

Abstract
Background and Purpose
Prolongation of the cardiac QRS complex is linked to increased mortality and may result from drug-induced inhibition of cardiac sodium channels (hNaV1.5). There has been no systematic evaluation of preclinical and marketed drugs for their additional potential to cause QRS prolongation via gap junction uncoupling.
Experimental Approach
Using the human cardiac gap junction connexin 43 (hCx43), a dye transfer ‘parachute’ assay to determine IC50 values for compound ranking was validated with compounds known to uncouple gap junctions. Uncoupling activity (and hNaV1.5 inhibition by automated patch clamp) was determined in a set of marketed drugs and preclinical candidate drugs, each with information regarding propensity to prolong QRS.
Key Results
The potency of known gap junction uncouplers to uncouple hCx43 was ranked (according to IC50) as phorbol ester>digoxin>meclofenamic acid>carbenoxolone>heptanol. Among the drugs associated with QRS prolongation, 29% were found to uncouple hCx43 (IC50 < 50 μM), whereas no uncoupling activity was observed in drugs not associated with QRS prolongation. In preclinical candidate drugs, hCx43 and hNaV1.5 IC50 values were similar (within threefold). No consistent margin over preclinical Cmax (free) was apparent for QRS prolongation associated with Cx43 inhibition. However, instances were found of QRS prolonging compounds that uncoupled hCx43 with significantly less activity at hNaV1.5.
Conclusion and Implications
These results demonstrate that off-target uncoupling activity is apparent in drug and drug-like molecules. Although the full ramifications of Cx inhibition remain to be established, screening for hCx43 off-target activity could reduce the likelihood of developing candidate drugs with a risk of causing QRS prolongation.
Introduction
Prolongation of the QRS interval of the ECG is associated with increased mortality in some patient groups (Kalahasti et al., 2003; Adesanya et al., 2008). The primary mechanism of drug-induced QRS prolongation is inhibition of the cardiac sodium channel (hNaV1.5; Alexander et al., 2013), which reduces the rate of depolarization during the cardiac action potential upstroke. Class I antiarrhythmic drugs block hNaV1.5 as their primary mechanism of action, and are associated with QRS prolongation, although off-target activity at hNaV1.5 is found in a variety of other drug classes, such as tricyclic antidepressants, antipsychotics and anticonvulsants (Harmer et al., 2011).
The QRS interval represents the time taken for the excitatory impulse to propagate throughout the ventricles, and conduction velocity is partly determined by the resistance of the intercellular connections between myocytes (Rohr, 2004). These connections are formed by gap junctions, which underlie impulse transmission and signalling molecule exchange and, in cardiomyocytes, are clustered at the intercalated disks linking individual myocytes (Severs et al., 2009; Rhett et al., 2012). Gap junctions themselves are formed from connexin proteins (Cx), a family of 20 or so mammalian subtypes (Cruciani and Mikalsen, 2006), with Cx isoform 43 (hCx43) constituting the predominant protein of the human ventricular cardiomyocyte gap junction (Severs et al., 2009). Although impulse propagation through a three-dimensional, non-uniform myocardial network is complex (Rohr, 2004), pharmacological inhibition of gap junctions using carbenoxolone has been shown to be associated with conduction slowing in human myocardium, without an alteration in the individual ionic currents underlying the action potential (de Groot et al., 2003; Kojodjojo et al., 2006). Genetic knockout of Cx43 in mice is associated with conduction slowing, QRS prolongation and increased susceptibility to ventricular arrhythmias (Thomas et al., 1998; Gutstein et al., 2001; Danik et al., 2004). Disturbances of atrioventricular (AV) conductance and the QRS waveform have also been observed in Cx40 knockout mice (Kirchhoff et al., 1998; Hagendorff et al., 1999). These observations implicate off-target Cx inhibition as a potential mechanism of QRS prolongation in drug-induced adverse events.
Cx have received little attention as drug targets, possibly due to the generally low Cx isoform selectivity of existing Cx inhibitors (Srinivas, 2009). In contrast to on-target selectivity, off-target activity at Cx could be an unrecognized mechanism of drug toxicity. Given the diversity of Cx function, and potential to unwittingly affect multiple members of the connexin family, off-target drug activity at Cx could potentially mediate multiple adverse drug events. A variety of serious human hereditary disorders have been linked to Cx gene mutations (Kelsell et al., 2001). However, only a few reports in the literature have linked drug side effects to Cx inhibition. The antidepressant bupropion has been linked to QRS prolongation via a Cx inhibition mechanism (Caillier et al., 2011), as has the cardiotoxic antipyschotic thioridazine (Matesic et al., 2006).
Establishing the molecular mechanisms of drug-induced cardiotoxicity and developing in vitro screening protocols have the potential to improve the safety profile of marketed drugs. For instance, the realization that drug-induced long QT syndrome could result from off-target activity at the hERG channel, and furthermore, that the ability of the channel to bind drug-like molecules might explain a significant portion of long QT adverse events, has enabled the development of effective screening strategies (Pollard et al., 2010). Although the scientific rationale relating gap junction conductance to impulse propagation and QRS duration has been established, a key piece of information missing is the frequency of off-target Cx inhibition among drug-like molecules, either as an isolated mechanism of QRS prolongation or, in combination with hNaV1.5 activity, as an increased risk of such deleterious effects (Lu et al., 2010; Harmer et al., 2011).
To investigate this relationship, we validated a variant of the well-established ‘parachute'-type hCx43 dye transfer assay, where dye-loaded donor cells expressing hCx43 are dispersed above, and allowed to ‘parachute’ onto, an acceptor cell monolayer also expressing hCx43. When gap junctions form between donor and acceptor cells, the extent of dye spread into the acceptor cell monolayer represents the amount of gap junction coupling between cells (Goldberg et al., 1995; Abbaci et al., 2008). A set of clinically-used drugs has been previously investigated by Harmer et al. (2011) with regard to their potential to prolong QRS. To investigate whether Cx uncoupling activity is present in these drugs and, if so, at what frequency, we validated the hCx43 parachute assay with known gap junction modulators and then tested a subset of these clinical QRS-annotated drugs in the hCx43 parachute assay. Secondly, a set of AstraZeneca's non-marketed compounds that had already undergone preclinical Safety Assessment studies (including known QRS interval effects and relevant plasma exposure levels) were tested in both hCx43 and hNaV1.5 assays to investigate the relationship between these mechanisms of QRS prolongation. These data provide an initial case study of the role of hCx43 uncoupling, in addition to hNaV1.5 inhibition, in drug-induced prolongation of the QRS interval.
Methods
BacMam virus production
Human Cx43 (SWISSPROT accession number P17302) residues 1–382 were made by gene synthesis (GeneArt®; Life Technologies, Paisley, UK) with attB1 and attB2 Gateway recombination sequences at the 5′ and 3′ ends respectively. This construct was then cloned by Gateway recombination into the pDONR221 entry vector, then by LR recombination into a Gateway-adapted pBacMam vector. Recombinant BacMam was amplified using sf21 cells in suspension culture as described (McCall et al., 2005). BacMam viral suspension was concentrated 100-fold. BacMam viral suspension (1 L) was aliquoted into 4 × 250 mL Beckman polypropylene centrifugation vessels. The suspension was centrifuged at 38 000× g for 2 h using an Avanti J-25I centrifuge (Beckman Coulter, High Wycombe, UK). The supernatant was removed and the pellet resuspended in 10 mL SF900II media.
Transduction of cells
HeLa cells were cultured in CellSTACKs (Corning Incorporated, Corning, NY, USA) in growth media (DMEM + 10% FCS + 2 mM glutamine). Cells were harvested, centrifuged at 250× g for 10 min and resuspended at 2.5 × 106 cells mL-1 in growth media. BacMam virus expressing hCx43 was added at a multiplicity of transduction of 100 and incubated in a shaking incubator at 37°C, 5% CO2, 140 rpm for 5 h in a 1 L Erlenmeyer flask. Cells were centrifuged at 250× g for 10 min and resuspended in DMEM + 20% FCS. An equal volume of cryopreservation media (FCS + 10% DMSO) was added and cells aliquoted into cryovials. Cells were cryopreserved using a controlled rate freezer (Planer Kryo 560-16, Planer PLC, Sunbury-On-Thames, UK) and stored in liquid nitrogen.
hCx43 Immunofluorescence
Cryopreserved HeLa cells expressing hCx43 (hCx43-HeLa) were thawed and plated in growth media at 104 cells per well in 96-well plates. Cells were incubated overnight at 37°C, 5% CO2 and then fixed in 4% formaldehyde solution at room temperature for 1 h. Blocking buffer (PBS, 1.1% BSA, 0.2% Triton X-100) was added to the cells for 1 h followed by anti-Cx43 primary antibody (1:400 in blocking buffer) overnight at 4°C. Cells were washed in PBS, incubated in blocking solution for 30 min and then secondary antibody (1:500 in blocking buffer) for 1 h. Cells were again washed before they were incubated with the nuclear label DRAQ-5 (1 μM). Immunofluorescence was detected using an InCell Analyzer (Biacore, GE Healthcare Bio-Sciences, Pittsburgh, PA, USA).
Parachute assay
hCx43-HeLa cells were thawed, washed in growth media (Phenol Red and HEPES-free DMEM + 10% FCS + 2 mM Glutamax™; Gibco/Life Technologies) and plated into 96-well ViewPlates at 2 × 104 cells per well (85 μL per well). An aliquot to provide donor cells was cultured in a standard TC flask. A bicarbonate rather than HEPES-buffered formulation of DMEM was used, as HEPES has been shown to inhibit some Cx isoforms (Bevans and Harris, 1999). Cells were maintained overnight in a 5% CO2 37°C atmosphere. The following day, the parachute assay was performed by incubating the cells with test compounds before the addition of parachute cells. Drugs were prepared as 10-point 1/2-log dilution series in DMSO from a top concentration of 50 mM. A digoxin (10 mM) reference series and controls to provide minimum and maximum signal (10 mM digoxin and DMSO, respectively) were included for addition to each cell plate. Intermediate dilution (1:10 or 1:50) plates were prepared in serum-free media from compound plates before a final compound dilution (1:20) into the plates containing hCx43-HeLa cells, to give a constant 0.5% or 0.1% final DMSO concentration. Parachute cells were loaded with dye by replacing flask media with a 1:200 dilution of Calcein-AM dye (1 mg·mL−1 DMSO stock) in media for 30 min. Parachute cells were dispersed with Accutase (Innovative Cell Technologies, San Diego, CA, USA), washed twice in media and 10 μL of 2 × 105 cells mL−1 (in media containing 10 μg·mL−1 Hoechst 33342 nuclear stain) added per well (final well volume 100 μL). Plates were returned to a 5% CO2, 37°C atmosphere for the required incubation time.
Imaging and analysis
Parachute assay plates were imaged on an ImageXpress Micro system (Molecular Devices, Sunnyvale, CA, USA) in the FITC channel using a 4× objective and six imaging sites per well. Image analysis was performed using the MetaXpress Count Nuclei algorithm to count green cells. Images were analysed twice, once to count total green cells including faintly-labelled acceptor cells and once to count bright parachute donor cells only. The labelled/donor cell parameter was calculated on a per well basis. To calculate IC50 values, four parameter logistic curves were fitted to the data (normalized to the minimum and maximum signal controls included on each plate). Nuclear staining was also imaged to ensure an intact acceptor cell monolayer.
hNaV1.5 IC50 determination
Compounds from AstraZeneca's internal library were tested for hNaV1.5 inhibition by automated patch clamp analysis, as described previously (Harmer et al., 2008).
Statistical analysis
Data are presented as mean ± SD or, for IC50 values, geometric mean. Statistical signifigance was determined using Student's t-tests while assay signal:noise discrimination was quantified using the Z'-factor statistic (Zhang et al., 1999).
Materials
Phenol Red and HEPES-free DMEM (Sigma D1145, Gillingham, UK), glutamine and Glutamax™ (Gibco/Life Technologies), Accutase (Innovative Cell Technologies), CellSTACKs (Corning #3271), 1 mL Matrix cryovials (Thermo Scientific #3741, Loughborough, UK) and 96-well, black clear-bottomed plates (ViewPlate, Perkin Elmer #6005182, Waltham, MA, USA) were used for cell culture. Immunofluorescence labelling was performed using rabbit anti-Cx43 (Sigma C6219), anti-rabbit-FITC antibody (Sigma F9887) and DRAQ-5 (Biostatus DR50050). Calcein-AM and Hoechst 33342 was obtained from Molecular Probes (Life Technologies C3099 and H3570). Drugs were obtained from the AstraZeneca compound collection or Sigma-Aldrich (UK) and prepared as 50 mM DMSO stock solution unless otherwise stated. Carbenoxolone was prepared as a aqueous stock solution or, for serial dilutions, as a DMSO : ethanol (3:2 by vol) stock solution. Heptanol was prepared at the required final concentration directly in DMEM media.
Results
Dye transfer is mediated by hCx43 gap junctions
Anti-Cx43 immunofluorescence microscopy confirmed robust expression of hCx43 in BacMam-transduced HeLa cells, whereas the parental line displayed little immunoreactivity (Figure 1A and B). To verify, using these cells, that the transfer of dye between donor and acceptor cells occurs via hCx43 gap junctions, the effects of the known gap junction uncoupling agents carbenoxolone and digoxin (De Mello, 1976; Davidson et al., 1986) were tested, as were untransduced parental HeLa cells. Using hCx43-HeLa cells, dye transfer from donor to acceptor cells was clearly apparent after 3 h incubation and this was reduced to levels apparent in untransduced cells by carbenoxolone or digoxin treatment (Figure 1). A ratio of 5–7 labelled hCx43-HeLa acceptor cells per donor cell was determined under these conditions. This is likely to be an underestimate of the true coupling ratio, as a portion of donor cells counted in the analysis never transferred dye to surrounding cells during the incubation period and those donor cells that did initiate dye transfer appeared at different time points. This variation in donor cell behaviour, together with the fact that several donor cells with different coupling behaviours might lie within the same area of labelled acceptor cells, made the spatial characteristics of dye spread unclear, especially when imaged at a single time point. Sophisticated methods to analyse dye tracer diffusion in order to estimate gap junction permeability have been published, for instance, following dye injection by micropipette where precise spatial information is available (e.g. Zimmerman and Rose, 1985; Mills and Massey, 1998). In the present study, the suitability of using a simple coupling ratio (in each image, the number of donor cells divided by the number of labelled acceptor cells) to determine approximate IC50 values was tested empirically.
Figure 1.
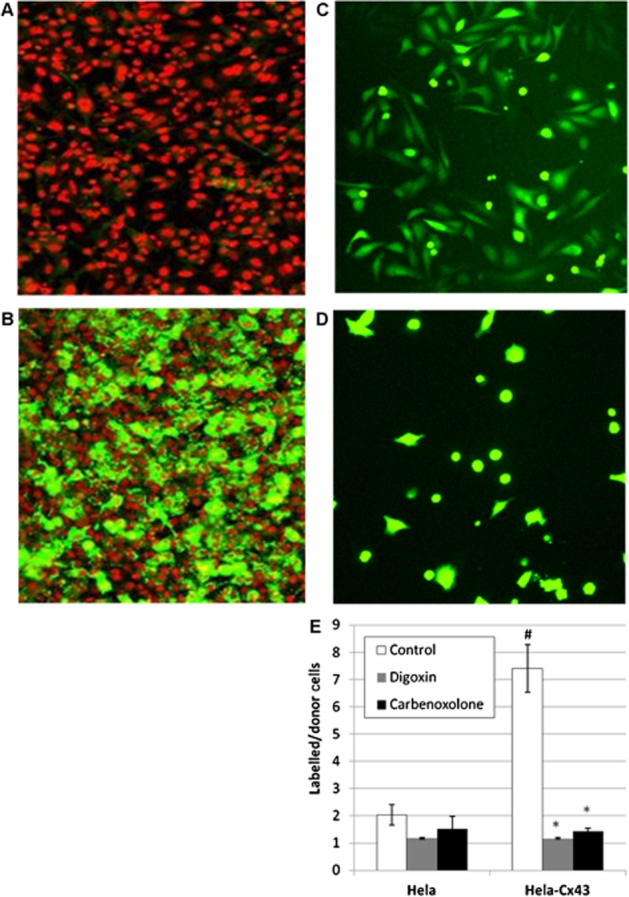
Calcein dye transfer in hCx43-HeLa ‘parachute’ assay is mediated by gap junctions. Immunofluorescence labelling of hCx43 protein in parental, untransduced HeLa cells (A) and hCx43-transduced HeLa cells (B), where green represents hCx43 immunoreactivity and red represents Drac5-stained nuclei. Representative images showing transfer of Calein dye from donor cells to acceptor cell monolayer after 3 h incubation (C) and in presence of 200 μM carbenoxolone (D). Quantification of dye transfer (see methods) in parental HeLa cells and hCx43-HeLa cells in presence of carbenoxolone (200 μM) and digoxin (10 μM) demonstrate that dye transfer is dependent on hCx43 and inhibited by known gap junction blockers (E). *P > 0.01 versus control; #P > 0.01 versus HeLa control (n = 4).
Determination of hCx43 uncoupling IC50
Transfer efficiency was relatively insensitive to cell density within the ranges 1 × 104–4 × 104 cells per well for acceptor cell density and 1 × 103–2 × 103 cells per well for donor parachute cells. Cell densities of 2 × 104 (acceptor) and 2 × 103 (donor) cells per well were used for all further experiments. The extent of dye transfer as a function of time was investigated. In the period 2.5 h–6 h significant dye transfer had occurred (Figure 2A). It has been argued that the size of the labelled patch of cells following diffusion from a ‘bolus’ source of a fixed amount of dye should be described by an asymptote and decline over time (Mills and Massey, 1998), similar to the situation observed in Figure 2A. Separation between minimum and maximum controls (i.e. wells treated with a maximally effective concentration of an uncoupling agent such as digoxin or carbenoxolone compared with wells treated with vehicle only), quantified using the Z'-factor statistic and eight data points per control (Zhang et al., 1999), typically gave values greater than 0.7 in this time period. Variation (%CV) across a single plate without drug treatment was less than 10%.
Figure 2.
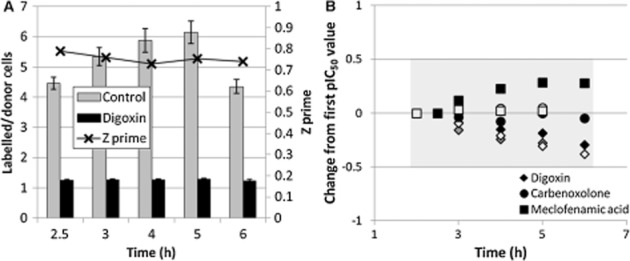
Effect of dye transfer time on signal-noise and IC50 value. (A) Representative result of five separate experiments showing time course of dye transfer under control and maximally inhibited (10 μM digoxin) conditions. Separation between minimum and maximum signals was quantified using the Z' factor statistic. (B) Effect of dye transfer time on calculated pIC50 values for Cx43 uncouplers digoxin, carbenoxolone and meclofenamic acid, shown as pIC50 change from first pIC50 value. Shaded area represents a <0.5 log unit change from initial value. Black, grey and white shading represent determinations in separate experiments (n = 3).
Although dye transfer occurring in the 3–6 h period (post-addition of donor cells) provided a robust signal, the repeatability or stability of IC50 measurements determined during this period was unknown. Repeated IC50 measurements for three gap junction uncouplers were made to assess this effect (Figure 2B). Digoxin showed a trend towards lower IC50 values over time, as might be expected given that the mechanism of gap junction uncoupling is believed to depend upon rundown of the Na+ gradient and a consequent increase in intracellular Ca2+ concentration (De Mello, 1976). However, the other two agents, carbenoxolone and meclofenamic acid, displayed stable or increasing IC50 values, suggesting that there is no consistent bias with increasing dye transfer time. Over the 3–6 h period, IC50 values did not change more than threefold (Figure 2B).
Whether the dye tracer molecule interacts with uncoupling agents is unknown. Antagonism, for instance at a shared binding site, was not apparent between the two, in that altering the concentration of dye in the donor cells (by reducing dye loading time) had no effect on the apparent IC50 values for carbenoxolone or digoxin (Figure 3A). The DMSO vehicle was found to affect dye transfer, with the inhibition caused by 2% DMSO being similar to a maximally effective concentration of carbenoxolone (Figure 3B). An approximate DMSO IC50 value of 0.8% (110 mM) was calculated. A vehicle concentration of 0.1% produced negligible inhibition of dye transfer and was considered optimum for compound testing, although concentrations of up to 0.5% allowed higher test concentrations if necessary.
Figure 3.
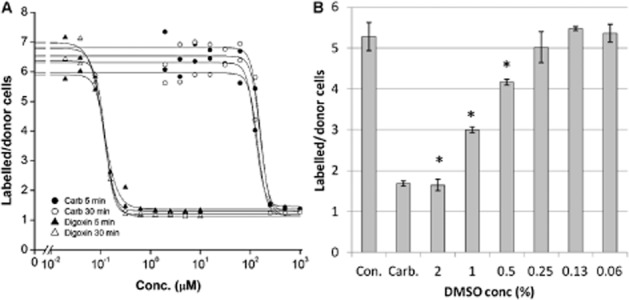
Effect of DMSO vehicle and effect of dye concentration on IC50 values. (A) Potential antagonistic effects of dye concentration on hCx43 uncoupling potency were tested by determining IC50 values (using a 1-in-2 dilution series) of digoxin and carbenoxolone (Carb.) using parachute cells loaded with low (5 min dye loading time) and high (30 min dye loading time) dye concentrations (representative of three separate experiments). (B) Effect of various concentrations of DMSO in comparison to control (Con.) and 200 μM carbenoxolone (Carb.) on dye transfer (representative of three separate experiments; *P < 0.05 vs. Con.).
IC50 values of known gap junction uncouplers
Using the conditions established above, IC50 values were determined for five well-characterized gap junction uncouplers (carbenoxolone, heptanol, meclofenamic acid, digoxin and PDBu). These were tested in a 10-point, 1/2-log serial dilution format at a final DMSO concentration of 0.5% (Figure 4). IC50 values of 210, 2208, 264, 0.136 and 0.023 μM (geometric mean of three determinations) were calculated for carbenoxolone, heptanol, meclofenamic acid, digoxin and PDBu respectively. Given the unusually short half-life of Cx proteins (estimated to be 1–2 h; Laird et al., 1991), agents affecting protein turnover were also tested. Brefeldin A, an inhibitor of protein forward trafficking, and cycloheximide, a protein synthesis inhibitor, were also tested but found to be inactive under these conditions.
Figure 4.
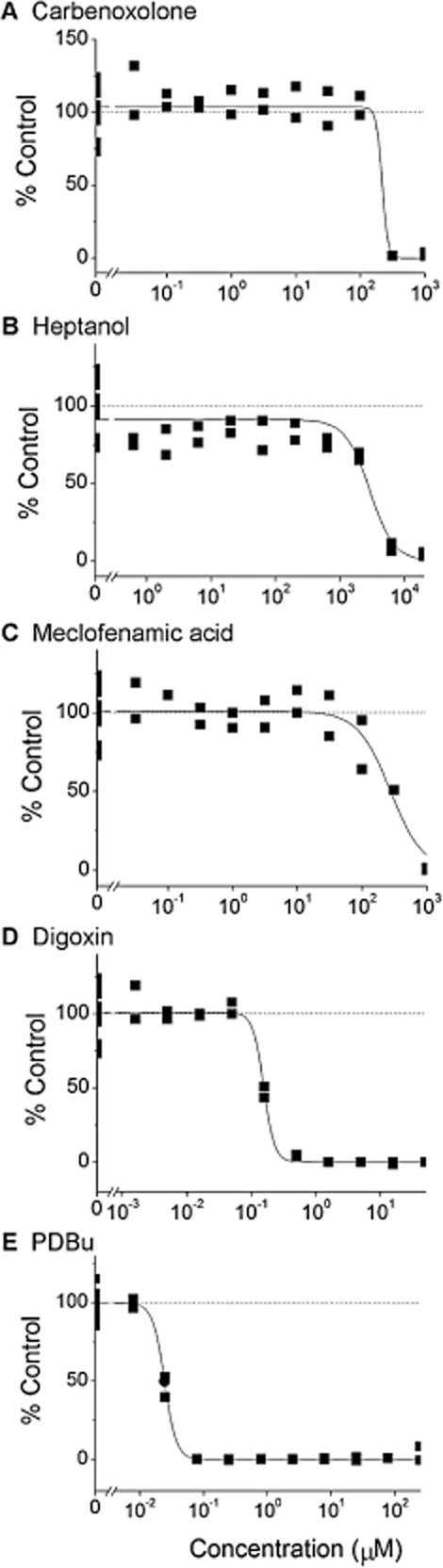
IC50 values determined for known gap junction uncouplers. Representative results from duplicate tests of known gap junction uncouplers from DMSO stock solutions in 10-point, 1/2-log dilution series with a final concentration of 0.5% DMSO.
hCx43 uncoupling activity of marketed drugs associated with QRS prolongation
Using the annotation of Harmer et al. (2011), 24 marketed drugs that are not class I antiarrhythmics but are associated with clinical reports of QRS prolongation were selected (amitriptyline, thioridazine, imipramine, nortriptyline, desipramine, maprotiline, fluoxetine, perhexiline, lamotrigine, bupropion, quinine, diphenhydramine, amiodarone, ropivacaine, risperidone, bupivacaine, amoxapine, venlafaxine, dolasetron, citalopram, diltiazem, amodiaquine, carbamazepine, mesoridazine). Ten drugs annotated to be devoid of clinical QRS prolongation (protriptyline, clomipramine, chlorpromazine, clozapine, verapamil, flunarizine, riluzole, propranolol, pyrimethamine, quinacrine) were also selected. These were tested for hCx43 uncoupling activity in a 10-point 1/2-log dilution series format at a top test concentration of 50 μM (0.1% final DMSO concentration). Digoxin was included as a reference compound on every plate and gave a mean IC50 value of 0.15 μM (pIC50 6.8 ± 0.2 SD; n = 15).
Ten drugs inhibited hCx43-mediated dye transfer with IC50 values <50 μM (n = 3; Table 1 together with hNaV1.5 activity taken from Harmer et al. 2011). Seven of these (thioridazine, nortriptyline, desipramine, maprotiline, fluoxetine, perhexiline, lamotrigine) were annotated with clinical QRS prolongation and three (protriptyline, clomipramine, chlorpromazine) were not. Using these criteria (i.e. hCx43 IC50 < 50 μM and the annotation of Harmer et al., 2011), the sensitivity and specificity for prediction of clinical QRS prolongation is 29% and 70% respectively. However, QRS prolongation in preclinical models has also been reported for protriptyline (Dumovic et al., 1977), clomipramine (Timour et al., 1990) and chlorpromazine (Nagamoto et al., 1976). Including these preclinical results would alter the sensitivity and specificity to 37% and 100%, respectively, although hNaV1.5 activity must also be taken into account. Of the 34 drugs tested, five are inactive at the hNaV1.5 channel (IC50 > 50 μM; lamotrigine, bupropion, venlafaxine, dolasetron, carbamazepine; data from Harmer et al., 2011) and, of these, one (20%; lamotrigine) was found to uncouple hCx43 (Figure 6).
Table 1.
Marketed drugs tested for hCx43 uncoupling activity
| Drug | hCx43 IC50 (μM) n = 3 | hNaV1.5 IC50 (μM)a | Categorya | Therapeutic free plasma concentration, highest (μM)a |
|---|---|---|---|---|
| Thioridazine | 14 | 3.5 | QRS | 0.98 |
| Perhexiline | 18 | 3.4 | QRS | 0.22 |
| Lamotrigine | 21 | 63.4 | QRS | 24 |
| Maprotiline | 25 | 2.8 | QRS | 0.24 |
| Nortriptyline | 28 | 2 | QRS | 0.036 |
| Fluoxetine | 35 | 4.9 | QRS | 0.093 |
| Desipramine | 45 | 2.4 | QRS | 0.36 |
| Amitriptyline | >50 | 1.6 | QRS | 0.049 |
| Bupropion | >50 | 76.2 | QRS | 0.095 |
| Imipramine | >50 | 3.6 | QRS | 0.13 |
| Quinine | >50 | 17.8 | QRS | 1.8 |
| Diphenhydramine | >50 | 16.4 | QRS | 0.034 |
| Amiodarone | >50 | 4.8 | QRS | 0.062 |
| Ropivacaine | >50 | 11.2 | QRS | 0.39 |
| Risperidone | >50 | 20.2 | QRS | 2.1 |
| Bupivacaine | >50 | 4.3 | QRS | 0.31 |
| Amoxapine | >50 | 5.8 | QRS | 0.19 |
| Venlafaxine | >50 | 90.1 | QRS | 1.1 |
| Dolasetron | >50 | 79.7 | QRS | 0.64 |
| Citalopram | >50 | 14.7 | QRS | 0.12 |
| Diltiazem | >50 | 14.2 | QRS | 1.1 |
| Amodiaquine | >50 | 9.7 | QRS | 0.058 |
| Carbamazepine | >50 | 152 | QRS | 13 |
| Mesoridazine | >50 | 7.2 | QRS | 0.13 |
| Clomipramine | 23 | 2.6 | Non-QRS (preclinical QRS) | 0.03 |
| Chlorpromazine | 28 | 3.1 | Non-QRS (preclinical QRS) | 0.065 |
| Protriptyline | 36 | 3.1 | Non-QRS (preclinical QRS) | 0.091 |
| Clozapine | >50 | 11.6 | Non-QRS | 0.092 |
| Verapamil | >50 | 9.3 | Non-QRS | 0.081 |
| Flunarizine | >50 | 2.6 | Non-QRS | 0.034 |
| Riluzole | >50 | 17.6 | Non-QRS | 0.26 |
| Propranolol | >50 | 7.5 | Non-QRS | 0.08 |
| Pyrimethamine | >50 | 24.6 | Non-QRS | 0.15 |
| Quinacrine | >50 | 20.2 | Non-QRS | 0.25 |
Taken from Harmer et al. (2011).
Figure 6.
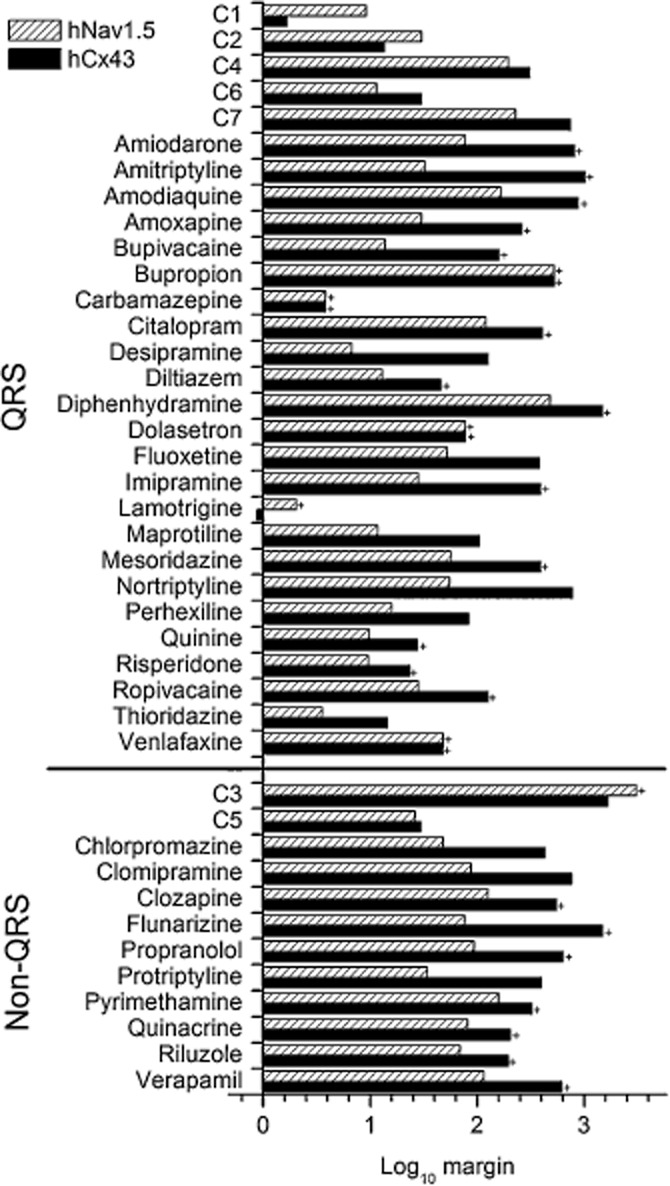
Comparison of hCx43 and hNaV1.5 IC50 values of all preclinical and marketed drugs assayed relative to therapeutic free plasma concentrations. The log10 ratio of IC50 : free Cmax was calculated using data presented in Tables 1 and 2. An IC50 value of 50 μM has been used to calculate the ratio where hNaV1.5 or hCx43 IC50 values were >50 μM (indicated by a + symbol).
hCx43 uncoupling activity of AstraZeneca compounds with preclinical study data
A set of compounds from AstraZeneca's internal library with preclinical safety assessment study data, including QRS duration and plasma exposure levels, were tested for hCx43 uncoupling activity. Of the 29 compounds known to cause QRS prolongation, together with 11 without such effects, seven were found to be hCx43 uncoupling agents (Table 2). Data from hNaV1.5 automated patch clamp analysis indicated that most of these seven compounds were also active at the hNaV1.5 channel (Table 2). Generally, hCx43 and hNaV1.5 IC50 values were similar (within threefold), giving no clear examples where QRS interval changes might be attributed to either mechanism alone, although compound C1 showed the most difference with a more than fivefold lower hCx43 IC50 value. A plot of hCx43 versus hNaV1.5 IC50 value is given in Figure 5, including both preclinical and marketed drugs annotated for QRS prolongation.
Table 2.
Preclinical compounds active (IC50 < 50 μM) in hCx43 assay together with preclinical assessment data
| Compound | hCx43 IC50 (μM) n = 3 | hNaV1.5 IC50 (μM) | hCx43 : hNaV1.5 ratioa | In vivo species | Highest Cmax (total) (μM) | Cmax (free) (μM) | IC50 : Cmax (free) ratiob | QRSc | |
|---|---|---|---|---|---|---|---|---|---|
| hCx43 | hNaV1.5 | ||||||||
| C1 | 9 | 49 | 5.4 | Guinea pig | 54 | 5.4 | 1.7 | 9.1 | Yes |
| C2 | 13 | 30 | 2.3 | Guinea pig | 34 | 1.0 | 13 | 30 | Yes |
| C3 | 27 | >50 | >1.9 | Dog | 0.23 | 0.016 | >100 | >100 | No |
| C4 | 31 | 20 | 0.6 | Dog | 6 | 0.1 | >100 | >100 | Yes |
| C5 | 32 | 29 | 0.9 | Dog | 10 | 1.1 | 29 | 26 | No |
| C6 | 33 | 13 | 0.4 | Dog | 5 | 1.1 | 30 | 12 | Yes |
| C7 | 37 | 11 | 0.3 | Dog | 16 | 0.05 | >100 | >100 | Yes |
hCx43 : hNaV1.5 ratio is the ratio between hCx43 and hNaV1.5 IC50 values.
IC50 : Cmax (free) ratio is the ratio between the hCx43 or hNaV1.5 IC50 values and the compound free plasma concentration [Cmax (free)].
Indicates whether QRS prolongation was noted in the in vivo study.
Figure 5.
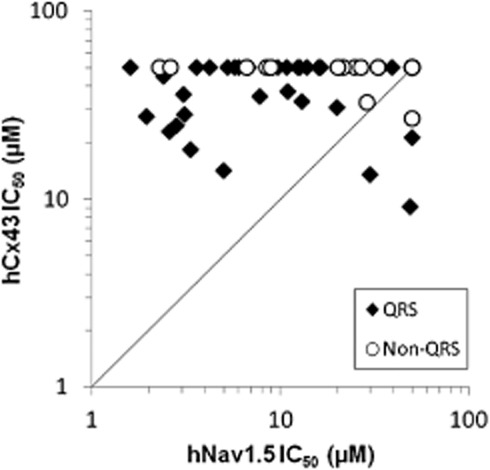
Comparison of hCx43 and hNaV1.5 activities of all assayed preclinical and marketed drugs. Combined data sets of all QRS prolongation-annotated compounds (preclinical and marketed) tested for hCx43 uncoupling IC50 plotted against hNaV1.5 IC50. Compounds assayed as inactive (>50 μM, non QRS) are plotted with a 50 μM IC50 value. Solid line has a slope of unity.
Total plasma exposure levels determined during the in vivo studies are given in Table 2, together with the free concentrations calculated using plasma protein binding data. Ratios between free plasma levels and IC50 values (hCx43 or hNaV1.5) are also presented and, for the purposes of this study, it was assumed that ratios greater than 100 would not be expected to produce any hCx43 or hNav1.5-mediated effect in vivo. On this basis, four compounds (C1, C2, C5 and C6) have been assessed at free exposure levels that might be expected, from their in vitro data, to produce hCx43 or hNaV1.5-mediated effects. Of these, the only compound (C5) not shown to produce QRS prolongation had hCx43 and hNaV1.5 IC50 values 29 and 26-fold, respectively, above free plasma levels. Two compounds known to cause QRS prolongation (C2 and C6) had IC50 values in both assays less than 30-fold above the free plasma levels. Compound C1, which displayed the greatest difference between IC50 values, caused QRS prolongation at a free plasma concentration 1.7-fold below the hCx43 IC50 value and 9.1-fold below the hNaV1.5 IC50 value (Figure 6).
Discussion and conclusions
Drug-induced QRS prolongation is a serious adverse event that may be prevented to some extent by screening for activity at the cardiac hNaV1.5 channel during drug development. Genetic and pharmacological evidence suggests that inhibition of cardiac gap junctions could also prolong the QRS interval; however, this association and its frequency have not been investigated systematically in drug and drug-like chemistry. In the present study, we demonstrated that off-target hCx43 uncoupling activity is clearly apparent in marketed drugs associated with QRS prolongation (37% of drugs based on a simple threshold classification). In candidate drugs where QRS prolongation in preclinical development contributed to their failure to reach the market, hCx43 uncoupling activity was also apparent. The IC50 values for hCx43 and hNaV1.5 activity were generally similar and it is possible that these are additive mechanisms for QRS prolongation. Although the combination of hCx43 IC50, hNaV1.5 IC50 and preclinical study plasma exposure data could not unambiguously attribute mechanism, examples were found (in marketed and preclinical drug) where compounds were more potent at hCx43 than hNaV1.5.
In vitro determination of hCx43 uncoupling IC50 value
Validation studies were performed to demonstrate that a parachute-type assay could produce robust IC50 values for compound ranking. Performance of the assay was good in terms of signal-to-noise (Z'-factor 0.7) under conditions where the majority of the plate well was imaged and analysed. A time period in excess of 3 h was established where plate imaging could be performed with acceptable signal size and repeatability of measured IC50 value. Imaging of nuclear staining ensured that the acceptor cell monolayer had not been disrupted by cytotoxic drug effects. Recently, another Cx uncoupling assay has been developed for compound screening (Haq et al., 2013); however, the current assay is a simpler system that may report fewer false positives. More complex assays utilizing neonatal cardiomyocytes rather than transfected HeLa cells have also been reported (Caillier et al., 2011) but are not scalable for compound profiling.
Concordance with studies of known gap junction uncoupling agents
As expected, known gap junction uncouplers were found to inhibit dye transfer in these experiments. IC50 values were in general agreement with those from other studies (Figure 4). Carbenoxolone concentrations as low as 3 μM have been shown to inhibit gap junctions in albumin-free conditions (Davidson et al., 1986; Davidson and Baumgarten, 1988), although 100–200 μM is more generally used (Srinivas, 2009). Heptanol concentrations of 1–2 mM inhibit electrical coupling between neonatal cardiomyocytes, via a mechanism thought to involve indirect actions on cholesterol-rich domains of the plasmalemma (Takens-Kwak et al., 1992; Bastiaanse et al., 1993). Meclofenamic acid (and other fenamates) has been shown to inhibit both electrical coupling and dye transfer mediated by hCx43, possibly by a direct interaction, with maximal inhibition occurring at approximately 100 μM in serum-free conditions (Harks et al., 2001). Nanomolar concentrations of cardiac glycosides such as digoxin are thought to uncouple gap junctions via an indirect action on intracellular Ca2+ (De Mello, 1976). Activation of PKC by nanomolar concentrations of phorbol ester in neonatal cardiomyocytes also inhibits dye transfer, presumably via an indirect effect on the phosphorylation cascade (Münster and Weingart, 1993; Kwak et al., 1995).
hCx43 uncoupling activity in marketed drugs
Ten marketed drugs were found to have hCx43 uncoupling activity in the present study (Table 1) and all are associated with preclinical or clinical data indicating a propensity to cause QRS prolongation. Six are antidepressants of the tricyclic (nortriptyline, desipramine, protriptyline and clomipramine), tetracyclic (maprotiline) or selective re-uptake inhibitor (fluoxetine) classes. Antipsychotics (thioridazine and chlorpromazine), anticonvulsants (lamotrigine) and antianginals (perhexiline) make up the remainder. Although gap junction modulation has been postulated as a potential anticonvulsant and antidepressant therapy (Carlen et al., 2000; Sun et al., 1992), very few drugs in clinical use primarily target Cx (for instance, the gap junction enhancer GAP-134 antiarrhythmic in phase I trials; De Vuyst et al., 2011). However, consistent with our data, off-target gap junction uncoupling activity has been shown previously for chlorpromazine (Orellana et al., 2006) and thioridazine (Matesic et al., 2006). Quinine, one of the few agents to show Cx isoform selectivity, was found to be inactive on hCx43, consistent with its previously published inhibitory profile (Srinivas et al., 2001). The difference in bupropion potency between the current study and that of Caillier et al. (2011) could be related to the use of alternative assay methodologies and drug-protein binding in the presence/absence of serum. In the present study, hNaV1.5 IC50 values were determined in the absence of serum proteins, which might bind compounds and lower their effective free concentrations, whereas the hCx43 IC50 value was determined in the presence of 10% serum. Highly protein-bound drugs, such as thioridazine (plasma protein binding 98%) and fluoxetine (94%) might be more potent under serum-free Cx43 assay conditions.
Significant hNaV1.5 activity was apparent in the set of marketed drugs, in both those associated with QRS prolongation and in those free from such effects. Drug-induced QRS prolongation often occurs outside the therapeutic plasma range, and reliable exposure data are scarce. Hence, interpretation of the marketed drug data set with regard to plasma exposures is complicated and, as an alternative, AstraZeneca internal compound data have been considered below in context with accurate plasma exposure measurements. For the validation compounds and marketed drug set, it is interesting to note that carbenoxolone (IC50 210 μM in the current study), at a concentration of 50 μM, has been shown to slow conduction velocity in rabbit Langendorff hearts but only uncouple a subset of electrically coupled cardiomyocyte cell pairs (de Groot et al., 2003). Heptanol (IC50 2208 μM in the current study) increases QRS duration in guinea pig Langendorff hearts by approximately 12 ms when used at 700 μM (Caillier et al., 2011). For the marketed drugs generally, examples were apparent where hCx43 and hNaV1.5 activities were of relatively similar potency and, in one case, the hCx43 activity was the more potent. These limited data suggest that changes to the ECG might occur at concentrations well below the IC50 value determined in the current study and, furthermore, that both hNaV1.5 and hCx43 mechanisms might contribute to QRS prolongation. However, larger data sets are required to complete a quantitative translation between in vitro and in vivo parameters.
Potential mechanisms of QRS prolongation in preclinical compounds
hCx43 uncoupling activity was also determined in a set of compounds from AstraZeneca's internal library that had known effects on QRS interval determined in preclinical safety assessment studies (Table 2). Hence, exposure data in the form of total plasma levels and calculated free concentrations were available and are presented in Table 2. Inhibition of hNaV1.5 was also determined for these compounds by automated patch clamp analysis. This allowed some comparison of the plasma exposures that produced QRS effects together with in vitro hCx43 and hNaV1.5 IC50 values, albeit using a small data set. A caveat is that the hCx43 IC50 was determined in the presence of 10% serum while the hNaV1.5 IC50 was determined in the absence of serum. Seven (of a total of 40 compounds) were found to uncouple hCx43. As described in the Results, four compounds were deemed to have applicable preclinical study data and one of these did not cause QRS prolongation at a free plasma exposure approximately 30-fold from the hCx43 IC50 value. The remaining three compounds were known to cause QRS prolongation, and displayed activity at both hCx43 and hNaV1.5, generally at exposures within 10- to 30-fold of in vitro IC50 values. While it has not been possible to distinguish compounds which might cause QRS prolongation through hCx43 uncoupling alone (given that the degree of uncoupling required to manifest the effect is unknown), nevertheless the data are consistent with both mechanisms contributing to the increase in QRS duration. Preclinical study data from two species have been combined for this analysis; however, this is a common situation in the drug development process where a cascade of increasingly complex in vitro assays and in vivo studies are performed to assess both efficacy and safety prior to human trials.
Cx uncoupling and other adverse events
Data from the Framingham Heart Study suggest that first-degree AV block (PR interval prolongation) is associated with increased mortality and AV conduction is highly dependent on Cx40 gap junction within the His–Purkinje conduction system (Kirchhoff et al., 1998; Severs et al., 2009; Cheng et al., 2009). Gap junctions also play a variety of other important physiological roles outside of the heart, including foetal development (hCx43 has been linked to cleft palate; Paznekas et al., 2003; Jugessur et al., 2011) and serious skin disorders (particularly hCx26; van Steensel, 2004). Lamotrigine, which uncoupled hCx43 in the present study, is associated with adverse events that include severe skin rash and teratogenicity, particularly cleft lip/palate, resulting in an FDA Alert to this effect in 2006 (Biton, 2006; Aurich-Barrera et al., 2010; FDA Website, 2012, see references). It is also noteworthy that bone marrow transplantations typically involve infusions of cells suspended in 10% DMSO cyroprotectant solutions and are associated with AV block and cardiac arrhythmias (Styler et al., 1992; Ruiz-Delgado et al., 2009). Given the IC50 value of 0.8% that we determined for DMSO, it is possible that localized cardiac DMSO concentrations are sufficient during infusion of a 10% solution to uncouple cardiac gap junctions. Whether off-target inhibition of gap junctions contributes to these significant adverse events is unknown; however, the possiblity that additional Cx isoforms mediate drug toxicities is intriguing.
Limitations of the study
Uncoupling activity demonstrated via an in vitro ‘parachute'-type assay does not necessarily imply a direct block of the junction pore by the drug molecule. It has also been suggested that the gating mechanisms for dye tracers and electrical coupling can be distinct, with electrical coupling potentially remaining intact when dye transfer is blocked (Bukauskas et al., 2006). Additionally, a drug's effect might vary considerably when measured over a period of hours (the conditions for dye transfer) or ms (QRS prolongation). The dye transfer experiments rely on both pre-existing gap junctions as well as the formation of new ones, a process that might not be relevant to myocardial gap junctions (although gap junction protein turnover is thought to be rapid; Laird et al., 1991).
The structural organization of the gap junction in a heterologous expression system may not fully represent the in vivo situation. Gap junctions are specialized areas on membrane structure and signalling. However, the present study did not aim to fully duplicate the cardiac myocyte, but to isolate a single mechanistic component (i.e. hCx43) of known importance to cardiac myocyte coupling. In this regard, it is known that other Cx isoforms (particularly Cx40; Kirchhoff et al., 1998; Hagendorff et al., 1999) are closely involved with impulse propagation and QRS waveform. Given the present demonstration of hCx43 uncoupling activity in compounds associated with QRS prolongation, examination of Cx40 and other isoforms is warranted.
Conclusions
We have systematically evaluated off-target gap junction uncoupling activity in marketed and preclinical drugs with the aim of understanding its potential role in drug toxicity. hCx43 uncoupling activity was clearly apparent in drugs associated with causing serious cardiac electrophysiological disturbances manifest as prolongation of the QRS waveform. Our data show a significant hit rate of up to 37% in drugs associated with QRS prolongation, together with coexistent hNaV1.5 activity. While the translation of in vitro hCx43 and hNaV1.5 data to ECG abnormalities observed in the clinic is an evolving area, the current data suggest that both activities are apparent at similar in vitro concentrations and hence QRS prolongation might result from both mechanisms in combination in some cases. Avoiding gap junction modulation in drug development could be a useful consideration in the prevention of off-target drug toxicity.
Conflicts of interest
None.
Glossary
- Cx
connexin
- PDBu
phorbol 12,13-dibutyrate
References
- Abbaci M, Barberi-Heyob M, Blondel W, Guillemin F, Didelon J. Advantages and limitations of commonly used methods to assay the molecular permeability of gap junctional intercellular communication. Biotechniques. 2008;45:33–52. doi: 10.2144/000112810. 56–62. [DOI] [PubMed] [Google Scholar]
- Adesanya CO, Yousuf KA, Co C, Gaur S, Ahmed S, Pothoulakis A, et al. Is wider worse? QRS duration predicts cardiac mortality in patients with right bundle branch block. Ann Noninvasive Electrocardiol. 2008;13:165–170. doi: 10.1111/j.1542-474X.2008.00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurich-Barrera B, Wilton L, Brown D, Shakir S. Paediatric postmarketing pharmacovigilance using prescription-event monitoring: comparison of the adverse event profiles of lamotrigine prescribed to children and adults in England. Drug Saf. 2010;33:751–763. doi: 10.2165/11536830-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Bastiaanse EM, Jongsma HJ, van der Laarse A, Takens-Kwak BR. Heptanol-induced decrease in cardiac gap junctional conductance is mediated by a decrease in the fluidity of membranous cholesterol-rich domains. J Membr Biol. 1993;136:135–145. doi: 10.1007/BF02505758. [DOI] [PubMed] [Google Scholar]
- Bevans CG, Harris AL. Regulation of connexin channels by pH. Direct action of the protonated form of taurine and other aminosulfonates. J Biol Chem. 1999;274:3711–3719. doi: 10.1074/jbc.274.6.3711. [DOI] [PubMed] [Google Scholar]
- Biton V. Pharmacokinetics, toxicology and safety of lamotrigine in epilepsy. Expert Opin Drug Metab Toxicol. 2:1009–1018. doi: 10.1517/17425255.2.6.1009. [DOI] [PubMed] [Google Scholar]
- Bukauskas FF, Bukauskiene A, Verselis VK. Conductance and permeability of the residual state of connexin43 gap junction channels. J Gen Physiol. 2006;119:171–185. doi: 10.1085/jgp.119.2.171. 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillier B, Pilote S, Castonguay A, Patoine D, Ménard-Desrosiers V, Vigneault P, et al. QRS widening and QT prolongation under bupropion: a unique cardiac electrophysiological profile. Fundam Clin Pharmacol. 2011;26:599–608. doi: 10.1111/j.1472-8206.2011.00953.x. [DOI] [PubMed] [Google Scholar]
- Carlen PL, Skinner F, Zhang L, Naus C, Kushnir M, Perez Velazquez JL. The role of gap junctions in seizures. Brain Res Brain Res Rev. 2000;32:235–241. doi: 10.1016/s0165-0173(99)00084-3. [DOI] [PubMed] [Google Scholar]
- Cheng S, Keyes MJ, Larson MG, McCabe EL, Newton-Cheh C, Levy D, et al. Long-term outcomes in individuals with prolonged PR interval or first-degree atrioventricular block. JAMA. 2009;301:2571–2577. doi: 10.1001/jama.2009.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani V, Mikalsen SO. The vertebrate connexin family. Cell Mol Life Sci. 2006;63:1125–1140. doi: 10.1007/s00018-005-5571-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danik SB, Liu F, Zhang J, Suk HJ, Morley GE, Fishman GI, et al. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res. 2004;95:1035–1041. doi: 10.1161/01.RES.0000148664.33695.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson JS, Baumgarten IM. Glycyrrhetinic acid derivatives: a novel class of inhibitors of gap-junctional intercellular communication. Structure-activity relationships. J Pharmacol Exp Ther. 1988;246:1104–1107. [PubMed] [Google Scholar]
- Davidson JS, Baumgarten IM, Harley EH. Reversible inhibition of intercellular junctional communication by glycyrrhetinic acid. Biochem Biophys Res Commun. 1986;134:29–36. doi: 10.1016/0006-291x(86)90522-x. [DOI] [PubMed] [Google Scholar]
- De Mello WC. Influence of the sodium pump on intercellular communication in heart fibres: effect of intracellular injection of sodium ion on electrical coupling. J Physiol. 1976;263:171–197. doi: 10.1113/jphysiol.1976.sp011627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vuyst E, Boengler K, Antoons G, Sipido KR, Schulz R, Leybaert L. Pharmacological modulation of connexin-formed channels in cardiac pathophysiology. Br J Pharmacol. 2011;163:469–483. doi: 10.1111/j.1476-5381.2011.01244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumovic P, Trethewie ER, Burrows G. The effect of tricyclic antidepressant drugs on the isolated perfused guinea-pig heart. Clin Exp Pharmacol Physiol. 1977;4:421–424. doi: 10.1111/j.1440-1681.1977.tb02679.x. [DOI] [PubMed] [Google Scholar]
- FDA website. 2012. Information for healthcare professionals: Lamotrigine (marketed as Lamictal). Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm126225.htm (accessed 20/11/ )
- Goldberg GS, Bechberger JF, Naus CC. A pre-loading method of evaluating gap junctional communication by fluorescent dye transfer. Biotechniques. 1995;18:490–497. [PubMed] [Google Scholar]
- de Groot JR, Veenstra T, Verkerk AO, Wilders R, Smits JP, Wilms-Schopman FJ, et al. Conduction slowing by the gap junctional uncoupler carbenoxolone. Cardiovasc Res. 2003;60:288–297. doi: 10.1016/j.cardiores.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagendorff A, Schumacher B, Kirchhoff S, Lüderitz B, Willecke K. Conduction disturbances and increased atrial vulnerability in connexin40-deficient mice analyzed by transesophageal stimulation. Circulation. 1999;99:1508–1515. doi: 10.1161/01.cir.99.11.1508. [DOI] [PubMed] [Google Scholar]
- Haq N, Grose G, Ward E, Chiu O, Tigue N, Dowell SJ, et al. A high-throughput assay for connexin 43 (Cx43, GJA1) gap junctions using codon-optimized aequorin. Assay Drug Dev Technol. 2013;11:93–100. doi: 10.1089/adt.2012.469. [DOI] [PubMed] [Google Scholar]
- Harks EG, de Roos AD, Peters PH, de Haan LH, Brouwer A, Ypey DL, et al. Fenamates: a novel class of reversible gap junction blockers. J Pharmacol Exp Ther. 2001;298:1033–1041. [PubMed] [Google Scholar]
- Harmer AR, Abi-Gerges N, Easter A, Woods A, Lawrence CL, Small BG, et al. Optimisation and validation of a medium-throughput electrophysiology-based hNav1.5 assay using IonWorks. J Pharmacol Toxicol Methods. 2008;57:30–41. doi: 10.1016/j.vascn.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Harmer AR, Valentin JP, Pollard CE. On the relationship between block of the cardiac Na+ channel and drug-induced prolongation of the QRS complex. Br J Pharmacol. 2011;164:260–273. doi: 10.1111/j.1476-5381.2011.01415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugessur A, Shi M, Gjessing HK, Lie RT, Wilcox AJ, Weinberg CR, et al. Fetal genetic risk of isolated cleft lip only versus isolated cleft lip and palate: a subphenotype analysis using two population-based studies of orofacial clefts in Scandinavia. Birth Defects Res A Clin Mol Teratol. 2011;91:85–92. doi: 10.1002/bdra.20747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalahasti V, Nambi V, Martin DO, Lam CT, Yamada D, Wilkoff BL, et al. QRS duration and prediction of mortality in patients undergoing risk stratification for ventricular arrhythmias. Am J Cardiol. 2003;92:798–803. doi: 10.1016/s0002-9149(03)00886-5. [DOI] [PubMed] [Google Scholar]
- Kelsell DP, Dunlop J, Hodgins MB. Human diseases: clues to cracking the connexin code? Trends Cell Biol. 2001;11:2–6. doi: 10.1016/s0962-8924(00)01866-3. [DOI] [PubMed] [Google Scholar]
- Kirchhoff S, Nelles E, Hagendorff A, Krüger O, Traub O, Willecke K. Reduced cardiac conduction velocity and predisposition to arrhythmias in connexin40-deficient mice. Curr Biol. 1998;8:299–302. doi: 10.1016/s0960-9822(98)70114-9. [DOI] [PubMed] [Google Scholar]
- Kojodjojo P, Kanagaratnam P, Segal OR, Hussain W, Peters NS. The effects of carbenoxolone on human myocardial conduction: a tool to investigate the role of gap junctional uncoupling in human arrhythmogenesis. J Am Coll Cardiol. 2006;48:1242–1249. doi: 10.1016/j.jacc.2006.04.093. [DOI] [PubMed] [Google Scholar]
- Kwak BR, van Veen TA, Analbers LJ, Jongsma HJ. TPA increases conductance but decreases permeability in neonatal rat cardiomyocyte gap junction channels. Exp Cell Res. 1995;220:456–463. doi: 10.1006/excr.1995.1337. [DOI] [PubMed] [Google Scholar]
- Laird DW, Puranam KL, Revel JP. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem J. 1991;273(Pt 1):67–72. doi: 10.1042/bj2730067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardizabal DV, Morris HH, Hovinga CA, Del Mar Carreño M. Tolerability and pharmacokinetics of oral loading with lamotrigine in epilepsy monitoring units. Epilepsia. 2003;44:536–539. doi: 10.1046/j.1528-1157.2003.46902.x. [DOI] [PubMed] [Google Scholar]
- Lu HR, Rohrbacher J, Vlaminckx E, Van Ammel K, Yan GX, Gallacher DJ. Predicting drug-induced slowing of conduction and pro-arrhythmia: identifying the ‘bad’ sodium current blockers. Br J Pharmacol. 2010;160:60–76. doi: 10.1111/j.1476-5381.2010.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall EJ, Danielsson A, Hardern I, Dartsch C, Hicks R, Wahlberg JM, et al. Improvements to the throughput of recombinant protein expression in the baculovirus/insect cell system. Protein Expr Purif. 2005;42:29–36. doi: 10.1016/j.pep.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Madias JE. Drug-induced QRS morphology and duration changes. Cardiol J. 2008;15:505–509. [PubMed] [Google Scholar]
- Matesic DF, Abifadel DN, Garcia EL, Jann MW. Effect of thioridazine on gap junction intercellular communication in connexin 43-expressing cells. Cell Biol Toxicol. 2006;22:257–268. doi: 10.1007/s10565-006-0047-7. [DOI] [PubMed] [Google Scholar]
- Mills SL, Massey SC. The kinetics of tracer movement through homologous gap junctions in the rabbit retina. Vis Neurosci. 1998;15:765–777. doi: 10.1017/s0952523898154159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münster PN, Weingart R. Effects of phorbol ester on gap junctions of neonatal rat heart cells. Pflugers Arch. 1993;423:181–188. doi: 10.1007/BF00374392. [DOI] [PubMed] [Google Scholar]
- Nagamoto Y, Arita M, Saikawa T. The prolongation of QRS-duration resulting from delayed recovery of ventricular excitability. A new mechanism for intraventricular conduction disturbance. A preliminary note. Jpn Heart J. 1976;17:760–761. doi: 10.1536/ihj.17.760. [DOI] [PubMed] [Google Scholar]
- Orellana JA, Palacios-Prado N, Sáez JC. Chlorpromazine reduces the intercellular communication via gap junctions in mammalian cells. Toxicol Appl Pharmacol. 2006;213:187–197. doi: 10.1016/j.taap.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard CE, Abi Gerges N, Bridgland-Taylor MH, Easter A, Hammond TG, Valentin JP. An introduction to QT interval prolongation and non-clinical approaches to assessing and reducing risk. Br J Pharmacol. 2010;159:12–21. doi: 10.1111/j.1476-5381.2009.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhett JM, Ongstad EL, Jourdan J, Gourdie RG. Cx43 associates with Nav1.5 in the cardiomyocyte perinexus. J Membrane Biol. 2012;245:411–422. doi: 10.1007/s00232-012-9465-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res. 2004;62:309–322. doi: 10.1016/j.cardiores.2003.11.035. [DOI] [PubMed] [Google Scholar]
- Ruiz-Delgado GJ, Mancías-Guerra C, Tamez-Gómez EL, Rodríguez-Romo LN, López-Otero A, Hernández-Arizpe A, et al. Dimethyl sulfoxide-induced toxicity in cord blood stem cell transplantation: report of three cases and review of the literature. Acta Haematol. 122:1–5. doi: 10.1159/000227267. [DOI] [PubMed] [Google Scholar]
- Severs NJ, Coppen SR, Dupont E, Yeh HI, Ko YS, Matsushita T. Gap junction alterations in human cardiac disease. Cardiovasc Res. 2009;62:368–377. doi: 10.1016/j.cardiores.2003.12.007. 2004. [DOI] [PubMed] [Google Scholar]
- Srinivas M. Pharmacology of connexin channels. In: Harris A, Locke D, editors. Connexins. New York: Humana Press; 2009. pp. 207–224. [Google Scholar]
- Srinivas M, Hopperstad MG, Spray DC. Quinine blocks specific gap junction channel subtypes. Proc Natl Acad Sci U S A. 2001;98:10942–10947. doi: 10.1073/pnas.191206198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel MA. Gap junction diseases of the skin. Am J Med Genet C Semin Med Genet. 2004;131C:12–19. doi: 10.1002/ajmg.c.30030. [DOI] [PubMed] [Google Scholar]
- Styler MJ, Topolsky DL, Crilley PA, Covalesky V, Bryan R, Bulova S, et al. Transient high grade heart block following autologous bone marrow infusion. Bone Marrow Transplant. 10:435–438. [PubMed] [Google Scholar]
- Sun JD, Liu Y, Yuan YH, Li J, Chen NH. Gap junction dysfunction in the prefrontal cortex induces depressive-like behaviors in rats. Neuropsychopharmacology. 1992;37:1305–1320. doi: 10.1038/npp.2011.319. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takens-Kwak BR, Jongsma HJ, Rook MB, Van Ginneken AC. Mechanism of heptanol-induced uncoupling of cardiac gap junctions: a perforated patch-clamp study. Am J Physiol. 1992;262(6 Pt 1):C1531–C1538. doi: 10.1152/ajpcell.1992.262.6.C1531. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Schuessler RB, Berul CI, Beardslee MA, Beyer EC, Mendelsohn ME, et al. Disparate effects of deficient expression of connexin43 on atrial and ventricular conduction: evidence for chamber-specific molecular determinants of conduction. Circulation. 1998;97:686–691. doi: 10.1161/01.cir.97.7.686. [DOI] [PubMed] [Google Scholar]
- Timour Q, Freysz M, Couzon P, Loufoua J, Bertrix L, Gerentes I, et al. Possible role of drug interactions in bupivacaine-induced problems related to intraventricular conduction disorders. Reg Anesth. 1990;15:180–185. [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zimmerman AL, Rose B. Permeability properties of cell-to-cell channels: kinetics of fluorescent tracer diffusion through a cell junction. J Membr Biol. 1985;84:269–283. doi: 10.1007/BF01871390. [DOI] [PubMed] [Google Scholar]