

Abstract
Background and Purpose
Astrocytic excitatory amino acid transporters (EAATs) regulate extracellular glutamate concentrations and play a role in preventing neuroexcitotoxicity. As the δ-opioid receptor (DOP receptor) is neuroprotective against excitotoxic injury, we determined whether DOP receptor activation up-regulates EAAT expression and function.
Experimental Approach
We measured mRNA and protein expression of EAAT1, EAAT2 and EAAT3 in cultured mouse astrocytes exposed to a specific DOP receptor agonist (UFP-512) with or without a DOP receptor antagonist, DOP receptor siRNA or inhibitors of PKC, PKA, PI3K, p38, MAPK, MEK and ERK, and evaluated the function of EAATs by measuring glutamate uptake.
Key Results
Astrocytic DOP receptor mRNA and protein were suppressed by DOP receptor siRNA knockdown. DOP receptor activation increased mRNA and protein expression of EAAT1 and EAAT2, but not EAAT3, thereby enhancing glutamate uptake of astrocytes. DOP receptor-induced EAAT1 and EAAT2 expression was largely reversed by DOP receptor antagonist naltrindole or by DOP receptor siRNA knockdown, and suppressed by inhibitors of MEK, ERK and p38. DOP receptor-accelerated glutamate uptake was inhibited by EAAT blockers, DOP receptor siRNA knockdown or inhibitors of MEK, ERK or p38. In contrast, inhibitors of PKA, PKC or PI3K had no significant effect on DOP receptor-induced EAAT expression.
Conclusions and Implications
DOP receptor activation up-regulates astrocytic EAATs via MEK-ERK-p38 signalling, suggesting a critical role for DOP receptors in the regulation of astrocytic EAATs and protection against neuroexcitotoxicity. As decreased EAAT expression contributes to pathophysiology in many neurological diseases, including amyotrophic lateral sclerosis, our findings present a new platform for potential treatments of these diseases.
Table of Links
| TARGETS | LIGANDS |
|---|---|
| δ receptor (DOP receptor) | Calphostin C |
| μ receptor (MOP receptor) | DPDPE |
| EAAT1 | DL-TBOA |
| EAAT2 | FR180204 |
| EAAT3 | L-glutamate |
| ERK | Naltrindole |
| MEK | SB203580 |
| p38MAPK (p38) | UCPH-101 |
| PI3K | U0126 |
| PKA | |
| PKC |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a,b,c,,).
Introduction
The role of excitatory amino acids in the brain is like a double-edged sword requiring a perfect harmony in the homeostasis for normal neural functioning. While their presence is paramount for normal functions, such as synaptic plasticity, learning and development under physiological conditions, an excess accumulation at toxic concentrations can result in neuronal death (Ganel and Rothstein, 1999). Glutamate-induced excitotoxicity has been implicated in various neurological pathologies, including ischaemia/hypoxia, inflammation, traumatic brain injury and neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) (Schwartz et al., 2003; Bruijn et al., 2004). It is still an important target in neurological research although many new strategies have been recently proposed for brain protection (van Leyen et al., 2006; Zhao et al., 2013).
Excessive exposure to excitatory amino acids accumulated in the extracellular space results from an imbalance in the interplay between the glutamate transport system and functional regulation in neurons and astrocytes. A family of sodium-dependent excitatory amino acid transporters (EAATs) orchestrates this regulation through glutamate uptake thereby buffering against excitotoxicity (Danbolt, 2001; Maragakis and Rothstein, 2004). To date, five EAATs (EAAT1–5) have been cloned from human and animal tissue (Arriza et al., 1994; Maragakis and Rothstein, 2004; Maragakis et al., 2004; Sheldon and Robinson, 2007). Of these, EAAT1 and EAAT2 are predominantly expressed in glial cells found in cerebellum, forebrain and hippocampus, EAAT3 and EAAT4 are typically localized in neurons of cerebellum and EAAT5 is located mainly in retinal ganglion cells (Danbolt, 2001).
Astrocytes play a significant role in regulating this precarious balance in extracellular excitatory amino acid concentrations via EAAT1 and EAAT2, thus preventing neuronal injury (Maragakis and Rothstein, 2004; Maragakis et al., 2004; Sheldon and Robinson, 2007; Gilley and Kernie, 2011; Kim et al., 2011). Astrocytic EAAT1 and EAAT2 dysfunction has been implicated in many neurological diseases. Indeed, the expression of astrocytic EAATs is diminished in patients with ALS (Gilley and Kernie, 2011). Thus, enhancing astrocytic EAAT expression presents a potential for developing novel treatment strategies.
We (Zhang et al., 2000; 2002,; Chao et al., 2007; 2008; 2012,,; Kang et al., 2009; Yang et al., 2009; Chao and Xia, 2010; He et al., 2013; Tian et al., 2013) and other investigators (Ke et al., 2009; Duan et al., 2011; Turner and Johnson, 2011; Yang et al., 2011) have previously demonstrated the role of δ-opioid receptor (DOP receptor) in neuroprotection against myriad neurological conditions and insults, including glutamate-induced neuronal injury. Because glutamate-induced excitotoxicity is a common pathway in the molecular pathogenesis of these diseases (Plaitakis, 1990; Bruijn et al., 2004; Liang et al., 2008), we conjectured a possible link between DOP receptor neuroprotection and astrocytic EAATs. However, it is unknown whether DOP receptor plays any role in the regulation of EAATs although astrocytes are known to express DOP receptor. We have therefore performed this study to address if astrocytic EAATs are involved in the mechanism for the DOP receptor protection. Specifically, we asked if activation of DOP receptor enhances the expression of EAAT1, EAAT2 and EAAT3 and alters the rate of glutamate uptake in astrocytes.
Methods
Cell culture
All experiments were approved by the Animal Care Committee of Yale University School of Medicine and The University of Texas Medical School at Houston, accredited by the American Association for Accreditation of Laboratory Animal Care.
Astrocyte-enriched cultures were prepared from the primary mixed glial cell cultures (obtained from newborn C57BL/6J mice brain) by three to four repetitions of trypsinization and replating. The astrocyte cultures were more than 95% pure when examined under indirect immunofluorescence with an anti-glial fibrillary acidic protein (GFAP) antibody staining (Liang et al., 2008). Astrocyte cultures were maintained in DMEM supplemented with 10% fetal calf serum (Sigma-Aldrich, St. Louis, MO, USA), 5 μg·mL−1 bovine insulin (Sigma-Aldrich) and 0.2% glucose. The medium was exchanged every 3 days until the cells reached confluence.
Drug treatments
The drug and molecular target nomenclature used was in accordance with the BJP's Concise Guide to PHARMACOLOGY (Alexander et al., 2013a,b,). To assess the signalling pathway of UFP-512-induced astrocytic EAAT expression, cells were pretreated with the indicated drugs for 1 h and then incubated with UFP-512 in the presence of each drug. UFP-512 is a specific and potent DOP receptor agonist (Balboni et al., 2002; Chao et al., 2007). The final concentrations used for each drug were as follows: naltrindole, DOP receptor antagonist, 10 μM; L-glutamate (Sigma-Aldrich), 100 μM; DL-threo-β-benzyloxyaspartic acid (TBOA) (Tocris Bioscience, Minneapolis, MN, USA), EAATs inhibitor, 100 μM; UCPH-101 (UCPH) (Tocris Bioscience), EAAT1 inhibitor, 10 μM; dihydrokainic acid (DHK) (Tocris Bioscience), EAAT2 inhibitor, 300 μM; calphostin C (Tocris Bioscience), PKC inhibitor, 500 nM; H89 (Tocris Bioscience), PKA inhibitor, 10 μM; LY294002 (Tocris Bioscience), PI3K inhibitor, 10 μM; SB203580 (Tocris Bioscience), p38 MAPK inhibitor, 10 μM; U0126 (Tocris Bioscience), mitogen-activated protein kinase kinase (MEK) inhibitor, 5 μM; FR180204 (Tocris Bioscience), ERK inhibitor, 500 nM.
siRNA transfection
Astrocytes were transfected with DOP receptor small interfering RNA (siRNA) constructs or control siRNA (Life Technologies, Grand Island, NY, USA) with Lipofectamine® RNAi MAX (Life Technologies) according to the manufacturer's instructions. Briefly, the siRNA construct was diluted in Opti-MEM and then mixed with an equal volume of Lipofectamine diluted in Opti-MEM (1:1 ratio Lipofectamine to siRNA) and allowed to incubate for 5 min. After incubation, the siRNA/Lipofectamine solution was added directly to cells. The final siRNA concentration was 15 nM. After 24 h, the siRNA was removed and cells were used for RNA extraction and other experiments.
RNA extraction and RT-PCR
RNA extraction and real-time (RT)-PCR were carried out as described previously (Liang et al., 2010). To examine mRNA expression by RT-PCR analysis, astrocytes were cultured at a concentration of 2 × 105 cells·mL−1 in 24-well culture plates (BD, Franklin Lakes, NJ, USA) in the presence or absence of 10 μM UFP-512 for 12 h. The cortex tissues were obtained from adult C57BL/6J mice. Total RNA was extracted using the guanidinium thiocyanate method (RNeasy Mini Kit; Qiagen, Valencia, CA, USA). cDNAs were generated using QuantiTect Rev. Transcription Kit (Qiagen). DOP receptor and GAPDH cDNAs were amplified using specific primers designed as previously described (Zhang et al., 2006; Liang et al., 2010). The sequences for DOP receptor and GAPDH derived were:
DOP receptor forward primer: 5′-TACATCTTCAATCTGGCT-3′
DOP receptor reverse primer: 5′-CGATGACGAAGATGTGGA-3′
GAPDH forward primer: 5′-TGTGTCCGTCGTGGATCTGA-3′
GAPDH reverse primer: 5′-CCTGCTTCACCACCTTCTTGA-3′
The reverse transcription product (1 μL) was amplified with a mixture of primers and 2.5 units of 50X Advantage® 2 Polymerase Mix (Clontech, Mountain View, CA, USA). Thermal cycling parameters for primer optimization were as follows: DOP receptor was activated at 95°C for 5 min followed by 33 cycles including 95°C for 30 s, 54°C for 30 s, 72°C for 1 min, then incubated at 72°C for 7 min. GAPDH was activated at 95°C for 5 min followed by 27 cycles including 95°C for 30 s, 58°C for 30 s, 72°C for 1 min, then incubated at 72°C for 7 min. Assays were carried out in at least three independent trials.
RNA extraction and RT-quantitative PCR
To ensure specificity and the absence of non-specific amplification, the primers were complementary to sequences present in different exons. The cDNA (2 μL) from the reverse transcription was mixed with a final concentration of SsoFast™ EvaGreen® Supermix (Bio-Rad, Hercules, CA, USA), 500 nM forward primer, 500 nM reverse primer and deionized water in a total volume of 10 μL. Thermal cycling parameters for primer optimization were as follows: activation at 95°C for 3 min followed by 50 cycles 95°C for 10 s and 60°C for 30 s. The mRNA level for each sample was normalized to GAPDH mRNA. The following specific primers for mouse GAPDH, EAAT1 and EAAT2 were used for quantitative PCR analysis:
GAPDH forward primer: 5′-TGTGTCCGTCGTGGATCTGA-3′
GAPDH reverse primer: 5′-CCTGCTTCACCACCTTCTTGA-3′
EAAT1 forward primer: 5′-TTGCAGCAAGGGGTCCGCAA-3′
EAAT1 reverse primer: 5′-GCAGTGACCGTGAGCAGAACGA-3′
EAAT2 forward primer: 5′-CAAGTCTGAGCTGGACACCA-3′
EAAT2 reverse primer: 5′-GTGTGCGGCATAGACACACT-3′
EAAT3 forward primer: 5′-GCCAGTTACATTCCGCTGTGCG-3′
EAAT3 reverse primer: 5′-TGCAGACCATGACCCGAGAGCA-3′
Western blot analysis
The total astrocytic proteins were collected with M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, Waltham, MA, USA). The whole brain and cortical tissues were obtained from adult C57BL/6J mice and tissue protein extraction was performed in a similar manner to that used in our previous work (Ma et al., 2005; Tian et al., 2013). Briefly, the tissue was homogenized with a Potter homogenizer in ice-cold hypotonic lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgClB2B, 0.1 mM EDTA, 0.1 mM EGTA (ethylene glycol tetraacetic acid), 1 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride, 2 μg·mL−1 leupeptin, 2 μg·mL−1 aprotinin, 0.5 mg·mL−1 benzamidine, 5 mM sodium fluoride, 2 mM sodium pyrophosphate, 1 mM sodium orthovanadate) and the lysates were incubated on ice for 30 min. After centrifugation at 14 000× g for 5 min, the supernatant was kept and stored at −80°C for protein assay. Western blotting was performed as described previously (Liang et al., 2008). The primary antibodies used were as follows: rabbit anti-DOP receptor antibody (1:2000, EMD Millipore, Billerica, MA, USA), rabbit polyclonal anti-EAAT1 antibody (1:200, Santa Cruz Biotechnology, Dallas, TX, USA), rabbit polyclonal anti-EAAT2 antibody (1:100, Santa Cruz Biotechnology), rabbit polyclonal anti-EAAT3 antibody (1:200, Santa Cruz Biotechnology), rabbit polyclonal Phospho-p38 MAPK antibody (1:1000, Cell Signaling Technology, Beverly, MA, USA), rabbit polyclonal p38 MAPK antibody (1:1000, Cell Signaling Technology) and mouse monoclonal anti-β-actin antibody (1:20 000, Sigma-Aldrich). After overnight incubation with primary antibodies at 4°C, each blot was probed with HRP-conjugated anti-rabbit IgG (1:5000, Santa Cruz Biotechnology) and anti-mouse IgG (1:5000, Sigma-Aldrich). The images of the Western blot were then developed with ECL Plus Western blotting detection reagents (GE Healthcare Life Sciences, Pittsburgh, PA, USA). Assays were carried out in at least three independent trials.
Immunocytochemistry
Rabbit anti-DOP receptor antibody (1:100, EMD Millipore, AB1560mi) and mouse monoclonal anti-GFAP antibody (1:500, Sigma-Aldrich) were used for immunostaining of astrocytic DOP receptor protein expression and astrocyte respectively. After fixation by 4% paraformaldehyde for 30 min at room temperature, cells were blocked with 5% normal goat serum and were then permeabilized with 0.05% Triton X-100 for 10 min. Cells were incubated with each primary antibody overnight at 4°C, and then stained with each secondary antibody conjugated Alexa dye (Alexa 488 Invitrogen 1:500 for DOP receptor; Alexa 647 Invitrogen 1:500 for GFAP, Grand Island, NY, USA) for 1 h at room temperature. Cells were counterstained with Hoechst 33342. Images were analysed with a deconvolution fluorescent microscope system (Nikon Eclipse Te2000-u, Nikon Instruments, Tokyo, Japan).
L-glutamate assay
We measured the astrocytic condition medium to verify if DOP receptor-induced expression of astrocytic EAAT1/EAAT2 is associated with a reduction in the glutamate concentration in the condition medium. Astrocytes were cultured at a concentration of 2 × 105 cells·mL−1 in 24-well culture plates (BD). Twelve groups of astrocytic culture were used for this experiment. The first group was kept in the medium for 25 h and then exposed to 100 μM glutamate for 30 min. The second group was kept in medium for 24 h and then exposed to 100 μM TBOA for 1 h, followed by exposure to 100 μM TBOA and 100 μM glutamate for 30 min. The third group was kept in medium with 10 μM UFP-512 for 25 h and then exposed to 100 μM glutamate for 30 min. The fourth group was kept in medium with 10 μM UFP-512 for 24 h and then exposed to 100 μM TBOA for 1 h, followed by exposure to 100 μM TBOA and 100 μM glutamate for 30 min. The fifth group was treated with DOP receptor siRNA for 24 h at the beginning, and then its medium was changed to one containing 10 μM UFP-512 and incubated for 25 h, followed by exposure to 100 μM glutamate for 30 min. The sixth group was kept in the medium for 24 h and then exposed to 10 μM UCPH for 1 h, followed by exposure to 10 μM UCPH and 100 μM glutamate for 30 min. The seventh group was kept in the medium with 10 μM UFP-512 for 24 h and then exposed to 10 μM UCPH for 1 h, followed by exposure to 10 μM UCPH and 100 μM glutamate for 30 min. The eighth group was kept in the medium for 24 h and then exposed to 300 μM DHK for 1 h, followed by exposure to 300 μM DHK and 100 μM glutamate for 30 min. The ninth group was kept in medium with 10 μM UFP-512 for 24 h and then exposed to 300 μM DHK for 1 h, followed by exposure to 300 μM DHK and 100 μM glutamate for 30 min. The tenth group was kept in the medium with 10 μM SB203580 for 1 h and then exposed to 10 μM UFP-512 for 25 h, followed by exposure to 100 μM glutamate for 30 min. The eleventh group was kept in the medium with 5 μM U0126 for 1 h and then exposed to 10 μM UFP-512 for 25 h, followed by exposure to 100 μM glutamate for 30 min. The twelfth group was kept in the medium with 500 nM FR180204 for 1 h and then exposed to 10 μM UFP-512 for 25 h, followed by exposure to 100 μM glutamate for 30 min. After the application of 100 μM glutamate to each group, 2 μL astrocyte-conditioned medium of each group was collected at 10, 20 and 30 min time points, respectively, which was used for the L-glutamate assay. We utilized the glutamate assay kit (BioVision, Milpitas, CA, USA) to measure extracellular glutamate concentrations according to the manufacturer's protocol. Absorbance at 450 nm was measured on a plate reader. Assays were performed in six independent trials.
Statistical analysis
All of the experiments were performed in triplicate and were repeated at least three times. The data are expressed as mean ± SD. Student's two-tailed t-test was used to compare paired data and one-way anova followed by Tukey's post hoc test for multiple comparisons were used to detect statistically significant differences between groups. Statistical significance was considered when P values were ≤0.05.
Results
The astrocytes express DOP receptor
We determined DOP receptor mRNA expression by RT-PCR with the mRNA of the cortex as a positive control as it is known to express DOP receptors (Xia and Haddad, 1991; 2001,). The astrocytes indeed expressed DOP receptor mRNA (Figure 1A). We observed that DOP receptor siRNA largely attenuated DOP receptor mRNA expression in these astrocytes (Figure 2A,B). The astrocytic DOP receptor expression was reaffirmed by Western blot with the proteins extracted from whole brain and cortical tissues as two positive controls (Figure 1B). As shown in Figure 1B, 36 kDa and 72 kDa DOP receptor proteins were expressed by naive astrocytes and UFP-512-treated astrocytes. In addition, the immunocytochemistry analysis demonstrated DOP receptor localization in the membrane, cytoplasm and nucleus (Figure 1C). Moreover, the Western blot and immunocytochemistry data showed that DOP receptor siRNA significantly reduced DOP receptor protein expression (Figure 2C,D).
Figure 1.
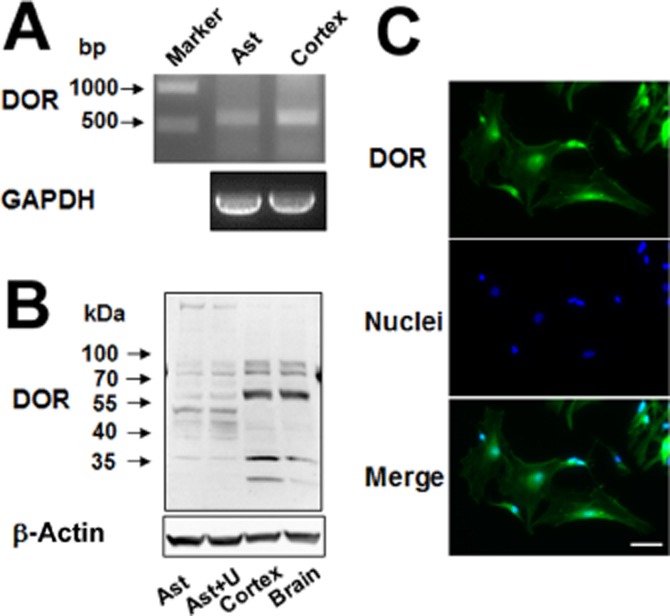
DOP receptor (DOR) expression in the astrocytes. (A) DOP receptor mRNA analysis by RT-PCR. Total mRNA was extracted from the astrocytes and cortical tissues. (B) DOP receptor protein detection by Western blot analysis. Total proteins were extracted from the astrocytes, UFP-512 treated astrocytes, cortex and whole brain. (C) Immunocytochemistry results showing that DOP receptor protein existed in the membrane, cytoplasma and nuclei of the astrocytes. Scale bar, 50 μm. Ast, astrocytes; U, astrocytes treated with 10 μM UFP-512 for 24 h. Note that the astrocytes expressed DOP receptor mRNA and 36 and 72 kDa DOP receptor protein.
Figure 2.
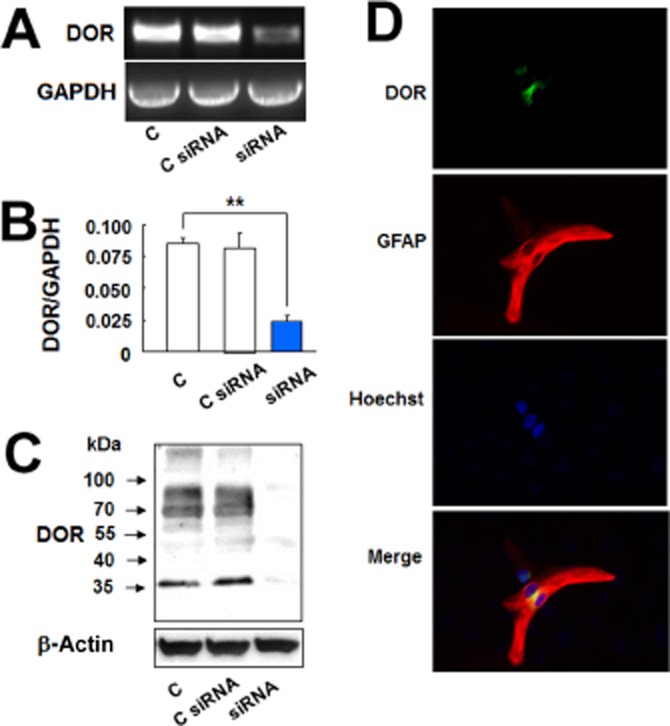
DOP receptor siRNA reduced DOP receptor (DOR) expression in the astrocytes. (A) DOP receptor mRNA analysis by RT-PCR. Total mRNA was extracted from the astrocytes and those treated with control siRNA or DOP receptor siRNA. (B) DOP receptor mRNA analysis by quantitative PCR. Total mRNA was extracted from the control astrocytes and those treated with control siRNA or DOP receptor siRNA. (C) DOP receptor protein detection by Western blot analysis. Total proteins were extracted from the control astrocytes and those treated with control siRNA or DOP receptor siRNA. (D) Astrocytes were treated with DOP receptor siRNA for 24 h and then were stained with DOP receptor and GFAP antibodies. Immunocytochemistry results showing DOP receptor siRNA markedly reduced astrocytic DOP receptor expression. C, non-treated astrocytes; C siRNA, astrocytes treated with control siRNA for 24 h; siRNA, astrocytes treated with DOP receptor siRNA for 24 h. **P < 0.01. Note that there was a significant reduction in astrocytic DOP receptor mRNA and protein expression after addition of DOP receptor siRNA.
DOP receptor activation enhances astrocytic expression of EAAT1 and EAAT2 and glutamate uptake
As the first step to determine if astrocytic EAATs play a role in DOP receptor-induced neuroprotection, we evaluated if DOP receptor activation can alter the expression of EAAT1 and EAAT2 in the astrocytes. The astrocytes were treated with UFP-512, a potent DOP receptor agonist (Balboni et al., 2002; Chao et al., 2007). After the treatment with UFP-512 (10 μM) for 12 h, both EAAT1 and EAAT2 mRNA levels significantly increased in the astrocytes (Figure 3A). After 24 h of UFP-512 treatment, the expression of EAAT1 and EAAT2 proteins also increased (Figure 3B). In contrast, neither the EAAT3 mRNA expression nor the protein levels were altered by DOP receptor activation by UFP-512 (Figure 3A,B). Additionally, the time-course experiment showed that DOP receptor-induced EAAT1 mRNA expression significantly increased at 6 h and reached peak at 12 h and DOP receptor-induced EAAT2 expression also peaked at 12 h (Figure 4A). Both EAAT1 and EAAT2 protein expressions were markedly up-regulated and reached peak at 24 h after UFP-512 stimulation (Figure 4B). Moreover, UFP-512 induced astrocytic EAAT1 and EAAT2 mRNA expression in a dose-dependent manner peaking at 10 μM concentration of UFP-512 (Figure 5A). The EAAT1 protein production also peaked at 10 μM, while EAAT2 protein reached a plateau between 10 and 100 μM concentrations (Figure 5B).
Figure 3.
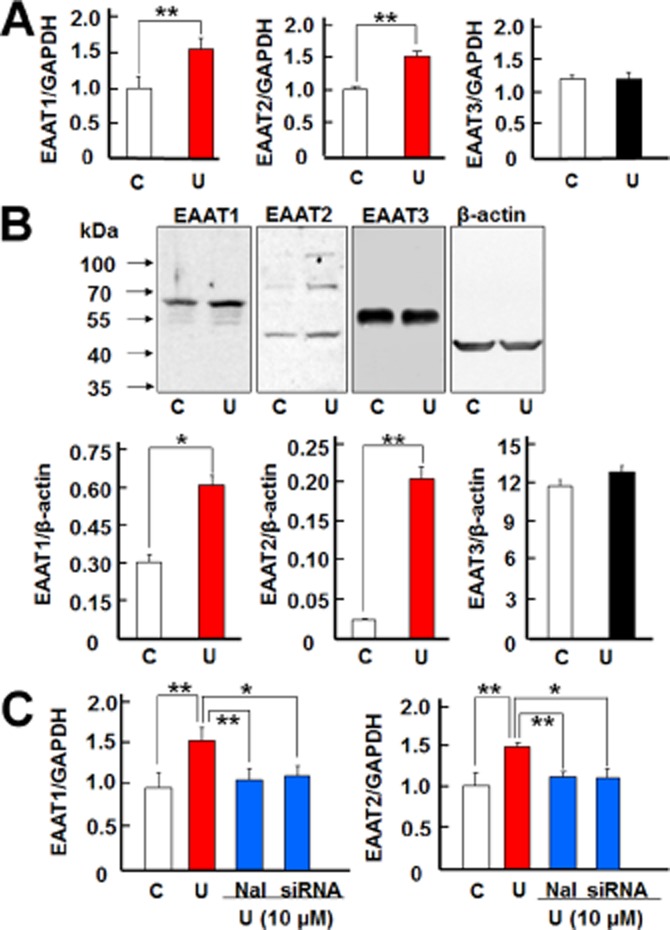
DOP receptors induced EAAT1 and EAAT2 expression in the astrocytes. (A) Quantitative PCR analysis showing that UFP-512 induced EAAT1 and EAAT2 mRNA expression, but did not change EAAT3 mRNA expression. Astrocytes were treated with 10 μM UFP-512 for 12 h and total mRNA was extracted for detection. (B) Western blot analysis showing that UFP-512 induced EAAT1 (65 kDa) and EAAT2 (70 kDa) protein expression, whereas it did not alter EAAT3 (57 kDa) protein expression. The astrocytes were treated with 10 μM UFP-512 for 24 h and total protein was extracted for measurements. (C) Effect of DOP receptor antagonism and siRNA knockdown on astrocytic EAATs. For DOP receptor antagonism, the astrocytes were pretreated with naltrindole for 1 h, and then treated with 10 μM UFP-512 and 10 μM naltrindole for 12 h. For DOP receptor siRNA knockdown, the astrocytes were pretreated with DOP receptor siRNA for 24 h and then treated with 10 μM UFP-512 for 12 h. C, non-treatment; Nal, 10 μM naltrindole, a DOP receptor antagonist; U, UFP-512, a specific DOP receptor agonist. *P < 0.05; **P < 0.01. Note that UFP-512 elevated EAAT1 and EAAT2 mRNA expression, while both naltrindole and DOP receptor siRNA abolished the UFP-512-induced increase in EAAT1 and EAAT2 expression.
Figure 4.
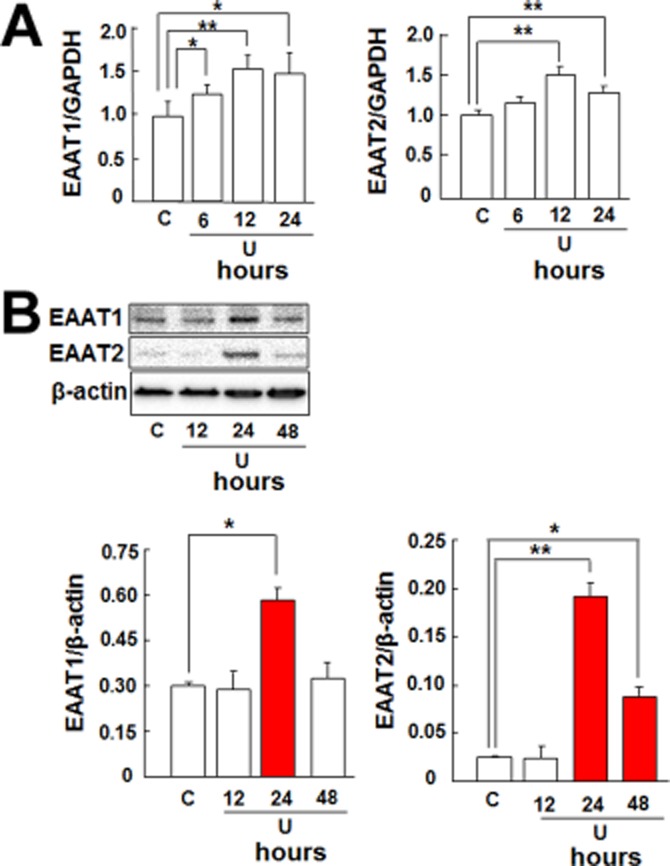
Time course of DOP receptor-induced EAAT1 and EAAT2 expression in the astrocytes. (A) Quantitative PCR analysis showing that UFP-512 induced EAAT1 and EAAT2 mRNA expression in a time-dependent manner with expression reaching peak at 12 h. (B) Western blot showing that UFP-512 induced EAAT1 and EAAT2 protein production in a time-dependent manner with production reaching peak at 24 h. C, non-treatment; U, 10 μM UFP-512. *P < 0.05; **P < 0.01. Note that UFP-512 induced both EAAT1 and EAAT2 mRNA as well as protein expression in a time-dependent manner.
Figure 5.
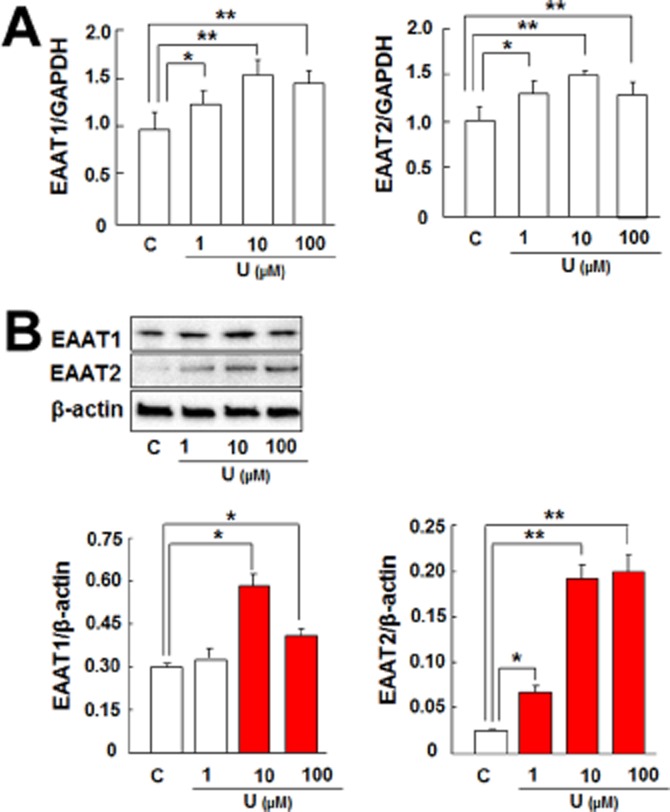
DOP receptor induced astrocytic EAAT1 and EAAT2 expression in a dose-dependent manner. (A) Astrocytes were treated with UFP-512 for 12 h. Then total mRNA was extracted for mRNA analysis. Quantitative PCR analysis showing that UFP-512 induced EAAT1 and EAAT2 mRNA expression in a dose-dependent manner with expression reaching peak at 10 μM. (B) Astrocytes were treated with UFP-512 for 24 h. Then total protein was extracted for protein analysis. Western blot showing that UFP-512 induced EAAT1 protein production in a dose-dependent manner with production reaching a peak at 10 μM and also up-regulated EAAT2 production, which reached a plateau at 10 μM and 100 μM. C, non-treatment; U, UFP-512. *P < 0.05; **P < 0.01. Note that UFP-512 induced EAAT1 and EAAT2 mRNA as well as protein expression in a time-dependent manner.
These observations were further verified by DOP receptor antagonism and knockdown of DOP receptor expression. Ten micromolar UFP-512 significantly increased EAAT1 and EAAT2 mRNA expression in the astrocytes (Figure 3C), while naltrindole, a DOP receptor antagonist, completely blocked such increase (Figure 3C). Furthermore, UFP-512 induced expression of astrocytic EAAT1 and EAAT2 mRNA was also significantly suppressed by DOP receptor siRNA (Figure 3C). The DOP receptor siRNA used in this study specifically knocked down the expression of DOP receptor because DOP receptor mRNA was remarkably reduced (Figure 2A), while the expression of GAPDH mRNA and β-actin protein had no significant change (Figure 2A,C) after the treatment with DOP receptor siRNA. Moreover, we observed a consistent decrease in 36 kDa and 72 kDa bands with no significant change in other non-DOP receptor bands.
Astrocytes play a crucial role in the maintenance of extracellular glutamate homeostasis and neuronal functioning. This function is carried out via EAAT1 and EAAT2 that are predominantly expressed in astrocytes. Thus, we investigated a possible connection between UFP-512 enhanced EAATs expression and astrocytic glutamate uptake. As illustrated in Figure 6, after 24 h treatment with UFP-512, the glutamate uptake accelerated in these astrocytes as opposed to those without UFP-512 treatment. Furthermore, this effect was completely eliminated by DOP receptor siRNA and TBOA, a potent inhibitor of all EAATs. Moreover, both DHK and UCPH-101 partially suppressed glutamate uptake in the astrocyte culture regardless of UFP-512 stimulation. However, DHK had a greater effect than UCPH-101 in blocking astrocytic glutamate uptake both in the presence and absence of UFP-512. Thus our results suggest that EAAT2 has a greater influence on the regulation of glutamate uptake compared with EAAT1.
Figure 6.
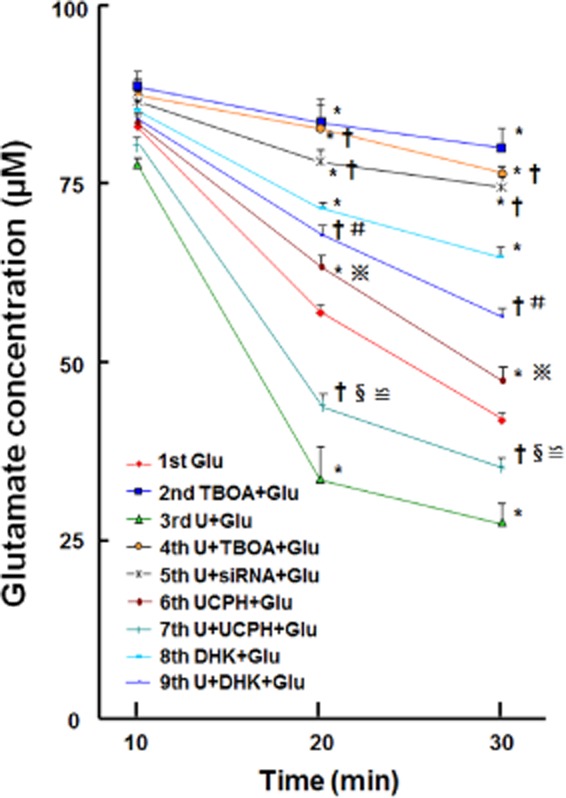
DOP receptor induced increase in astrocytic glutamate uptake. Nine groups of astrocytic culture were used for this set of experiments. Each group of cultured astrocytes was treated as detailed in the Methods section. Two microlitres of the treated astrocytic-conditioned media were collected from each group at 10, 20 and 30 min time points for extracellular glutamate analysis with the L-glutamate assay. *P < 0.05 versus Glu. †P < 0.05 versus U + Glu. §P < 0.05 U + UCPH + Glu versus UCPH + Glu. #P < 0.05 U + DHK + Glu versus DHK + Glu. ≌ P < 0.05 U + UCPH + Glu versus U + DHK + Glu. P < 0.05 UCPH + Glu versus DHK + Glu. Glu, 100 μM glutamate; DHK, 300 μM dihydrokainic acid, a potent EAAT2 inhibitor; siRNA, DOP receptor siRNA; TBOA, 100 μM threo-ß-benzyloxyaspartate, a potent blocker of all subtypes of the excitatory amino acid transporters; U, 10 μM UFP-512; UCPH, 10 μM UCPH-101, a specific EAAT1 inhibitor. Note that UFP-512 enhanced astrocytic glutamate uptake and this enhancement was completely blocked by the glutamate inhibitor TBOA as well as DOP receptor siRNA. Both DHK and UCPH-101 partially suppressed glutamate uptake in astrocyte culture in the presence or absence of UFP-512 stimulation. DHK had a greater effect than UCPH-101, suggesting that EAAT2 has a greater effect than EAAT1.
PKA, PKC and PI3K inhibitors do not affect DOP receptor-induced expression of astrocytic EAAT1 and EAAT2
We have previously demonstrated that PKC activation is required for DOP receptor-mediated neuroprotection against hypoxic insults in neurons (Ma et al., 2005; Chao et al., 2012). Therefore, we questioned if PKC pathways affect the DOP receptor-induced expression of astrocytic EAAT1 and EAAT2 as well. We tested it by utilizing Calphostin C, a potent PKC inhibitor. Contrary to our expectations, Calphostin C did not affect EAAT1 and EAAT2 expression (Figures 7 and 8) following UFP-512 treatment. This observation suggests that DOP receptor activation employs a different pathway in the astrocytes compared with the neurons.
Figure 7.
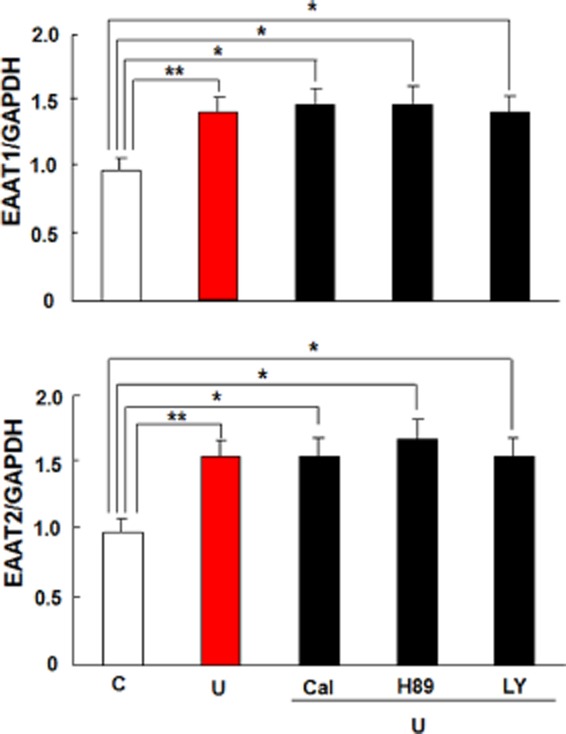
Effects of PKC, PKA or PI3K inhibitors on DOP receptor-induced EAAT1 and EAAT2 mRNA expression in the astrocytes. Astrocytes were pretreated with the indicated inhibitor for 1 h, followed by 10 μM UFP-512 and the inhibitor for 12 h. Total mRNA was extracted for quantitative PCR analysis. C, non-treatment; Cal, 500 nM calphostin C, a PKC inhibitor; H89, a PKA inhibitor, 10 μM; LY, 10 μM LY294002, a PI3K inhibitor; U: 10 μM UFP-512. *P < 0.05; **P < 0.01. The values are the means ± SD. At least three independent trials were performed for each assay. Note that the inhibitors of PKC, PKA or PI3K did not suppress astrocytic EAAT1 and EAAT2 mRNA expression induced by DOP receptor activation.
Figure 8.
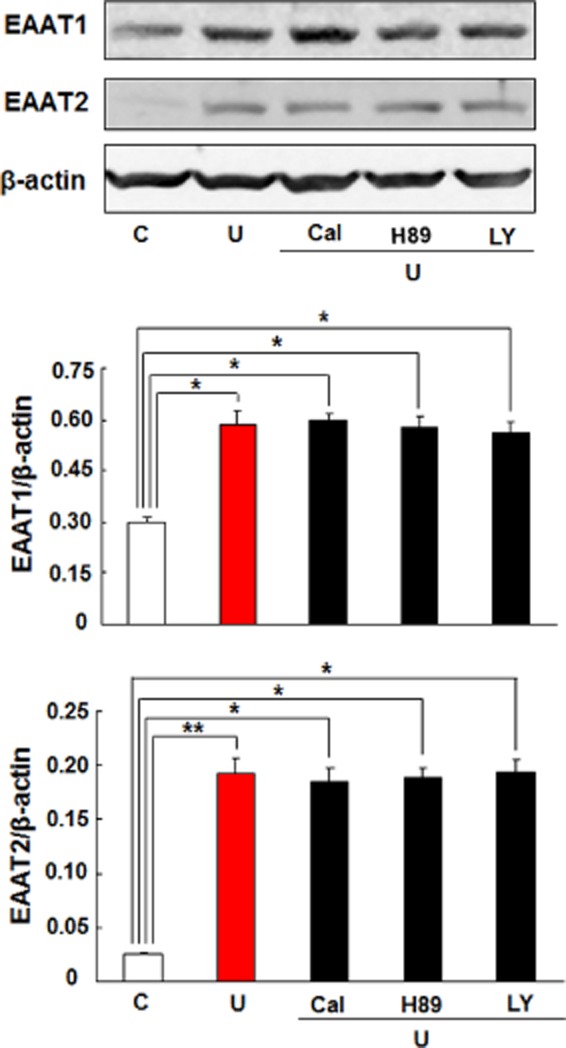
Effects of PKC, PKA or PI3K inhibitors on DOP receptor-induced EAAT1 and EAAT2 protein expression in the astrocytes. The astrocytes were pretreated with the indicated inhibitor for 1 h, followed by 10 μM UFP-512 and the inhibitor for 24 h. C, non-treatment; Cal, 500 nM calphostin C, PKC inhibitor; H89, a PKA inhibitor, 10 μM; LY, 10 μM LY294002, a PI3K inhibitor; U, 10 μM UFP-512. *P < 0.05; **P < 0.01. Note that the inhibitors of PKC, PKA or PI3K did not alter astrocytic EAAT1 and EAAT2 protein expression induced by DOP receptor activation.
PKA and PI3K are two important signalling proteins involved in a multitude of cellular functions. Several studies have repeatedly demonstrated a role of the kinase pathways including PKA and PI3K in DOP receptor activity (Fryer et al., 2001; Narita et al., 2006). There is also evidence suggesting that these signalling pathways are associated with EAAT1 and EAAT2 expressions (Pignataro et al., 2011; Lee et al., 2012). Thus, we investigated if these kinases were involved in UFP-512-induced expression of astrocytic EAAT1 and EAAT2. However, neither PKA inhibitor (H89) nor P13K inhibitor (LY294002) could suppress the mRNA and protein expression of EAAT1 and EAAT2 in the astrocytes treated with UFP-512 (Figures 7 and 8). We also verified that these inhibitors did not alter EAAT1 and EAAT2 expression even in the absence of UFP-512 treatment (Figure 9).
Figure 9.
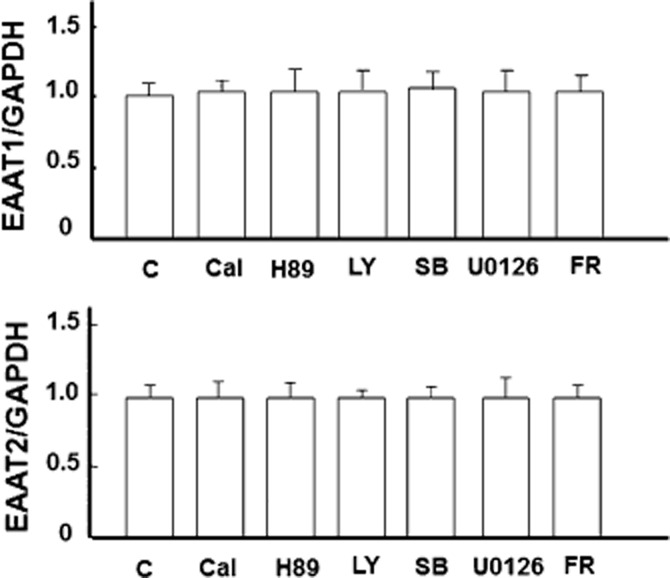
No appreciable effect of signal pathway inhibitors on EAAT1 and EAAT2 expression in the absence of UFP-512 stimulation. Astrocytes were treated with indicated inhibitor for 12 h. Then total mRNA was extracted for mRNA analysis. Quantitative PCR analysis showing that EAAT1 and EAAT2 expression was not affected. C, non-treatment; Cal: 500 nM calphostin C, a PKC inhibitor; FR: 500 nM FR180204, ERK inhibitor; H89, a PKA inhibitor, 10 μM; LY, 10 μM LY294002, a PI3K inhibitor; SB, 10 μM SB203580, p38 inhibitor; U0126, an MEK inhibitor, 5 μM. Note that the inhibitors had no effect on astrocytic EAAT1 and EAAT2 expression.
DOP receptor activation induces EAAT1 and EAAT2 expression through MEK-ERK-p38 interaction
We have previously demonstrated that DOP receptor-mediated neuroprotection against hypoxia depends on an increase in ERK and Bcl-2 activity, which counteracts hypoxic increase in p38 activity and cytochrome c release (Ma et al., 2005). Therefore, we further determined whether the ERK and p38 signalling is involved in the mechanism for the DOP receptor-induced EAAT1 and EAAT2 expression in the astrocytes.
The astrocytes were pretreated with U0126, an inhibitor of MEK1 and MEK2 both of which are kinases upstream to ERK (Ma et al., 2005), for 1 h before applying UFP-512. As shown in Figures 10 and 11, U0126 completely abrogated UFP-512-induced EAAT1 and EAAT2 expression at both mRNA and the protein levels. This effect was further confirmed using FR180204, a potent inhibitor of ERK (Figures 10 and 11).
Figure 10.
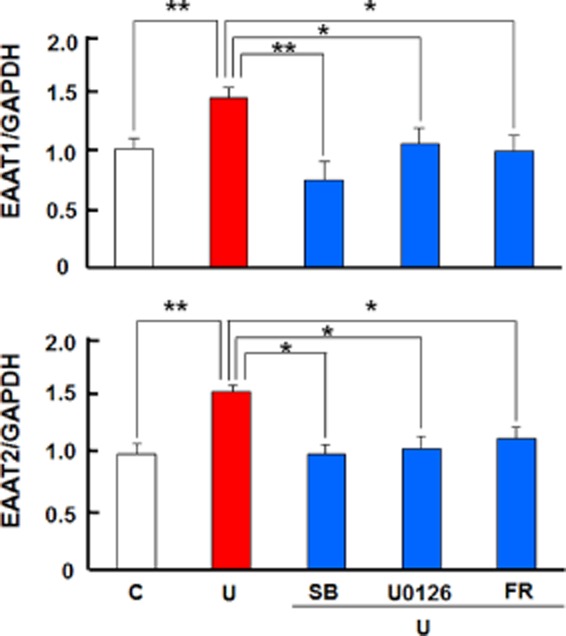
DOP receptor-induced expression of EAAT1 and EAAT2 mRNAs via MEK-ERK-p38 signalling in the astrocytes. The astrocytes were pretreated with the indicated inhibitor for 1 h, followed by the inhibitor plus 10 μM UFP-512 for 12 h. C, non-treatment; FR: 500 nM FR180204, ERK inhibitor; SB, 10 μM SB203580, p38 inhibitor; U, 10 μM UFP-512; U0126, an MEK inhibitor, 5 μM. *P < 0.05; **P < 0.01. Note that the inhibitors of MEK, ERK and p38 MAPKs blocked astrocytic EAAT1 and EAAT2 mRNA expression induced by DOP receptor activation with UFP-512.
Figure 11.
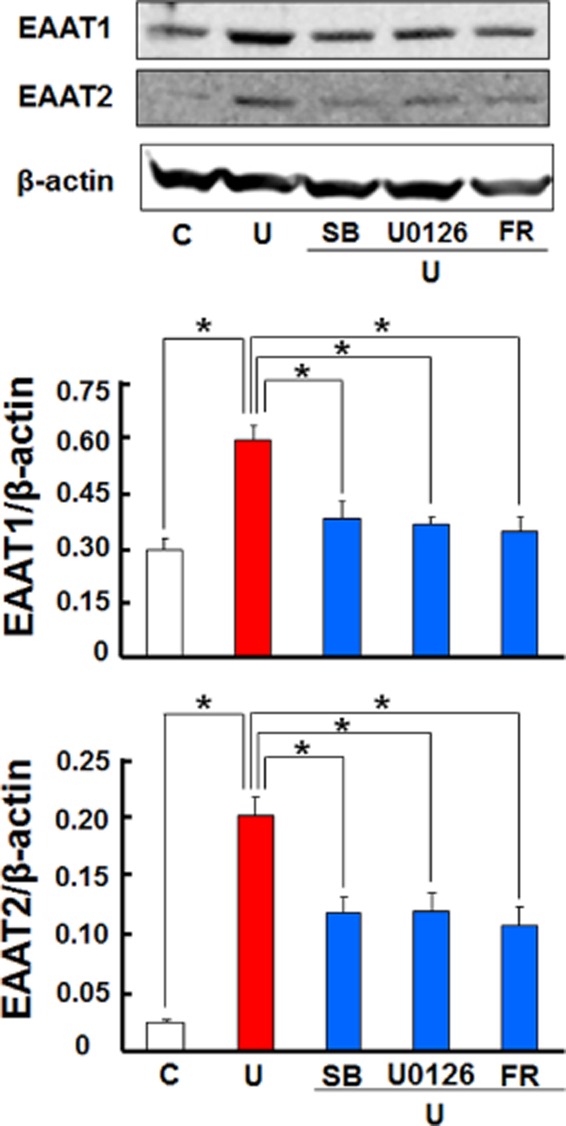
DOP receptor induced expression of EAAT1 and EAAT2 proteins through MEK-ERK-p38 signalling in the astrocytes. The astrocytes were pretreated with the indicated inhibitor for 1 h, followed by the inhibitor plus 10 μM UFP-512 for 24 h. C, non-treatment; FR: 500 nM FR180204, an ERK inhibitor; SB, 10 μM SB203580, p38 inhibitor; U, 10 μM UFP-512; U0126, an MEK inhibitor, 5 μM. *P < 0.05; **P < 0.01. Note that the inhibitors of MEK, ERK and p38 blocked astrocytic EAAT1 and EAAT2 protein expression induced by DOP receptor activation with UFP-512.
P38 MAPK is a key intracellular transduction pathway that has been implicated in many stress- and disease-related states. Increased p38 activity plays an important role in cell death under stressful conditions and offsets ERK activation in neurons (Ma et al., 2005). In this study, however, we observed that p38 blockade with SB203580 significantly abolished DOP receptor-induced EAAT1 and EAAT2 expression (Figures 10 and 11). The glutamate uptake experiments also revealed that the inhibitors for MEK, ERK and p38 significantly reduced DOP receptor-accelerated glutamate uptake in astrocyte (Figure 12). Furthermore, we found that UFP-512 up-regulated p38 phosphorylation in the astrocytes, which was almost completely suppressed by the inhibitors of MEK and ERK (Figure 13). In addition, the inhibitors did not affect EAATs expression and p38 activation without UFP-512 stimulation (Figures 9 and 14). These observations suggest a synergic interaction between ERK and p38 in the astrocytes and DOP receptor-induced up-regulation of astrocytic EAAT1 and EAAT2 is via a MEK-ERK-p38 signalling pathway.
Figure 12.
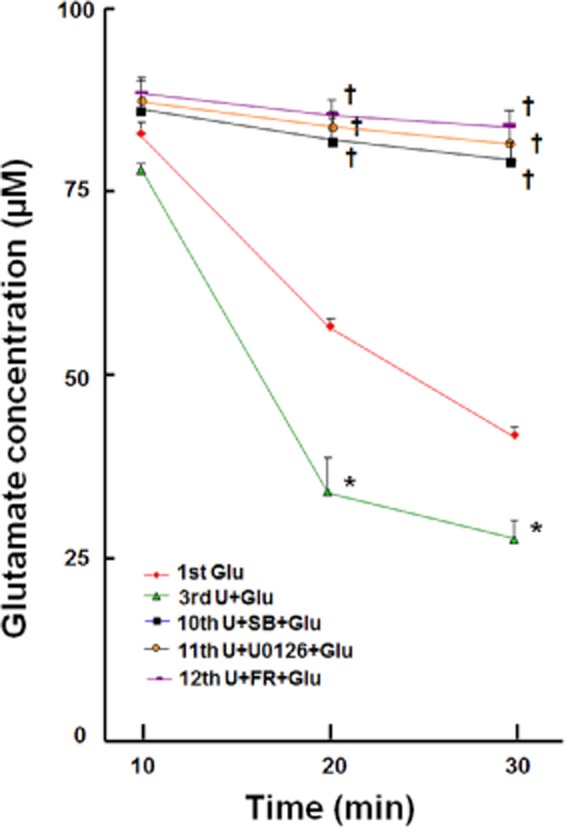
Effects of MEK, ERK and p38 inhibitors on DOP receptor-accelerated glutamate uptake in the astrocyte. Each group of cultured astrocytes was treated as detailed in the Methods section. After these treatments, 2 μL of astrocytic condition medium were collected from each group at 10, 20 and 30 min time points for extracellular glutamate analysis with the L-glutamate assay. FR, 500 nM FR180204, an ERK inhibitor; SB, 10 μM SB203580, p38 inhibitor; U0126, an MEK inhibitor, 5 μM. *P < 0.05 versus Glu. †P < 0.05 versus U + Glu. Note that UFP-512 enhanced astrocytic glutamate uptake was completely blocked by inhibitors for MEK, ERK and p38.
Figure 13.
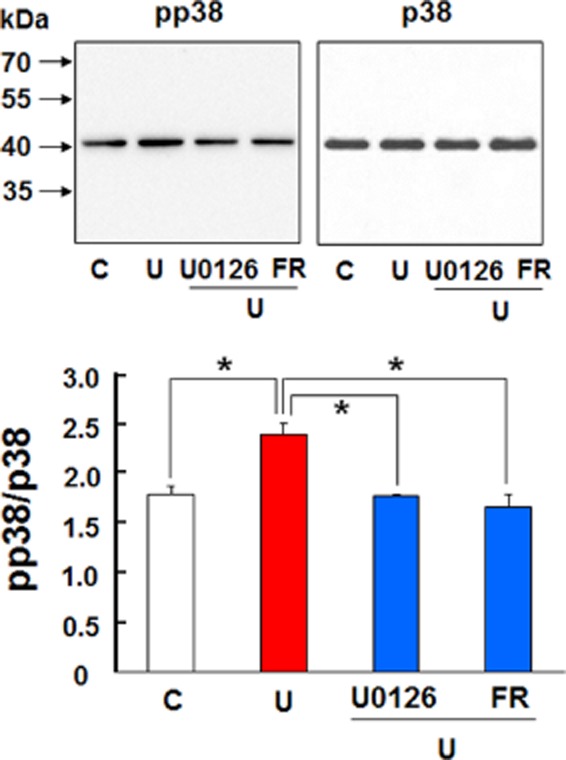
Effects of MEK and ERK inhibitors on DOP receptor-induced p38 phosphorylation in the astrocytes. The astrocytes were pretreated with the indicated inhibitor for 1 h, followed by the inhibitor plus 10 μM UFP-512 for 24 h. C, non-treatment; FR, 500 nM FR180204, an ERK inhibitor; pp38: phosphorylated p38; U, 10 μM UFP-512; U0126, an MEK inhibitor, 5 μM. *P < 0.05. Note that the inhibitors of MEK and ERK suppressed UFP-512-induced p38 phosphorylation.
Figure 14.
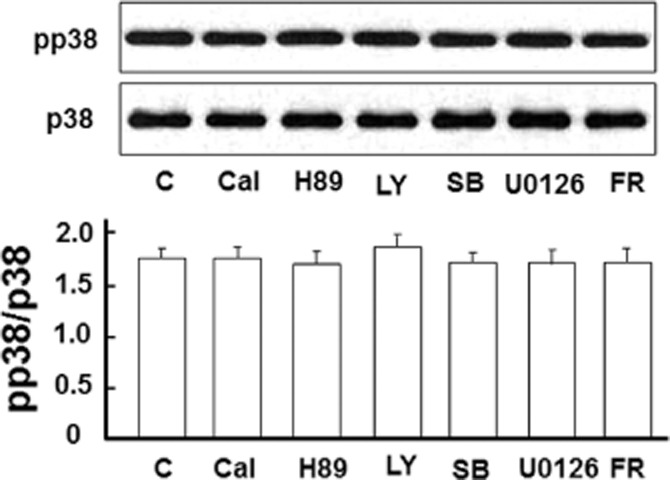
No appreciable change in p38 activation after the treatment with PK and MAPK inhibitors in the absence of UFP-512 stimulation. Astrocytes were treated with indicated inhibitor for 24 h. Then total protein was extracted for Western blot. C, non-treatment; Cal, 500 nM calphostin C, a PKC inhibitor; FR, 500 nM FR180204, ERK inhibitor; H89, a PKA inhibitor, 10 μM; LY, 10 μM LY294002, a PI3K inhibitor; pp38, phosphorylated p38; SB, 10 μM SB203580, p38 inhibitor; U0126, an MEK inhibitor, 5 μM. Note that the inhibitors had no effect on p38 activation.
Discussion and conclusion
In this work, we have demonstrated for the first time that DOP receptor activation induces astrocytic expression of EAAT1 and EAAT2, but not EAAT3, an effect that is completely reversed by DOP receptor antagonist or siRNA knockdown. Furthermore, we observed an increased glutamate uptake in the astrocytes treated with UFP-512, an effect completely blocked by an EAAT inhibitor or DOP receptor knockdown. PKA, PKC and PI3K inhibitors did not, while MEK, ERK or p38 inhibitors did, significantly alter DOP receptor-induced EAAT1 and EAAT2 expression. These observations strongly suggest that DOP receptor activation up-regulates astrocytic EAAT expression and function through a MEK-ERK-p38 signalling, thereby promoting neuroprotection against excitotoxicity.
The realm of modern neurovascular biology is undergoing a paradigm shift with a gradual acceptance of the concepts of multiple targets in addition to neurons and looking beyond the neuronal signalling to glial pathways as the probable underlying mechanisms. Our observations from the current study corroborate a novel mechanism for DOP receptor-mediated regulation of extracellular excitatory activity.
DOP receptors, EAATs and astrocytes
The DOP receptor protein exists mainly in three forms of varying sizes, namely, 36, 48 and 72 kDa, in terms of Western blot detection. In addition, a 65 kDa band was detected in certain experimental conditions (Persson et al., 2005; Kabli and Cahill, 2007; Zambelli et al., 2014). Our previous studies as well as this work have shown that although the detectable bands of DOP receptor varies in different tissue and cell types in various conditions, 36 and 72 kDa bands are commonly detected in most conditions. The 72 kDa DOP receptor is a possible dimeric form of a 36 kDa DOP receptor post-translationally truncated at the C-terminus (Persson et al., 2005). We have therefore measured their density and compared them among various groups in this work. Immunocytochemical staining (Figure 1C) demonstrated DOP receptor localization in the cytosolic and nuclear regions, in addition to the membrane. In fact, it is known that the DOP receptor protein is produced by membrane-bound ribosome in endoplasmic reticulum and post-translationally modified by endoplasmic reticulum and Golgi apparatus (Leskela et al., 2007). Our current observations are consistent with previous studies that demonstrate the astrocytic expression of DOP receptor as well as other opioid receptors (Ruzicka et al., 1995; Stiene-Martin et al., 1998).
Our studies and those of others have well demonstrated that DOP receptor activation induces neuroprotection in many neurological conditions including glutamate-induced injury (Zhang et al., 2000; 2002,; Chao et al., 2007; 2008; 2012,,; Kang et al., 2009; Ke et al., 2009; Yang et al., 2009; 2011,; Chao and Xia, 2010; Duan et al., 2011; Turner and Johnson, 2011; He et al., 2013; Tian et al., 2013). In the present work, we have established a novel link between DOP receptor activation and EAAT expression as well as function in the astrocytes. This is specific to EAAT1 and EAAT2. Although astrocytes express EAAT3 (also named EAAC1), it is mainly localized in the perinuclear region of the cell and has less function in glutamate uptake (Dallas et al., 2007). The present study demonstrates that DOP receptor activation does not alter EAAT3 mRNA and protein expression in the astrocytes. This is different from an earlier study showing an inhibitory effect of DOP receptor on EAAT3 in the oocytes (Xia et al., 2006), suggesting a differential role of DOP receptor in the regulation of EAAT3 in different tissues/cells. Although microglia also express EAATs, the level of EAAT expression is much lower compared with the astrocytes, and microglial EAATs has little function in glutamate uptake and neuroprotection as we demonstrated previously (Liang et al., 2008). Thus, we believe that microglia should not have a considerable effect on the results from the present study. However, we will further clarify this issue in future work.
Our finding is not consistent with the results reported by Thorlin et al. (1998). They studied the effect of 10 μM [D-pen2, D-pen5]-enkephalin (DPDPE) on astrocytic EAAT2 mRNA and glutamate uptake. The specificity of DPDPE is much lower compared with that of UFP-512. Because the EC50 of DPDPE for DOP receptor is only 5.2 nM (Chandrakumar et al., 1992), 10 μM may also activate μ-opioid receptors (Han, 2003) or other receptors, thus inducing a complex effect or even opposite outcome (Zhang et al., 2000; 2002,; Chao et al., 2007). Also, it is unclear as to whether EAAT2 protein level changed in their study. Furthermore, they observed glutamate uptake after 48 h of DPDPE treatment. Our study shows that after DOP receptor activation, astrocytic glutamate transporter production reached a peak at 24 h and dramatically decreased by 48 h (Figure 4B). This might be due to several reasons, such as desensitization of DOP receptor and loss of activity of the added agonist. Therefore, comparing their data with ours may not be a fair comparison on many different levels. Moreover, we have used multiple approaches, including DOP receptor antagonist and siRNA, to verify our results.
Our data from various sets of experiments with DOP receptor activation, DOP receptor antagonism and knockdown of DOP receptor expression consistently demonstrated that DOP receptor activation up-regulates EAAT1 and EAAT2 expression and in turn enhances the glutamate uptake in the astrocytes in vitro. This novel finding sheds a new light on the mechanism of DOP receptor neuroprotection. However, it needs further verification if this mechanism plays a critical role in the control of glutamate homeostasis in the in vivo brain model.
Potential pathways for DOP receptor-mediated regulation of astrocytic EAATs
Our previous studies (Ma et al., 2005; Chao and Xia, 2010; Chao et al., 2012; He et al., 2013) and those of other groups (Narita et al., 2006; Peng et al., 2009; Chao and Xia, 2010; He et al., 2013) suggest the involvement of PKA, PKC and PI3K in DOP receptor signalling. In the current study, however, we observed that PKA, PKC and PI3K inhibitors did not alter DOP receptor-induced EAAT1 and EAAT2 expression in the astrocytes, suggesting that DOP receptor-induced expression of astrocytic EAATs does not involve PKA, PKC and PI3K signals. This may represent a major difference between neurons and astrocytes in intracellular signalling. More studies are worthy to be conducted for exploring in-depth mechanisms.
The MAPKs play a pivotal role in the mediation of cellular responses to a variety of cellular and environmental events. There was also a study suggesting a possible connection between glutamate signalling and the MAPK cascade (Abe and Saito, 2001). In the present study, the blockade of p38 with SB203580 significantly reduced DOP receptor-induced EAAT expression, suggesting the involvement of p38 in the DOP receptor-mediated up-regulation of EAAT expression. Indeed, our direct evidence further demonstrates that DOP receptor activation promotes p38 phosphorylation. Again, this notion is different from our finding in the neurons where DOP receptor activation attenuates p38 activity (Ma et al., 2005). More interestingly, the DOP receptor-mediated up-regulation of p38 phosphorylation could be largely suppressed by the inhibitors of MEK and ERK in the astrocytes. The synergic interaction between ERK and p38 is opposite to the antagonism between them in neurons where DOP receptor-mediated protection against hypoxia depends on an increase in ERK that counteracts the increase in p38 activity (Ma et al., 2005). Indeed, a recent study reported an involvement of p38 and ERK activity in protection of astrocytes against C2-ceramide- and staurosporine-induced cell death (Li and Reiser, 2011). The authors concluded that p38 and ERK contributes to astrocytic cytoprotection against apoptosis as evidenced by the inhibition of this effect following application of p38 and ERK inhibitors. Taken together, our current findings suggest that p38 activation induces cytoprotection in the astrocytes by promoting EAAT1 and EAAT2 expression. Its activity is likely regulated by MEK1-MEK2-ERK as their inhibitors completely abrogated the DOP receptor-induced expression of astrocytic EAAT1 and EAAT2. Further studies to reaffirm the differential regulation of MAPKs in neurons versus glia and the underlying mechanism are, however, warranted.
Implication of DOP receptor-mediated up-regulation of astrocytic EAATs
Our findings may provide a clue for new therapeutic strategies against hypoxic/ischaemic injury as an enhanced capacity for astrocytic uptake of excitatory amino acids helps reduce neuroexcitotoxicity in hypoxic/ischaemic conditions. To this end, we have started a new study on astrocytic DOP receptor versus EAAT under hypoxic conditions to obtain direct evidence for this point.
Moreover, neuroexcitotoxicity has been attributed to pathophysiological changes in many neurodegenerative disorders such as ALS. ALS is an adult motor neuron disease clinically characterized by a progressive muscular weakness, muscle atrophy, spasticity and eventually respiratory compromise. This brought forth the existing clinical heterogeneity of the disease and the postulation that the underlying pathophysiological mechanisms are multifactorial. Although the aetiology and pathogenesis of this malady remains essentially unclear, glutamatergic dysfunction has been implicated in the development of ALS besides the genetic involvement (Beleza-Meireles and Al-Chalabi, 2009) since 1990 when Plaitakis first hypothesized a defective excitatory amino acid metabolism as the cause of ALS (Plaitakis, 1990). Glutamate is required for normal functioning, while abnormally high levels lead to excitotoxicity that offsets neuronal degeneration and implicated in ALS. Removal of excess glutamate from the extracellular space is rapidly achieved by EAATs, a high-affinity sodium-dependent transporter system highly expressed in astrocytes (Arriza et al., 1994; Liang et al., 2008). Abundant evidence suggests that pathognomonic defect in ALS is a dysfunction of the astrocytic EAATs that is also implicated in other neurodegenerative diseases such as epilepsy, Huntington's disease, Alzheimer's disease and ischaemic stroke injury (Tanaka et al., 1997; Shin et al., 2005; Yamada et al., 2006; Yamanaka et al., 2008). Many studies have demonstrated the neuroprotective role of astrocytic EAAT expression in in vitro and in vivo models of ischaemic injury, motor neuron degeneration (Rothstein et al., 2005) and Huntington's disease (Shin et al., 2005). Some investigators have shown that transplanting the normal astrocytes to mouse model of ALS enhances the EAAT2 expression and declines disease progression (Sheldon and Robinson, 2007; Kim et al., 2011), while others have demonstrated that the loss or mutation of EAAT1 elevates extracellular glutamate levels and induces neurological disorders (Rothstein et al., 1996; Watanabe et al., 1999). Thus, up-regulation of astrocytic EAATs expression might offer a prime therapeutic target for acute and chronic neurological diseases, especially for ALS. Results from our present study are novel and have the potential to open grounds for discovering new treatment options although needs more research for verification and validation of our findings.
In conclusion, our study illustrates several exciting findings: (i) DOP receptor activation enhances astrocytic EAAT1 and EAAT2 expressions at both mRNA and protein levels; (ii) DOP receptor activation accelerates astrocytic glutamate uptake; and (iii) DOP receptor up-regulation of astrocytic EAATs relies on MEK/ERK-p38 signalling, but is independent of the PKA, PKC and PI3K pathways. Although further research is required to endorse these findings especially with respect to their clinical application, the novel association between DOP receptor and astrocytic EAATs presented here reconceptualizes and provides new opportunities to develop novel therapies for hypoxic/ischaemic brain injury and neurodegenerative disorders such as ALS.
Acknowledgments
This work was supported by NIH (HD-034852 and AT-004422) and The Vivian L Smith Neurologic Foundation.
Glossary
- ALS
amyotrophic lateral sclerosis
- DHK
dihydrokainic acid
- DOP receptor
δ-opioid receptor
- DPDPE
[D-pen2, D-pen5]-enkephalin
- EAATs
excitatory amino acid transporters
- GFAP
glial fibrillary acidic protein
- MEK
mitogen-activated protein kinase kinase
- RT-PCR
real-time PCR
- siRNA
small interfering RNA
- TBOA
DL-threo-β-benzyloxyaspartic acid
- UCPH
UCPH-101
Author contributions
Y. X. and J. L. designed the research; J. L. performed the research; Y. X., D. H. K., Y. Y., L. Z. and G. B. contributed research reagents and analytic tools; J. L., D. C. and Y. X. analysed the data; and Y. X., J. L., S. H. K. and D. C. wrote the paper.
Conflict of interest
None.
References
- Abe K, Saito H. Possible linkage between glutamate transporter and mitogen-activated protein kinase cascade in cultured rat cortical astrocytes. J Neurochem. 2001;76:217–223. doi: 10.1046/j.1471-4159.2001.00062.x. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013b;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balboni G, Salvadori S, Guerrini R, Negri L, Giannini E, Jinsmaa Y, et al. Potent delta-opioid receptor agonists containing the Dmt-Tic pharmacophore. J Med Chem. 2002;45:5556–5563. doi: 10.1021/jm020336e. [DOI] [PubMed] [Google Scholar]
- Beleza-Meireles A, Al-Chalabi A. Genetic studies of amyotrophic lateral sclerosis: controversies and perspectives. Amyotroph Lateral Scler. 2009;10:1–14. doi: 10.1080/17482960802585469. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Chandrakumar NS, Stapelfeld A, Beardsley PM, Lopez OT, Drury B, Anthony E, et al. Analogs of the delta opioid receptor selective cyclic peptide [2-D-penicillamine,5-D-penicillamine]-enkephalin: 2′,6′-dimethyltyrosine and Gly3-Phe4 amide bond isostere substitutions. J Med Chem. 1992;35:2928–2938. doi: 10.1021/jm00094a002. [DOI] [PubMed] [Google Scholar]
- Chao D, Xia Y. Ionic storm in hypoxic/ischemic stress: can opioid receptors subside it? Prog Neurobiol. 2010;90:439–470. doi: 10.1016/j.pneurobio.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao D, Bazzy-Asaad A, Balboni G, Xia Y. delta-, but not mu-, opioid receptor stabilizes K(+) homeostasis by reducing Ca(2+) influx in the cortex during acute hypoxia. J Cell Physiol. 2007;212:60–67. doi: 10.1002/jcp.21000. [DOI] [PubMed] [Google Scholar]
- Chao D, Bazzy-Asaad A, Balboni G, Salvadori S, Xia Y. Activation of DOR attenuates anoxic K + derangement via inhibition of Na + entry in mouse cortex. Cereb Cortex. 2008;18:2217–2227. doi: 10.1093/cercor/bhm247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao D, He X, Yang Y, Bazzy-Asaad A, Lazarus LH, Balboni G, et al. DOR activation inhibits anoxic/ischemic Na + influx through Na + channels via PKC mechanisms in the cortex. Exp Neurol. 2012;236:228–239. doi: 10.1016/j.expneurol.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas M, Boycott HE, Atkinson L, Miller A, Boyle JP, Pearson HA, et al. Hypoxia suppresses glutamate transport in astrocytes. J Neurosci. 2007;27:3946–3955. doi: 10.1523/JNEUROSCI.5030-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Duan YL, Wang SY, Zeng QW, Su DS, Li W, Wang XR, et al. Astroglial reaction to delta opioid peptide [D-Ala2, D-Leu5] enkephalin confers neuroprotection against global ischemia in the adult rat hippocampus. Neuroscience. 2011;192:81–90. doi: 10.1016/j.neuroscience.2011.06.067. [DOI] [PubMed] [Google Scholar]
- Fryer RM, Wang Y, Hsu AK, Gross GJ. Essential activation of PKC-delta in opioid-initiated cardioprotection. Am J Physiol Heart Circ Physiol. 2001;280:H1346–H1353. doi: 10.1152/ajpheart.2001.280.3.H1346. [DOI] [PubMed] [Google Scholar]
- Ganel R, Rothstein J. Glutamate transporter dysfunction and neuronal death. In: Monyer H, Adelmann GA, Jonas P, editors. Ionotropic Glutamate Receptors in the CNS. Berlin: Springer; 1999. pp. 472–493. In: (eds). [Google Scholar]
- Gilley JA, Kernie SG. Excitatory amino acid transporter 2 and excitatory amino acid transporter 1 negatively regulate calcium-dependent proliferation of hippocampal neural progenitor cells and are persistently upregulated after injury. Eur J Neurosci. 2011;34:1712–1723. doi: 10.1111/j.1460-9568.2011.07888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS. Acupuncture: neuropeptide release produced by electrical stimulation of different frequencies. Trends Neurosci. 2003;26:17–22. doi: 10.1016/s0166-2236(02)00006-1. [DOI] [PubMed] [Google Scholar]
- He X, Sandhu HK, Yang Y, Hua F, Belser N, Kim DH, et al. Neuroprotection against hypoxia/ischemia: δ-opioid receptor-mediated cellular/molecular events. Cell Mol Life Sci. 2013;70:2291–2303. doi: 10.1007/s00018-012-1167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabli N, Cahill CM. Anti-allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain. 2007;127:84–93. doi: 10.1016/j.pain.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Kang X, Chao D, Gu Q, Ding G, Wang Y, Balboni G, et al. delta-Opioid receptors protect from anoxic disruption of Na + homeostasis via Na + channel regulation. Cell Mol Life Sci. 2009;66:3505–3516. doi: 10.1007/s00018-009-0136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Dian-san S, Xiang-rui W. Delta opioid agonist [D-Ala2, D-Leu5] enkephalin (DADLE) reduced oxygen-glucose deprivation caused neuronal injury through the MAPK pathway. Brain Res. 2009;1292:100–106. doi: 10.1016/j.brainres.2009.06.104. [DOI] [PubMed] [Google Scholar]
- Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK, Dash R, et al. Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol. 2011;226:2484–2493. doi: 10.1002/jcp.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Sidoryk-Wegrzynowicz M, Wang N, Webb A, Son DS, Lee K, et al. GPR30 regulates glutamate transporter GLT-1 expression in rat primary astrocytes. J Biol Chem. 2012;287:26817–26828. doi: 10.1074/jbc.M112.341867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskela TT, Markkanen PM, Pietila EM, Tuusa JT, Petaja-Repo UE. Opioid receptor pharmacological chaperones act by binding and stabilizing newly synthesized receptors in the endoplasmic reticulum. J Biol Chem. 2007;282:23171–23183. doi: 10.1074/jbc.M610896200. [DOI] [PubMed] [Google Scholar]
- van Leyen K, Kim HY, Lee SR, Jin G, Arai K, Lo EH. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke. 2006;37:3014–3018. doi: 10.1161/01.STR.0000249004.25444.a5. [DOI] [PubMed] [Google Scholar]
- Li R, Reiser G. Phosphorylation of Ser45 and Ser59 of αB-crystallin and p38/extracellular regulated kinase activity determine αB-crystallin-mediated protection of rat brain astrocytes from C2-ceramide- and staurosporine-induced cell death. J Neurochem. 2011;118:354–364. doi: 10.1111/j.1471-4159.2011.07317.x. [DOI] [PubMed] [Google Scholar]
- Liang J, Takeuchi H, Doi Y, Kawanokuchi J, Sonobe Y, Jin S, et al. Excitatory amino acid transporter expression by astrocytes is neuroprotective against microglial excitotoxicity. Brain Res. 2008;1210:11–19. doi: 10.1016/j.brainres.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Liang J, Takeuchi H, Jin S, Noda M, Li H, Doi Y, et al. Glutamate induces neurotrophic factor production from microglia via protein kinase C pathway. Brain Res. 2010;1322:8–23. doi: 10.1016/j.brainres.2010.01.083. [DOI] [PubMed] [Google Scholar]
- Ma MC, Qian H, Ghassemi F, Zhao P, Xia Y. Oxygen-sensitive {delta}-opioid receptor-regulated survival and death signals: novel insights into neuronal preconditioning and protection. J Biol Chem. 2005;280:16208–16218. doi: 10.1074/jbc.M408055200. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Glutamate transporters: animal models to neurologic disease. Neurobiol Dis. 2004;15:461–473. doi: 10.1016/j.nbd.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Dietrich J, Wong V, Xue H, Mayer-Proschel M, Rao MS, et al. Glutamate transporter expression and function in human glial progenitors. Glia. 2004;45:133–143. doi: 10.1002/glia.10310. [DOI] [PubMed] [Google Scholar]
- Narita M, Kuzumaki N, Miyatake M, Sato F, Wachi H, Seyama Y, et al. Role of delta-opioid receptor function in neurogenesis and neuroprotection. J Neurochem. 2006;97:1494–1505. doi: 10.1111/j.1471-4159.2006.03849.x. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng PH, Huang HS, Lee YJ, Chen YS, Ma MC. Novel role for the delta-opioid receptor in hypoxic preconditioning in rat retinas. J Neurochem. 2009;108:741–754. doi: 10.1111/j.1471-4159.2008.05807.x. [DOI] [PubMed] [Google Scholar]
- Persson AI, Thorlin T, Eriksson PS. Comparison of immunoblotted delta opioid receptor proteins expressed in the adult rat brain and their regulation by growth hormone. Neurosci Res. 2005;52:1–9. doi: 10.1016/j.neures.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Capone D, Polichetti G, Vinciguerra A, Gentile A, Di Renzo G, et al. Neuroprotective, immunosuppressant and antineoplastic properties of mTOR inhibitors: current and emerging therapeutic options. Curr Opin Pharmacol. 2011;11:378–394. doi: 10.1016/j.coph.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Plaitakis A. Glutamate dysfunction and selective motor neuron degeneration in amyotrophic lateral sclerosis: a hypothesis. Ann Neurol. 1990;28:3–8. doi: 10.1002/ana.410280103. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Ruzicka BB, Fox CA, Thompson RC, Meng F, Watson SJ, Akil H. Primary astroglial cultures derived from several rat brain regions differentially express mu, delta and kappa opioid receptor mRNA. Brain Res Mol Brain Res. 1995;34:209–220. doi: 10.1016/0169-328x(95)00165-o. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Shaked I, Fisher J, Mizrahi T, Schori H. Protective autoimmunity against the enemy within: fighting glutamate toxicity. Trends Neurosci. 2003;26:297–302. doi: 10.1016/S0166-2236(03)00126-7. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiene-Martin A, Zhou R, Hauser KF. Regional, developmental, and cell cycle-dependent differences in mu, delta, and kappa-opioid receptor expression among cultured mouse astrocytes. Glia. 1998;22:249–259. [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Thorlin T, Roginski RS, Choudhury K, Nilsson M, Ronnback L, Hansson E, et al. Regulation of the glial glutamate transporter GLT-1 by glutamate and delta-opioid receptor stimulation. FEBS Lett. 1998;425:453–459. doi: 10.1016/s0014-5793(98)00288-9. [DOI] [PubMed] [Google Scholar]
- Tian X, Guo J, Zhu M, Li M, Wu G, Xia Y. δ-Opioid receptor activation rescues the functional TrkB receptor and protects the brain from ischemia-reperfusion injury in the rat. PLoS ONE. 2013;8:e69252. doi: 10.1371/journal.pone.0069252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner SM, Johnson SM. Delta-opioid receptor activation prolongs respiratory motor output during oxygen-glucose deprivation in neonatal rat spinal cord in vitro. Neuroscience. 2011;187:70–83. doi: 10.1016/j.neuroscience.2011.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Morimoto K, Hirao T, Suwaki H, Watase K, Tanaka K. Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain Res. 1999;845:92–96. doi: 10.1016/s0006-8993(99)01945-9. [DOI] [PubMed] [Google Scholar]
- Xia P, Pei G, Schwarz W. Regulation of the glutamate transporter EAAC1 by expression and activation of delta-opioid receptor. Eur J Neurosci. 2006;24:87–93. doi: 10.1111/j.1460-9568.2006.04897.x. [DOI] [PubMed] [Google Scholar]
- Xia Y, Haddad GG. Ontogeny and distribution of opioid receptors in the rat brainstem. Brain Res. 1991;549:181–193. doi: 10.1016/0006-8993(91)90457-7. [DOI] [PubMed] [Google Scholar]
- Xia Y, Haddad GG. Major difference in the expression of delta- and mu-opioid receptors between turtle and rat brain. J Comp Neurol. 2001;436:202–210. [PubMed] [Google Scholar]
- Yamada T, Kawahara K, Kosugi T, Tanaka M. Nitric oxide produced during sublethal ischemia is crucial for the preconditioning-induced down-regulation of glutamate transporter GLT-1 in neuron/astrocyte co-cultures. Neurochem Res. 2006;31:49–56. doi: 10.1007/s11064-005-9077-4. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang H, Shah K, Karamyan VT, Abbruscato TJ. Opioid receptor agonists reduce brain edema in stroke. Brain Res. 2011;1383:307–316. doi: 10.1016/j.brainres.2011.01.083. [DOI] [PubMed] [Google Scholar]
- Yang Y, Xia X, Zhang Y, Wang Q, Li L, Luo G, et al. delta-Opioid receptor activation attenuates oxidative injury in the ischemic rat brain. BMC Biol. 2009;7:55. doi: 10.1186/1741-7007-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambelli VO, Fernandes AC, Gutierrez VP, Ferreira JC, Parada CA, Mochly-Rosen D, et al. Peripheral sensitization increases opioid receptor expression and activation by crotalphine in rats. PLoS ONE. 2014;9:e90576. doi: 10.1371/journal.pone.0090576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Haddad GG, Xia Y. delta-, but not mu- and kappa-, opioid receptor activation protects neocortical neurons from glutamate-induced excitotoxic injury. Brain Res. 2000;885:143–153. doi: 10.1016/s0006-8993(00)02906-1. [DOI] [PubMed] [Google Scholar]
- Zhang J, Gibney GT, Zhao P, Xia Y. Neuroprotective role of delta-opioid receptors in cortical neurons. Am J Physiol Cell Physiol. 2002;282:C1225–C1234. doi: 10.1152/ajpcell.00226.2001. [DOI] [PubMed] [Google Scholar]
- Zhang J, Qian H, Zhao P, Hong SS, Xia Y. Rapid hypoxia preconditioning protects cortical neurons from glutamate toxicity through delta-opioid receptor. Stroke. 2006;37:1094–1099. doi: 10.1161/01.STR.0000206444.29930.18. [DOI] [PubMed] [Google Scholar]
- Zhao LR, Piao CS, Murikinati SR, Gonzalez-Toledo ME. The role of stem cell factor and granulocyte-colony stimulating factor in treatment of stroke. Recent Pat CNS Drug Discov. 2013;8:2–12. doi: 10.2174/1574889811308010002. [DOI] [PMC free article] [PubMed] [Google Scholar]