

Abstract
Malaria remains a global public health concern and current treatment options are suboptimal in some clinical settings. For effective chemotherapy, antimalarial drug concentrations must be sufficient to remove completely all of the parasites in the infected host. Optimized dosing therefore requires a detailed understanding of the time course of antimalarial response, whilst simultaneously considering the parasite life cycle and host immune elimination. Recently, the World Health Organization (WHO) has recommended the development of mathematical models for understanding better antimalarial drug resistance and management. Other international groups have also suggested that mechanistic pharmacokinetic (PK) and pharmacodynamic (PD) models can support the rationalization of antimalarial dosing strategies. At present, artemisinin-based combination therapy (ACT) is recommended as first line treatment of falciparum malaria for all patient groups. This review summarizes the PK–PD characterization of artemisinin derivatives and other partner drugs from both preclinical studies and human clinical trials. We outline the continuous and discrete time models that have been proposed to describe antimalarial activity on specific stages of the parasite life cycle. The translation of PK–PD predictions from animals to humans is considered, because preclinical studies can provide rich data for detailed mechanism-based modelling. While similar sampling techniques are limited in clinical studies, PK–PD models can be used to optimize the design of experiments to improve estimation of the parameters of interest. Ultimately, we propose that fully developed mechanistic models can simulate and rationalize ACT or other treatment strategies in antimalarial chemotherapy.
Keywords: antimalarial chemotherapy, malaria, mechanism based model, pharmacodynamic, pharmacokinetics
Introduction
Malaria has an annual incidence of approximately 220 million cases and remains a major health concern in countries where the disease is endemic [1]. However, despite recent advances in antimalarial chemotherapy [2–4], current dosing regimens are generally empirical and suboptimal in some clinical settings [4–6]. For effective treatment, antimalarials must deliver sufficient drug concentrations over time to eradicate all of the parasites in the infected host. An understanding of drug pharmacokinetics (PK) and pharmacodynamics (PD) is therefore an essential step towards the development of optimized dosing guidelines in malaria therapy.
Mathematical models can provide a quantitative description of the dose–concentration (PK) and concentration–effect (PD) relationship [7–9]. Population PK–PD analysis is particularly useful because these models incorporate between-patient variability in the time course of drug response and can therefore assist with the dose individualization of medicines [10]. This approach has been used to improve the attainment of therapeutic endpoints in patient subpopulations with infectious disease [11]. Recently, the World Health Organization (WHO) has recommended the development of mathematical models for better understanding of antimalarial drug resistance and management [1]. Other international groups have also suggested that mechanistic PK–PD models can support the rationalization of antimalarial dosing strategies [4,6]. These models must relate antimalarial drug concentrations to the parasite–time profile, as well as consider the parasite life cycle and host immune response [12,13]. Incorporation of the parasite life cycle in the red blood cell (ring, trophozoite and schizont) is important, due to differences in the stage-specific actions of antimalarial agents [14]. Once developed, malaria-specific PK–PD models can simulate and rationalize the design of effective mono- or combination therapy in humans [15]. A further benefit of population PK–PD analysis is the ability to identify the sources of variability in pregnant women and infants, who are most susceptible to malaria infection [1].
In this review, we present the development of antimalarial PK–PD in preclinical evaluation, and then describe the approaches used for the mathematical modelling of antimalarial response in human clinical settings. Finally, we outline the translation of preclinical dose predictions to humans and discuss the experimental challenges for PK–PD modelling of antimalarial drugs.
Models in preclinical evaluation
In vitro techniques for the investigation of antimalarial drugs have been refined since the pioneering research to establish methods for the continuous culture of Plasmodium falciparum [16]. These studies were used to quantify efficacy according to the drug concentration causing 50% inhibition of parasites (IC50), determined by fitting a sigmoid equation to the concentration–response curve [17]. Culture-adapted P. falciparum (0.2–2% parasitaemia) is commonly used in drug discovery and the matrix typically comprises human erythrocytes (2–5% haematocrit) suspended in a buffered tissue culture medium, supplemented with serum/albumin and incubated at 37°C [18–21]. However, as the in vitro culture milieu is markedly different from the physiological environment in a malaria-infected human host, not least in relation to immune responses, these studies have limited application in clinically relevant PD models [18,20,22]. Nevertheless, in vitro isobologram analyses [19,23,24] have been used to demonstrate potential in vivo outcomes for artemisinin-based combination therapy (ACT) and other antimalarial drug combinations [1,25–29]. Isobolograms identify whether fractional inhibitory concentrations of two drugs are antagonistic, additive or synergistic and have become a screening tool for potentially successful drug combinations [19,29–31]. A recent illustration of the value of isobologram analyses is the translation of in vitro and murine studies to a simian model confirming that mefloquine was the best partner drug for artemisone [29,32].
Animal models of malaria infection have the potential to provide rich PK and PD data for sophisticated modelling of single- or multiple-dose regimens of mono- or combination therapy. Murine malaria models using P. berghei are well established for studies of disease pathology and drug efficacy, because parasite morphology and development are comparable with human malaria infections [33–35]. However, as there are physiological differences between rodents and humans, limitations in regard to disease pathogenesis, immunity and the use of murine-specific parasite species should be recognized [36,37]. The latter includes variations in the effects of artemisinin drugs against P. berghei, relative to that in other human malaria infections. Nonetheless, murine models are particularly useful because all erythrocytic stages of the P. berghei parasite life cycle are observable in thin blood films, and can be differentiated by light microscopy [33,34].
The two most widely used methods of evaluating murine antimalarial efficacy are the ‘Peters 4 day test’ and modified versions of the ‘Thompson’ and ‘Rane’ tests, although others have been reviewed previously [18,34,35,37,38]. The Peters 4 day test is a multiple dose test of malaria suppression, with parasite inoculation on the first day and concurrent drug treatment given as four doses on consecutive days. Data are analyzed by determining the dose at which 50% (ED50) or 90% (ED90) suppression of parasitaemia occurs, compared with untreated control mice, thus providing a simple measure of efficacy [37,39]. The Rane test comprises escalating single-dose administration to mice with a patent malaria infection, typically established 2–4 days after parasite inoculation. Drug efficacy, expressed as the minimum effective dose (MED), is generally based on the survival of treated mice [18,35,37]. The Thompson test monitors the progress of infection by light microscopy examination of daily blood films, to determine the rate, extent and duration of effect following single-dose administration [18,40–44]. Contemporary adaptations of the Rane and Thompson tests may be regarded as ‘onset/recrudescence’ or ‘treatment’ investigations of antimalarial efficacy. A modified Thompson method compares the efficacy of a fixed total dose administered via different regimens [43,44] and is a plausible approach for investigating ACT regimens [45]. A variation of this method comprises a range of doses to mice over a 3 day period, commencing 3 days after inoculation [46–48]. Well-designed studies based on this method, with rich PK and PD data, could be used to develop mechanism based PK–PD models of individual and combination drug regimens. While previous studies have generally used destructive measures of sampling (several mice per time point for different time points), small volume collection of multiple samples is also plausible in mice and rats [49,50]. Nonetheless, a useful advantage of population PK modelling is the ability to analyze both sparse and rich data of drug concentration.
Animal models have been used in lead optimization [39,51] or antimalarial activity studies in rodents [52–55] and monkeys [32,56]. Batty et al. have detailed the murine parasite time course after dihydroartemisinin [57,58], chloroquine [45] or piperaquine treatment [45,59,60], and have demonstrated enhanced magnitude and duration of antimalarial killing following combination therapy or multiple dosing. The disposition of dihydroartemisin in mice has been described by a one compartment model, with a short elimination half-life of ∼0.3 h [57,61]. A substantial increase in this elimination half-life (to ∼30 h) has been reported for the emerging ozonide OZ439 in murine PK studies [52]. For the quinoline compounds, two [45,59] and three compartment models [62] have characterized their disposition, with longer elimination half-lives of ∼30–300 h for piperaquine and ∼99 h for chloroquine.
Despite the potential for detailed PK–PD modelling in animal malaria models, research in this area is limited. A discrete time model has been constructed, with the 24 h erythrocytic cycle of P. berghei divided into 24 compartments, each representing 1 h of parasite growth [57]. This model accounted for the progression of P. berghei through the asexual life cycle, parasite multiplication and elimination rates after dihydroartemisinin dosing. Recently, Patel et al. [61] developed a more elaborate mechanism-based model that simultaneously described all erythrocytic stages of P. berghei growth, immune elimination and the parasiticidal effect of dihydroartemisinin. This model also incorporated the delay between antimalarial concentration and onset of parasite killing, therefore demonstrating the potential for translating the results from preclinical evaluation to human clinical trials.
Models in human trials
The WHO currently recommends ACTs as first line treatment of falciparum malaria for all patient groups [25]. ACT involves the combination of artemisinin derivatives (artesunate, artemether and dihydroartemisinin) with another antimalarial such as mefloquine, amodiaquine, lumefantrine and piperaquine. While the artemisinins are rapidly eliminated (elimination half-life ∼1 h) [63–65], compared with other available antimalarials they produce a significantly greater reduction in the number of parasites in the first 24 to 48 h following treatment [64,66,67]. The partner drugs, co-administered with the artemisinin derivatives generally have a slower onset but longer duration of action. These drugs are important for eliminating the residual biomass and attenuating the recrudescence associated with short term artemisinin monotherapy [66,68].
The population PK of dihydroartemisinin, which is the active metabolite of artesunate and artemether, has been characterized by one compartment models [69–74]. For the partner drugs, PK profiles have been described by one [75–78] and two compartment models [79–81] for mefloquine and lumefantrine, two [82,83] and three compartment models [74,84,85] for piperaquine and one [86] and two compartment models [87] for both sulphadoxine and pyrimethamine. In addition, two compartment models have characterized the disposition of amodiaquine and/or its active metabolite desethylamodiaquine [72,88,89]. A parent-metabolite model has also been developed with a two compartment model for amodiaquine followed by a three compartment disposition of desethylamodiaquine [90]. Figure 1 presents the population mean PK profiles for each of the antimalarials, dihydroartemisinin, mefloquine, lumefantrine and piperaquine, where large apparent volumes of distribution are observed for lumefantrine and piperaquine. A summary of mean population PK estimates is provided in Table 1.
Figure 1.
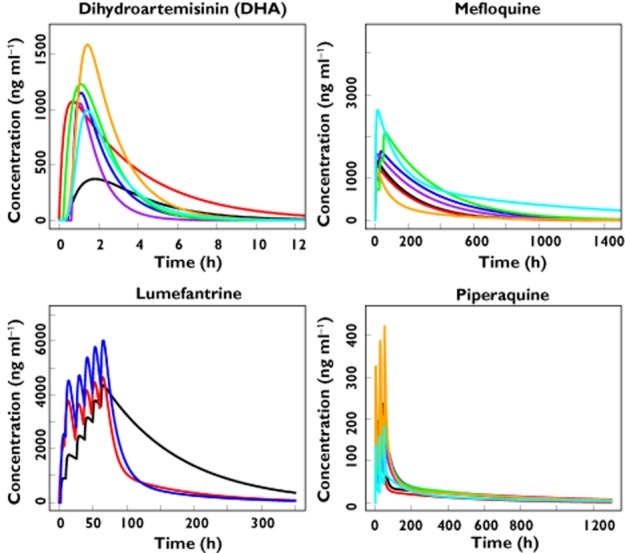
Population mean pharmacokinetic profiles reported in the literature for dihydroartemisinin [69–72,74] >(  , McGready et al. [70], pregnant women;
, McGready et al. [70], pregnant women;  , Stepniewska et al. [72], children;
, Stepniewska et al. [72], children;  , Morris et al. [71], non-pregnant women;
, Morris et al. [71], non-pregnant women;  , Morris et al. [71], pregnant women;
, Morris et al. [71], pregnant women;  , Jamsen et al. [69], non-pregnant adults;
, Jamsen et al. [69], non-pregnant adults;  , Tarning et al. [74], non-pregnant women;
, Tarning et al. [74], non-pregnant women;  , Tarning et al. [74], pregnant women); mefloquine [75,77,80,108] (
, Tarning et al. [74], pregnant women); mefloquine [75,77,80,108] ( , Nabangchang et al. [108], non-pregnant women;
, Nabangchang et al. [108], non-pregnant women;  , Nabangchang et al. [108], pregnant women;
, Nabangchang et al. [108], pregnant women;  , Simpson et al. [77], split dose;
, Simpson et al. [77], split dose;  , Simpson et al. [77], single dose;
, Simpson et al. [77], single dose;  , Ashley et al. [75], non-pregnant adults;
, Ashley et al. [75], non-pregnant adults;  , Svensson et al. [80], non-pregnant adults RS;
, Svensson et al. [80], non-pregnant adults RS;  , Svensson et al. [80], non-pregnant adults SR), lumefantrine [76,79,81] (
, Svensson et al. [80], non-pregnant adults SR), lumefantrine [76,79,81] (  , Hietala et al. [76], children;
, Hietala et al. [76], children;  , Ezzet et al. [79], non-pregnant adults;
, Ezzet et al. [79], non-pregnant adults;  , Tarning et al. [81], pregnant women) and piperaquine [74,82–85]. (
, Tarning et al. [81], pregnant women) and piperaquine [74,82–85]. ( , Hung et al. [82], non-pregnant adults;
, Hung et al. [82], non-pregnant adults;  , Hung et al. [82], children;
, Hung et al. [82], children;  , Tarning et al. [83], non-pregnant adults;
, Tarning et al. [83], non-pregnant adults;  , Tarning et al. [85], non-pregnant women;
, Tarning et al. [85], non-pregnant women;  , Tarning et al. [85], pregnant women;
, Tarning et al. [85], pregnant women;  , Tarning et al. [85], children;
, Tarning et al. [85], children;  , Hoglund et al. [84], pregnant and non-pregnant women)
, Hoglund et al. [84], pregnant and non-pregnant women)
Table 1.
Population mean pharmacokinetic parameters for antimalarial drugs in human clinical trials
| Antimalarial | Gender | Pregnant | Malaria | Model | ka | tlag | CL/F | V1/F | Q/F | V2/F | Q2/F | V3/F | Reference. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (M/F) | (Y/N) | (falci/viv) | (1 h−1) | (h) | (l h−1) | (l) | (l h−1) | (l) | (l h−1) | (l) | |||
| Pharmacokinetics in adults | |||||||||||||
| Dihydroartemisinin | M/F | N | falci | 1-comp. | 0.820 | 0.210 | 47.5 | 32.1 | – | – | – | – | [66] |
| F | Y | falci | 1-comp. | 1.19 | 0.420 | 88.5* | 232* | – | – | – | – | [67] | |
| F | Y | falci | 1-comp. | 4.59 | 0.627 | 91.6 | 91.4 | – | – | – | – | [68] | |
| F | N | falci | 1-comp. | 4.59 | 0.627 | 64.0 | 91.4 | – | – | – | – | [68] | |
| F | Y | falci | 1-comp. | 8.15† | – | 78.0 | 129 | – | – | – | – | [71] | |
| F | N | falci | 1-comp. | 8.15† | – | 78.0 | 129 | – | – | – | – | [71] | |
| Mefloquine | M/F | N | falci | 1-comp. | 0.290 | – | 1.40* | 453* | – | – | – | – | [72] |
| (RS enantiomer) | M/F | N | falci | 2-comp. | 0.198 | – | 3.51 | 559 | 2.54 | 395 | – | – | [77] |
| (SR enantiomer) | M/F | N | falci | 2-comp. | 0.255 | – | 0.602 | 261 | 0.942 | 287 | – | – | [77] |
| F | N | falci | 1-comp. | – | – | 1.87* | 440* | – | – | – | – | [106] | |
| F | Y | falci | 1-comp. | – | – | 2.38* | 549* | – | – | – | – | [106] | |
| Lumefantrine | M/F | N | falci | 2-comp. | 0.170 | – | 7.04 | 103 | 4.08 | 272 | – | – | [76] |
| F | Y | falci | 2-comp. | 0.0588 | 1.67 | 6.11 | 20.2 | 1.82 | 160 | – | – | [78] | |
| Piperaquine | F | N | falci | 3-comp. | 2.88† | – | 60.2 | 3070 | 427 | 4440 | 160 | 31400 | [71] |
| F | Y | falci | 3-comp. | 2.88† | – | 87.3 | 3070 | 427 | 4440 | 160 | 31400 | [71] | |
| M/F | N | falci/viv | 2-comp. | 0.083 | 0.490 | 42.3* | 682* | 129* | 23122* | – | – | [79] | |
| M/F | N | falci | 2-comp. | 0.717 | – | 66.0 | 8660 | 131 | 24000 | – | – | [80] | |
| F | Y/N | falci | 3-comp. | 2.35† | – | 44.6 | 1820 | 47.7 | 15900 | 352 | 7520 | [81] | |
| Pyrimethamine | F | Y | falci | 2-comp. | 1.87 | – | 1.04 | 174 | 0.439 | 184 | – | – | [84] |
| F | N | falci | 2-comp. | 1.69 | – | 0.707 | 108 | 0.402 | 67.4 | – | – | [84] | |
| Sulphadoxine | F | Y | falci | 2-comp. | 0.475 | – | 0.0690 | 12.3 | 0.0041 | 0.860 | – | – | [84] |
| F | N | falci | 2-comp. | 0.754 | – | 0.0470 | 10.6 | 0.0039 | 0.810 | – | – | [84] | |
| Amodiaquine† | F | Y | viv | 2-comp. | 0.515 | 0.395 | 2530 | 4850 | 2750 | 29000 | – | – | [87] |
| Desethylamodiaquine† | F | Y | viv | 3-comp. | – | – | 34.3 | 197 | 161 | 2670 | 26.0 | 5700 | [87] |
| Pharmacokinetics in children | |||||||||||||
| Dihydroartemisinin | M/F | N | falci | 1-comp. | 4.27 | – | 8.27* | 29.7* | – | – | – | – | [69] |
| Lumefantrine | M/F | N | falci | 1-comp. | 0.820 | 1.92 | 1.08* | 125* | – | – | – | – | [73] |
| Mefloquine | M/F | N | falci | 1-comp. | 0.290 | – | 1.28* | 375* | – | – | – | – | [74] |
| Mefloquine (split dose) | M/F | N | falci | 1-comp. | 0.290 | – | 1.03* | 350* | – | – | – | – | [74] |
| Piperaquine | M/F | N | falci/viv | 2-comp. | 0.075 | – | 29.6* | 315* | 104* | 10507* | – | – | [79] |
| M/F | N | falci | 3-comp. | 1.4† | – | 7.50 | 247 | 13.1 | 254 | 10.8 | 3340 | [82] | |
| Pyrimethamine | M/F | N | falci | 1-comp. | 1.48 | – | 0.258 | 46.0 | – | – | – | – | [83] |
| Sulphadoxine | M/F | N | falci | 1-comp. | 0.630 | – | 0.0151 | 3.09 | – | – | – | – | [83] |
| Amodiaquine‡ | M/F | N | falci | 2-comp. | 0.13 | – | 224* | 187* | 272* | 4976* | – | – | [85] |
| Desethylamodiaquine‡ | M/F | N | falci | 2-comp. | – | – | 10.5* | 205* | 20.8* | 998* | – | – | [85] |
Falci is uncomplicated falciparum malaria, viv is uncomplicated vivax malaria, comp. compartment; ka is the rate constant for first order absorption, tlag is the lag time in oral absorption, CL/F is the apparent central compartment clearance, V1/F is the apparent central volume of distribution, Q/F is the apparent inter-compartmental clearance from the shallow peripheral compartment, V2/F is the apparent shallow peripheral volume of distribution, Q2/F is the apparent inter-compartmental clearance from the deep peripheral compartment, V3/F is the apparent deep peripheral volume of distribution.
Calculated for mean or median bodyweight of patient population in the study.
Models in which the kinetics of drug absorption are described by a series of transit compartments. The number of transit compartments was seven for dihydroartemisinin and five for piperaquine ([71]), three for piperaquine ([81]) and two for piperaquine ([82]).
Describes parent (amodiaquine) and metabolite (desethylamodiaquine) models.
The change in parasite count (within the human host) in the presence of drug treatments results from an interplay between parasite growth and the rate at which the parasites are killed by the antimalarials. Early literature described the parasite time course in a simplistic manner using a single ordinary differential equation (ODE; see Equation 1 below), in which the antimalarial killing rate is assumed to be related to the drug concentration via a sigmoid Emax model [91–93].
| (1) |
where P(t) represents total parasite burden at time t, a is the parasite growth rate constant and kdrug is the killing rate constant of the drug. Equation 1 assumes that the drug only kills the parasite during the log-growth phase and does not consider elimination because of either natural or acquired immunity of the human host.
Others extended the single ODE to characterize the life cycle of P. falciparum, and consequently account for the stage specific antimalarial activity (e.g. dihydroartemisinin is active against all erythrocytic stages [14,94]). Initially, a model for severe falciparum malaria was developed with two ODEs to represent the circulating and sequestering parasites [95]. This was followed by a model for uncomplicated malaria with four ODEs, each representing 12 h of the 48 h life cycle [80]. Extensions of the model included another ODE (i.e. an additional compartment) for damaged circulating parasites that are removed by the spleen [76,96].
In addition to the continuous time PK–PD models above, discrete time models to include the stage specific activity of the drug have been proposed [13,97,98]. For these models the life cycle is divided into intervals and expressed by the following equations:
where Pi(a,t) represents the expected number of parasites in patient i aged a hours at time t and depends on the number at the previous time point and age interval, as well as the killing effect of drug (defined as function S above). Parasite multiplication factor (PMF) represents the number of merozoites per schizont that successfully invade red blood cells, that is, the measure of parasite growth at the end of each 48 h cycle. Figure 2 presents simulated mean PK and PD profiles based on the discrete time model in Saralamba et al. [97] for falciparum malaria patients receiving artesunate-mefloquine, dihydroartemisinin-piperaquine or artemether-lumefantrine.
Figure 2.
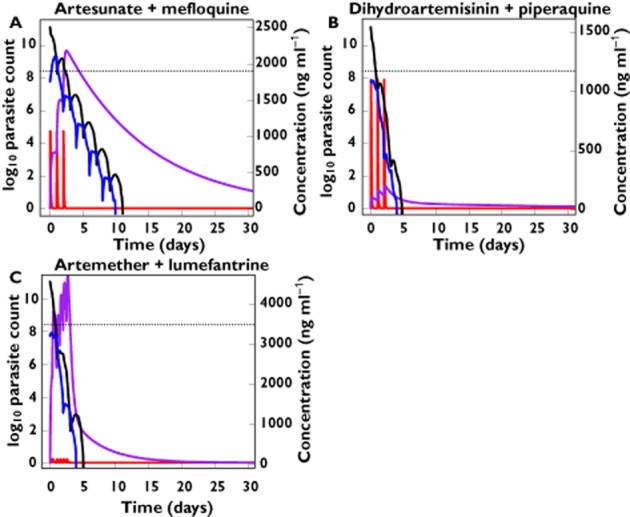
Predicted total number of circulating and sequestered parasites (log10 units – left y-axis) over time (solid black and blue lines, respectively) and concentration (ng ml−1 units – right y-axis vs. time profiles (solid red and purple lines) for the artemisinin-based combination therapies: artesunate-mefloquine (A), dihydroartemisinin-piperaquine (B) and artemether-lumefantrine (C). Dashed lines indicate the limit of parasite detection. Predicted profiles are based on the mean PK and PD parameter values presented in Zaloumis S et al. [22]. A)  , log10 circulating parasite count;
, log10 circulating parasite count;  , log10 sequestered parasite count;
, log10 sequestered parasite count;  , DHA;
, DHA;  , MQ;
, MQ;  , limit of detection. B)
, limit of detection. B)  , log10 circulating parasite count;
, log10 circulating parasite count;  , log10 sequestered parasite count;
, log10 sequestered parasite count;  , DHA;
, DHA;  , PQ
, PQ  , limit of detection. C)
, limit of detection. C)  , log10 circulating parasite count;
, log10 circulating parasite count;  , log10 sequestered parasite count;
, log10 sequestered parasite count;  , ART;
, ART;  , LF
, LF  , limit of detection
, limit of detection
Population PK–PD modelling of current antimalarials has contributed to clinical dosing strategies. Findings from Simpson et al. [93], together with population PK modelling comparing split and single-dose mefloquine [77], led to the adoption of split dosing, currently administered as 8.3 mg kg−1 day–1 over 3 days with artesunate. The absorption of lumefantrine was observed to increase if co-administered with fat [99] and is recommended to be taken immediately after food. More recently, higher doses of piperaquine have been suggested for children [85], but this recommendation has not yet translated to changes in the WHO treatment policy guidelines (Joel Tarning, personal communication).
Current WHO treatment guidelines for ACTs recommend a ‘one dosing regimen fits all’ approach, with adjustment of the dose based only on bodyweight. In the field, outpatient clinics are often in remote locations and personalized dosing is not possible. However, it would be feasible to recommend different dosing regimens for patient groups, such as infants, children, pregnant women and patients with other co-morbidities (e.g. HIV infection). This identification of important covariate effects is an area that can benefit significantly from population PK–PD modelling. Recent reports from western Cambodia and Thailand [28,100,101] have shown reduced susceptibility to artesunate (the most widely used artemisinin derivative), and another study observed that high doses of artesunate (oral dose of 6 mg kg−1) were associated with an increased risk of neutropenia [102]. In the context of emerging resistance and dose-dependent toxicity of artesunate, it is critical that the recommended dose and frequency of administration are carefully assessed and optimized according to their PK–PD. An understanding of the relationship between antimalarial concentration and therapeutic response was identified as a high priority research area by The Malaria Eradication Research Agenda (malERA) Consultative group on Drugs [4].
Translating animal models to humans
Application of physiological principles is required to translate PK–PD model predictions from animal models to humans. This is potentially beneficial because rich sampling and stage specific parasitaemia data can be obtained from preclinical studies and can assist with mechanism-based modelling [57,59,61]. The scaling of antimalarial drug concentrations is achievable by considering established relationships that account for body size or maturation [103], while covariate effects can differentiate categorical differences between groups (e.g. pregnant vs. non-pregnant women). For the parasite–time profile, the main difference is the time to complete asexual schizogony, which takes 18–24 h for P. berghei in rodents [34,37,104] and approximately 48 h for P. falciparum in humans [16,18,25,66]. While the translation of findings from animal studies to humans is currently polarized in the malaria community, these issues relate primarily to murine models of cerebral malaria. However, there is a long history of animal studies, especially in regard to antimalarial drug investigations [36,37].
Challenges in PK–PD analysis
In preclinical evaluation, the Peters 4 day test is well established for the purpose of screening and evaluating antimalarial drugs. However, data generated by this technique are not suitable for PK–PD modelling and it is unlikely that any adaptations of the test could be utilized in such a manner. The relatively crude data from total parasite density of daily blood films is an impediment to development of PK–PD models, for which rich sampling and stage-specific parasitaemia is required.
In clinical studies, collection of blood samples by venepuncture or finger prick for the measurement of antimalarial concentrations and parasite burden is limited because of ethical and logistic constraints. Therefore, sampling designs of population PK–PD studies must be optimized and evaluated to ensure precise estimation of the PK and PD parameters of interest are achieved [69,105–107]. Further, the parasite density estimated by microscopy in humans with falciparum malaria only represents the number of parasites circulating in the blood (parasites aged ∼1–26 h) and not the total burden, and the limit of detection of the circulating parasites is approximately 5 × 108 parasites. Thus, commonly, parasites are only detected for patients in the first 48 h post treatment (see Figure 2 for simulated parasite vs. time profiles for circulating and sequestered parasites). It is also currently unknown as to how the minimum inhibitory concentration (MIC) in vivo relates to in vitro measures generated from parasite isolates. In this regard, defining this in vivo to in vitro relationship could be considered as a primary objective in phase II clinical trials, and will improve the inputs of population PK–PD models [66].
Conclusions
Effective eradication of malaria requires optimized dosing regimens that consider the parasite life cycle, host immune elimination and the time course of drug parasiticidal effects. Each of these complex processes can be simultaneously incorporated in mechanistic PK–PD models that describe the relationship between antimalarial drug concentrations and parasite killing. In this review, we have summarized the development of antimalarial PK and PK–PD models from preclinical and human clinical studies. However, these models have predominantly been used to characterize the data, despite the ability for simulating effective combination treatment strategies. We propose that animal models can provide detailed data that allow for the description of drug PK–PD against each of the intra-erythrocytic stages of parasite growth. Once appropriately translated, these predictions can then assist with the identification of useful drug combinations in clinical development. Alternatively, phase II determinations of MIC in vivo could be calibrated against in vitro measures of the concentration–effect relationship to predict dosing strategies that ensure cure. Established PK–PD models can then explore covariate effects during phase III clinical trials or post-registration, to allow for the individual adjustment of mono or combination therapy dosing regimens. In addition, PK–PD models can optimize experimentation, by facilitating the design of studies that maximize the information gained whilst reducing the burden of financial and ethical constraints. This is relevant to developing countries where malaria infection is highly prevalent. Overall, proficient application of mechanistic PK–PD models will improve the development of ACT and other strategies for antimalarial chemotherapy.
Competing Interests
KP, CK and KB have no conflicts of interests related to this submitted work. JAS and SZ received funding for a separate project from the Medicines for Malaria Venture in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
SZ was supported by the Victorian Centre for Biostatistics (ViCBiostat) which is funded by the National Health and Medical Research Centre of Australia (NHMRC) Centre of Research Excellence 1035261, and by NHMRC Project Grant 1025319.
References
- 1.World Health Organization. Global Malaria Programme: World Malaria Report. Geneva, Switzerland: World Health Organization; 2012. [Google Scholar]
- 2.Wells TN, Alonso PL, Gutteridge WE. New medicines to improve control and contribute to the eradication of malaria. Nat Rev Drug Discov. 2009;8:879–891. doi: 10.1038/nrd2972. [DOI] [PubMed] [Google Scholar]
- 3.Baird JK. Effectiveness of antimalarial drugs. N Engl J Med. 2005;352:1565–1577. doi: 10.1056/NEJMra043207. [DOI] [PubMed] [Google Scholar]
- 4.The malERA Consultative Group on Drugs. A research agenda for malaria eradication: drugs. PLoS Med. 2011;8:e1000402. doi: 10.1371/journal.pmed.1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnes KI, Watkins WM, White NJ. Antimalarial dosing regimens and drug resistance. Trends Parasitol. 2008;24:127–134. doi: 10.1016/j.pt.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Barnes KI, Lindegardh N, Ogundahunsi O, Olliaro P, Plowe CV, Randrianarivelojosia M, Gbotosho GO, Watkins WM, Sibley CH, White NJ. World Antimalarial Resistance Network (WARN) IV: clinical pharmacology. Malar J. 2007;6:122. doi: 10.1186/1475-2875-6-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holford NHG, Sheiner LB. Understanding the dose-effect relationship – clinical application of pharmacokinetic-pharmacodynamic models. Clin Pharmacokin. 1981;6:429–453. [Google Scholar]
- 8.Al-Sallami HS, Pavan Kumar VV, Landersdorfer CB, Bulitta JB, Duffull SB. The time course of drug effects. Pharm Stat. 2009;8:176–185. doi: 10.1002/pst.393. [DOI] [PubMed] [Google Scholar]
- 9.Wright DF, Winter HR, Duffull SB. Understanding the time course of pharmacological effect: a PKPD approach. Br J Clin Pharmacol. 2011;71:815–823. doi: 10.1111/j.1365-2125.2011.03925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duffull SB, Wright DF, Winter HR. Interpreting population pharmacokinetic-pharmacodynamic analyses – a clinical viewpoint. Br J Clin Pharmacol. 2011;71:807–814. doi: 10.1111/j.1365-2125.2010.03891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts JA, Kirkpatrick CM, Lipman J. Monte Carlo simulations: maximizing antibiotic pharmacokinetic data to optimize clinical practice for critically ill patients. J Antimicrob Chemother. 2011;66:227–231. doi: 10.1093/jac/dkq449. [DOI] [PubMed] [Google Scholar]
- 12.Hoshen MB, Heinrich R, Stein WD, Ginsburg H. Mathematical modelling of the within-host dynamics of Plasmodium falciparum. Parasitology. 2000;121:227–235. doi: 10.1017/s0031182099006368. [DOI] [PubMed] [Google Scholar]
- 13.Hoshen MB, Na-Bangchang K, Stein WD, Ginsburg H. Mathematical modelling of the chemotherapy of Plasmodium falciparum malaria with artesunate: postulation of ‘dormancy’, a partial cytostatic effect of the drug, and its implication for treatment regimens. Parasitology. 2000;121:237–246. doi: 10.1017/s0031182099006332. [DOI] [PubMed] [Google Scholar]
- 14.ter Kuile F, White NJ, Holloway P, Pasvol G, Krishna S. Plasmodium falciparum: in vitro studies of the pharmacodynamic properties of drugs used for the treatment of severe malaria. Exp Parasitol. 1993;76:85–95. doi: 10.1006/expr.1993.1010. [DOI] [PubMed] [Google Scholar]
- 15.Winter K, Hastings IM. Development, evaluation, and application of an in silico model for antimalarial drug treatment and failure. Antimicrob Agents Chemother. 2011;55:3380–3392. doi: 10.1128/AAC.01712-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 17.Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother. 1979;16:710–718. doi: 10.1128/aac.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov. 2004;3:509–520. doi: 10.1038/nrd1416. [DOI] [PubMed] [Google Scholar]
- 19.Fivelman QL, Adagu IS, Warhurst DC. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob Agents Chemother. 2004;48:4097–4102. doi: 10.1128/AAC.48.11.4097-4102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LeRoux M, Lakshmanan V, Daily JP. Plasmodium falciparum biology: analysis of in vitro versus in vivo growth conditions. Trends Parasitol. 2009;25:474–481. doi: 10.1016/j.pt.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 21.Noedl H, Wongsrichanalai C, Wernsdorfer WH. Malaria drug-sensitivity testing: new assays, new perspectives. Trends Parasitol. 2003;19:175–181. doi: 10.1016/s1471-4922(03)00028-x. [DOI] [PubMed] [Google Scholar]
- 22.Zaloumis S, Humberstone A, Charman SA, Price RN, Moehrle J, Gamo-Benito J, McCaw J, Jamsen KM, Smith K, Simpson JA. Assessing the utility of an anti-malarial pharmacokinetic-pharmacodynamic model for aiding drug clinical development. Malar J. 2012;11:303–317. doi: 10.1186/1475-2875-11-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berenbaum MC. A method for testing for synergy with any number of agents. J Infect Dis. 1978;137:122–130. doi: 10.1093/infdis/137.2.122. [DOI] [PubMed] [Google Scholar]
- 24.Chawira AN, Warhurst DC. The effect of artemisinin combined with standard antimalarials against chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum in vitro. J Trop Med Hyg. 1987;90:1–8. [PubMed] [Google Scholar]
- 25.World Health Organization. Guidelines for the Treatment of Malaria. 2nd edn. Geneva, Switzerland: World Health Organization; 2011. [Google Scholar]
- 26.Nosten F, White NJ. Artemisinin-based combination treatment of falciparum malaria. Am J Trop Med Hyg. 2007;77:181–192. [PubMed] [Google Scholar]
- 27.Kremsner PG, Krishna S. Antimalarial combinations. Lancet. 2004;364:285–294. doi: 10.1016/S0140-6736(04)16680-4. [DOI] [PubMed] [Google Scholar]
- 28.Noedl H, Se Y, Sriwichai S, Schaecher K, Teja-Isavadharm P, Smith B, Rutvisuttinunt W, Bethell D, Surasri S, Fukuda MM, Socheat D, Chan Thap L. Artemisinin resistance in Cambodia: a clinical trial designed to address an emerging problem in Southeast Asia. Clin Infect Dis. 2010;51:e82–89. doi: 10.1086/657120. [DOI] [PubMed] [Google Scholar]
- 29.Vivas L, Rattray L, Stewart LB, Robinson BL, Fugmann B, Haynes RK, Peters W, Croft SL. Antimalarial efficacy and drug interactions of the novel semi-synthetic endoperoxide artemisone in vitro and in vivo. J Antimicrob Chemother. 2007;59:658–665. doi: 10.1093/jac/dkl563. [DOI] [PubMed] [Google Scholar]
- 30.Noedl H, Krudsood S, Leowattana W, Tangpukdee N, Thanachartwet W, Looareesuwan S, Miller RS, Fukuda M, Jongsakul K, Yingyuen K, Sriwichai S, Ohrt C, Knirsch C. In vitro antimalarial activity of azithromycin, artesunate, and quinine in combination and correlation with clinical outcome. Antimicrob Agents Chemother. 2007;51:651–656. doi: 10.1128/AAC.01023-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelly JX, Smilkstein MJ, Brun R, Wittlin S, Cooper RA, Lane KD, Janowsky A, Johnson RA, Dodean RA, Winter R, Hinrichs DJ, Riscoe MK. Discovery of dual function acridones as a new antimalarial chemotype. Nature. 2009;459:270–273. doi: 10.1038/nature07937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obaldia N, 3rd, Kotecka BM, Edstein MD, Haynes RK, Fugmann B, Kyle DE, Rieckmann KH. Evaluation of artemisone combinations in Aotus monkeys infected with Plasmodium falciparum. Antimicrob Agents Chemother. 2009;53:3592–3594. doi: 10.1128/AAC.00471-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackenstedt U, Brockelman CR, Mehlhorn H, Raether W. Comparative morphology of human and animal malaria parasites. I. Host-parasite interface. Parasitol Res. 1989;75:528–535. doi: 10.1007/BF00931161. [DOI] [PubMed] [Google Scholar]
- 34.Peters W. Chemotherapy and Drug Resistance in Malaria. London: Academic Press; 1970. [Google Scholar]
- 35.Ager ALJ. Rodent malaria models. In: Peters W, Richards WHG, editors. Antimalarial Drugs 1: Biological Background, Experimental Methods, and Drug Resistance 2: Current Antimalarials and New Drug Developments. Berlin: Springer-Verlag; 1984. pp. 225–265. [Google Scholar]
- 36.Langhorne J, Buffet P, Galinski M, Good M, Harty J, Leroy D, Mota MM, Pasini E, Renia L, Riley E, Stins M, Duffy P. The relevance of non-human primate and rodent malaria models for humans. Malar J. 2011;10:23. doi: 10.1186/1475-2875-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters W, Robinson BL. Handbook of Animal Models of Infection. San Diego, CA: Academic Press; 1999. [Google Scholar]
- 38.Thompson PE, Werbel LM. Antimalarial Agents: Chemistry and Pharmacology. New York: Academic Press; 1972. [Google Scholar]
- 39.Gujjar R, El Mazouni F, White KL, White J, Creason S, Shackleford DM, Deng X, Charman WN, Bathurst I, Burrows J, Floyd DM, Matthews D, Buckner FS, Charman SA, Phillips MA, Rathod PK. Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J Med Chem. 2011;54:3935–3949. doi: 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Posner GH, Paik IH, Chang W, Borstnik K, Sinishtaj S, Rosenthal AS, Shapiro TA. Malaria-infected mice are cured by a single dose of novel artemisinin derivatives. J Med Chem. 2007;50:2516–2519. doi: 10.1021/jm070149m. [DOI] [PubMed] [Google Scholar]
- 41.Rottmann M, McNamara C, Yeung BK, Lee MC, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, Gonzalez-Paez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FC, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, McIntosh K, Padmanilayam M, Santo Tomas J, Scheurer C, Scorneaux B, Tang Y, Urwyler H, Wittlin S, Charman WN. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430:900–904. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 43.Thompson PE, Bayles A, Olszewski B. Antimalarial activity of 2,4-diamino-6-[(3,4-dichlorobenzyl) nitros-amino] quinazoline (CI-679 base) and CI-679 acetate. Laboratory studies in mice and rhesus monkeys. Am J Trop Med Hyg. 1970;19:12–26. doi: 10.4269/ajtmh.1970.19.12. [DOI] [PubMed] [Google Scholar]
- 44.Slack RD, Mott BT, Woodard LE, Tripathi A, Sullivan D, Nenortas E, Girdwood SC, Shapiro TA, Posner GH. Malaria-infected mice are completely cured by one 6 mg/kg oral dose of a new monomeric trioxane sulfide combined with mefloquine. J Med Chem. 2012;55:291–296. doi: 10.1021/jm201214d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore BR, Page-Sharp M, Stoney JR, Ilett KF, Jago JD, Batty KT. Pharmacokinetics, pharmacodynamics, and allometric scaling of chloroquine in a murine malaria model. Antimicrob Agents Chemother. 2011;55:3899–3907. doi: 10.1128/AAC.00067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Batty KT, Law AS, Stirling V, Moore BR. Pharmacodynamics of doxycycline in a murine malaria model. Antimicrob Agents Chemother. 2007;51:4477–4479. doi: 10.1128/AAC.00529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Opsenica I, Opsenica D, Smith KS, Milhous WK, Solaja BA. Chemical stability of the peroxide bond enables diversified synthesis of potent tetraoxane antimalarials. J Med Chem. 2008;51:2261–2266. doi: 10.1021/jm701417a. [DOI] [PubMed] [Google Scholar]
- 48.Zaknoon F, Wein S, Krugliak M, Meir O, Rotem S, Ginsburg H, Vial H, Mor A. Antiplasmodial properties of acyl-lysyl oligomers in culture and animal models of malaria. Antimicrob Agents Chemother. 2011;55:3803–3811. doi: 10.1128/AAC.00129-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meesters RJ, Hooff GP. State-of-the-art dried blood spot analysis: an overview of recent advances and future trends. Bioanalysis. 2013;5:2187–2208. doi: 10.4155/bio.13.175. [DOI] [PubMed] [Google Scholar]
- 50.Rahavendran SV, Vekich S, Skor H, Batugo M, Nguyen L, Shetty B, Shen Z. Discovery pharmacokinetic studies in mice using serial microsampling, dried blood spots and microbore LC-MS/MS. Bioanalysis. 2012;4:1077–1095. doi: 10.4155/bio.12.85. [DOI] [PubMed] [Google Scholar]
- 51.Dong Y, Wittlin S, Sriraghavan K, Chollet J, Charman SA, Charman WN, Scheurer C, Urwyler H, Santo Tomas J, Snyder C, Creek DJ, Morizzi J, Koltun M, Matile H, Wang X, Padmanilayam M, Tang Y, Dorn A, Brun R, Vennerstrom JL. The structure-activity relationship of the antimalarial ozonide arterolane (OZ277) J Med Chem. 2010;53:481–491. doi: 10.1021/jm901473s. [DOI] [PubMed] [Google Scholar]
- 52.Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, Chiu FC, Chollet J, Craft JC, Creek DJ, Dong Y, Matile H, Maurer M, Morizzi J, Nguyen T, Papastogiannidis P, Scheurer C, Shackleford DM, Sriraghavan K, Stingelin L, Tang Y, Urwyler H, Wang X, White KL, Wittlin S, Zhou L, Vennerstrom JL. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci U S A. 2011;108:4400–4405. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delves M, Plouffe D, Scheurer C, Meister S, Wittlin S, Winzeler EA, Sinden RE, Leroy D. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS Med. 2012;9:e1001169. doi: 10.1371/journal.pmed.1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peters W, Fleck SL, Robinson BL, Stewart LB, Jefford CW. The chemotherapy of rodent malaria. LX. The importance of formulation in evaluating the blood schizontocidal activity of some endoperoxide antimalarials. Ann Trop Med Parasitol. 2002;96:559–573. doi: 10.1179/000349802125001744. [DOI] [PubMed] [Google Scholar]
- 55.Janse CJ, Waters AP, Kos J, Lugt CB. Comparison of in vivo and in vitro antimalarial activity of artemisinin, dihydroartemisinin and sodium artesunate in the Plasmodium berghei-rodent model. Int J Parasitol. 1994;24:589–594. doi: 10.1016/0020-7519(94)90150-3. [DOI] [PubMed] [Google Scholar]
- 56.Edstein MD, Kotecka BM, Ager AL, Smith KS, DiTusa CA, Diaz DS, Kyle DE, Schiehser GA, Jacobus DP, Rieckmann KH, O'Neil MT. Antimalarial pharmacodynamics and pharmacokinetics of a third-generation antifolate – JPC2056 – in cynomolgus monkeys using an in vivo in vitro model. J Antimicrob Chemother. 2007;60:811–818. doi: 10.1093/jac/dkm280. [DOI] [PubMed] [Google Scholar]
- 57.Gibbons PL, Batty KT, Barrett PH, Davis TM, Ilett KF. Development of a pharmacodynamic model of murine malaria and antimalarial treatment with dihydroartemisinin. Int J Parasitol. 2007;37:1569–1576. doi: 10.1016/j.ijpara.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 58.Moore BR, Jago JD, Batty KT. Plasmodium berghei: parasite clearance after treatment with dihydroartemisinin in an asplenic murine malaria model. Exp Parasitol. 2008;118:458–467. doi: 10.1016/j.exppara.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 59.Moore BR, Batty KT, Andrzejewski C, Jago JD, Page-Sharp M, Ilett KF. Pharmacokinetics and pharmacodynamics of piperaquine in a murine malaria model. Antimicrob Agents Chemother. 2008;52:306–311. doi: 10.1128/AAC.00878-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moore BR, Ilett KF, Page-Sharp M, Jago JD, Batty KT. Piperaquine pharmacodynamics and parasite viability in a murine malaria model. Antimicrob Agents Chemother. 2009;53:2707–2713. doi: 10.1128/AAC.00056-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patel K, Batty KT, Moore BR, Gibbons PL, Bulitta JB, Kirkpatrick CM. Mechanism-based model of parasite growth and dihydroartemisinin pharmacodynamics in murine malaria. Antimicrob Agents Chemother. 2013;57:508–516. doi: 10.1128/AAC.01463-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tarning J, Lindegardh N, Sandberg S, Day NJ, White NJ, Ashton M. Pharmacokinetics and metabolism of the antimalarial piperaquine after intravenous and oral single doses to the rat. J Pharm Sci. 2008;97:3400–3410. doi: 10.1002/jps.21226. [DOI] [PubMed] [Google Scholar]
- 63.Ilett K, Batty KT. Artemisinin and its derivatives. In: Yu VL, Edwards G, Mckinnon PS, Peloquin CA, Morse G, editors. Antimicrobial Therapy and Vaccines. Pittsburgh, PA: ESun Technologies; 2005. pp. 981–1002. [Google Scholar]
- 64.Price RN, Simpson JA, Davis TE. Artemisinins. In: Grayson ML, editor. Kucers' the Use of Antibiotics: A Clinical Review of Antibacterial, Antifungal, Antiparasitic and Antiviral Drugs. London: Hodder Arnold; 2010. pp. 2090–2104. [Google Scholar]
- 65.Morris CA, Duparc S, Borghini-Fuhrer I, Jung D, Shin CS, Fleckenstein L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar J. 2011;10:263. doi: 10.1186/1475-2875-10-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White NJ. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob Agents Chemother. 1997;41:1413–1422. doi: 10.1128/aac.41.7.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.German PI, Aweeka FT. Clinical pharmacology of artemisinin-based combination therapies. Clin Pharmacokinet. 2008;47:91–102. doi: 10.2165/00003088-200847020-00002. [DOI] [PubMed] [Google Scholar]
- 68.White NJ. The assessment of antimalarial drug efficacy. Trends Parasitol. 2002;18:458–464. doi: 10.1016/s1471-4922(02)02373-5. [DOI] [PubMed] [Google Scholar]
- 69.Jamsen KM, Duffull SB, Tarning J, Lindegardh N, White NJ, Simpson JA. Optimal designs for population pharmacokinetic studies of oral artesunate in patients with uncomplicated falciparum malaria. Malar J. 2011;10:181. doi: 10.1186/1475-2875-10-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McGready R, Stepniewska K, Ward SA, Cho T, Gilveray G, Looareesuwan S, White NJ, Nosten F. Pharmacokinetics of dihydroartemisinin following oral artesunate treatment of pregnant women with acute uncomplicated falciparum malaria. Eur J Clin Pharmacol. 2006;62:367–371. doi: 10.1007/s00228-006-0118-y. [DOI] [PubMed] [Google Scholar]
- 71.Morris CA, Onyamboko MA, Capparelli E, Koch MA, Atibu J, Lokomba V, Douoguih M, Hemingway-Foday J, Wesche D, Ryder RW, Bose C, Wright L, Tshefu AK, Meshnick S, Fleckenstein L. Population pharmacokinetics of artesunate and dihydroartemisinin in pregnant and non-pregnant women with malaria. Malar J. 2011;10:114. doi: 10.1186/1475-2875-10-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stepniewska K, Taylor W, Sirima SB, Ouedraogo EB, Ouedraogo A, Gansane A, Simpson JA, Morgan CC, White NJ, Kiechel JR. Population pharmacokinetics of artesunate and amodiaquine in African children. Malar J. 2009;8:200. doi: 10.1186/1475-2875-8-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tarning J, Kloprogge F, Piola P, Dhorda M, Muwanga S, Turyakira E, Nuengchamnong N, Nosten F, Day NP, White NJ, Guerin PJ, Lindegardh N. Population pharmacokinetics of artemether and dihydroartemisinin in pregnant women with uncomplicated Plasmodium falciparum malaria in Uganda. Malar J. 2012;11:293. doi: 10.1186/1475-2875-11-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tarning J, Rijken MJ, McGready R, Phyo AP, Hanpithakpong W, Day NP, White NJ, Nosten F, Lindegardh N. Population pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and nonpregnant women with uncomplicated malaria. Antimicrob Agents Chemother. 2012;56:1997–2007. doi: 10.1128/AAC.05756-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ashley EA, Stepniewska K, Lindegardh N, McGready R, Hutagalung R, Hae R, Singhasivanon P, White NJ, Nosten F. Population pharmacokinetic assessment of a new regimen of mefloquine used in combination treatment of uncomplicated falciparum malaria. Antimicrob Agents Chemother. 2006;50:2281–2285. doi: 10.1128/AAC.00040-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hietala SF, Martensson A, Ngasala B, Dahlstrom S, Lindegardh N, Annerberg A, Premji Z, Farnert A, Gil P, Bjorkman A, Ashton M. Population pharmacokinetics and pharmacodynamics of artemether and lumefantrine during combination treatment in children with uncomplicated falciparum malaria in Tanzania. Antimicrob Agents Chemother. 2010;54:4780–4788. doi: 10.1128/AAC.00252-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simpson JA, Price R, ter Kuile F, Teja-Isavatharm P, Nosten F, Chongsuphajaisiddhi T, Looareesuwan S, Aarons L, White NJ. Population pharmacokinetics of mefloquine in patients with acute falciparum malaria. Clin Pharmacol Ther. 1999;66:472–484. doi: 10.1016/S0009-9236(99)70010-X. [DOI] [PubMed] [Google Scholar]
- 78.Martin C, Gimenez F, Bangchang KN, Karbwang J, Wainer IW, Farinotti R. Whole blood concentrations of mefloquine enantiomers in healthy Thai volunteers. Eur J Clin Pharmacol. 1994;47:85–87. doi: 10.1007/BF00193485. [DOI] [PubMed] [Google Scholar]
- 79.Ezzet F, van Vugt M, Nosten F, Looareesuwan S, White NJ. Pharmacokinetics and pharmacodynamics of lumefantrine (benflumetol) in acute falciparum malaria. Antimicrob Agents Chemother. 2000;44:697–704. doi: 10.1128/aac.44.3.697-704.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Svensson US, Alin H, Karlsson MO, Bergqvist Y, Ashton M. Population pharmacokinetic and pharmacodynamic modelling of artemisinin and mefloquine enantiomers in patients with falciparum malaria. Eur J Clin Pharmacol. 2002;58:339–351. doi: 10.1007/s00228-002-0485-y. [DOI] [PubMed] [Google Scholar]
- 81.Tarning J, McGready R, Lindegardh N, Ashley EA, Pimanpanarak M, Kamanikom B, Annerberg A, Day NP, Stepniewska K, Singhasivanon P, White NJ, Nosten F. Population pharmacokinetics of lumefantrine in pregnant women treated with artemether-lumefantrine for uncomplicated Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2009;53:3837–3846. doi: 10.1128/AAC.00195-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hung TY, Davis TM, Ilett KF, Karunajeewa H, Hewitt S, Denis MB, Lim C, Socheat D. Population pharmacokinetics of piperaquine in adults and children with uncomplicated falciparum or vivax malaria. Br J Clin Pharmacol. 2004;57:253–262. doi: 10.1046/j.1365-2125.2003.02004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tarning J, Ashley EA, Lindegardh N, Stepniewska K, Phaiphun L, Day NP, McGready R, Ashton M, Nosten F, White NJ. Population pharmacokinetics of piperaquine after two different treatment regimens with dihydroartemisinin-piperaquine in patients with Plasmodium falciparum malaria in Thailand. Antimicrob Agents Chemother. 2008;52:1052–1061. doi: 10.1128/AAC.00955-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hoglund RM, Adam I, Hanpithakpong W, Ashton M, Lindegardh N, Day NP, White NJ, Nosten F, Tarning J. A population pharmacokinetic model of piperaquine in pregnant and non-pregnant women with uncomplicated Plasmodium falciparum malaria in Sudan. Malar J. 2012;11:398. doi: 10.1186/1475-2875-11-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tarning J, Zongo I, Some FA, Rouamba N, Parikh S, Rosenthal PJ, Hanpithakpong W, Jongrak N, Day NP, White NJ, Nosten F, Ouedraogo JB, Lindegardh N. Population pharmacokinetics and pharmacodynamics of piperaquine in children with uncomplicated falciparum malaria. Clin Pharmacol Ther. 2012;91:497–505. doi: 10.1038/clpt.2011.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bell DJ, Nyirongo SK, Mukaka M, Molyneux ME, Winstanley PA, Ward SA. Population pharmacokinetics of sulfadoxine and pyrimethamine in Malawian children with malaria. Clin Pharmacol Ther. 2011;89:268–275. doi: 10.1038/clpt.2010.297. [DOI] [PubMed] [Google Scholar]
- 87.Karunajeewa HA, Salman S, Mueller I, Baiwog F, Gomorrai S, Law I, Page-Sharp M, Rogerson S, Siba P, Ilett KF, Davis TM. Pharmacokinetic properties of sulfadoxine-pyrimethamine in pregnant women. Antimicrob Agents Chemother. 2009;53:4368–4376. doi: 10.1128/AAC.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hietala SF, Bhattarai A, Msellem M, Roshammar D, Ali AS, Stromberg J, Hombhanje FW, Kaneko A, Bjorkman A, Ashton M. Population pharmacokinetics of amodiaquine and desethylamodiaquine in pediatric patients with uncomplicated falciparum malaria. J Pharmacokinet Pharmacodyn. 2007;34:669–686. doi: 10.1007/s10928-007-9064-2. [DOI] [PubMed] [Google Scholar]
- 89.Jullien V, Ogutu B, Juma E, Carn G, Obonyo C, Kiechel JR. Population pharmacokinetics and pharmacodynamic considerations of amodiaquine and desethylamodiaquine in Kenyan adults with uncomplicated malaria receiving artesunate-amodiaquine combination therapy. Antimicrob Agents Chemother. 2010;54:2611–2617. doi: 10.1128/AAC.01496-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tarning J, Chotsiri P, Jullien V, Rijken MJ, Bergstrand M, Cammas M, McGready R, Singhasivanon P, Day NP, White NJ, Nosten F, Lindegardh N. Population pharmacokinetic and pharmacodynamic modeling of amodiaquine and desethylamodiaquine in women with Plasmodium vivax malaria during and after pregnancy. Antimicrob Agents Chemother. 2012;56:5764–5773. doi: 10.1128/AAC.01242-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hoshen MB, Stein WD, Ginsburg H. Modelling the chloroquine chemotherapy of falciparum malaria: the value of spacing a split dose. Parasitology. 1998;116:407–416. doi: 10.1017/s0031182098002480. [DOI] [PubMed] [Google Scholar]
- 92.Hoshen MB, Stein WD, Ginsburg H. Mathematical modelling of malaria chemotherapy: combining artesunate and mefloquine. Parasitology. 2002;124:9–15. doi: 10.1017/s0031182001008952. [DOI] [PubMed] [Google Scholar]
- 93.Simpson JA, Watkins ER, Price RN, Aarons L, Kyle DE, White NJ. Mefloquine pharmacokinetic-pharmacodynamic models: implications for dosing and resistance. Antimicrob Agents Chemother. 2000;44:3414–3424. doi: 10.1128/aac.44.12.3414-3424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Skinner TS, Manning LS, Johnston WA, Davis TM. In vitro stage-specific sensitivity of Plasmodium falciparum to quinine and artemisinin drugs. Int J Parasitol. 1996;26:519–525. doi: 10.1016/0020-7519(96)89380-5. [DOI] [PubMed] [Google Scholar]
- 95.Gravenor MB, van Hensbroek MB, Kwiatkowski D. Estimating sequestered parasite population dynamics in cerebral malaria. Proc Natl Acad Sci U S A. 1998;95:7620–7624. doi: 10.1073/pnas.95.13.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gordi T, Xie R, Jusko WJ. Semi-mechanistic pharmacokinetic/pharmacodynamic modelling of the antimalarial effect of artemisinin. Br J Clin Pharmacol. 2005;60:594–604. doi: 10.1111/j.1365-2125.2005.02508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Saralamba S, Pan-Ngum W, Maude RJ, Lee SJ, Tarning J, Lindegardh N, Chotivanich K, Nosten F, Day NP, Socheat D, White NJ, Dondorp AM, White LJ. Intrahost modeling of artemisinin resistance in Plasmodium falciparum. Proc Natl Acad Sci U S A. 2011;108:397–402. doi: 10.1073/pnas.1006113108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith T, Dietz K, Vounatsou P, Muller I, English M, Marsh K. Bayesian age-stage modelling of Plasmodium falciparum sequestered parasite loads in severe malaria patients. Parasitology. 2004;129:289–299. doi: 10.1017/s003118200400575x. [DOI] [PubMed] [Google Scholar]
- 99.Ezzet F, Mull R, Karbwang J. Population pharmacokinetics and therapeutic response of CGP 56697 (artemether + benflumetol) in malaria patients. Br J Clin Pharmacol. 1998;46:553–561. doi: 10.1046/j.1365-2125.1998.00830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Phyo AP, Nkhoma S, Stepniewska K, Ashley EA, Nair S, McGready R, ler Moo C, Al-Saai S, Dondorp AM, Lwin KM, Singhasivanon P, Day NP, White NJ, Anderson TJ, Nosten F. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet. 2012;379:1960–1966. doi: 10.1016/S0140-6736(12)60484-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bethell D, Se Y, Lon C, Socheat D, Saunders D, Teja-Isavadharm P, Khemawoot P, Darapiseth S, Lin J, Sriwichai S, Kuntawungin W, Surasri S, Lee SJ, Sarim S, Tyner S, Smith B, Fukuda MM. Dose-dependent risk of neutropenia after 7-day courses of artesunate monotherapy in Cambodian patients with acute Plasmodium falciparum malaria. Clin Infect Dis. 2010;51:e105–114. doi: 10.1086/657402. [DOI] [PubMed] [Google Scholar]
- 103.Anderson BJ, Holford NH. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet. 2009;24:25–36. doi: 10.2133/dmpk.24.25. [DOI] [PubMed] [Google Scholar]
- 104.Janse C, Waters AP. The Plasmodium Berghei Research Model of Malaria. Leiden, The Netherlands: Leiden University Medical Centre, Leiden University Malaria Research Group (LMRG); 2002. Available at https://www.lumc.nl/con/1040/81028091348221/810281121192556/811070740182556/ (last accessed 17 December 2013) [Google Scholar]
- 105.World Health Organization. Methods and Techniques for Assessing Exposure to Antimalarial Drugs in Clinical Field Studies. Geneva, Switzerland: World Health Organization; 2010. [Google Scholar]
- 106.Simpson JA, Jamsen KM, Price RN, White NJ, Lindegardh N, Tarning J, Duffull SB. Towards optimal design of anti-malarial pharmacokinetic studies. Malar J. 2009;8:189. doi: 10.1186/1475-2875-8-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jamsen KM, Duffull SB, Tarning J, Lindegardh N, White NJ, Simpson JA. Optimal designs for population pharmacokinetic studies of the partner drugs co-administered with artemisinin derivatives in patients with uncomplicated falciparum malaria. Malar J. 2012;11:143. doi: 10.1186/1475-2875-11-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Na Bangchang K, Davis TM, Looareesuwan S, White NJ, Bunnag D, Karbwang J. Mefloquine pharmacokinetics in pregnant women with acute falciparum malaria. Trans R Soc Trop Med Hyg. 1994;88:321–323. doi: 10.1016/0035-9203(94)90101-5. [DOI] [PubMed] [Google Scholar]