

Abstract
Background and Purpose
The most common mutation in cystic fibrosis (CF), F508del, causes defects in trafficking, channel gating and endocytosis of the CF transmembrane conductance regulator (CFTR) protein. Because CF is an orphan disease, therapeutic strategies aimed at improving mutant CFTR functions are needed to target the root cause of CF.
Experimental Approach
Human CF airway epithelial cells were treated with roscovitine 100 μM for 2 h before CFTR maturation, expression and activity were examined. The mechanism of action of roscovitine was explored by recording the effect of depleting endoplasmic reticulum (ER) Ca2+ on the F508del-CFTR/calnexin interaction and by measuring proteasome activity.
Key Results
Of the cyclin-dependent kinase (CDK) inhibitors investigated, roscovitine was found to restore the cell surface expression and defective channel function of F508del-CFTR in human CF airway epithelial cells. Neither olomoucine nor (S)-CR8, two very efficient CDK inhibitors, corrected F508del-CFTR trafficking demonstrating that the correcting effect of roscovitine was independent of CDK inhibition. Competition studies with inhibitors of the ER quality control (ERQC) indicated that roscovitine acts on the calnexin pathway and on the degradation machinery. Roscovitine was shown (i) to partially inhibit the interaction between F508del-CFTR and calnexin by depleting ER Ca2+ and (ii) to directly inhibit the proteasome activity in a Ca2+-independent manner.
Conclusions and Implications
Roscovitine is able to correct the defective function of F508del-CFTR by preventing the ability of the ERQC to interact with and degrade F508del-CFTR via two synergistic but CDK-independent mechanisms. Roscovitine has potential as a pharmacological therapy for CF.
Table of Links
| TARGETS | LIGANDS |
|---|---|
| CDK1 | 2-APB |
| CDK2 | ATP |
| CDK5 | Calmodulin |
| CDK9 | Curcumin |
| CFTR | DIDS |
| CK1 | DPC |
| CLK3 | Forskolin |
| DYRK1A | Genistein |
| ERK2 | Glibenclamide |
| GSK-3 | HSP90 |
| IP3 receptor | KN62 |
| KV1.3 | Miglustat |
| KV2.1 | NO |
| KV4.3 | Roscovitine |
| KV11.1 | Sildenafil |
| L-type Ca channels | Thapsigargin |
| NaV1.5 | Vardenafil |
This Table lists key protein targets and ligands in this document, which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a,b).
Introduction
Protein misfolding contributes to a large number of diseases, which include diabetes mellitus, atherosclerosis, sickle cell anaemia and a number of neurodegenerative disorders such as Alzheimer's, Parkinson's or Huntington's diseases (for review see Herczenik and Gebbink, 2008). In many cases, misfolded proteins accumulate and aggregate within pathological cells. In others, the misfolded protein is rapidly degraded and cleared from the cell, leading to a loss-of-function phenotype; this being an example of the most common form of cystic fibrosis (CF). CF is an autosomal recessive disorder caused by mutations in the CF transmembrane conductance regulator (CFTR) gene (for review see Becq, 2010; Lukacs and Verkman, 2012). CFTR is a cAMP-activated, ATP-gated chloride channel (Riordan, 2008). With some variability, the F508del mutation is observed in most CF individuals worldwide. The loss of phenylalanine at position 508 in the CFTR protein results in protein misfolding and rapid degradation by the ubiquitin-proteasome system through a process referred to as endoplasmic reticulum (ER)-associated degradation (ERAD) (Lukacs and Verkman, 2012).
Cellular chaperones play a key role in helping the intracellular processing of proteins, including that of CFTR. A host of chaperones, enzymes and regulatory proteins direct the folding, complex assembly and ultimately the exit of secretory proteins, which can be viewed as the clients of this ER machinery (Ellgaard and Helenius, 2003). Among the strategies for CF treatments, approaches to correct the dysfunction of mutant protein biogenesis have been encouraged by two important observations. Firstly, the F508del mutant is temperature-sensitive (culturing CF cells for 24 h at 27°C facilitates some protection of F508del-CFTR from ERAD and delivery to the apical surface, presumably by promoting correct folding of F508del-CFTR) and, secondly, it retains some biological activity despite a clear gating defect (Denning et al., 1992). Based on the dissection of the molecular machinery controlling the biogenesis of misfolded proteins, there have been significant efforts to discover small molecules that can mimic the effect of low temperature or chemical chaperones (Becq, 2010). To reverse the defective trafficking of F508del-CFTR, a number of methods have been employed including the use of proteostasis regulators, sarcoplasmic-reticulum Ca2+-ATPase inhibitors, such as thapsigargin (Egan et al., 2002; Norez et al., 2006a), nitric oxide donors (Zaman et al., 2001), the heat shock protein (HSP) modulator sodium 4-phenylbutyrate (Rubenstein and Zeitlin, 2000), the glucosidase inhibitor miglustat (Lubamba et al., 2009; Norez et al., 2009), the type V phosphodiesterase inhibitors sildenafil and vardenafil (Dormer et al., 2005) and histone deacetylase inhibitors (Hutt et al., 2010). Another strategy is to use pharmacological chaperones, that is small molecules directly interacting with the CFTR protein, that promote its folding (Pedemonte et al., 2014; Van et al., 2006; Norez et al., 2008). However, despite these advances, therapies for CF have not improved, possibly due to insufficient restoration of CFTR function and/or poor selectivity for the processing of CFTR compared with other proteins and off-target effects. VX-809 represents a class of CFTR correctors, distinguished from previously described CFTR correctors by the magnitude of its increase in F508del-CFTR-mediated chloride transport in cultures of F508del-HBE and its selectivity for CFTR compared to other normal and misfolded proteins (Van et al., 2011). The potentiator Kalydeco™ (also known as Ivacaftor or VX-770), developed by Vertex Pharmaceuticals (Cambridge, MA, USA), has recently been approved by the United States Food and Drug Administration and the European Medicines Agency for the treatment of CF patients carrying at least one CFTR allele with the G551D mutation (2–5% of all patients). In contrast, VX-809 is still in clinical trials. Because of multiple defects caused by the F508del mutation, it is probable that correction of the mutant protein will require combined treatment with drugs having different mechanisms of action (for review see Galietta, 2013). At present, despite extensive research efforts since cloning of the CF gene, the medications prescribed to CF patients still target the secondary manifestations of the disease rather than its cause. Therefore, new molecular targets and small pharmacological ligands that overcome the processing defect of F508del-CFTR and traffic the mutant protein to the apical membrane are needed. Collectively, these molecules are termed CFTR correctors because they restore the cell surface expression of the mutant protein. The design of small-molecule inhibitors of protein kinases is currently an area of intense interest in medicine; thus we screened a small library of protein kinase inhibitors, comprising inhibitors of cyclin-dependent kinases (CDK). Although the search for CDK inhibitors was initially directed towards treatments for cancers, they are also being evaluated for other indications such as polycystic kidney disease (Bukanov et al., 2006) and Alzheimer's disease (for review see Knockaert et al., 2002; Wesierska-Gadek et al., 2009; Galons et al., 2010). Thus, increasing attention has been devoted to identifying additional targets for CDK inhibitors. During our drug screening campaign, we identified a new corrector of F508del-CFTR, roscovitine, a CDK inhibitor already in clinical trials as a potential cancer therapy (Meijer et al., 1997; Meijer and Raymond, 2003; Bettayeb et al., 2008; 2010,). Our results show that this correction, resulting from an inhibitory effect on the ability of the ER quality control (ERQC) to interact with and degrade F508del-CFTR proteins, occurs through a CDK-independent mechanism.
Methods
Kinase preparations and assays
Buffer A: 10 mM MgCl2, 1 mM EGTA, 1 mM DTT, 25 mM Tris-HCl pH 7.5, 50 μg heparin·mL−1. Buffer C: 60 mM ß-glycerophosphate, 15 mM p-nitrophenylphosphate, 25 mM 3-(N-morpholino)propanesulfonic acid (Mops) (pH 7.2), 5 mM EGTA, 15 mM MgCl2, 1 mM DTT, 1 mM sodium vanadate, 1 mM phenylphosphate. Kinase activities were assayed in Buffer A or C, at 30°C, at a final ATP concentration of 15 μM. Blank values were subtracted and activities were expressed as % of the maximal activity, that is in the absence of inhibitors. Controls were performed with appropriate dilutions of dimethylsulfoxide (DMSO). CDK1/cyclin B (M phase starfish oocytes, native), CDK2/cyclin A and CDK5/p25 (human, recombinant) were prepared as previously described (Bettayeb et al., 2008). Their kinase activity was assayed in buffer C, with 1 mg histone H1 mL−1, in the presence of 15 μM [γ-33P]-ATP (3000 Ci mmol−1; 10 mCi·mL−1) in a final volume of 30 μL. After 30 min of incubation at 30°C, 25 μL aliquots of supernatant were spotted onto 2.5 × 3 cm pieces of Whatman P81 phosphocellulose paper (VWR International, Fontenay-sous-Bois, France), and 20 s later, the filters were washed five times (for at least 5 min each time) in a solution of 10 mL phosphoric acid L−1 water. The wet filters were counted in the presence of 1 mL ACS (Amersham, VWR International) scintillation fluid. CDK9/cyclin T (human, recombinant, expressed in insect cells) was assayed as described for CDK1/cyclin B, but using a pRB fragment (a.a.773–928) (3.5 μg per assay) as a substrate. GSK-3α/β (porcine brain, native) was assayed as described for CDK1 but in buffer A and using a GSK-3-specific substrate (GS-1: YRRAAVPPSPSLSRHSSPHQpSEDEEE) (pS stands for phosphorylated serine) (Bach et al., 2005). GS-1 was synthesized by Millegen (Labège, France). CK1 (porcine brain, native) was assayed as described for CDK1 but using the CK1-specific peptide substrate RRKHAAIGpSAYSITA (Reinhardt et al., 2007) obtained from Millegen. Erk2 (rat, recombinant) was assayed as described for CDK1 but using the specific substrate Ets1 (amino acids 1–138) in buffer A. DYRK1A [rat, recombinant, expressed in Escherichia coli as glutathione-S-transferase (GST) fusion protein] was purified by affinity chromatography on glutathione-agarose and assayed as described for CDK1/cyclin B using Woodtide (KKISGRLSPIMTEQ) (1.5 μg per assay) as a substrate. CLK3 (human, recombinant, expressed in E. coli as GST fusion proteins) was assayed in buffer A (+0.15 mg BSA·mL−1) with RS peptide (GRSRSRSRSRSR) (1 μg per assay).
Cell culture
In this study, we used the human nasal airway epithelial cell line JME/CF15, derived from a CF patient homozygous for the F508del mutation (Jefferson et al., 1990). The culture conditions are as described previously (Jefferson et al., 1990; Norez et al., 2009). For the D1ER experiments, cells were grown until 70–80% confluence and were transiently transfected with 2 μg cDNA encoding the D1ER construct. D1ER is a commercial plasmid pcDNA-D1ER which is retained in the ER with a C-terminal KDEL sequence (Palmer and Tsien, 2006). D1ER is a second generation cameleon (calcium sensor) targeted to ER. CFBE41o− airway epithelial cell lines were obtained from Pr Gruenert (Department of Otolaryngology–Head and Neck Surgery, UCSF, San Francisco, CA, USA) (Bruscia et al., 2002). Cell culture media and supplements were purchased from Lonza (Basel, Switzerland) and PAA (Pasching, Austria). The CFBE41 cell line used in experiments was cultured as described previously (Bebok et al., 2005). HEK293T cells were grown in high glucose DMEM (Biowhittaker, Lonza, Basel, Switzerland) supplemented with FBS (10%), L-glutamine (2 mM), penicillin (100 U·mL−1) and streptomycin (10 mg·mL−1). The cells were incubated in a 5% CO2 humidified atmosphere after being transfected with 2 μg human pcDNA-SCN5A L325R (this is a plasmid code for the α-subunit of the ER-arrested L325 mutant forms of the sodium channel Nav1.5; it is kindly provided by Dr H. Abriel, University of Bern, Bern, Switzerland) and 2 μg human β1-subunit (this is a plasmid code for the β1- subunit of sodium channel Nav1.5 and is a generous gift from M. Chahine, Laval University, Québec, Canada) using the calcium-phosphate method as previously described (Margolskee et al., 1993). The human sodium channel β1-subunit and CD8 were inserted in the pIRES bicistronic vector in the form of pCD8-IRES-β1. Before the patch-clamp experiments were performed, 2-day post-transfection cells were incubated for 5 min in a medium containing anti-CD8 coated beads (Dynabeads CD8, Invitrogen, Dynal AS, Oslo, Norway). The unattached beads were removed by washing with extracellular solution. Cells expressing CD8 were distinguished from non-transfected cells by visualizing beads fixed on the cell membrane by light microscopy. Using this strategy, transfected cells that bound beads also expressed the β1-subunit protein.
The pretreatments of 2, 24 or 48 h with several corrector compounds were performed at 37°C in a 5% CO2 humidified atmosphere. Experiments were then realized at room temperature (the duration of experiment did not exceed 30 min to avoid any effect of the temperature on the recorded response).
Immunofluorescence study
CF15 cells were incubated with monoclonal anti-human CFTR antibody (1:100, IgG2a clone 24-1; R&D Systems, Minneapolis, MN, USA) overnight at 4°C. Cells were then incubated with the FluoProbes 488 (1:400; FluoProbes, Interchim, France) secondary antibody. Nuclei were stained red with TO-PRO-3 iodide (Invitrogen, Carlsbad, CA, USA) for 15 min at room temperature (1:1000 in Tris-buffered saline). Fluorescence was examined with a spectral confocal station FV 1000 installed on an inverted microscope IX-81 (Olympus, Tokyo, Japan). For more details, see Norez et al. (2006a).
Immunoprecipitation and Western blot analysis
For lysates, total protein was quantified using the Bradford (1976) protein assay reagent (Bio-Rad S.A, Marnes-la-Coquette, France), and 50 μg of protein was loaded onto an SDS-PAGE apparatus. For immunoprecipitation, CF15 cell lysates were incubated with monoclonal anti-CFTR antibody (2 μg, IgG2a clone 24-1; R&D Systems). Immunoblots were probed with polyclonal rabbit anti-calnexin antibody (2 μL·mL−1, SPA-860; Assay Designs, Ann Arbor, MI, USA). The protein levels were quantified by densitometry and expressed as a % of controls. Other details appear elsewhere (Norez et al., 2006a).
Iodide efflux
The F508del-CFTR Cl− channel activity was assayed by measuring the rate of iodide (125I) efflux from CF15 cells as described previously (Norez et al., 2006a). Time-dependent rates of 125I efflux were calculated from the following: ln (125I t1/125It2)/(t1 − t2), where 125It is the intracellular 125I at time t, and t1 and t2 were successive time points. Curves were constructed by plotting rates of 125I versus time. All comparisons were based on maximal values for the time-dependent rates (k = peak rates, min−1), excluding the points used to establish the baseline (k peak-k basal, min−1) (for other details, see Norez et al., 2006a).
Patch-clamp experiments
CFTR chloride currents
Perforated whole-cell patch-clamp experiments were performed on CF15 cells. Importantly, in these experiments, the composition of intrapipette solution allowed the recording of Cl− currents only and the simultaneous presence in the bath solution of calixarene and 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) prevented activation of non-CFTR Cl− currents as previously described (Norez et al., 2006b). The mean access resistance and whole-cell capacitance were 12 ± 0.6 MΩ and 35 ± 4.3 pF (Norez et al., 2006b). I–V relationships were obtained by clamping the membrane potential to −40 mV and by pulses from −80 to +80 mV (20 mV increments). Other details can be found in Norez et al. (2006b).
Voltage-gated sodium currents
Whole-cell configuration of the patch-clamp technique was used on transiently transfected HEK293 cells (20.03 ± 1.39 pF, n = 27). Sodium currents were generated by clamping the cell membrane from a holding potential of −140 mV to potentials ranging from −100 to 40 mV for 50 ms in 10 mV increments with 5 s stimulus intervals. The patch pipettes were filled with (mM): 35 NaCl, 105 CSF, 10 EGTA and 10 HEPES. The pH was adjusted to 7.4 using CsOH. The bath solution contained (mM): 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2, 10 glucose and 10 HEPES. The pH was adjusted to 7.4 using NaOH. A −7 mV correction of the liquid junction potential between the patch pipette and the bath solutions was performed. Other details can be found in Mercier et al. (2012)
Recording calcium signals
Fluo-4 experiments
CF15 cells were loaded with 3 μM Fluo-4 acetoxymethyl (AM)ester (FluoProbes®, Montluçon, France) for 20 min at room temperature in buffer solution containing (in mM): 130 NaCl, 5.4 KCl, 2.5 CaCl2, 0.8 MgCl2, 5.6 glucose, 10 HEPES, pH 7.4 (adjusted with Tris base), rinsed and allowed to equilibrate for 5–10 min. Ca2+ activity was recorded by confocal laser scanning microscopy using Bio-Rad MRC 1024 equipped with 15 mW Ar/Kr gas laser (Bio-Rad, Hemel Hempstead, UK). Maximal resolution was obtained with Olympus plan apo X60 oil, 1.4 NA, objective lens (Olympus). Fluorescence signal collection was performed through the control software Lasersharp 3.2 (Bio-Rad). The resolution time was 30 s for the protocol in which cells are incubated for 2 h in the presence of a given pharmacological agent. Raw fluorescence values were converted into Ca2+ concentrations by applying the self-ratio method, assuming the KD of fluo-4 for Ca2+ was 396 nM and for resting [Ca2+] it was assumed to be 62 nM at the beginning of each experiment. All the experiments were performed on at least two different cell passages. Other details can be found in Norez et al. (2006a).
ER Ca2+ experiments
JME/CF15 cells were transiently transfected with 2 μg cDNA encoding the ER-targeted cameleon Ca2+ indicator D1ER construct (Palmer and Tsien, 2006) 48 h before the experiments, using lipofectamine 2000 (Invitrogen). For dual emission imaging of cameleon D1ER constructs, cells were illuminated at 440 nm (440AF21; Omega Optical, Brattleboro, VT, USA) and emission was collected through a 455DRLP dichroic mirror, alternatively at 480 nm (480AF30; Omega Optical) and 535 nm (535AF26; Omega Optical). When required, photobleaching was corrected. Other details in can be found in Antigny et al. (2012).
Proteasome activity assay
Proteasome enzymatic activity was measured as described by Canu et al. (2000) and following the manufacturer's protocol (20S proteasome activity assay kit; Millipore Bioscience Research Reagents, Temecula, CA, USA). In brief, this assay is based on the detection of the fluorophore 7-amino-4-methylcoumarin (AMC) after cleavage from the labelled substrate LLVY-AMC by the proteasome machinery. The free AMC fluorescence was quantified using a 380/460 nm filter set. The intracellular proteasome activity was detected in cells after 2 h of treatment with roscovitine, whereas the inhibitory effect of roscovitine on purified proteasome enzyme was evaluated after incubation of the 20S proteasome-positive control for 30 min according to the assay instructions. Other experimental details are provided in Norez et al. (2008).
Statistics
Results are expressed as means ± SEM of n observations. Sets of data were compared with either anova or Student's t-test. Differences were considered statistically significant when P < 0.05; ns, non-significant difference; *P < 0.05, **P < 0.01, ***P < 0.001. All statistical tests were performed using GraphPad Prism version 4.0 for Windows (GraphPad Software, San Diego, CA, USA) and Origin version 5.0 (RITME Informatique, Paris, France).
Chemicals
(R)-roscovitine (termed roscovitine throughout the manuscript), olomoucine, thapsigargin, forskolin and genistein were from LC Laboratories (PKC Pharmaceuticals, Woburn, MA, USA). VX809 and VX-770 were from Vertex Pharmaceutics. Corr4a was from Rosalind Franklin University (North Chicago, IL, USA). The CFTR inhibitor CFTRinh-172 (3-[(3-trifluoromethyl)-phenyl]-5-[(4-carboxyphenyl)methylene]-2-thioxo-4-thiazolidinone) was from VWR International. All other chemicals were from Sigma (Saint Quentin Fallavier, France). Miglustat was obtained from IsoLab.
(S)-roscovitine, (S)-CR8, metabolite M3 were synthesized as described in Meijer et al. (1997) and Oumata et al. (2009). O-benzyl-roscovitine and N6-methyl-(R)-roscovitine were synthesized as described in Tang et al. (2005).
Results
Evaluation of the effects of protein kinase inhibitors on the function of endogenous F508del-CFTR
We took advantage of our screening facilities dedicated to the search for CFTR modulators and trafficking correctors to evaluate the effect of several protein kinase inhibitors on the function of F508del-CFTR in the human airway epithelial CF15 cells. To that end, CF15 cells were incubated for 2 h at 37°C with the drug candidate and the activity of F508del-CFTR was measured as the iodide efflux stimulated by a mixture of the adenylate cyclase activator forskolin plus the isoflavone genistein (denoted genistein). Among the kinase inhibitors, we selected (R)-roscovitine (1) and olomoucine (3) (Figure 1A), two selective agents for CDKs 1, 2, 5, 7 and 9 (Meijer et al., 1997), LY294002 as an inositol-3 kinase inhibitor, KN62, H89, W7 and chelerythrin chloride as inhibitors of Ca2+/calmodulin-dependent protein kinase II, PKA, calmodulin and PKC (Meijer et al., 1997; Yurko-Mauro and Reenstra, 1998) respectively. The results indicate that neither olomoucine, LY294002, KN62, H89, W7 nor chelerythrin significantly restored F508del-CFTR activity in CF15 cells (Figure 1B). In contrast, roscovitine was found to be potent at restoring F508del-CFTR activity in CF15 cells (Figure 1B).
Figure 1.

Functional restoration of F508del-CFTR activity in CF cells by roscovitine via CDK-independent pathway. (A) Structure of roscovitine and its derivatives used in this study. (B) Effect of incubating CF15 cells for 2 h with different kinase inhibitors on iodide efflux. Concentrations used are 100 μM for roscovitine, olomoucine, LY294002, (S)-CR8, M3, O-benzyl-(R)-roscovitine, N6-Methyl-(R)-roscovitine and the main metabolite of roscovitine, M3; 1 μM for KN 62; 10 μM for H89; 1 μM for chelerythrin chloride and 50 μM for W7. (C) Examples of efflux curves obtained in untreated CF15 cells, CF15 cells incubated for 24 h at 27°C (denoted low temperature) and CF15 cells incubated for 2 h at 37°C with roscovitine or miglustat and stimulated by forskolin/genistein (Fsk/Gst) as indicated by the horizontal bar above the traces. For the low-temperature protocol, cells were maintained in the culture medium at 27°C for 24 h. (D) Concentration-response curves with CF15 cells incubated for 2 h with roscovitine at different concentrations obtained with the iodide efflux technique. (E) Effect of chloride channel inhibitors on iodide efflux in roscovitine-corrected cells (100 μM, 2 h) stimulated by forskolin/genistein in the absence or presence of the following inhibitors: GPinh5a (1 μM) calixarene (100 nM), CFTRinh-172 (10 μM), DIDS (500 μM), DPC (500 μM) or glibenclamide (500 μM). Basal corresponds to unstimulated roscovitine-corrected cells. Data are expressed as % of CFTR maximal activity. (B–E) The activity of F508del-CFTR was stimulated by a cocktail composed of the adenylate cyclase agonist forskolin to stimulate the production of intracellular cAMP and of the CFTR activator genistein. n = 4 for each condition. ***P < 0.001; ns, not significant.
In elucidate the molecular targets and mechanism of action of roscovitine in its ability to restore F508del-CFTR activity in CF15 cells, we tested a series of roscovitine analogue; their structures and effects on kinases are presented Figure 1A and Table 1. These included: (S)-CR8 (4), a derivative of roscovitine which is slightly more active on the kinase targets than roscovitine, but much more (100 fold) potent at inducing cell death (Bettayeb et al., 2008); the main roscovitine metabolite (M3) (5), which is essentially inactive on kinases; and two other kinase dead roscovitine analogues: O6-benzyl-roscovitine (6) and N6-methyl-roscovitine (7) (Tang et al., 2005). Among these compounds, only the metabolite M3 (5) had a significant restorative effect on F508del-CFTR activity (Figure 1B). Despite lower or higher inhibitory activities, respectively olomoucine (3) and (S)-CR8 (4) were unable to restore the activity of F508del-CFTR (Figure 1B). Moreover, the two kinase-inactive derivatives of roscovitine (6, 7) were also unable to correct F508del-CFTR (Figure 1B). The lack of effect of compounds (6) and (7) demonstrates that the N6 nitrogen may be involved in key H-bonds, as it is in the interaction of roscovitine with its CDK targets (Meijer et al., 1997; Bettayeb et al., 2010). Altogether, these results suggest that the restorative activity of roscovitine is unlikely to be due to inhibition of its kinase targets.
Table 1.
Kinase inhibitory activities of roscovitine and its derivatives
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|
| CDK1/cyclin B | 0.48 | 0.82 | 5.2 | 0.24 | 45 | 100 | 63 |
| CDK2/cyclin A | 0.20 | 0.22 | 3.3 | 0.13 | 23 | 120 | 60 |
| CDK5/p25 | 0.32 | 0.65 | 5.2 | 0.28 | 48 | 200 | 110 |
| CDK9/cyclin T | 0.85 | 0.94 | 1.2 | 0.18 | 51 | >1000 | 150 |
| CK1 | 5.1 | 6.8 | 83 | 1.4 | 80 | 80 | 180 |
| CLK3 | 34 | 14 | −(100) | 27 | 81 | −(1000) | >1000 |
| DYRK1A | 2.3 | 0.79 | 74 | 0.84 | 18 | 47 | 120 |
| ERK2 | 20 | 8.2 | 33 | 6, 7 | >100 | >1000 | −(1000) |
| GSK-3 | 81 | >100 | 98 | >100 | −(100) | −(1000) | >1000 |
Compounds were tested on nine purified kinases. IC50 values, calculated from the dose-response curves, are reported in μM. Key: 1, (R)-roscovitine; 2, (S)-roscovitine; 3, olomoucine; 4, (S)-CR8; 5, metabolite M3; 6, O-benzyl-(R)roscovitine; 7, N6-Methyl-(R)-roscovitine. −, no inhibition at highest dose tested (in parentheses).
Correction of the defective trafficking and function of endogenous F508del-CFTR by roscovitine
We focused our investigations on the pharmacological properties of roscovitine to elucidate its mechanism of action. Whereas no stimulation occurred in the presence of forskolin/genistein in untreated CF15 cells, an increased rate of iodide efflux (kpeak-kbasal = 0.115 ± 0.020 min−1) was observed following treatment of CF cells with roscovitine (100 μM, 2 h, 37°C). The correcting effect of roscovitine was compared with that obtained in CF15 cells treated with miglustat (100 μM, 2 h, 37°C), a glucosidase inhibitor known to promote the delivery of F508del-CFTR to the cell surface (Lubamba et al., 2009; Norez et al., 2009), and with CF15 cells cultured at low temperature (27°C for 24 h) (Denning et al., 1992). Interestingly, the restorative effects on the function of the mutated channel were similar for roscovitine, miglustat and low temperature (Figure 1C). Next we determined the EC50 for roscovitine on F508del-CFTR function. For CF15 cells incubated for 2 h at 37°C with increasing concentrations of roscovitine, we calculated an EC50 of 38 ± 0.5 μM (Figure 1D). Using similar experimental procedures, the S-isomer of roscovitine (2) was found to have an EC50 of 32 ± 1 μM (n = 4, data not shown).
The effect of a range of known Cl− channel blockers on the forskolin/genistein-stimulated iodide efflux in roscovitine-treated cells was determined. Glibenclamide and diphenylamine-2-carboxylic acid (DPC) are two non-specific inhibitors of Cl− channels. The α-aminoazaheterocycle-methylglyoxal adducts GPinh5a and the thiazolidinone compounds CFTRinh-172 have been developed as selective CFTR blockers. Other Cl− channel inhibitors were tested such as DIDS and TS-TM calix[4]arene, two inhibitors of outwardly rectifying Cl− channels but not of CFTR. The efflux induced in CF15 cells pretreated with roscovitine (100 μM, 2 h, 37°C) and stimulated with forskolin/genistein was completely inhibited by GPinh5a, CFTRinh-172, glibenclamide and DPC but not affected by either DIDS or TS-TM calix[4]arene (Figure 1E). This pharmacological profile of inhibition is in agreement with the expected signature of CFTR (Sheppard and Welsh, 1993; Schultz et al., 1999; Li et al., 2004; Wang et al., 2004). Finally, because CDK inhibitors have antiproliferative properties associated with apoptosis-inducing activity, the cytotoxicity of roscovitine in CF15 cells was determined. We found no cellular toxicity of roscovitine in the range of concentrations (100 nM to 1 mM) tested after 2 h of incubation at 37°C (data not shown).
In the next series of experiments, F508del proteins were localized in cells before and after treatment with roscovitine. Firstly, we found F508del-CFTR proteins mostly restricted around the nucleus in CF15 cells maintained at 37°C (Figure 2A, panels a–d). After treatment with roscovitine (100 μM, 2 h, 37°C), F508del-CFTR was redirected towards the plasma membrane and also throughout the cells (Figure 2A, panels e–h). Figure 2, panel h, is a merge of fluorescence image (Figure 2A, panels e, f) with the corresponding light micrograph (Figure 2A, panel g) showing plasma membrane and cytosol location of F508del-CFTR proteins.
Figure 2.

Effect of roscovitine on the cellular localization and Cl− channel function of F508del-CFTR in CF15 cells. (A) Confocal images showing the localization of F508del-CFTR in untreated cells (a–d) or after 2 h of incubation with 100 μM roscovitine (e–h). d and h are a merge of respectively a, b and c and e, f and g. Scale bars are 10 μm. F508del-CFTR is stained in green and nucleus in red. Images are representative images of three independent experiments for each condition. (B) Perforated whole-cell patch-clamp experiments in roscovitine-corrected CF15 cells (100 μM, 2 h). This example illustrates the complete sequence of an experiment from a single cell. The current was recorded in the basal condition (upper traces), following addition of forskolin 10 μM + genistein 30 μM (Fsk/Gst; middle traces) and finally after adding 100 μM glibenclamide (glib; bottom right traces). (C) Curves of current densities (pA/pF) as a function of potential (mV). n = 3 for each condition.
We also performed whole-cell patch-clamp experiments to record CFTR currents in CF15 cells after 2 h of incubation at 37°C with roscovitine. As expected, the cocktail forskolin+genistein had no effect in untreated CF15 (data not shown, see also Norez et al., 2006b). In contrast, a linear voltage-independent Cl− current was recorded from cells incubated with roscovitine (Figure 2B, middle traces and C). The Cl− current was totally inhibited by glibenclamide (Figure 2B, bottom traces and C) to a level of current not significantly different from the basal level (Figure 2B, upper traces and C).
Dynamic and reversible effect of roscovitine
To explore the mechanism of action of roscovitine, we first analysed the effect of modifying the duration of treatment of CF cells at 37°C with 100 μM of the drug and measured the functional rescue of F508del-CFTR. We observed a time-dependent correcting effect with a maximal effect after only 2–4 h of treatment and then a progressive decline after 6 h (Figure 3A). Secondly, in a separate series of experiments, we incubated CF cells at 37°C with 100 μM roscovitine for 2 h, then washed the drug out and analysed the forskolin+genistein iodide efflux response. In that case, the effect on F508del-CFTR function was maximal during the first 2 h after compound washout and then progressively declined (Figure 3B). Thus the correction of F508del-CFTR function by roscovitine appears to be a dynamic and reversible process.
Figure 3.

Dynamics of the roscovitine-dependent restoration of F508del-CFTR activity in CF15 cells. (A) Time dependence of roscovitine correction. CF15 cells incubated for different durations with roscovitine and stimulated by forskolin/genistein. (B) Persistence of correction. CF15 cells were treated for 2 h with 100 μM roscovitine and F508del-CFTR activity was assayed at different times after washout. CFTR-dependent response was induced by forskolin 10 μM + genistein 30 μM. n = 4 for each condition. ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant.
Comparisons of efficacy of F508del-CFTR correctors on CFBE cells
The in vitro efficacy of roscovitine was then compared with several previously described CFTR correctors and low-temperature correction using cultured CFBE cells. These included drugs approved for non-CF indications (e.g. miglustat) (Norez et al., 2006b; da Cruz et al., 2011) and compounds identified through high-throughput screening (HTS) (e.g. corr-4a, VRT-809) (Wang et al., 2007; Van et al., 2011). The CFBE cells were pre-incubated with each compound at the maximally effective concentration and treatment duration (Figure 4A). Roscovitine, VX-809, miglustat and IsoLAB induced slightly but not significantly different, effects on F508del-CFTR but were more efficacious than Corr-4a (Figure 4A). To enhance chloride transport through F508del-CFTR corrected by roscovitine, the CFTR potentiator VX-770 (ivacaftor) was added to maximize the Po of the CFTR channel. Acute application of 10 μM VX-770 increased forskolin-stimulated chloride transport in CFBE pretreated with roscovitine for 2 h (Figure 4B). Interestingly, acute application of VX-770 activated the roscovitine-mediated correcting effect on F508del-CFTR suggesting a potential combination of these two compounds in CF therapeutics.
Figure 4.

Comparisons of efficacy of F508del-CFTR correctors on CFBE cells. (A) Comparison of effect of roscovitine with that induced by other F508del-CFTR correctors. CFBE cells incubation conditions are indicated below each bar and the cells were stimulated with forskolin/genistein. (B) Synergistic effect of roscovitine with the CFTR potentiator VX-770. CFBE cells were treated for 2 h with 100 μM roscovitine and F508del-CFTR activity was induced by different stimulations as indicated below each bar. Fsk : forskolin 10 μM. Gst : genistein 30 μM. VX-770 10 μM. n = 4 for each condition. ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant.
Roscovitine showed selectivity for its correcting effect on F508del-CFTR processing
Misfolding of Nav1.5 appears to be one of the mechanisms underlying Brugada syndrome (Keller et al., 2005), as is the case for KvLQT1 and hERG channels in congenital long QT syndrome (Gong et al., 2006) and CFTR channels in CF. Incubation of cells expressing L325R-Nav1.5 channels or F508del-CFTR channels for 24 h at 28°C partially corrected the mutant channels (Denning et al., 1992; Keller et al., 2005), but did not improve the processing of the normal or mutant forms of hERG (Keller et al., 2005). To assess the selectivity of roscovitine's action, we compared the ability of this compound to correct ER-arrested L325R mutant forms of Nav1.5 in L325R-Nav1.5 heterologously expressed in HEK293 cells. Voltage-gated sodium currents were recorded using the whole-cell configuration of the patch-clamp technique (Figure 5A and B). As expected, a low-temperature treatment restored Nav1.5 activity in L325R-Nav1.5 transfected cells (Figure 5C). In contrast, roscovitine did not improve the processing of the mutant form (Figure 5). We concluded that roscovitine selectively addresses the underlying processing defect in F508del-CFTR.
Figure 5.

Roscovitine failed to restore the activity of L325R trafficking-defective mutant of Nav1.5. (A) Example of current density traces of L325R-Nav1.5 mutant expressed in HEK293T cells treated or not by roscovitine (100 µM). (B) Current density relationship of L325R-Nav1.5 mutant expressed in HEK293T cells. Cells were treated with either DMSO (vehicle, n = 8) or roscovitine 100 μM (n = 6) for 2 to 4 h before the experiments. The inset shows the voltage-clamp protocol applied. (C) Comparison between the effect of roscovitine on L325R-Nav1.5 mutants and its functional restoration by low-temperature treatment. Histograms show the peak current density recorded at −30 mV normalized to the control condition. The cells were treated as in (A) or incubated for 24 h at 28°C before the experiments. n = 6–13 cells. **P < 0.01; ns, not significant.
Synergistic effects of ERQC modulators with roscovitine
To further investigate the mechanism of action of roscovitine, we studied its effect in competition with several inhibitors of the biosynthetic pathway of CFTR and of the ERQC machinery in CF cells (see schematic drawing in Figure 6). Brefeldin A, a vesicular ER/Golgi-intermediate compartment traffic inhibitor, completely inhibited the effect of roscovitine (Figure 6A). This suggests that F508del-CFTR proteins follow conventional intracellular traffic when restored by roscovitine.
Figure 6.

Competition and synergistic effects of roscovitine and inhibitors of ERQC. Histograms showing the correction of macroscopic iodide efflux in CF15 cells treated with roscovitine, brefeldin A (BFA), roscovitine+BFA (A), geldanamycin, roscovitine+geldanamycin (B), thapsigargin (TG), roscovitine+thapsigargin (C) or MG132, roscovitine+MG132 (D). In A–D, roscovitine was used as a positive control and the corresponding bar represents separate quadruplicate results. The condition denoted as untreated corresponds to cells at 37°C without any corrector. CF15 cells were treated for 2 h with roscovitine (100 μM), thapsigargin (10 μM), geldanamycin (2.5 μg mL−1), MG132 (20 μM) or with brefeldin A (BFA, 20 μM). Stimulation was by forskolin (10 μM) + genistein (30 μM). n = 4 for each condition. ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant. The scheme illustrates the ERQC inhibitors used.
The HSP90 inhibitor geldanamycin has been shown to stabilize F508del-CFTR by preventing the interaction between the chaperone and the mutant protein. In the presence of geldanamycin, we observed a significant (P < 0.001) potentiation of the effect of roscovitine on F508del-CFTR function (Figure 6B), whereas geldanamycin by itself was unable to restore the function of F508del-CFTR.
The sarcoendoplasmic reticulum calcium ATPase pump inhibitor thapsigargin is a partial corrector of F508del-CFTR (Egan et al., 2002; Norez et al., 2006a) and is known to alter the F508del-CFTR/calnexin interaction (Egan et al., 2002; Norez et al., 2006a). After incubation of CF cells with thapsigargin, we confirmed partial correction of F508del-CFTR (Figure 6C) and also observed a small but significant (P < 0.05) potentiation of the effect of roscovitine on F508del-CFTR function (Figure 6C).
Interestingly, no potentiation of the effect of roscovitine in CF cells treated with MG132, a proteasome inhibitor (Figure 6D), was observed. MG132 by itself was not able to restore F508del-CFTR function (Figure 6D). These results suggest that the biogenesis route used by F508del-CFTR in the presence of the corrector roscovitine is brefeldin A-sensitive and can be boosted following HSP90 inhibition and to a lesser extent by ER calcium ATPase pump inhibition. However, inhibiting the cellular proteasome by MG132 had no additive effect.
Roscovitine prevents interactions between calnexin and F508del-CFTR
We next tested the hypothesis that the restoration of F508del-CFTR function induced by roscovitine might be due to perturbation of the calnexin/F508del-CFTR interaction and/or to alterations in the proteasome-dependent degradation pathway. We tested the first hypothesis with co-immunoprecipitation experiments on CF cells. Two control experiments were carried out using CF15 lysate and mouse IgG (Figure 7A, lanes 1 and 2). Following immunoprecipitation with CFTR antibody, anti-calnexin Western blot showed a decreasing intensity of the calnexin band after 2 h of thapsigargin treatment (41% inhibition, lane 6 Figure 7A and B). Importantly, roscovitine prevented (53% inhibition, lane 5 Figure 7A and B) the F508del-CFTR/calnexin interaction as compared with CF15 at 37°C (lane 3 Figure 7A and B). The vehicle alone, DMSO, was used as negative control (lane 4 Figure 7A and B). To ascertain that the effect of roscovitine was not due to a direct effect on the production of calnexin itself, we performed Western blots with the anti-calnexin antibody using lysates of CF15 incubated following the same experimental conditions as mentioned earlier. We observed no differences in intensity between the molecular form of calnexin without (lane 1 Figure 7C) or after treatment with thapsigargin (lane 2 Figure 7C), roscovitine (lane 3 Figure 7C) or DMSO (lane 4 Figure 7C). This observation was confirmed by densitometry analysis of the ratio of calnexin/β-tubulin used as control (Figure 7D). Taken together, these results show that in the human airway epithelial CF15 cells, roscovitine partially inhibits the F508del-CFTR/calnexin interaction, and this probably contributes to its ability to correction the function of F508del-CFTR.
Figure 7.
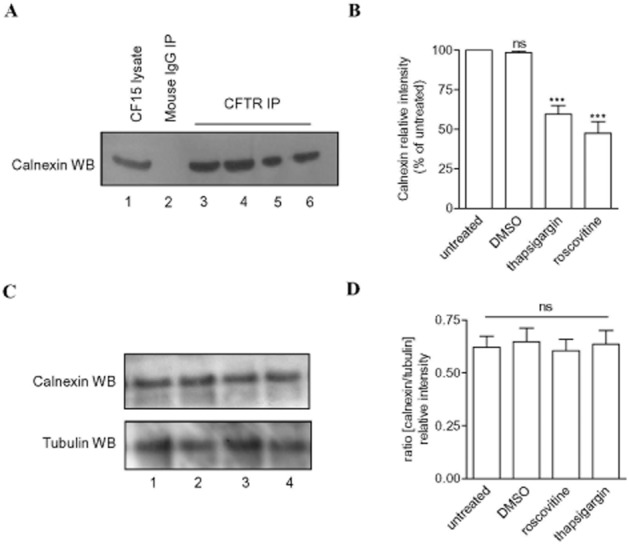
Roscovitine partially prevents the interaction of F508del-CFTR with calnexin. (A) Co-immunoprecipitation of F508del-CFTR with calnexin in untreated CF15 cells (lane 3) or cells treated with DMSO (lane 4, DMSO used as the vehicle of compounds), roscovitine (lane 5) or thapsigargin (lane 6). CF15 lysates (50 μg of total protein, lane 1) and non-immune mouse IgG (lane 2) were used as positive and negative controls respectively. In F508del-CFTR immunoprecipitations (CFTR IP) following calnexin Western blot revealed a principal band corresponding to the calnexin immunoprecipitated with F508del-CFTR. (B) Bar graphs show the chaperone intensity band expressed as % of the F508del-CFTR IP/calnexin Western blot from untreated cells. (C) Expression of CFTR or tubulin in CF15 cells untreated (lane 1) or treated with thapsigargin (lane 2), roscovitine (lane 3) or DMSO (lane 4). Fifty microgram of total protein was loaded onto an SDS-PAGE apparatus. (D) Bar graphs show the ratio densitometry between intensity bands of calnexin and tubulin. A–D treatments correspond to 2 h of cell incubation with 100 μM of compound, except for DMSO 1/1000. n = 3 for each condition. ***P < 0.001; ns, not significant.
Effect of roscovitine on the ER calcium-dependent biosynthetic pathway of F508del-CFTR
It is known that calnexin requires calcium for optimal activity and that thapsigargin inhibits the release of Ca2+ from the ER mediated by calnexin/F508del-CFTR (Egan et al., 2002; Norez et al., 2006a). Additional experiments were thus conducted to explore the cellular mechanism leading to correction of endogenous F508del-CFTR trafficking by roscovitine. To that end, we first measured the effect of roscovitine on global Ca2+ stores in Fluo-4 loaded CF cells. We recorded an increased cytosolic Ca2+ level following application of 100 μM roscovitine (Figure 8A). In contrast, olomoucine, which is not an F508del-CFTR corrector (Figure 1B), had no effect on the global Ca2+ response (data not shown). After 10 min of incubation in buffer solution, which allowed refilling of ER calcium stores, 100 μM 2-aminoethyoxydiphenyl borate (2-APB), an IP3 receptor inhibitor, was applied in the presence or absence of roscovitine. Treatment with 2-APB completely abolished the Ca2+ response recorded in the presence of roscovitine (Figure 8A). At the end of the protocol, adding roscovitine to the bath perfusing the cells reversed the inhibition (see the end of the exemplar calcium recording, Figure 8A). In order to eliminate the Ca2+ influx, the Ca2+ response was quantified in the absence of extracellular Ca2+ (Figure 8B). The mean responses induced by roscovitine or with the different agents are expressed as normalized AUC in comparison with the response induced by ATP for 5 min (Figure 8B). These results suggest that roscovitine increased Ca2+ mobilization in a reversible manner and this effect was independent of extracellular calcium but inhibited by 2-APB. To investigate the effect of roscovitine on calcium mobilization, we next measured more precisely the ER Ca2+ content with a FRET-based D1ER indicator (Palmer and Tsien, 2006). D1ER is a second generation cameleon (calcium sensor) targeted to ER. We found that roscovitine induced, in the absence of extracellular calcium, Ca2+ depletion of the ER in CF15 cells transiently transfected with cDNA encoding the genetics D1ER Ca2+ (Figure 8C). This ER Ca2+ release was reversible (Figure 8C) and totally abolished by 2-APB (Figure 8D). Then, we reasoned that if the roscovitine-induced correction of F508del-CFTR trafficking is correlated to the level of calcium in the ER, 2-APB would be expected to inhibit the roscovitine-mediated correction of F508del-CFTR. Surprisingly, roscovitine/2-APB co-treatment only partially prevented the correcting effect of roscovitine, as shown by the decrease in stimulating effect induced by forskolin/genistein of the CFTR-dependent iodide efflux in CF cells at 37°C (Figure 9A). To further confirm that 2-APB is unable to completely prevent the effect of roscovitine, we repeated these experiments with different concentrations of 2-APB (Figure 9B). These experiments revealed only a maximum inhibition of 50%, suggesting that roscovitine corrects the abnormal function of F508del-CFTR through two processes, a Ca2+-dependent and also a Ca2+-independent mechanism of action.
Figure 8.

Mobilization of ER calcium stores by roscovitine. (A) Typical traces of general Ca2+ mobilization induced by roscovitine in the presence or absence of 2-APB in CF15 cells; results were obtained using Fluo-4. (B) Bar graphs show the AUC measured after 5 min exposure to 100 μM roscovitine (n = 83) or ATP (n = 33) or 10 μM thapsigargin (n = 36) in the absence of extracellular Ca2+. (C) Typical traces of ER Ca2+ mobilization induced by roscovitine in the absence or presence of extracellular Ca2+depletion using the cameleon calcium sensor D1ER. JME/CF15 cells were transiently transfected with the ER Ca2+ probe D1ER. (D) Quantification of the ER Ca2+ depletion induced by 100 μM roscovitine (n = 57), ATP (100 μM, n = 28), roscovitine + 2APB (100 μM, n = 34) or 1 μM thapsigargin (n = 28); ***P < 0.001; **P < 0.01; ns, not significant.
Figure 9.

Calcium-independent restoration of F508del-CFTR activity. (A) Quantification of F508del-CFTR activity was assayed with iodide efflux experiments in the presence of forskolin (10 μM) + genistein (30 μM) on CF15 cells incubated for 2 h with roscovitine (100 μM) in the presence or absence of 2-APB (100 μM). (B) Concentration-response curve obtained using iodide efflux technique for measuring CFTR activity in CF15 cells treated for 2 h with 100 μM roscovitine in presence of several concentrations of 2-APB and stimulated by forskolin + genistein. (A and B) n = 4 for each condition or concentration. ***P < 0.001; **P < 0.01; ns, not significant.
Effect of roscovitine on the intracellular degradation machinery
To explore the molecular basis of the Ca2+-independent effect of roscovitine, we evaluated its effects on proteasome activity in CF15 cells. Indeed, as shown in Figure 4D, we found that the proteasome inhibitor MG132 was unable to potentiate the effect of roscovitine. Thus, we evaluated the effect of roscovitine on the proteasome activity of CF cells using a proteasome activity assay kit. The proteasome activity was measured in CF15 cells treated for 2 h with roscovitine, M132 or lactacystin [two known proteasome inhibitors (Tsubuki et al., 1993with; Fenteany et al., 1995)], or co-treated with roscovitine and 2-APB (Figure 0A). Quantification of these results revealed ∼47% inhibition by roscovitine and ∼90% by MG132 or lactacystin (Figure 0A). The proteasome inhibitory activity induced by roscovitine was similar in the absence and presence of 2-APB (Figure 0A), suggesting that inhibition of the proteasome activity by roscovitine is Ca2+-independent. Finally, we performed a similar experiment directly on purified 20S proteasome. Again, roscovitine significantly decreased the activity of the purified 20S proteasome (Figure 0B). 2-APB was without effect on the inhibition of the purified 20S proteasome induced by roscovitine (Figure 0B). Altogether, these results demonstrate that roscovitine inhibits the proteasome activity directly via a Ca2+-independent mechanism of action.

Inhibition of the proteasome activity by roscovitine. Histograms showing the effect of 100 μM roscovitine in the presence or absence of 2-APB on the proteasome activity of CF15 cells (A) and on purified proteasome enzyme denoted as 20S positive control (B). Cells were treated for 2 h and 20S positive control was treated for 30 min with roscovitine (100 μM), 2-APB (100 μM) lactacystin (20 μM) or MG132 (20 μM). (A and B) 3 < n < 9 for each condition. ***P < 0.001; **P < 0.01; ns, not significant.
Discussion and conclusions
We demonstrated that (i) roscovitine is a specific pharmacological corrector of F508del-CFTR with an EC50 of 38 μM, (ii) roscovitine corrects the F508del-CFTR trafficking defect via a complex mechanism of action that is independent of its effects on CDK1, CDK2, CDK7, CDK9 and probably on kinases in general, (iii) roscovitine prevents the interaction of F508del-CFTR with the ER resident calnexin by decreasing the ER calcium stores, (iv) roscovitine directly inhibits the proteasome activity in CF15 cells via a Ca2+-independent mechanism of action. Taking these results together, we propose that roscovitine corrects the function of misprocessed proteins by modulating the proteostasis of F508del-CFTR and in particular ERQC and ERAD in CF cells via two independent but complementary pathways.
The 2,6,9-trisubstituted purine roscovitine is a CDK inhibitor that competes with ATP for its binding site on the enzyme (Meijer et al., 1997; Meijer and Raymond, 2003). Pharmacological inhibitors of CDKs are currently being evaluated for their therapeutic use against cancer, polycystic kidney disease, neurodegenerative and cardiovascular disorders, and viral infections (Knockaert et al., 2002). R-roscovitine is currently undergoing phase 2 clinical trials as an anticancer drug (Aldoss et al., 2009; Hsieh et al., 2009) due to its ability to affect the cell cycle, CDK7/9-dependent transcription and induce apoptosis of cancer cells (Meijer and Raymond, 2003; Bettayeb et al., 2010). CDK2, CDK3, CDK4, CDK6 and CDK7 have been shown to have essential functions in cell cycle control. However, CDKs are involved in other processes such as apoptosis (CDK1, CDK5), neuronal activity (CDK5, CDK11) and transcription (CDK7, CDK8, CDK9). Among the first inhibitors to be discovered were 6-dimethylaminopurine and then isopentenyladenine, from which more potent and selective inhibitors were optimized, such as olomoucine (Vesely et al., 1994), roscovitine (Meijer et al., 1997; Meijer and Raymond, 2003) and purvalanol (Gray et al., 1998). Roscovitine and olomoucine are both potent inhibitors of CDK1/CDK2 with IC50 values of 0.7 and 7 μM respectively.
Interestingly, R-roscovitine enhanced the resolution of inflammation by promoting inflammatory cell apoptosis in several models including lung inflammation, by reducing the concentrations of the anti-apoptotic protein Mcl-1 (Rossi et al., 2006). The effects of roscovitine on polymorphonuclear neutrophils (PNM) have also been evaluated (Moriceau et al., 2010) because CF as a chronic inflammatory lung disease is characterized by airway inflammation dominated by PNM (Downey et al., 2009). In particular, PNM apoptosis is delayed in CF patients (Moriceau et al., 2009) but roscovitine restored normal levels of CF PNM apoptosis in a dose-dependent manner (from 0.5 to 20 μM) (Moriceau et al., 2010).
Roscovitine has also been shown to affect the activity of ionic channels though a CDK-independent process, as in the present study for CFTR channels. For example, roscovitine binds to L-type calcium channels (Yarotskyy et al., 2010) and blocks various voltage-dependent potassium channels (Kv1.3, Kv2.1, Kv4.3 and Kv11.1) by an open-channel mechanism (Ganapathi et al., 2009). Interestingly, unlike other hERG channel blockers, roscovitine is not trapped by hERG channel closing leading to the absence of arrhythmias (Ganapathi et al., 2009). This property could explain the lack of arrhythmias reported in clinical trials for cancer using roscovitine (Benson et al., 2007). Indeed, roscovitine was recently shown to have anti-arrhythmic properties (Yazawa et al., 2011). In the present work, we identified an additional CDK-independent effect of roscovitine as a pharmacological corrector of F508del-CFTR abnormal trafficking in epithelial cells. We demonstrated that treatment with other CDK inhibitors, less (olomoucine) or more efficient ((S)-CR8) than roscovitine or other kinase inhibitors (i.e. inhibitors of Ca2+/calmodulin-dependent protein kinase II, PKA, calmodulin and PKC) were ineffective at correcting F508del-CFTR whereas the roscovitine's main metabolite M3, inactive as a CDK inhibitor, partially corrected F508del-CFTR activity in CF cells. All these observations indicate a CDK-independent mechanism of action of roscovitine and its metabolite M3.
Although the mechanism of action of roscovitine on ion channel activity and trafficking remains unclear, two molecular targets of the ERQC and ERAD were highlighted by our study. Our data suggest that roscovitine, by disturbing the calcium concentration in the ER, alters the interaction of the nascent protein with cellular chaperones. In epithelial cells, CFTR protein interacts directly and/or indirectly with a variety of molecular chaperones and co-chaperones assisting the protein folding assembly, disassembly and translocation (Wang et al., 2006). In contrast, in CF cells, some of the interactions could be targeted to redirect F508del-CFTR away from ERAD and towards the trafficking pathway (Wang et al., 2006). Some of the pharmacological chaperones described so far, including miglustat and thapsigargin, alter the recognition of the mutant protein by calnexin (Gong et al., 2006; Norez et al., 2006a; 2009). In all these cases, the complex pharmacological chaperone/traffic-deficient protein appeared to be stabilized in an intermediate state in its folding path that more closely resembled the native state of the wild-type protein (Bernier et al., 2004). However, these results are not generally confirmed (Loo et al., 2004) nor has the influence on the interaction between calnexin and the F508del-CFTR (Farinha and Amaral, 2005), but it is possible that these conflicting results are a consequence of different durations of treatment. Our data highlighted the importance of both concentration (Figure 1C) and duration of incubation (Figure 3) to observe an optimal restoration of F508del-CFTR trafficking.
In the present study, we showed that roscovitine affected the release of F508del-CFTR from the ER stores. We hypothesize that roscovitine, by depleting ER Ca2+, disturbs the calnexin/F508del-CFTR interaction, which then allows the delivery of F508del-CFTR to the plasma membrane. Previous reports have already indicated that molecules decreasing and maintaining low levels of calcium in the ER disturb the recognition of F508del-CFTR by the Ca2+-dependent ER lectin, principally calnexin (Norez et al., 2006a), allowing translocation of F508del-CFTR to the cell surface. Furthermore, it has been reported that roscovitine increases intracellular calcium release and capacitative calcium entry in PC12 cells (Choi and Chung, 2010) and enhances the calcium influx in primary cultured hippocampal neurons (Tomizawa et al., 2002).
Another contribution to the corrector effect of roscovitine relates to its effect on the degradation machinery, which might favour proper folding and maturation of F508del-CFTR. ERAD has important consequences for protein folding, transport and degradation (Meusser et al., 2005) and is a central element of the secretory pathway. We showed that roscovitine directly inhibits the proteasome machinery. However, previous studies have demonstrated that direct inhibition of the proteasome with lactacystin and MG132 did not promote F508del-CFTR translocation to the cell surface (Jensen et al., 1995; Ward et al., 1995; Johnson et al., 1998). We also found that MG132 was unable to restore the function of F508del-CFTR in CF15 cells following incubation and iodide efflux assay (this study and Norez et al., 2009). However, the proteasome inhibitor bortezomib (Velcade) has been shown to increase HSP70, down-regulate valosin-containing protein expression and partially restore the function of mature F508del-CFTR in IB3-1 cells and human CF cells (Vij et al., 2006). Interestingly, both bortezomib and roscovitine are proteostasis regulators with a dual effect on proteasome activity and chaperoning. In dysferlinopathies, autosomal recessive progressive muscular dystrophies, the protein dysferlin encoded by missense alleles is rapidly degraded by the cellular ERQC but functionally restored by bortezomib (Azakir et al., 2012). Other F508del-CFTR correctors, called MPBs, have been shown to prevent the recognition of F508del-CFTR by the proteasome machinery allowing restoration of F508del-CFTR to the cell surface (Norez et al., 2008). Altogether, the results suggest that inhibition of the proteosomal degradation pathway does not enhance F508del-CFTR folding, but rather leads to a potentiation of another corrector effect. We found that roscovitine mimicked the Ca2+-dependent mechanism of action of thapsigargin on F508del-CFTR but induced a higher correction than that obtained with thapsigargin and was not totally affected by 2-APB co-treatment. Our hypothesis is, therefore, that the calnexin-dependent correction of F508del-CFTR induced by roscovitine is potentiated by its own action on the degradation machinery.
Numerous potential candidates are being selected by HTS for their ability to restore F508del-CFTR function and are being progressively developed towards clinical trials. However, these correctors are only modestly effective and have incompletely understood mechanisms. For example, 4-phenylbutyrate, which at mM concentrations improves F508del-CFTR trafficking in CF epithelial cells (Rubenstein and Zeitlin, 1998) or the Ca-ATPase pump inhibitors, thapsigargin and curcumin, which normalize both nasal epithelial chloride and sodium transport in F508del-CFTR mice (Egan et al., 2002; 2004,). Another example is the quinazoline compound VX-809, which was identified using a protocol in which cells were pre-incubated for 48 h with 10 μM of the drugs (Van et al., 2011). However, although VX-809 appeared to be a promising investigational corrector of F508del-CFTR misprocessing, it was found to have limited clinical benefit and an incompletely understood mechanism has hampered drug development. VX-770 (Ivacaftor), the only clinically approved potentiator drug, substantially enhances the channel function of several mutants. Although VX-809-induced changes in F508-CFTR activity at the plasma membrane are potentiated twofold by VX-770 in preclinical settings (Van et al., 2011), an additional improvement in corrector efficacy seems to be required to achieve substantial clinical benefit in CF (Clancy et al., 2012). Here, we demonstrated that roscovitine corrects F508del-CFTR trafficking in CF cells to a level similar to VX-809 after only 2 h of treatment and is like the potentiator VX-770. We consider that these results are a good indicator for the use of roscovitine as a potential pharmacological therapy for CF patients.
In conclusion, roscovitine increases the amount of functional F508del-CFTR at the plasma membrane of CF15 cells by a dual, CDK-independent mechanism of action. This appeared to be mainly due to a combination of two effects, inhibition of the calnexin/F508del-CFTR interaction strengthened by a direct inhibitory effect on the proteasome machinery. These results indicate that roscovitine (or analogues) may be a valuable proteostasis regulator for future clinical evaluation in CF patients.
Acknowledgments
This work was supported by grants from the French association ‘Vaincre la Mucoviscidose’ (F. B., C. N. and L. M.). This study has benefited from the facilities and expertise of ImageUP platform (http://sfa.univ-poitiers.fr/imageup/). Authors thank Dr A. Cantereau for her technical assistance. Thanks to Drs A. Palmer and R.Y. Tsien for providing the D1ER construct and Dr M. Frieden for her expertise. F. A. was supported by the Swiss National Science Foundation and the foundation Carlos and Elsie de Reuter.
Glossary
- CDK
cyclin-dependent kinase
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum-associated degradation
- ERQC
endoplasmic reticulum quality control
Author contributions
C. N. conceived and designed the biological experiments (iodide efflux, immunolabelling, Western blotting and co-immunoprecipitation) and drafted the manuscript. C. V., J. B., S. N., F. A., A. C. and P. B. performed calcium measurements with the Fluo4 probe (C. V.) or the D1ER construct (F. A.), experiments on CFBE cells and proteasome activity measurements (J. B.), CFTR activity (S. N.) or NaV1.5 activity (A. C. and P. B.) measurement by patch-clamp experiments. N. O. and H. G. designed and synthesized the compounds and E. D. performed the kinase assays. L. M. and F. B. were the coordinators of this project. All authors approved the final version of the manuscript.
Conflict of interest
L. M. is co-inventor of the roscovitine patent. L. M. and F. B. are co-inventors of the roscovitine and CF patent. L. M. and H. G. are co-founders of ManRos Therapeutics. L. M. is CEO and CSO of ManRos Therapeutics.
References
- Aldoss IT, Tashi T, Ganti AK. Seliciclib in malignancies. Expert Opin Investig Drugs. 2009;18:1957–1965. doi: 10.1517/13543780903418445. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14:Ion channels. Br J Pharmacol. 2013a;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antigny F, Girardin N, Frieden M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J Biol Chem. 2012;287:5917–5927. doi: 10.1074/jbc.M111.295733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azakir BA, Di FS, Kinter J, Sinnreich M. Proteasomal inhibition restores biological function of mis-sense mutated dysferlin in patient derived muscle cells. J Biol Chem. 2012;287:10344–10354. doi: 10.1074/jbc.M111.329078. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bach S, Knockaert M, Reinhardt J, Lozach O, Schmitt S, Baratte B, et al. Roscovitine targets, protein kinases and pyridoxal kinase. J Biol Chem. 2005;280:31208–31219. doi: 10.1074/jbc.M500806200. [DOI] [PubMed] [Google Scholar]
- Bebok Z, Collawn JF, Wakefield J, Parker W, Li Y, Varga K, et al. Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. J Physiol. 2005;569(Pt 2):601–615. doi: 10.1113/jphysiol.2005.096669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becq F. Cystic fibrosis transmembrane conductance regulator modulators for personalized drug treatment of cystic fibrosis: progress to date. Drugs. 2010;70:241–259. doi: 10.2165/11316160-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Benson C, White J, De BJ, O'Donnell A, Raynaud F, Cruickshank C, et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br J Cancer. 2007;96:29–37. doi: 10.1038/sj.bjc.6603509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier V, Bichet DG, Bouvier M. Pharmacological chaperone action on G-protein-coupled receptors. Curr Opin Pharmacol. 2004;4:528–533. doi: 10.1016/j.coph.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Bettayeb K, Oumata N, Echalier A, Ferandin Y, Endicott JA, Galons H, et al. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene. 2008;27:5797–5807. doi: 10.1038/onc.2008.191. [DOI] [PubMed] [Google Scholar]
- Bettayeb K, Baunbæk D, Delehouze C, Loaëc N, Hole AJ, Baumli S, et al. CDK inhibitors roscovitine and CR8 trigger Mcl-1 down-regulation and apoptotic cell death in neuroblastoma cells. Genes Cancer. 2010;1:369–380. doi: 10.1177/1947601910369817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bruscia E, Sangiuolo F, Sinibaldi P, Goncz KK, Novelli G, Gruenert DC. Isolation of CF cell lines corrected at DeltaF508-CFTR locus by SFHR-mediated targeting. Gene Ther. 2002;9:683–685. doi: 10.1038/sj.gt.3301741. [DOI] [PubMed] [Google Scholar]
- Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949–952. doi: 10.1038/nature05348. [DOI] [PubMed] [Google Scholar]
- Canu N, Barbato C, Ciotti MT, Serafino A, Dus L, Calissano P. Proteasome involvement and accumulation of ubiquitinated proteins in cerebellar granule neurons undergoing apoptosis. J Neurosci. 2000;20:589–599. doi: 10.1523/JNEUROSCI.20-02-00589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HS, Chung SH. Roscovitine increases intracellular calcium release and capacitative calcium entry in PC12 cells. Neurosci Lett. 2010;469:141–144. doi: 10.1016/j.neulet.2009.11.061. [DOI] [PubMed] [Google Scholar]
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67:12–18. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz FP, Newberry S, Jenkinson SF, Wormald MR, Butters TD, Alonzi DS, et al. 4-C-Me-DAB and 4-C-Me-LAB – enantiomeric alkyl-branched pyrrolidine iminosugars – are specific and potent α-glucosidase inhibitors; acetone as the sole protecting group. Tetrahedron Lett. 2011;52:219–223. doi: 10.1016/j.tetlet.2010.10.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Dormer RL, Harris CM, Clark Z, Pereira MM, Doull IJ, Norez C, et al. Sildenafil (Viagra) corrects DeltaF508-CFTR location in nasal epithelial cells from patients with cystic fibrosis. Thorax. 2005;60:55–59. doi: 10.1136/thx.2003.019778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey DG, Bell SC, Elborn JS. Neutrophils in cystic fibrosis. Thorax. 2009;64:81–88. doi: 10.1136/thx.2007.082388. [DOI] [PubMed] [Google Scholar]
- Egan ME, Glockner-Pagel J, Ambrose C, Cahill PA, Pappoe L, Balamuth N, et al. Calcium-pump inhibitors induce functional surface expression of Delta F508-CFTR protein in cystic fibrosis epithelial cells. Nat Med. 2002;8:485–492. doi: 10.1038/nm0502-485. [DOI] [PubMed] [Google Scholar]
- Egan ME, Pearson M, Weiner SA, Rajendran V, Rubin D, Glockner-Pagel J, et al. Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science. 2004;304:600–602. doi: 10.1126/science.1093941. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Farinha CM, Amaral MD. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol Cell Biol. 2005;25:5242–5252. doi: 10.1128/MCB.25.12.5242-5252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- Galietta LJ. Managing the underlying cause of cystic fibrosis: a future role for potentiators and correctors. Paediatr Drugs. 2013;15:393–402. doi: 10.1007/s40272-013-0035-3. [DOI] [PubMed] [Google Scholar]
- Galons H, Oumata N, Meijer L. Cyclin-dependent kinase inhibitors: a survey of recent patent literature. Expert Opin Ther Pat. 2010;20:377–404. doi: 10.1517/13543770903524284. [DOI] [PubMed] [Google Scholar]
- Ganapathi SB, Kester M, Elmslie KS. State-dependent block of HERG potassium channels by R-roscovitine: implications for cancer therapy. Am J Physiol Cell Physiol. 2009;296:C701–C710. doi: 10.1152/ajpcell.00633.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q, Jones MA, Zhou Z. Mechanisms of pharmacological rescue of trafficking-defective hERG mutant channels in human long QT syndrome. J Biol Chem. 2006;281:4069–4074. doi: 10.1074/jbc.M511765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NS, Wodicka L, Thunnissen AM, Norman TC, Kwon S, Espinoza FH, et al. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science. 1998;281:533–538. doi: 10.1126/science.281.5376.533. [DOI] [PubMed] [Google Scholar]
- Herczenik E, Gebbink MF. Molecular and cellular aspects of protein misfolding and disease. FASEB J. 2008;22:2115–2133. doi: 10.1096/fj.07-099671. [DOI] [PubMed] [Google Scholar]
- Hsieh WS, Soo R, Peh BK, Loh T, Dong D, Soh D, et al. Pharmacodynamic effects of seliciclib, an orally administered cell cycle modulator, in undifferentiated nasopharyngeal cancer. Clin Cancer Res. 2009;15:1435–1442. doi: 10.1158/1078-0432.CCR-08-1748. [DOI] [PubMed] [Google Scholar]
- Hutt DM, Herman D, Rodrigues AP, Noel S, Pilewski JM, Matteson J, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol. 2010;6:25–33. doi: 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson DM, Valentich JD, Marini FC, Grubman SA, Iannuzzi MC, Dorkin HL, et al. Expression of normal and cystic fibrosis phenotypes by continuous airway epithelial cell lines. Am J Physiol. 1990;259(6 Pt 1):496–505. doi: 10.1152/ajplung.1990.259.6.L496. [DOI] [PubMed] [Google Scholar]
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- Johnson LG, Mewshaw JP, Ni H, Friedmann T, Boucher RC, Olsen JC. Effect of host modification and age on airway epithelial gene transfer mediated by a murine leukemia virus-derived vector. J Virol. 1998;72:8861–8872. doi: 10.1128/jvi.72.11.8861-8872.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller DI, Rougier JS, Kucera JP, Benammar N, Fressart V, Guicheney P, et al. Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc Res. 2005;67:510–519. doi: 10.1016/j.cardiores.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Knockaert M, Greengard P, Meijer L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol Sci. 2002;23:417–425. doi: 10.1016/s0165-6147(02)02071-0. [DOI] [PubMed] [Google Scholar]
- Li H, Findlay IA, Sheppard DN. The relationship between cell proliferation, Cl- secretion, and renal cyst growth: a study using CFTR inhibitors. Kidney Int. 2004;66:1926–1938. doi: 10.1111/j.1523-1755.2004.00967.x. [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, Clarke DM. Thapsigargin or curcumin does not promote maturation of processing mutants of the ABC transporters CFTR, and P-glycoprotein. Biochem Biophys Res Commun. 2004;325:580–585. doi: 10.1016/j.bbrc.2004.10.070. [DOI] [PubMed] [Google Scholar]
- Lubamba B, Lebacq J, Lebecque P, Vanbever R, Leonard A, Wallemacq P, et al. Airway delivery of low-dose miglustat normalizes nasal potential difference in F508del cystic fibrosis mice. Am J Respir Crit Care Med. 2009;179:1022–1028. doi: 10.1164/rccm.200901-0049OC. [DOI] [PubMed] [Google Scholar]
- Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol Med. 2012;18:81–91. doi: 10.1016/j.molmed.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolskee RF, McHendry-Rinde B, Horn R. Panning transfected cells for electrophysiological studies. Biotechniques. 1993;15:906–911. [PubMed] [Google Scholar]
- Meijer L, Raymond E. Roscovitine and other purines as kinase inhibitors. From starfish oocytes to clinical trials. Acc Chem Res. 2003;36:417–425. doi: 10.1021/ar0201198. [DOI] [PubMed] [Google Scholar]
- Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Mercier A, Clément R, Harnois T, Bourmeyster N, Faivre JF, Findlay I, et al. The β1-subunit of Na(v)1.5 cardiac sodium channel is required for a dominant negative effect through α-α interaction. PLoS ONE. 2012;7:e48690. doi: 10.1371/journal.pone.0048690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- Moriceau S, Kantari C, Mocek J, Davezac N, Gabillet J, Guerrera IC, et al. Coronin-1 is associated with neutrophil survival and is cleaved during apoptosis: potential implication in neutrophils from cystic fibrosis patients. J Immunol. 2009;182:7254–7263. doi: 10.4049/jimmunol.0803312. [DOI] [PubMed] [Google Scholar]
- Moriceau S, Lenoir G, Witko-Sarsat V. In cystic fibrosis homozygotes and heterozygotes, neutrophil apoptosis is delayed and modulated by diamide or roscovitine: evidence for an innate neutrophil disturbance. J Innate Immun. 2010;2:260–266. doi: 10.1159/000295791. [DOI] [PubMed] [Google Scholar]
- Norez C, Antigny F, Becq F, Vandebrouck C. Maintaining low Ca2+ level in the endoplasmic reticulum restores abnormal endogenous F508del-CFTR trafficking in airway epithelial cells. Traffic. 2006a;7:562–573. doi: 10.1111/j.1600-0854.2006.00409.x. [DOI] [PubMed] [Google Scholar]
- Norez C, Noel S, Wilke M, Bijvelds M, Jorna H, Melin P, et al. Rescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the alpha-glucosidase inhibitor miglustat. FEBS Lett. 2006b;580:2081–2086. doi: 10.1016/j.febslet.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Norez C, Bilan F, Kitzis A, Mettey Y, Becq F. Proteasome-dependent pharmacological rescue of cystic fibrosis transmembrane conductance regulator revealed by mutation of glycine 622. J Pharmacol Exp Ther. 2008;325:89–99. doi: 10.1124/jpet.107.134502. [DOI] [PubMed] [Google Scholar]
- Norez C, Antigny F, Noel S, Vandebrouck C, Becq F. A cystic fibrosis respiratory epithelial cell chronically treated by miglustat acquires a non-cystic fibrosis-like phenotype. Am J Respir Cell Mol Biol. 2009;41:217–225. doi: 10.1165/rcmb.2008-0285OC. [DOI] [PubMed] [Google Scholar]
- Oumata N, Ferandin Y, Meijer L, Galons H. Practical synthesis of roscovitine and CR8. Org Process Res Dev. 2009;13:641–644. [Google Scholar]
- Palmer AE, Tsien RY. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat Protoc. 2006;1:1057–1065. doi: 10.1038/nprot.2006.172. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N, Diena T, Caci E, Nieddu E, Mazzei M, Ravazzolo R, et al. Antihypertensive 1,4-dihydropyridines as correctors of the cystic fibrosis transmembrane conductance regulator channel gating defect caused by cystic fibrosis mutations. Mol Pharmacol. 2014;68:1736–1746. doi: 10.1124/mol.105.015149. 2005. [DOI] [PubMed] [Google Scholar]
- Reinhardt J, Ferandin Y, Meijer L. Purification of CK1 by affinity chromatography on immobilised axin. Protein Expr Purif. 2007;54:101–109. doi: 10.1016/j.pep.2007.02.020. [DOI] [PubMed] [Google Scholar]
- Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056–1064. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am J Respir Crit Care Med. 1998;157:484–490. doi: 10.1164/ajrccm.157.2.9706088. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of DeltaF508-CFTR. Am J Physiol Cell Physiol. 2000;278:C259–C267. doi: 10.1152/ajpcell.2000.278.2.C259. [DOI] [PubMed] [Google Scholar]
- Schultz BD, Frizzell RA, Bridges RJ. Rescue of dysfunctional deltaF508-CFTR chloride channel activity by IBMX. J Membr Biol. 1999;170:51–66. doi: 10.1007/s002329900537. [DOI] [PubMed] [Google Scholar]
- Sheppard DN, Welsh MJ. Inhibition of the cystic fibrosis transmembrane conductance regulator by ATP-sensitive K + channel regulators. Ann N Y Acad Sci. 1993;707:275–284. doi: 10.1111/j.1749-6632.1993.tb38058.x. [DOI] [PubMed] [Google Scholar]
- Tang L, Li MH, Cao P, Wang F, Chang WR, Bach S, et al. Crystal structure of pyridoxal kinase in complex with roscovitine and derivatives. J Biol Chem. 2005;280:31220–31229. doi: 10.1074/jbc.M500805200. [DOI] [PubMed] [Google Scholar]
- Tomizawa K, Ohta J, Matsushita M, Moriwaki A, Li ST, Takei K, et al. Cdk5/p35 regulates neurotransmitter release through phosphorylation and downregulation of P/Q-type voltage-dependent calcium channel activity. J Neurosci. 2002;22:2590–2597. doi: 10.1523/JNEUROSCI.22-07-02590.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubuki S, Kawasaki H, Saito Y, Miyashita N, Inomata M, Kawashima S. Purification and characterization of a Z-Leu-Leu-Leu-MCA degrading protease expected to regulate neurite formation: a novel catalytic activity in proteasome. Biochem Biophys Res Commun. 1993;196:1195–1201. doi: 10.1006/bbrc.1993.2378. [DOI] [PubMed] [Google Scholar]
- Van GF, Straley KS, Cao D, González J, Hadida S, Hazlewood A, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- Van GF, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesely J, Havlicek L, Strnad M, Blow JJ, Donella-Deana A, Pinna L, et al. Inhibition of cyclin-dependent kinases by purine analogues. Eur J Biochem. 1994;224:771–786. doi: 10.1111/j.1432-1033.1994.00771.x. [DOI] [PubMed] [Google Scholar]
- Vij N, Fang S, Zeitlin PL. Selective inhibition of endoplasmic reticulum-associated degradation rescues DeltaF508-cystic fibrosis transmembrane regulator and suppresses interleukin-8 levels: therapeutic implications. J Biol Chem. 2006;281:17369–17378. doi: 10.1074/jbc.M600509200. [DOI] [PubMed] [Google Scholar]
- Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- Wang XF, Reddy MM, Quinton PM. Effects of a new cystic fibrosis transmembrane conductance regulator inhibitor on Cl- conductance in human sweat ducts. Exp Physiol. 2004;89:417–425. doi: 10.1113/expphysiol.2003.027003. [DOI] [PubMed] [Google Scholar]
- Wang Y, Loo TW, Bartlett MC, Clarke DM. Additive effect of multiple pharmacological chaperones on maturation of CFTR processing mutants. Biochem J. 2007;406:257–263. doi: 10.1042/BJ20070478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Wesierska-Gadek J, Chamrad I, Krystof V. Novel potent pharmacological cyclin-dependent kinase inhibitors. Future Med Chem. 2009;1:1561–1581. doi: 10.4155/fmc.09.110. [DOI] [PubMed] [Google Scholar]
- Yarotskyy V, Gao G, Du L, Ganapathi SB, Peterson BZ, Elmslie KS. Roscovitine binds to novel L-channel (CaV1.2) sites that separately affect activation and inactivation. J Biol Chem. 2010;285:43–53. doi: 10.1074/jbc.M109.076448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurko-Mauro KA, Reenstra WW. Prostaglandin F2alpha stimulates CFTR activity by PKA- and PKC-dependent phosphorylation. Am J Physiol. 1998;275(3 Pt 1):C653–C660. doi: 10.1152/ajpcell.1998.275.3.C653. [DOI] [PubMed] [Google Scholar]
- Zaman K, McPherson M, Vaughan J, Hunt J, Mendes F, Gaston B, et al. S-nitrosoglutathione increases cystic fibrosis transmembrane regulator maturation. Biochem Biophys Res Commun. 2001;284:65–70. doi: 10.1006/bbrc.2001.4935. [DOI] [PubMed] [Google Scholar]