitazone acts at peroxisome proliferator-activated receptor-gamma to rapidly inhibit neuropathic pain in rats. J Pain. 2008;9:639–649. doi: 10.1016/j.jpain.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccocioppo R, de Guglielmo G, Melis M, De Luca MA, Li H, Cippitelli A, et al. Activation of PPARgamma by the anti-diabetic agent pioglitazone reduces opiate reinforcement and opiate-induced activation of the mesolimbic dopamine system. New Orleans, USA. Neuroscience. 2012;2012:668.16. [Google Scholar]
- Contet C, Filliol D, Matifas A, Kieffer BL. Morphine-induced analgesic tolerance, locomotor sensitization and physical dependence do not require modification of mu opioid receptor, cdk5 and adenylate cyclase activity. Neuropharmacology. 2008;54:475–486. doi: 10.1016/j.neuropharm.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Davis ME, Akera T, Brody TM. Reduction of opiate binding to brainstem slices associated with the development of tolerance to morphine in rats. J Pharmacol Exp Ther. 1979;211:112–119. [PubMed] [Google Scholar]
- Diaz A, Pazos A, Florez J, Hurle MA. Autoradiographic mapping of mu-opioid receptors during opiate tolerance and supersensitivity in the rat central nervous system. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:101–109. doi: 10.1007/s002100000258. [DOI] [PubMed] [Google Scholar]
- Fan F, Compton DR, Ward S, Melvin L, Martin BR. Development of cross-tolerance between delta 9-tetrahydrocannabinol, CP 55,940 and WIN 55,212. J Pharmacol Exp Ther. 1994;271:1383–1390. [PubMed] [Google Scholar]
- Fehrenbacher JC, Loverme J, Clarke W, Hargreaves KM, Piomelli D, Taylor BK. Rapid pain modulation with nuclear receptor ligands. Brain Res Rev. 2009;60:114–124. doi: 10.1016/j.brainresrev.2008.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bueno B, Madrigal JL, Lizasoain I, Moro MA, Lorenzo P, Leza JC. Peroxisome proliferator-activated receptor gamma activation decreases neuroinflammation in brain after stress in rats. Biol Psychiatry. 2005;57:885–894. doi: 10.1016/j.biopsych.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Gillies PS, Dunn CJ. Pioglitazone. Drugs. 2000;60:333–343. doi: 10.2165/00003495-200060020-00009. , discussion 344–335. [DOI] [PubMed] [Google Scholar]
- Gofflot F, Chartoire N, Vasseur L, Heikkinen S, Dembele D, Le Merrer J, et al. Systematic gene expression mapping clusters nuclear receptors according to their function in the brain. Cell. 2007;131:405–418. doi: 10.1016/j.cell.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. ScientificWorldJournal. 2007;7:98–111. doi: 10.1100/tsw.2007.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Coats BD, Lewis SS, Zhang Y, Sprunger DB, Rezvani N, et al. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav Immun. 2008a;22:1178–1189. doi: 10.1016/j.bbi.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Northcutt AL, Chao LW, Kearney JJ, Zhang Y, Berkelhammer DL, et al. Minocycline suppresses morphine-induced respiratory depression, suppresses morphine-induced reward, and enhances systemic morphine-induced analgesia. Brain Behav Immun. 2008b;22:1248–1256. doi: 10.1016/j.bbi.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Lewis SS, Coats BD, Skyba DA, Crysdale NY, Berkelhammer DL, et al. Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast) Brain Behav Immun. 2009;23:240–250. doi: 10.1016/j.bbi.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev. 2011;63:772–810. doi: 10.1124/pr.110.004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inturrisi CE. Clinical pharmacology of opioids for pain. Clin J Pain. 2002;18(4 Suppl):S3–S13. doi: 10.1097/00002508-200207001-00002. [DOI] [PubMed] [Google Scholar]
- Iwai S, Maeda T, Kiguchi N, Kobayashi Y, Fukazawa Y, Ozaki M, et al. Pioglitazone attenuates tactile allodynia and microglial activation in mice with peripheral nerve injury. Drug Discov Ther. 2008;2:353–356. [PubMed] [Google Scholar]
- Johnson EE, Chieng B, Napier I, Connor M. Decreased mu-opioid receptor signalling and a reduction in calcium current density in sensory neurons from chronically morphine-treated mice. Br J Pharmacol. 2006;148:947–955. doi: 10.1038/sj.bjp.0706820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston IN, Westbrook RF. Inhibition of morphine analgesia by LPS: role of opioid and NMDA receptors and spinal glia. Behav Brain Res. 2005;156:75–83. doi: 10.1016/j.bbr.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Jones JR, Shelton KD, Guan Y, Breyer MD, Magnuson MA. Generation and functional confirmation of a conditional null PPARgamma allele in mice. Genesis. 2002;32:134–137. doi: 10.1002/gene.10042. [DOI] [PubMed] [Google Scholar]
- Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci. 2008;13:1813–1826. doi: 10.2741/2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch T, Hollt V. Role of receptor internalization in opioid tolerance and dependence. Pharmacol Ther. 2008;117:199–206. doi: 10.1016/j.pharmthera.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Kreek MJ. Drug addictions. Molecular and cellular endpoints. Ann N Y Acad Sci. 2001;937:27–49. [PubMed] [Google Scholar]
- Landreth GE, Heneka MT. Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer's disease. Neurobiol Aging. 2001;22:937–944. doi: 10.1016/s0197-4580(01)00296-2. [DOI] [PubMed] [Google Scholar]
- Le Bars D, Gozariu M, Cadden SW. Animal models of nociception. Pharmacol Rev. 2001;53:597–652. [PubMed] [Google Scholar]
- MacRae JR, Siegel S. Extinction of tolerance to the analgesic effect of morphine: intracerebroventricular administration and effects of stress. Behav Neurosci. 1987;101:790–796. doi: 10.1037//0735-7044.101.6.790. [DOI] [PubMed] [Google Scholar]
- Maguma H, Taylor DA. The effect of chronic opioid vs. cannabinoid exposure on the expression of tolerance to morphine- or WIN-55,212-2-induced analgesia and hypothermia in the guinea pig. Eur J Pharmacol. 2011;660:334–340. doi: 10.1016/j.ejphar.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Maldonado R. Study of cannabinoid dependence in animals. Pharmacol Ther. 2002;95:153–164. doi: 10.1016/s0163-7258(02)00254-1. [DOI] [PubMed] [Google Scholar]
- Martini L, Whistler JL. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr Opin Neurobiol. 2007;17:556–564. doi: 10.1016/j.conb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006;58:726–741. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Grisel JE, Hayward MD, Bales JR, Rubinstein M, Belknap JK, et al. Disparate spinal and supraspinal opioid antinociceptive responses in beta-endorphin-deficient mutant mice. Neuroscience. 2000;101:709–717. doi: 10.1016/s0306-4522(00)00422-x. [DOI] [PubMed] [Google Scholar]
- Moreno S, Farioli-Vecchioli S, Ceru MP. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience. 2004;123:131–145. doi: 10.1016/j.neuroscience.2003.08.064. [DOI] [PubMed] [Google Scholar]
- Morgenweck J, Griggs RB, Donahue RR, Zadina JE, Taylor BK. PPARgamma activation blocks development and reduces established neuropathic pain in rats. Neuropharmacology. 2013;70:236–246. doi: 10.1016/j.neuropharm.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napimoga MH, Vieira SM, Dal-Secco D, Freitas A, Souto FO, Mestriner FL, et al. Peroxisome proliferator-activated receptor-gamma ligand, 15-deoxy-Delta12,14-prostaglandin J2, reduces neutrophil migration via a nitric oxide pathway. J Immunol. 2008;180:609–617. doi: 10.4049/jimmunol.180.1.609. [DOI] [PubMed] [Google Scholar]
- Narita M, Miyatake M, Suzuki M, Kuzumaki N, Suzuki T. Role of astrocytes in rewarding effects of drugs of abuse. Nihon Shinkei Seishin Yakurigaku Zasshi. 2006;26:33–39. [PubMed] [Google Scholar]
- Necela BM, Su W, Thompson EA. Toll-like receptor 4 mediates cross-talk between peroxisome proliferator-activated receptor gamma and nuclear factor-kappaB in macrophages. Immunology. 2008;125:344–358. doi: 10.1111/j.1365-2567.2008.02849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE, Kendall DA, Randall MD. Further characterization of the time-dependent vascular effects of delta9-tetrahydrocannabinol. J Pharmacol Exp Ther. 2006;317:428–438. doi: 10.1124/jpet.105.095828. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Stevenson LA, Griffin G. Cross-tolerance between delta-9-tetrahydrocannabinol and the cannabimimetic agents, CP 55,940, WIN 55,212-2 and anandamide. Br J Pharmacol. 1993;110:1483–1490. doi: 10.1111/j.1476-5381.1993.tb13989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. 2002;22:9980–9989. doi: 10.1523/JNEUROSCI.22-22-09980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp T, Delaney KA. Inadequate analgesia in emergency medicine. Ann Emerg Med. 2004;43:494–503. doi: 10.1016/j.annemergmed.2003.11.019. [DOI] [PubMed] [Google Scholar]
- Sarruf DA, Yu F, Nguyen HT, Williams DL, Printz RL, Niswender KD, et al. Expression of peroxisome proliferator-activated receptor-gamma in key neuronal subsets regulating glucose metabolism and energy homeostasis. Endocrinology. 2009;150:707–712. doi: 10.1210/en.2008-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage SR, Kirsh KL, Passik SD. Challenges in using opioids to treat pain in persons with substance use disorders. Addict Sci Clin Pract. 2008;4:4–25. doi: 10.1151/ascp08424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shavit Y, Wolf G, Goshen I, Livshits D, Yirmiya R. Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance. Pain. 2005;115:50–59. doi: 10.1016/j.pain.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Sim LJ, Selley DE, Dworkin SI, Childers SR. Effects of chronic morphine administration on mu opioid receptor-stimulated [35S]GTPgammaS autoradiography in rat brain. J Neurosci. 1996;16:2684–2692. doi: 10.1523/JNEUROSCI.16-08-02684.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith U. Pioglitazone: mechanism of action. Int J Clin Pract Suppl. 2001;121:13–18. [PubMed] [Google Scholar]
- Stopponi S, Somaini L, Cippitelli A, Cannella N, Braconi S, Kallupi M, et al. Activation of nuclear PPARgamma receptors by the antidiabetic agent pioglitazone suppresses alcohol drinking and relapse to alcohol seeking. Biol Psychiatry. 2011;69:642–649. doi: 10.1016/j.biopsych.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Stopponi S, de Guglielmo G, Somaini L, Cippitelli A, Cannella N, Kallupi M, et al. Activation of PPARgamma by pioglitazone potentiates the effects of naltrexone on alcohol drinking and relapse in msP rats. Alcohol Clin Exp Res. 2013;37:1351–1360. doi: 10.1111/acer.12091. [DOI] [PubMed] [Google Scholar]
- Sun Y, Alexander SP, Kendall DA, Bennett AJ. Cannabinoids and PPARalpha signalling. Biochem Soc Trans. 2006;34(Pt 6):1095–1097. doi: 10.1042/BST0341095. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Deguchi Y, Kaneto H. Blockade of the development of analgesic tolerance to morphine by concurrent treatment with opioid- but not non-opioid-mediated stress in mice. Jpn J Pharmacol. 1988;46:1–5. doi: 10.1254/jjp.46.1. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Hasegawa-Moriyama M, Sakurai T, Inada E. The macrophage-mediated effects of the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuate tactile allodynia in the early phase of neuropathic pain development. Anesth Analg. 2011;113:398–404. doi: 10.1213/ANE.0b013e31821b220c. [DOI] [PubMed] [Google Scholar]
- Tao PL, Law PY, Loh HH. Decrease in delta and mu opioid receptor binding capacity in rat brain after chronic etorphine treatment. J Pharmacol Exp Ther. 1987;240:809–816. [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- Tureyen K, Kapadia R, Bowen KK, Satriotomo I, Liang J, Feinstein DL, et al. Peroxisome proliferator-activated receptor-gamma agonists induce neuroprotection following transient focal ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J Neurochem. 2007;101:41–56. doi: 10.1111/j.1471-4159.2006.04376.x. [DOI] [PubMed] [Google Scholar]
- Vigano D, Rubino T, Vaccani A, Bianchessi S, Marmorato P, Castiglioni C, et al. Molecular mechanisms involved in the asymmetric interaction between cannabinoid and opioid systems. Psychopharmacology (Berl) 2005;182:527–536. doi: 10.1007/s00213-005-0114-4. [DOI] [PubMed] [Google Scholar]
- Xing B, Liu M, Bing G. Neuroprotection with pioglitazone against LPS insult on dopaminergic neurons may be associated with its inhibition of NF-kappaB and JNK activation and suppression of COX-2 activity. J Neuroimmunol. 2007;192:89–98. doi: 10.1016/j.jneuroim.2007.09.029. [DOI] [PubMed] [Google Scholar]
- Yamashiro O, Takahashi M, Kaneto H. Role of vasopressin in the blockade of the development of morphine tolerance by footshock and psychological stress. Arch Int Pharmacodyn Ther. 1990;307:60–70. [PubMed] [Google Scholar]
- Zachariou V, Bolanos CA, Selley DE, Theobald D, Cassidy MP, Kelz MB, et al. An essential role for DeltaFosB in the nucleus accumbens in morphine action. Nat Neurosci. 2006;9:205–211. doi: 10.1038/nn1636. [DOI] [PubMed] [Google Scholar]