

Abstract
Background and Purpose
Neurosteroids potentiate responses of the GABAA receptor to the endogenous agonist GABA. Here, we examined the ability of neurosteroids to potentiate responses to the allosteric activators etomidate, pentobarbital and propofol.
Experimental Approach
Electrophysiological assays were conducted on rat α1β2γ2L GABAA receptors expressed in HEK 293 cells. The sedative activity of etomidate was studied in Xenopus tadpoles and mice. Effects of neurosteroids on etomidate-elicited inhibition of cortisol synthesis were determined in human adrenocortical cells.
Key Results
The neurosteroid 5β-pregnan-3α-ol-20-one (3α5βP) potentiated activation of GABAA receptors by GABA and allosteric activators. Co-application of 1 μM 3α5βP induced a leftward shift (almost 100-fold) of the whole-cell macroscopic concentration–response relationship for gating by etomidate. Co-application of 100 nM 3α5βP reduced the EC50 for potentiation by etomidate of currents elicited by 0.5 μM GABA by about three-fold. In vivo, 3α5βP (1mg kg-1) reduced the dose of etomidate required to produce loss of righting in mice (ED50) by almost 10-fold. In tadpoles, the presence of 50 or 100 nM 3α5βP shifted the EC50 for loss of righting about three- or ten-fold respectively. Exposure to 3α5βP did not influence inhibition of cortisol synthesis by etomidate.
Conclusions and Implications
Potentiating neurosteroids act similarly on orthosterically and allosterically activated GABAA receptors. Co-application of neurosteroids with etomidate can significantly reduce dosage requirements for the anaesthetic, and is a potentially beneficial combination to reduce undesired side effects.
Tables of Links
| TARGETS | LIGANDS | |
|---|---|---|
| GABAA receptors | Alphaxalone | Pentobarbital |
| Steroid 11β-hydroxylase (CYP11B2) | Etomidate | Propofol |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a,b,).
Introduction
The GABAA receptor is the major inhibitory ionotropic transmitter-gated ion channel in the brain. In mature neurons, activation of GABAA receptor results in influx of Cl– leading to hyperpolarization of the cell or reduction of the effects of excitatory channels. These receptors can be activated by a variety of drugs. One class of agonists that includes GABA interacts with the orthosteric transmitter-binding sites, located in the extracellular domain of the receptor at the interfaces between the β and α subunits (Miller and Smart, 2010). The second, diverse class of agonists is termed allosteric ligands. These include neuroactive steroids (such as 5β-pregnan-3α-ol-20-one, 3α5βP), barbiturates (such as pentobarbital) and other i.v. anaesthetics (such as etomidate and propofol). The allosteric ligands interact with their individual binding sites that do not overlap with the orthosteric transmitter-binding site (Hosie et al., 2006; Li et al., 2006; Chiara et al., 2013; Yip et al., 2013). The allosteric ligands also act as modulators, potentiating responses to GABA. The potentiating effect may (Hosie et al., 2007) or may not (Stewart et al., 2008) be mediated by binding sites distinct from the sites responsible for direct activation.
The interaction between allosteric and orthosteric agents, and between two allosteric agents, is an important problem, both in terms of fundamental insights into the properties of the GABAA receptor and, in more physiological and clinical contexts, in terms of the establishment of the overall level of inhibitory influence in the CNS. We focus here on the interactions of neuroactive steroids, particularly the endogenous steroid 3α5βP, with other allosteric agents. Endogenous steroids are reported to play a role in modulating the magnitude of inhibitory synaptic events (Belelli and Herd, 2003; Belelli et al., 2003), whereas changes in the levels of endogenous steroids may underlie premenstrual dysphoria (Smith, 2001). In addition, exogenous steroids have been found to enhance responses to the allosteric agonist pentobarbital (Peters et al., 1988) and to enhance the anaesthetic potency of etomidate (Richards and White, 1981).
Potentiation or activation of the GABAA receptor underlies the behavioural actions of allosteric ligands, many of which are in clinical use as anaesthetics, anticonvulsants, anxiolytics or sedatives (Franks, 2006; 2008,). One such drug, etomidate, is commonly used to induce sedation (Criado et al., 1980; Ray and McKeown, 2012). It is a preferred anaesthetic induction agent in situations where reduced blood pressure is not clinically tolerable. As many other imidazole-containing drugs, etomidate also acts to suppress synthesis of adrenocortical steroids (Wagner et al., 1984; Ayub and Levell, 1989). The adrenocortical suppressant effects of etomidate, with the potential for a delayed hypotensive response, have limited the clinical use of etomidate, especially in patients with severe sepsis (Hunter and Kirschner, 2013).
Here, we have shown that neuroactive steroids enhance activation and modulation of the α1β2γ2L GABAA receptor by allosteric drugs, focusing on the i.v. anaesthetic etomidate and the endogenous neurosteroid 3α5βP. In electrophysiological assays, the application of 3α5βP shifted the etomidate concentration–response relationship to lower concentrations. The concentration–response relationship for potentiation of GABA-activated receptors by etomidate was also shifted to lower concentrations in the presence of the steroid. In behavioural assays, exposure to 3α5βP reduced the dose of etomidate required to produce loss of righting. Neurosteroids could thus allow GABAA receptor-mediated anaesthesia to be achieved with lower doses of etomidate.
Methods
Molecular biology and receptor expression
The experiments on heterologously expressed receptors were conducted on rat wild-type and mutated α1β2γ2L GABAA receptors. The α1(Q241L) and α1(T236I) mutations were generated using QuikChange (Agilent Technologies, Santa Clara, CA, USA). We have previously characterized the physiological consequences of the α1(Q241L) mutation (Akk et al., 2008). The complementary DNAs for the subunits were subcloned into the pcDNA3 expression vector (Life Technologies,Carlsbad, CA, USA) in the T7 orientation.
The electrophysiological recordings were conducted on receptors expressed in HEK 293 cells (ATCC CRL-1573). The cells were plated at a density of ∼200 000 cells per 35 mm dish. Transfection was carried out 1 day after plating, using a calcium phosphate precipitation-based transient transfection technique. A total of 3 μg of complementary DNA in the ratio of 1:1:1 (α : β : γ) was mixed with 12.5 μL of 2.5 M CaCl2, and distilled H2O to a final volume of 125 μL. The solution was added slowly, without mixing, to an equal volume of BES buffer (50mM N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES), 280 mM NaCl, 1.5 mM Na2HPO4; pH 6.95). The combined mixture was incubated at room temperature for 10 min followed by mixing the contents and an additional 15 min incubation. The precipitate was added to the cells in a 35 mm dish for overnight incubation at 37°C, followed by replacement of medium in the dish. The experiments were conducted in the course of the next 2 days after changing the medium.
Electrophysiological recordings and data analysis
HEK 293 cells expressing high levels of GABAA receptors were identified using a bead-binding technique. The amino terminus of the α1 subunit has been tagged with the FLAG epitope (Ueno et al., 1996). Surface expression of the FLAG epitope was verified using a mouse monoclonal antibody to the FLAG epitope (M2, Sigma-Aldrich, St. Louis, MO, USA), which had been adsorbed to immunobeads with a covalently attached goat anti-mouse IgG antibody (Life Technologies).
The experiments were conducted using the standard whole-cell voltage clamp technique. The bath solution contained (in mM): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 D-glucose and 10 HEPES; pH 7.4. The pipette solution contained (in mM): 140 CsCl, 4 NaCl, 4 MgCl2, 0.5 CaCl2, 5 EGTA, 10 HEPES, pH 7.4.
The drugs were applied onto the cells using an SF-77B fast perfusion stepper system (Warner Instruments, Hamden, CT, USA). The cells were clamped at −60 mV. The currents were recorded using an Axopatch 200B amplifier (Molecular Devices, Union City, CA, USA), low-pass filtered at 2 kHz and digitized with a Digidata 1320 series interface (Molecular Devices) at 10 kHz. The analysis of current traces was conducted using pClamp 9.0 software (Molecular Devices).
The experiments consisted of applying a given concentration of an allosteric or orthosteric agonist in the absence or presence of a modulator. The effect of the modulator was evaluated from the ratio of the response in the presence of a modulator to that in the absence of the modulator, that is, the response ratio.
Behavioural assays
Animals
All animal care and experimental procedures were conducted according to protocols approved by the Washington University Animal Studies Committee. We have complied with the ARRIVE guidelines for work involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 276 animals (240 Xenopus tadpoles and 36 BALB/c mice) were used in the experiments described here.
In Xenopus laevis tadpole behavioural assays, etomidate, 3α5βP or etomidate + 3α5βP were added to beakers containing 100 mL of oxygenated Tadpole Ringer's solution to yield known final concentrations of the drugs. The Tadpole Ringer's solution contained 5.8 mM NaCl, 67 μM KCl, 34 μM Ca(NO3)2, 83 μM MgSO4, 419 μM Tris-HCl, 80 μM Tris-base; pH 7.5. Ten tadpoles (obtained from Nasco, Fort Atkinson, WI, USA) were distributed into each beaker and allowed to equilibrate in the Tadpole Ringer's solution for 3 h. In the end of the equilibration period, the loss of righting reflex (LRR) was measured by turning the tadpole over using a hooked glass rod. LRR was defined as the inability of a tadpole to right itself within 5 s on its back, for three consecutive trials. The tests were conducted in the presence of 0.1–10 μM etomidate, in the absence and presence of 50 or 100 nM 3α5βP. The tadpole extracellular fluid readily equilibrates with the Ringer's solution in which they swim, so the data obtained represent concentration–response rather than dose–response relationships.
Additional behavioural assays were conducted on 7–8-week-old male BALB/c mice (22–25 g each), obtained from Harlan Laboratories (Indianapolis, IN, USA). After 4 min under a moderate heat source (lamp), each mouse was placed into a commercially available 3.2 cm wide restraining device (Braintree Scientific, Inc., Braintree, MA, USA). The mice were injected i.v. with etomidate or etomidate + 1 mg·kg−1 of 3α5βP. Control experiments were conducted with 1 mg·kg−1 of 3α5βP alone, or with 35% propylene glycol (solubilization agent for etomidate). Injections were administered via tail vein at a volume of 5 μL·g−1 body mass. Each mouse was then placed on its back inside an observation cage under the moderate heat source. LRR was defined as inability of a mouse to right itself within 5 s of being placed onto its back in six consecutive trials (Stastna et al., 2011).
In vitro cortisol synthesis assay
Cortisol synthesis was measured in the human adrenocortical cell line (ATCC CRL-2128) using the approach described previously (Cotten et al., 2010). The cells were initially grown in 12-well culture plates in 2 mL of growth medium containing DMEM/F12 supplemented with 2.5% NuSerum (BD Biosciences, San Jose, CA, USA), 1% ITS+ premix supplements (BD Biosciences) and penicillin – streptomycin (10 IU·mL−1 to 10 μg·mL−1; Mediatech, Inc., Herndon, VA, USA). Once the cells reached 95% confluence, the growth medium was replaced with assay medium containing DMEM/F12 supplemented with 0.1% ITS+ premix supplements, penicillin – streptomycin (10 IU·mL−1 to 10 μg·mL−1), and 20 μM forskolin (R&D Systems, Minneapolis, MN, USA). The assay medium also contained 3α5βP and/or etomidate. After incubation for 48 h, 1.2 mL of assay medium was collected from each well and centrifuged to remove cells and debris. Cortisol concentration in the supernatant was determined using a competitive antibody binding assay according to the manufacturer's instructions (R&D Systems).
Data analysis
Response ratios in the text are shown as X-fold changes (means ± SD). Statistical analysis was performed using the STATA software package (StataCorp, College Station, TX, USA) or Excel (Microsoft, Redmond, WA, USA), to compare the observed ratio with 1 (no effect) using a two-tailed paired t-test (Excel). This test is equivalent to a one-sample t-test to a hypothetical value of 1. This test is designed to determine whether the drug has a significant effect.
Concentration–response curves for 3α5βP were generated by applying a series of steroid concentrations in the presence of 0.5 μM GABA or 4 μM etomidate. The data at a given concentration of 3α5βP were averaged across all cells, and the averaged data were fit. Etomidate concentration–response curves were generated by applying a series of etomidate concentrations in the absence or presence of 0.5 μM GABA and/or 3α5βP. The data at a given etomidate concentration were averaged across all cells for a given agonist–modulator combination, and the averaged data were fit. The concentration–response curves were fitted to the equation: Y([variable]) = Ymin + (Ymax − Ymin) [variable]n/([variable]n + EC50n), where Ymin is the low-concentration offset, Ymax is the maximal potentiating effect, [variable] is the concentration of 3α5βP or etomidate, EC50 is the concentration producing the half-maximal effect and n is the Hill coefficient. The fitting was conducted using the program NFIT (The University of Texas Medical Branch at Galveston). The fitting program returns uncertainty estimates on the best-fitting parameter values.
To equalize the apparent maximal effect when steroid potentiation of currents elicited by GABA was compared with potentiation of currents elicited by etomidate, the data were plotted in a normalized form. Normalization was conducted through the following equation:
Normalized potentiation = [(peak current in the presence of 3α5βP/peak current in the absence of 3α5βP) − 1]/[(maximal peak current in the presence of 3α5βP/peak current in the absence of 3α5βP) − 1]. For the normalized responses, a value of 0 indicates no effect and 1 the maximal effect.
Materials
All the compounds used in the study were obtained from Sigma-Aldrich or R&D Systems. The stock solution of GABA (at 500 mM) was made in bath solution, and stored at −20°C. The stock solution of pentobarbital was made at 1 mM in bath solution, and stored at +4°C. The stock solution of propofol was made at 200 mM in DMSO, and stored at room temperature. For use in electrophysiological and tadpole behavioural assays, the stock solution of etomidate (20 mM) was made in DMSO, and stored at +4°C, and the stock solutions of 3α5βP or of alphaxalone (10 mM) were made in DMSO, and stored at room temperature. The stock solutions were further diluted on the day of experiment. For use in mouse behavioural assays, 3α5βP was dissolved in a 45% (w/v in water) (2-hydroxypropyl)-β-cyclodextrin solution at a complexation ratio of 1:4. Etomidate (Bedford Laboratories, Bedford, OH, USA) was supplied at a concentration of 2 mg·mL−1 (in 35% propylene glycol), and was further diluted for administration in the same vehicle.
Results
Steroid-mediated potentiation of allosteric activation
Co-application of potentiating steroids enhances current responses from allosterically activated GABAA receptors. Peak responses to 4 μM etomidate (a concentration eliciting approximately 2% of the maximal response to GABA) from rat α1β2γ2L receptors were potentiated by 12 ± 11-fold (mean ± SD; 1 = no effect; seven cells, P < 0.05 vs. no effect) in the presence of 10 μM 3α5βP. A typical recording is shown in Figure 1A. Currents elicited by etomidate in the presence of 3α5βP were blocked by 100 μM picrotoxin (data not shown). Both etomidate and 3α5βP at these concentrations are efficacious potentiators of currents elicited by GABA. Accordingly, to avoid contamination the experiments were conducted on cells with no prior exposure to GABA.
Figure 1.
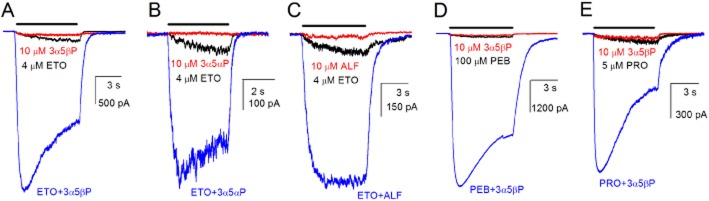
The effects of steroids on receptors activated by allosteric agonists. Sample current traces from HEK 293 cells expressing rat α1β2γ2L GABAA receptors. The receptors were activated by 4 μM etomidate (ETO) in the absence or presence of 10 μM 3α5βP (A), 3α5αP (B) or alphaxalone (ALF; C). Panels D and E show the ability of 10 μM 3α5βP to enhance gating by two other allosteric agonists, 100 μM pentobarbital (PEB) and 5 μM propofol (PRO). Currents resulting from exposure to steroid alone are also shown. Bars on top of current traces indicate the duration of drug application. The data indicate that co-application of steroids with the allosteric activator etomidate, pentobarbital or propofol results in a strong enhancement of the peak response.
The modulatory effect was also observed in the presence of other potentiating steroids, such as the endogenous isomer of 3α5βP, 5α-pregnan-3α-ol-20-one (3α5αP) and the synthetic steroid alphaxalone. The application of 10 μM 3α5αP potentiated peak responses to 4 μM etomidate by 31 ± 25-fold (six cells, P < 0.05). Co-application of 10 μM alphaxalone with etomidate enhanced the peak response by 33 ± 28-fold (11 cells, P < 0.01). Typical current traces are shown in Figure 1B and C.
Potentiating effects of steroids were observed on currents elicited by other allosteric agonists. Responses to 100 μM pentobarbital were potentiated by 45 ± 33-fold (five cells, P < 0.05) by 10 μM 3α5βP. When the receptors were activated by 5 μM propofol, 10 μM 3α5βP potentiated the peak response by 16 ± 7-fold (five cells, P < 0.01). Sample traces are shown in Figure 1D and E.
In further experiments, we focused on the endogenous steroid 3α5βP and the allosteric activator etomidate. The goals of the experiments were to characterize the interactions between 3α5βP and etomidate acting at the GABAA receptor, and to determine whether these interactions are reflected in behavioural responses and/or in a well-characterized adverse effect of etomidate.
Properties of potentiation of etomidate-elicited currents by 3α5βP
To determine if the nature of the agonist affects the potency of 3α5βP, we compared steroid concentration–response relationships in receptors activated by etomidate or GABA. The concentrations of the two agonists were selected to elicit similar fractional responses. The concentration of GABA was 0.5 μM, which elicited 1 ± 1% (six cells) of the response to saturating GABA. Etomidate was employed at 4 μM, a concentration that elicited 2 ± 1% (five cells) of the response to saturating GABA. We found that the EC50 values (shown as best-fit ± uncertainty estimate) for 3α5βP were similar for both agonists: 442 ± 19 and 395 ± 78 nM for receptors activated by GABA and etomidate respectively (Figure 2A). Maximal potentiating effects were also similar (67 ± 1 and 50 ± 4-fold in the presence of GABA and etomidate respectively). Representative current traces are shown in Figure 2B.
Figure 2.

Properties of steroid-mediated potentiation of receptors activated by etomidate. (A) Concentration–response curves for 3α5βP. The wild-type α1β2γ2L receptors were activated by 0.5 μM GABA or 4 μM etomidate in the absence and presence of 30–3000 nM 3α5βP. The data points give mean ± SEM from six (GABA) or five cells (etomidate). The data are plotted in normalized form (see Methods for details). The EC50 estimates are 442 ± 19 and 395 ± 78 nM for receptors activated by 0.5 μM GABA and 4 μM etomidate respectively. The fitted values for Ymax were 67 ± 1 and 50 ± 4 (best-fitting values ± calculated uncertainty) for receptors activated by GABA and etomidate respectively. (B) Representative traces for receptors activated by 0.5 μM GABA or 4 μM etomidate, and modulated by 30–3000 nM 3α5βP. (C and D) Sample whole-cell recordings from HEK 293 cells expressing rat α1(Q241L)β2γ2L or α1(T236I)β2γ2L GABAA receptors. The receptors were activated by 30 μM etomidate in the absence and presence of 10 μM 3α5βP. The α1(Q241L) but not the α1(T236I) mutation abolished potentiation of etomidate-activated receptors by the steroid 3α5βP. Overall, the data indicate that 3α5βP similarly modulates receptors activated by GABA and the allosteric activator etomidate. Bars on top of current traces indicate the duration of drug application.
Mutations to the first membrane-spanning domain in the α1 subunit can affect modulation of GABA-elicited currents by potentiating steroids. In particular, the α1(Q241L) mutation abolishes potentiation by 3α5βP, possibly by eliminating a critical hydrogen bond within the receptor or with the steroid molecule (Hosie et al., 2006; Akk et al., 2008). To gain further insight into similarities in steroid-mediated potentiation of currents elicited by allosteric agonists and the transmitter GABA, we probed the effect of the α1(Q241L) mutation on potentiation of currents elicited by etomidate. To reliably record control electrophysiological responses, we employed a higher concentration of etomidate (30 μM, EC4). We found that the α1(Q241L) mutation abolished potentiation of etomidate-activated receptors by 10 μM 3α5βP (Figure 2C). The peak response in the presence of the steroid was 96 ± 26% of control (seven cells, P > 0.6). As shown in Figure 3A, for wild-type receptors, there was a marked potentiation of responses to 30 μM etomidate. These data suggest that the mechanisms of potentiation of receptors activated by allosteric agonists are similar to the mechanisms for receptors activated by the transmitter GABA.
Figure 3.
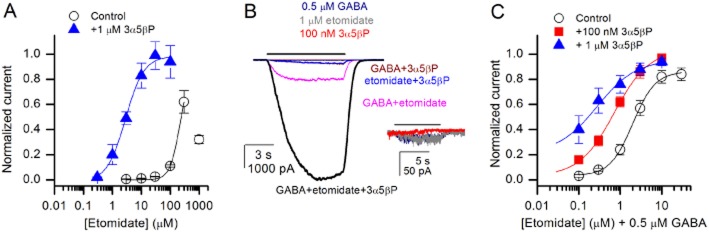
3α5βP affects etomidate concentration–response properties in the absence and presence of GABA. (A) Concentration–response curves for gating by etomidate in the absence and presence of 1 μM 3α5βP. The data were normalized to the response to a saturating concentration (100 μM) of GABA in the same cell. The data points give mean ± SEM from five cells in each condition. The data were fitted as described in Methods. The results (best fit±uncertainty estimate) from the fits are as follows. Control: EC50 = 218 ± 5 μM, Hill coefficient, n = 2.5 ± 0.1, Ymax was constrained to an arbitrarily chosen value of 0.9 and Ymin was constrained to 0. The data point at 1000 μM is skewed by channel block, and was not included in the fit. Etomidate +1 μM 3α5βP: EC50 = 2.8 ± 0.5 μM, n = 1.4 ± 0.3, Ymax = 0.98 ± 0.04, Ymin = −0.01 ± 0.07. (B) Sample current response traces from HEK 293 cells expressing rat α1β2γ2L GABAA receptors activated by 0.5 μM GABA, 1 μM etomidate or 100 nM 3α5βP, or by combinations of these drugs. The bar on top of current traces indicates the duration of drug application. The data show that the application of GABA + etomidate + 3α5βP potentiates the current responses beyond what is seen with applications of single drugs or any combination of two drugs. The inset shows current traces in the presence of GABA, etomidate or 3α5βP, at a higher resolution. (C) Concentration–response curves for etomidate in the presence of 0.5 μM GABA, GABA + 100 nM 3α5βP or GABA + 1 μM 3α5βP. The data were normalized to the response to 100 μM GABA. The data points give mean ± SEM from five cells under each condition. The data were fitted as described in Methods. The results from the fits are as follows. Control (etomidate + 0.5 μM GABA): EC50 = 2.0 ± 0.1 μM, n = 1.6 ± 0.2, Ymax = 0.86 ± 0.02, Ymin = 0.03 ± 0.02. Etomidate + 0.5 μM GABA + 100 nM (1 μM) 3α5βP: EC50 = 0.71 ± 0.05 μM, n = 1.0 ± 0.1, Ymax = 1.04 ± 0.06, Ymin was constrained to the response obtained in the presence of 100 nM 3α5βP + 0.5 μM GABA (0.03). Etomidate + 0.5 μM GABA + 100 nM (1 μM) 3α5βP: EC50 = 0.28 ± 0.06 μM, n = 0.9 ± 0.2, Ymax = 0.96 ± 0.04, Ymin was constrained to the normalized response obtained in the presence of 1 μM 3α5βP + 0.5 μM GABA (0.21).
Indistinguishable potentiation of orthosterically (GABA) and allosterically (etomidate) activated receptors by 3α5βP suggests that in both cases the steroid is the potentiating agent, and GABA or etomidate acts as the agonist component. To provide additional confirmation, we examined the effect of the α1(T236I) mutation that reduces direct gating by several steroids, including 3α5βP (Hosie et al., 2006). We found that receptors containing the α1(T236I) mutation retained their ability to be potentiated during co-application of 30 μM etomidate and 10 μM 3α5βP (8 ± 6-fold, eight cells, P < 0.05 vs. no effect). Thus, these data support the idea that when the two agonists are combined the major interaction between 3α5βP and etomidate is for the steroid to enhance response to etomidate, rather than for etomidate to enhance direct activation by steroid. Sample current traces are shown in Figure 2D.
Effect of steroid on the concentration–response properties for gating and potentiation by etomidate
Next, we examined the effect of 3α5βP on the etomidate concentration–response relationship. Cells expressing wild-type α1β2γ2L receptors were exposed to etomidate alone or to etomidate in the presence of 1 μM 3α5βP. The presence of steroid shifted the etomidate concentration–response curve to the left. The EC50 for direct gating by etomidate was 218 ± 5 μM in the absence of steroid, and 2.8 ± 0.5 μM in the presence of 1 μM 3α5βP (Figure 3A). Etomidate is a relatively high-efficacy agonist, with a maximal response of well over 60% of that observed in the presence of saturating GABA. In the presence of 3α5βP, the peak response to saturating etomidate reached that observed in the presence of saturating GABA (Figure 3A).
Under physiological conditions, neurons are tonically exposed to low (0.5–1.0 μM) concentrations of GABA in the CSF (Eckstein et al., 2008). The synaptic α1β2γ2L subtype responds weakly to such low levels of GABA (less than 2% of maximal response). We observed that co-application of 0.5 μM GABA with a mix of low concentrations of a steroid (100 nM 3α5βP) and etomidate (1 μM) strongly potentiated the peak response. The average response to 0.5 μM GABA was increased by 9 ± 3-fold by 100 nM 3α5βP, 37 ± 21-fold by 1 μM etomidate and 210 ± 103-fold (mean ± SD, six cells) by the combination of 3α5βP and etomidate. A sample recording is shown in Figure 3B. To learn more about how the presence of low GABA affects currents elicited by the combination of anaesthetic + steroid, we conducted concentration–response relationship measurements for etomidate in the presence of a fixed low concentration of GABA (0.5 μM) and a range of concentrations of 3α5βP (0, 0.1 and 1 μM). The data demonstrate that the concentration of etomidate required to elicit inward currents is significantly lower when GABA is added to the solution bathing the receptors. With no steroid in the solution, the addition of 0.5 μM GABA shifted the EC50 for etomidate from 218 to 2.0 ± 0.1 μM. The addition of 3α5βP further shifted the concentration–response curves (Figure 3C). In the presence of GABA + 100 nM 3α5βP, the EC50 for etomidate was 0.71 ± 0.05 μM, and in the presence of GABA + 1 μM 3α5βP, the EC50 for etomidate was 0.28 ± 0.06 μM.
The application of 3α5βP reduces the dose of etomidate required to produce loss of righting
GABAA receptor-mediated currents underlie the ability of etomidate to induce or maintain sedation (Franks, 2006; Solt and Forman, 2007). The data obtained on recombinant receptors thus suggest that etomidate dose–response curves for sedation may be left-shifted in the presence of 3α5βP. We probed the effect of steroid on the anaesthetic properties of etomidate in two animal models. First, we examined the tadpole LRR in the presence of etomidate and various concentrations of 3α5βP. Loss of righting can be considered a surrogate for sedation in animal models (Halsey et al., 1986; Franks, 2008). Our data show that the etomidate EC50 for LRR was reduced from 1.6 to 0.5 or 0.1 μM in the presence of 50 or 100 nM 3α5βP respectively (Figure 4). We note that the findings for etomidate EC50 are in good agreement with previous data (2.3 μM; Belelli et al., 2003). In control experiments, 50–100 nM 3α5βP applied in the absence of etomidate were unable to elicit LRR (data not shown).
Figure 4.
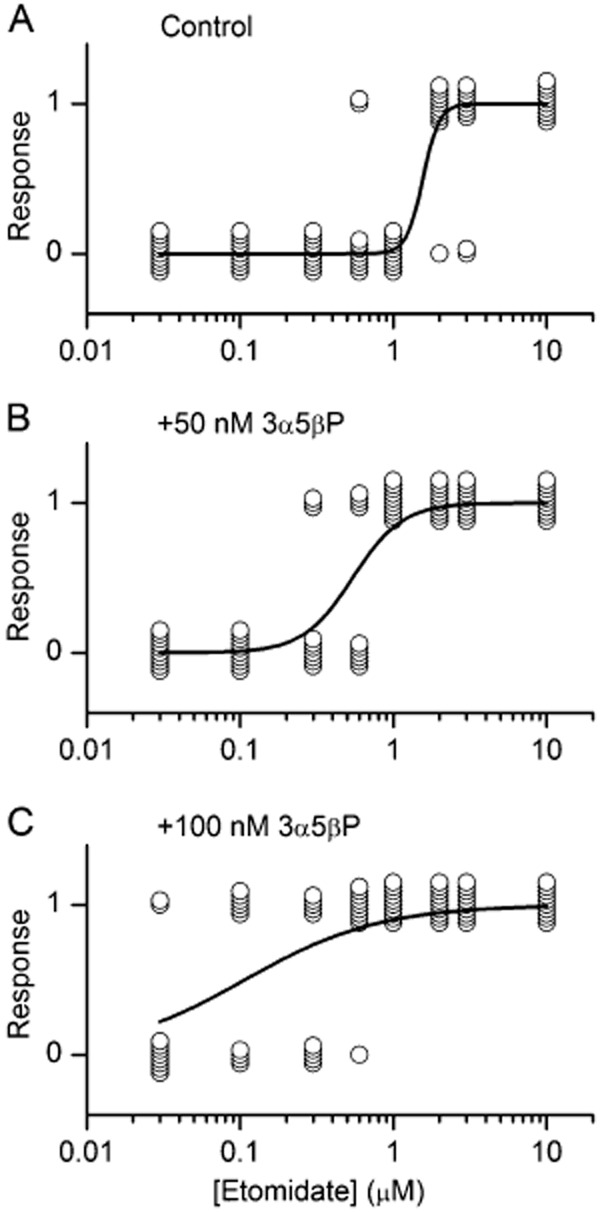
Effect of 3α5βP on etomidate-induced loss of righting (LRR) in tadpoles. (A) Exposure to etomidate causes LRR in Xenopus tadpoles. The data are plotted as quantal dose–response relationship where each symbol corresponds to one tadpole. The ordinate gives the response to etomidate that could manifest as no effect (0) or LRR (1). (B and C) Co-application of 50 or 100 nM 3α5βP shifts the etomidate LRR curve to lower concentrations. The curves were fitted with the following equation : Y([etomidate]) = [etomidate]n/([etomidate]n + EC50n), where EC50 is the concentration producing the half-maximal effect, and n is the Hill coefficient. The concentrations of etomidate producing half-maximal effect were as follows. Control: EC50 = 1.6 ± 0.2 μM. Etomidate + 50 nM 3α5βP: EC50 = 0.5 ± 0.1 μM. Etomidate + 100 nM 3α5βP: EC50 = 0.11 ± 0.03 μM.
In the second experiment, we examined the effect of a subanaesthetic dose of 3α5βP on the etomidate ED50 for LRR in mice. The dose of etomidate producing half-maximal effect was 0.19 mg·kg−1 under control conditions, and 0.02 mg·kg−1 in the presence of 1 mg·kg−1 3α5βP (Figure 5). Assuming uniform distribution of the drug, these doses correspond to 0.8 and 0.04 μmol·kg−1 etomidate respectively. Injections of 1 mg·kg−1 of 3α5βP alone, or 35% propylene glycol (solvent), did not result in LRR (data not shown; but see Covey et al., 2000).
Figure 5.
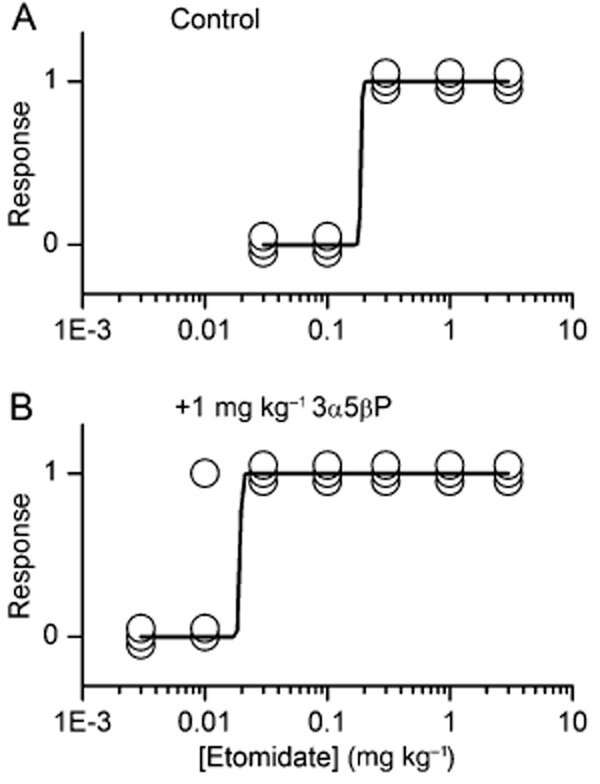
Effect of 3α5βP on etomidate-induced loss of righting (LRR) in mice. (A) Mice were exposed to etomidate alone. Data are presented and analyzed as in Figure 4. Half-maximal effect was produced by 0.19 mg·kg−1 etomidate. (B) Mice were exposed to etomidate in the presence of 1 mg·kg−1 3α5βP. Half-maximal effect was produced by 0.02 mg·kg−1 etomidate.
Co-application of steroid with etomidate is not accompanied by changes in inhibition of adrenal steroid synthesis
Etomidate potently inhibits steroid 11β-hydroxylase (CYP11B2), a crucial enzyme in the adrenocortical steroid synthesis pathway (Dorr et al., 1984; de Jong et al., 1984). As a result, etomidate exposure can be associated with adrenocortical suppression that can continue for several days following initial exposure and may lead to increased mortality (Wagner et al., 1984; Ray and McKeown, 2012; Hunter and Kirschner, 2013). The observed reduced etomidate requirement for producing LRR in the presence of 3α5βP raises the possibility that sedation could be achieved at doses where adverse effects are manifested less strongly.
In order to assess the effect of 3α5βP on cortisol synthesis, we used an elisa to measure cortisol release from human adrenocortical cells, employing a commercially available cortisol antibody binding assay. This assay uses a prolonged exposure to drugs (48 h) and has been used to examine the potential for etomidate and analogues to inhibit cortisol synthesis (Cotten et al., 2009; 2010,).
The cells were exposed to 0.1–100 μM etomidate in the absence and presence of 100 nM 3α5βP. The data suggest that the etomidate-induced reduction of cortisol release is not affected by 100 nM 3α5βP. In three experiments examining the effect of etomidate alone on release of cortisol, the IC50 for reduction of cortisol release was 9 ± 8 nM. In three experiments examining the effect of etomidate in the presence of 100 nM 3α5βP, the IC50 was 18 ± 10 nM. The difference between means was not statistically significant (P > 0.28; t-test). Exposure to 100 nM 3α5βP in the absence of etomidate did not affect baseline release of cortisol (not shown). A plot showing the averaged data on the effect of etomidate on cortisol release is given in Figure 6.
Figure 6.
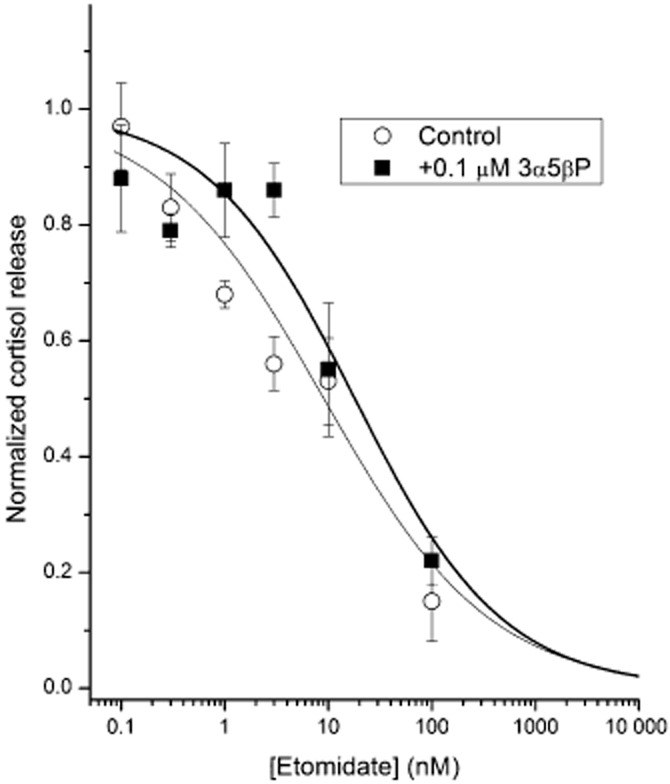
Effect of 3α5βP on etomidate-induced reduction of cortisol release. Exposure to etomidate reduces cortisol release in human adrenocortical cells (H295R cell line). The results of elisa give mean ± SEM for normalized cortisol release from three experiments at each condition. Cells were exposed to 0.1–100 nM etomidate in the absence (Control) or presence of 0.1 μM 3α5βP. The mean data were fitted with the following equation: Y([etomidate]) = [steroid]n/([steroid]n + IC50n), where IC50 is the concentration producing the half-maximal effect, and n is the Hill coefficient. The IC50 values (best fit±SD) and Hill coefficients (from three experiments under each condition) are as follows. Control: IC50 = 9 ± 8 nM, n = 0.54 ± 0.14. Etomidate + 0.1 μM 3α5βP: IC50 = 18 ± 10 nM, n = 0.61 ± 0.15. The IC50 estimates were not statistically different (t-test, P > 0.05).
Discussion and conclusions
We have shown that potentiating neurosteroids positively modulate the potency of both orthosteric and allosteric agonists as activators of recombinant α1β2γ2L GABAA receptors. Specifically, current responses to submaximal concentrations of etomidate, pentobarbital or propofol were enhanced in the presence of the potentiating steroid 3α5βP. The steroid also enhanced responses to the combination of etomidate and GABA, and shifted the concentration–response curves for etomidate to lower concentrations, both in the absence and presence of GABA. The behavioural outcome of co-application of 3α5βP with etomidate is that loss of righting in Xenopus tadpoles or mice occurs at lower doses of etomidate. The presence of the neuroactive steroid had no effect on adrenocortical steroid synthesis, indicating that lower effective doses of anaesthetics in the presence of steroid may be accompanied by reduced side effects.
Electrophysiological data indicate that a similar mechanism underlies the interaction between 3α5βP and orthosteric or allosteric activators. The α1(Q241L) mutation abolished potentiation of receptors activated by GABA (Hosie et al., 2006) or etomidate (Figure 2A). Comparison of concentration–response properties for 3α5βP demonstrates that the apparent affinity of the steroid was the same for receptors activated by low concentrations of GABA or by etomidate (Figure 2C).
It is likely that potentiation of GABAA receptors containing the α1 and γ2 subunits, and either the β2 or β3 subunit, underlies or contributes to many anaesthetic end points. Knock-in mice containing mutations that render the α1 subunit resistant to isoflurane require higher doses of isoflurane to elicit loss of righting (Sonner et al., 2007). Introduction of the α1H101R mutation, which renders the receptor insensitive to diazepam, leads to loss of sedative and amnesic actions of diazepam in mice harbouring this mutation (Rudolph et al., 1999). The β2 subunit has been implicated in the sedative actions of etomidate. In behavioural tests examining loss of pedal withdrawal reflex or spontaneous or forced locomotor activity in the presence of etomidate, mice harbouring the N265S mutation in the β2 subunit show significant changes compared with wild-type littermates (Reynolds et al., 2003). We therefore consider that the effects seen in electrophysiological data obtained on recombinant α1β2γ2L receptors underlie or at least contribute to the behavioural effects observed in tadpole and mouse assays. In future experiments, it will be important to test the sensitivity of other subtypes of the GABAA receptor, especially those contributing to tonic responses, to combinations of anaesthetics and steroids. Furthermore, targets besides the GABAA receptor may be relevant. For example, nicotinic receptors are inhibited by barbiturates at clinically relevant concentrations (Downie et al., 2000; Coates et al., 2001). It is not clear how co-application of steroids with barbiturates affects this type of modulatory effect.
A major adverse effect of etomidate is inhibition of steroid 11β-hydroxylase, leading to inhibition of synthesis of adrenocortical steroids (Wagner et al., 1984; Ayub and Levell, 1989). Chemical modifications of the etomidate structure have been made with the goal of reducing this side effect (Cotten et al., 2009; 2010,). An alternative approach is to determine conditions that enhance the effectiveness of etomidate and thereby lower the required etomidate dosage for anaesthesia. Our data demonstrate that co-application of 3α5βP lowers the dosage requirement for etomidate. The presence of 1 mg·kg−1 3α5βP shifted the etomidate-induced loss of righting curve by almost 10-fold in mice. The presence of steroid was without effect on cortisol synthesis in the human adrenocortical cell assay, indicating that the potential to reduce cortisol synthesis was not altered. This suggests that reduced etomidate dosage in the presence of steroids may be accompanied by a reduction in this off-target effect. Because steroids and analogues are known to differ greatly in potency and efficacy (Akk et al., 2007), a more significant effect on etomidate-induced anaesthesia may be achieved using other endogenous or novel synthetic steroids or analogues.
The plasma concentration of etomidate in surgical patients is 0.6 μM (Hebron et al., 1983), which is comparable with the concentration estimated from the dose producing LRR in mice (Figure 5; 0.8 μM) or the EC50 for LRR in tadpoles (Figure 4; 1.6 μM). It is perhaps fortuitous that the estimated EC50 for etomidate potentiation of responses of α1β2γ2L receptors to 0.5 μM GABA is also 2 μM (Figure 3C). However, even 100 nM 3α5βP produced a significant left-shift in the concentration–potentiation relationship for etomidate (Figure 3C). At a concentration of etomidate of 0.6 μM, the presence of 100 nM 3α5αP increased the response to 0.5 μM GABA about fivefold over the response in the presence of GABA plus etomidate alone.
The estimates for baseline brain concentration of potentiating steroids are in tens of nanomoles (Weill-Engerer et al., 2002), and our data indicate that both 3α5αP and 3α5βP can enhance responses to etomidate, leading to the possibility that several endogenous steroids may contribute in vivo. Higher (micromolar) concentrations of steroids can elicit loss of righting on their own (Akk et al., 2007). Thus, there is a window of steroid concentrations where a leftward shift in etomidate actions is observed without the steroid itself acting as an anaesthetic agent.
Functional interactions between GABAergic drugs have been observed previously. The steroid 3α5βP has been shown to enhance currents elicited by pentobarbital in bovine adrenomedullary chromaffin cells (Peters et al., 1988). Richards and White (1981) found that the steroid alphaxalone enhanced the anaesthetic effect of etomidate in rats. Synergistic interactions (loosely defined as more than additive effects of combinations of drugs) in electrophysiological assays have previously been noted for benzodiazepines and barbiturates (DeLorey et al., 1993), or benzodiazepines and propofol (Reynolds and Maitra, 1996), and benzodiazepines show synergy with propofol with respect to some anaesthetic end points (Reynolds and Maitra, 1996; Wilder-Smith et al., 2001). An additive effect of the actions of propofol and sevoflurane was found using recombinant GABAA receptors (Sebel et al., 2006). A recent critical review re-analysing previously published data found both synergy and simple additivity with respect to different end points in the actions of propofol and thiopental (Hendrickx et al., 2008). We have previously shown that the benzodiazepine diazepam potentiates peak currents from α1β2γ2L GABAA receptors elicited by pentobarbital, propofol or etomidate (Li et al., 2013). Although these studies support the idea that a variety of GABAergic agents interact at the levels of the receptor and the whole organism, further studies are needed to determine the kinetic mechanism of potentiation of etomidate-activated receptors by neuroactive steroids, and whether the effect can be considered synergistic.
In summary, the available data indicate considerable interplay among GABAergic drugs. It was surprising that the synaptic class of GABAA receptors generated a significant response to a low concentration (0.5 μM) of GABA in the combined presence of etomidate and 3α5βP. This might indicate that endogenous levels of GABA could significantly activate the synaptic population of receptors under certain conditions. Our data also demonstrate that both endogenous (3α5βP and 3α5αP) and exogenous (alphaxalone) neuroactive steroids can potentiate the action of etomidate, and at least 3α5βP potentiates the actions of a barbiturate and propofol. This then raises the possibility that endogenous potentiating steroids, including 3α5βP, can fine-tune the clinical actions of anaesthetics and that physiological changes in the levels of steroids may affect the doses of anaesthetics required to produce the desired end point. Brain levels of GABAergic potentiating steroids depend on age and gender, and can change following stress or alcohol use, or in disease states (Smith, 2001; Backstrom et al., 2003; Schumacher et al., 2003; Morrow et al., 2009). We hypothesize that all these factors can influence the induction and maintenance of general anaesthesia. An increase in the levels of progesterone (a precursor of both 3α5βP and 3α5αP) in CSF (Datta et al., 1986) may underlie the reduction in minimal anaesthetic concentration for some inhaled anaesthetics during pregnancy (Palahniuk et al., 1974).
Acknowledgments
Supported by grant P01GM47969 (to Dr Steinbach and Dr Evers) from the National Institutes of Health (Bethesda, Maryland). J. H. S. is the Russell and Mary Shelden Professor of Anethesiology.
Glossary
- 3α5αP
5α-pregnan-3α-ol-20-one
- 3α5βP
5β-pregnan-3α-ol-20-one (pregnanolone)
- LRR
loss of righting reflex
Author contributions
S. M., J. H. S., A. S. E. and G. A. conceived the project and designed the experiments. P. L., J. R. B., B. D. M. and S. M. conducted the experiments. J. H. S., A. S. E. and G. A. wrote the manuscript.
Conflict of interest
A. S. E. has served as an advisor to and has an equity interest in Sage Therapeutics, a company that is developing neurosteroids as sedative agents. Sage had no input into this work and did not provide any financial support to this work or to any of the laboratories involved in this study.
References
- Akk G, Covey DF, Evers AS, Steinbach JH, Zorumski CF, Mennerick S. Mechanisms of neurosteroid interactions with GABAA receptors. Pharmacol Ther. 2007;116:35–57. doi: 10.1016/j.pharmthera.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Li P, Bracamontes J, Reichert DE, Covey DF, Steinbach JH. Mutations of the GABAA receptor α1 subunit M1 domain reveal unexpected complexity for modulation by neuroactive steroids. Mol Pharmacol. 2008;74:614–627. doi: 10.1124/mol.108.048520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to pharmacology 2013/14: Ligand-gated ion channels. Br J Pharmacol. 2013a;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayub M, Levell MJ. Inhibition of human adrenal steroidogenic enzymes in vitro by imidazole drugs including ketoconazole. J Steroid Biochem. 1989;32:515–524. doi: 10.1016/0022-4731(89)90384-1. [DOI] [PubMed] [Google Scholar]
- Backstrom T, Andersson A, Andree L, Birzniece V, Bixo M, Bjorn I, et al. Pathogenesis in menstrual cycle-linked CNS disorders. Ann N Y Acad Sci. 2003;1007:42–53. doi: 10.1196/annals.1286.005. [DOI] [PubMed] [Google Scholar]
- Belelli D, Herd MB. The contraceptive agent Provera enhances GABAA receptor-mediated inhibitory neurotransmission in the rat hippocampus: evidence for endogenous neurosteroids? J Neurosci. 2003;23:10013–10020. doi: 10.1523/JNEUROSCI.23-31-10013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Muntoni AL, Merrywest SD, Gentet LJ, Casula A, Callachan H, et al. The in vitro and in vivo enantioselectivity of etomidate implicates the GABAA receptor in general anaesthesia. Neuropharmacology. 2003;45:57–71. doi: 10.1016/s0028-3908(03)00144-8. [DOI] [PubMed] [Google Scholar]
- Chiara DC, Jayakar SS, Zhou X, Zhang X, Savechenkov PY, Bruzik KS, et al. Specificity of intersubunit general anesthetic-binding sites in the transmembrane domain of the human α1β3γ2 γ-aminobutyric acid type A (GABAA) receptor. J Biol Chem. 2013;288:19343–19357. doi: 10.1074/jbc.M113.479725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates KM, Mather LE, Johnson R, Flood P. Thiopental is a competitive inhibitor at the human α7 nicotinic acetylcholine receptor. Anesth Analg. 2001;92:930–933. doi: 10.1097/00000539-200104000-00026. [DOI] [PubMed] [Google Scholar]
- Cotten JF, Husain SS, Forman SA, Miller KW, Kelly EW, Nguyen HH, et al. Methoxycarbonyl-etomidate: a novel rapidly metabolized and ultra-short-acting etomidate analogue that does not produce prolonged adrenocortical suppression. Anesthesiology. 2009;111:240–249. doi: 10.1097/ALN.0b013e3181ae63d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten JF, Forman SA, Laha JK, Cuny GD, Husain SS, Miller KW, et al. Carboetomidate: a pyrrole analog of etomidate designed not to suppress adrenocortical function. Anesthesiology. 2010;112:637–644. doi: 10.1097/ALN.0b013e3181cf40ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covey DF, Nathan D, Kalkbrenner M, Nilsson KR, Hu Y, Zorumski CF, et al. Enantioselectivity of pregnanolone-induced γ-aminobutyric acidA receptor modulation and anesthesia. J Pharmacol Exp Ther. 2000;293:1009–1016. [PubMed] [Google Scholar]
- Criado A, Maseda J, Navarro E, Escarpa A, Avello F. Induction of anaesthesia with etomidate: haemodynamic study of 36 patients. Br J Anaesth. 1980;52:803–806. doi: 10.1093/bja/52.8.803. [DOI] [PubMed] [Google Scholar]
- Datta S, Hurley RJ, Naulty JS, Stern P, Lambert DH, Concepcion M, et al. Plasma and cerebrospinal fluid progesterone concentrations in pregnant and nonpregnant women. Anesth Analg. 1986;65:950–954. [PubMed] [Google Scholar]
- DeLorey TM, Kissin I, Brown P, Brown GB. Barbiturate-benzodiazepine interactions at the γ-aminobutyric acidA receptor in rat cerebral cortical synaptoneurosomes. Anesth Analg. 1993;77:598–605. doi: 10.1213/00000539-199309000-00030. [DOI] [PubMed] [Google Scholar]
- Dorr HG, Kuhnle U, Holthausen H, Bidlingmaier F, Knorr D. Etomidate: a selective adrenocortical 11β-hydroxylase inhibitor. Klin Wochenschr. 1984;62:1011–1013. doi: 10.1007/BF01711722. [DOI] [PubMed] [Google Scholar]
- Downie DL, Franks NP, Lieb WR. Effects of thiopental and its optical isomers on nicotinic acetylcholine receptors. Anesthesiology. 2000;93:774–783. doi: 10.1097/00000542-200009000-00027. [DOI] [PubMed] [Google Scholar]
- Eckstein JA, Ammerman GM, Reveles JM, Ackermann BL. Analysis of glutamine, glutamate, pyroglutamate, and GABA in cerebrospinal fluid using ion pairing HPLC with positive electrospray LC/MS/MS. J Neurosci Methods. 2008;171:190–196. doi: 10.1016/j.jneumeth.2008.02.019. [DOI] [PubMed] [Google Scholar]
- Franks NP. Molecular targets underlying general anaesthesia. Br J Pharmacol. 2006;147(Suppl. 1):S72–S81. doi: 10.1038/sj.bjp.0706441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–386. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- Halsey MJ, Wardley-Smith B, Wood S. Pressure reversal of alphaxalone/alphadolone and methohexitone in tadpoles: evidence for different molecular sites for general anesthesia. Br J Pharmacol. 1986;89:299–305. doi: 10.1111/j.1476-5381.1986.tb10260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebron BS, Edbrooke DL, Newby DM, Mather SJ. Pharmacokinetics of etomidate associated with prolonged i.v. infusion. Br J Anaesth. 1983;55:281–287. doi: 10.1093/bja/55.4.281. [DOI] [PubMed] [Google Scholar]
- Hendrickx JF, Eger EI, 2nd, Sonner JM, Shafer SL. Is synergy the rule? A review of anesthetic interactions producing hypnosis and immobility. Anesth Analg. 2008;107:494–506. doi: 10.1213/ane.0b013e31817b859e. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HM, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444:486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, Smart TG. Neurosteroid binding sites on GABAA receptors. Pharmacol Ther. 2007;116:7–19. doi: 10.1016/j.pharmthera.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Hunter BR, Kirschner J. In patients with severe sepsis, does a single dose of etomidate to facilitate intubation increase mortality? Ann Emerg Med. 2013;61:571–572. doi: 10.1016/j.annemergmed.2012.12.024. [DOI] [PubMed] [Google Scholar]
- de Jong FH, Mallios C, Jansen C, Scheck PA, Lamberts SW. Etomidate suppresses adrenocortical function by inhibition of 11β-hydroxylation. J Clin Endocrinol Metab. 1984;59:1143–1147. doi: 10.1210/jcem-59-6-1143. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci. 2006;26:11599–11605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Eaton MM, Steinbach JH, Akk G. The benzodiazepine diazepam potentiates tresponses of α1β2γ2L γ-aminobutyric acid Type A receptors activated by either γ-aminobutyric acid or allosteric agonists. Anesthesiology. 2013;118:1417–1425. doi: 10.1097/ALN.0b013e318289bcd3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS, Smart TG. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci. 2010;31:161–174. doi: 10.1016/j.tips.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Morrow AL, Biggio G, Serra M, Becker HC, Lopez MF, Porcu P, et al. The role of neuroactive steroids in ethanol/stress interactions: proceedings of symposium VII at the Volterra conference on alcohol and stress. Alcohol. 2009;43:521–530. doi: 10.1016/j.alcohol.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palahniuk RJ, Shnider SM, Eger EI., 2nd Pregnancy decreases the requirement for inhaled anesthetic agents. Anesthesiology. 1974;41:82–83. doi: 10.1097/00000542-197407000-00021. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JA, Kirkness EF, Callachan H, Lambert JJ, Turner AJ. Modulation of the GABAA receptor by depressant barbiturates and pregnane steroids. Br J Pharmacol. 1988;94:1257–1269. doi: 10.1111/j.1476-5381.1988.tb11646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray DC, McKeown DW. Etomidate for critically ill patients. Pro: yes we can use it. Eur J Anaesthesiol. 2012;29:506–510. doi: 10.1097/EJA.0b013e32835819b0. [DOI] [PubMed] [Google Scholar]
- Reynolds DS, Rosahl TW, Cirone J, O'Meara GF, Haythornthwaite A, Newman RJ, et al. Sedation and anesthesia mediated by distinct GABAA receptor isoforms. J Neurosci. 2003;23:8608–8617. doi: 10.1523/JNEUROSCI.23-24-08608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JN, Maitra R. Propofol and flurazepam act synergistically to potentiate GABAA receptor activation in human recombinant receptors. Eur J Pharmacol. 1996;314:151–156. doi: 10.1016/s0014-2999(96)00527-4. [DOI] [PubMed] [Google Scholar]
- Richards CD, White AE. Additive and non-additive effects of mixtures of short-acting intravenous anaesthetic agents and their significance for theories of anaesthesia. Br J Pharmacol. 1981;74:161–170. doi: 10.1111/j.1476-5381.1981.tb09969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Benke D, Brunig I, Benson JA, Fritschy JM, et al. Benzodiazepine actions mediated by specific γ-aminobutyric acidA receptor subtypes. Nature. 1999;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- Schumacher M, Weill-Engerer S, Liere P, Robert F, Franklin RJ, Garcia-Segura LM, et al. Steroid hormones and neurosteroids in normal and pathological aging of the nervous system. Prog Neurobiol. 2003;71:3–29. doi: 10.1016/j.pneurobio.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Sebel LE, Richardson JE, Singh SP, Bell SV, Jenkins A. Additive effects of sevoflurane and propofol on γ-aminobutyric acid receptor function. Anesthesiology. 2006;104:1176–1183. doi: 10.1097/00000542-200606000-00012. [DOI] [PubMed] [Google Scholar]
- Smith SS. Pre-menstrual steroids. Cell Mol Life Sci. 2001;58:1263–1275. doi: 10.1007/PL00000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt K, Forman SA. Correlating the clinical actions and molecular mechanisms of general anesthetics. Curr Opin Anaesthesiol. 2007;20:300–306. doi: 10.1097/ACO.0b013e32816678a5. [DOI] [PubMed] [Google Scholar]
- Sonner JM, Werner DF, Elsen FP, Xing Y, Liao M, Harris RA, et al. Effect of isoflurane and other potent inhaled anesthetics on minimum alveolar concentration, learning, and the righting reflex in mice engineered to express α1 γ-aminobutyric acid type A receptors unresponsive to isoflurane. Anesthesiology. 2007;106:107–113. doi: 10.1097/00000542-200701000-00019. [DOI] [PubMed] [Google Scholar]
- Stastna E, Krishnan K, Manion BD, Taylor A, Rath NP, Chen ZW, et al. Neurosteroid analogues. 16. A new explanation for the lack of anesthetic effects of Δ(16)-alphaxalone and identification of a Δ(17(20)) analogue with potent anesthetic activity. J Med Chem. 2011;54:3926–3934. doi: 10.1021/jm2002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart D, Desai R, Cheng Q, Liu A, Forman SA. Tryptophan mutations at azi-etomidate photo-incorporation sites on α1 or β2 subunits enhance GABAA receptor gating and reduce etomidate modulation. Mol Pharmacol. 2008;74:1687–1695. doi: 10.1124/mol.108.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Zorumski C, Bracamontes J, Steinbach JH. Endogenous subunits can cause ambiguities in the pharmacology of exogenous γ-aminobutyric acidA receptors expressed in human embryonic kidney 293 cells. Mol Pharmacol. 1996;50:931–938. [PubMed] [Google Scholar]
- Wagner RL, White PF, Kan PB, Rosenthal MH, Feldman D. Inhibition of adrenal steroidogenesis by the anesthetic etomidate. N Engl J Med. 1984;310:1415–1421. doi: 10.1056/NEJM198405313102202. [DOI] [PubMed] [Google Scholar]
- Weill-Engerer S, David JP, Sazdovitch V, Liere P, Eychenne B, Pianos A, et al. Neurosteroid quantification in human brain regions: comparison between Alzheimer's and nondemented patients. J Clin Endocrinol Metab. 2002;87:5138–5143. doi: 10.1210/jc.2002-020878. [DOI] [PubMed] [Google Scholar]
- Wilder-Smith OH, Ravussin PA, Decosterd LA, Despland PA, Bissonnette B. Midazolam premedication reduces propofol dose requirements for multiple anesthetic endpoints. Can J Anaesth. 2001;48:439–445. doi: 10.1007/BF03028305. [DOI] [PubMed] [Google Scholar]
- Yip GM, Chen ZW, Edge CJ, Smith EH, Dickinson R, Hohenester E, et al. A propofol binding site on mammalian GABAA receptors identified by photolabeling. Nat Chem Biol. 2013;9:715–720. doi: 10.1038/nchembio.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]