

Abstract
Background and Purpose
Chemokines are involved in neuroinflammation and contribute to chronic pain processing. The new chemokine prokineticin 2 (PROK2) and its receptors (PKR1 and PKR2) have a role in inflammatory pain and immunomodulation. In the present study, we investigated the involvement of PROK2 and its receptors in neuropathic pain.
Experimental Approach
Effects of single, intrathecal, perineural and s.c. injections of the PKR antagonist PC1, or of 1 week s.c. treatment, on thermal hyperalgesia and tactile allodynia was evaluated in mice with chronic constriction of the sciatic nerve (CCI). Expression and localization of PROK2 and of its receptors at peripheral and central level was evaluated 10 days after CCI, following treatment for 1 week with saline or PC1. IL-1β and IL-10 levels, along with glia activation, were evaluated.
Key Results
Subcutaneous, intrathecal and perineural PC1 acutely abolished the CCI-induced hyperalgesia and allodynia. At 10 days after CCI, PROK2 and its receptor PKR2 were up-regulated in nociceptors, in Schwann cells and in activated astrocytes of the spinal cord. Therapeutic treatment with PC1 (s.c., 1 week) alleviated established thermal hyperalgesia and allodynia, reduced the injury-induced overexpression of PROK2, significantly blunted nerve injury-induced microgliosis and astrocyte activation in the spinal cord and restored the physiological levels of proinflammatory and anti-inflammatory cytokines in periphery and in spinal cord.
Conclusion and Implications
The prokineticin system contributes to pain modulation via neuron–glia interaction. Sustained inhibition of the prokineticin system, at peripheral or central levels, blocked both pain symptoms and some events underlying disease progression.
Table of Links
| TARGETS | LIGANDS |
| Prokineticin receptor PKR1 | Prokineticin 1, PROK1 |
| Prokineticin receptor PKR2 | Prokineticin 2, PROK2 |
This Table lists the protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Neuropathic pain, resulting from damage to or dysfunction of the nervous system, is a chronic pain largely resistant to treatment mainly because the underlying mechanisms are still poorly understood. Increasing evidence now suggests that potent neuromodulators as proinflammatory cytokines and chemokines are involved in neuroinflammation at different anatomical locations, including the injured nerve, dorsal root ganglion (DRG), spinal cord and brain, and contribute to chronic pain processing (Abbadie et al., 2009; Gao and Ji, 2010a).
The cytokine prokineticin 2 (PROK2 or mammalian Bv8), which belongs to a new family of chemokines, displays a major role in triggering inflammatory pain by acting on two GPCRs, the prokineticin receptors PKR1 and PKR2 (see Negri et al., 2007; Negri and Lattanzi, 2012; receptor nomenclature follows Alexander et al., 2013), localized on peripheral nociceptors also expressing the ion channels, TRPV1 and TRPA1 (Negri et al., 2006b; Vellani et al., 2006). Prokr1 gene deletion or pretreatment with the prokineticin receptor antagonist, PC1, markedly reduced the inflammation-induced hypersensitivity and the up-regulation of Bv8/PROK2 (Balboni et al., 2008; Giannini et al., 2009; Negri and Lattanzi, 2011). Apart from the central and peripheral nervous system (Cheng et al., 2006; Hu et al., 2006; Negri et al., 2006a), PROK2 is constitutively expressed also in the bone marrow and in the peripheral blood cells, but it is strongly up-regulated in inflammatory conditions, associated with infiltrating cells (LeCouter et al., 2004; Giannini et al., 2009).
In vivo and in vitro experiments from our and other groups demonstrated potent chemotactic and immunomodulatory activities of the prokineticins, able to induce a proinflammatory phenotype of macrophages and to skew the Th1/Th2 balance towards a Th1 response mainly through PKR1 activation (Dorsch et al., 2005; Martucci et al., 2006; Franchi et al., 2008). As the prokineticins and their receptors, usually found in neurons and in immuno-inflammatory cells, are also expressed in glia cells (Koyama et al., 2006; Cheng et al., 2012) and Bv8/PROK2 is involved in both nociception and immunoregulation, the prokineticin system appears to be a pivotal candidate for mediating the neuroimmune interactions in neuropathic pain.
This possibility was confirmed by our results demonstrating that PROK2 and PKR2 were overexpressed in sciatic nerve, DRG and spinal cord of mice with chronic constriction (CCI) of the sciatic nerve and that therapeutic treatment with the prokineticin receptor antagonist PC1 alleviated thermal hyperalgesia and prevented development of allodynia. The fact that such treatment also prevented activation of glia and restored pro-inflammatory and anti-inflammatory cytokines to physiological levels, suggests that blocking of the prokineticin system might offer a new opportunity for disease modification in inflammatory states.
Methods
Animal preparation
All animal care and experimental procedures complied with the International Association for the Study of Pain and European Community (E.C.L358/118/12/86) guidelines and were approved by the Animal Care and Use Committee of the Italian Ministry of Health. All efforts were made to minimize animal suffering and to reduce the number of animals used. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 140 animals were used in the experiments described here.
Experiments were carried out in male CD1 mice (25–30 g, Harlan Laboratories, San Pietro al Natisone, Udine, Italy). Animals were housed individually in cages, under conditions of optimum light, temperature and humidity (12:12 h light/dark cycles, 22 ± 2°C, 50–60%) with food and water ad libitum and acclimatized to the environment for 4–5 days before surgery or pharmacological treatment. Mononeuropathy was induced by the CCI of the sciatic nerve (Bennett and Xie, 1988) in CD1 mice anaesthetized with ketamine-xylazine (60 mg kg−1 + 10 mg kg−1, i.p.). Three loose ligatures with 4-0 silk suture thread were made around the nerve with a 1.0–1.5 mm interval between each of them. In sham-operated mice, an identical dissection was performed on the same side, except that the sciatic nerve was not tied.
Nociceptive behavioural tests
Behavioural experiments were carried out by researchers unaware of the treatments, between 1000 h and 1400 h, in a reserved quiet temperature-controlled room. For testing mechanical sensitivity, animals were put in boxes on an elevated metal mesh floor and allowed 30 min for habituation before examination. The plantar surface of each hindpaw was stimulated with a series of von Frey hairs with logarithmically incrementing stiffness (0.04–2.0 g, 2Biological Instruments, Besozzo, Varese, Italy), presented perpendicular to the plantar surface (7–8 s for each hair). The 50% paw withdrawal threshold (PWT) was determined using Dixon's up-down method (Chaplan et al., 1994). For testing heat sensitivity, animals were put in plastic boxes and allowed 30 min for habituation before examination. Heat sensitivity was tested by radiant heat using a Hargreaves apparatus (Ugo Basile, Comerio, Varese, Italy) and expressed as paw withdrawal latency (PWL). The radiant heat intensity was adjusted so that basal PWL was between 10 and 12 s with a cut-off of 20 s to prevent tissue damage.
Experimental design
The PKR antagonist PC1 is a triazine-guanidine derivative that, in vitro blocks Bv8-induced intracellular calcium increase, in CHO cells transfected with PKR1 and PKR2,. It shows an affinity 30 times higher for PKR1 than for PKR2 and, in vivo, antagonizes hyperalgesia induced by the intraplantar, s.c. or intrathecal administration of Bv8, being ineffective against other pro-algesic mediators such as bradykinin, PGE2 and ATP (Balboni et al., 2008; Miele et al., 2010). PC1 was injected in CD1 mice intrathecally (i.t., 5 µL per mice) with a 30G needle inserted between the L5 and L6 level (Hylden and Wilcox, 1980), by perineural injection (p.n., 10 µL per mice) with a 30G needle in the region surrounding the sciatic nerve at high thigh level of the limb in which the sciatic nerve was ligated (Kiguchi et al., 2010) or s.c. (50 µL for 10 g body weight) into the flank region of the mouse. Single bolus i.t. injections of PC1 (1, 10, 100 ng per mice, n = 15) and single bolus p.n. injections (5, 15, 50 ng per mice, n = 15) were performed, in different group of mice, on day 3 after CCI. Single bolus systemic (s.c.) injection of PC1 (30, 75 and 150 µg kg−1 s.c.) was performed on day 3 and on day 17 after CCI (n = 15). Then we chose the highest, more effective dose (150 µg kg−1 s.c.) for chronic treatment in therapeutic schedules: groups of mice were divided as follows: (i) sham-operated mice (n = 5); (ii) CCI mice treated with saline from day 3 to day 9 after sciatic nerve ligation (CCI/saline; n = 8); (iii) CCI mice treated with PC1 150 µg kg−1 s.c., twice a day from day 3 to day 9 after sciatic nerve ligation (CCI/PC1, n = 8); (iv) CCI mice treated with PC1 150 µg kg−1 s.c., twice a day from day 17 to day 20 after sciatic nerve ligature (n = 8). In sham mice, in CCI/saline and in CCI/PC1 animals, tactile allodynia and thermal hyperalgesia were assessed daily before and on day 1 to day 42 after CCI, in the following sequence: von Frey stimulation, plantar test.
Histochemical and biochemical evaluation
The expression (RT-PCR) and the distribution (immunohistochemistry) of PROK2, PKR1 and PKR2 in the spinal cord, lumbar DRG and sciatic nerve, the activation of microglia and astrocytes in the spinal cord, the mRNA expression and the protein amount of IL-1β and IL-10 in the spinal cord and in the sciatic nerve were evaluated in different groups of sham, CCI/saline and CCI/PC1 mice killed 10 days after CCI induction. Each group consisted of at least five animals.
RNA extraction and real-time PCR
Total RNA was extracted from sciatic nerves, pooled from two mice (six samples, i.e. 12 mice per experimental group), from L4, L5 DRG and from dorsal spinal cord at L4–L6 level using TRIzol reagent (Invitrogen, Life Technologies, San Giuliano Milanese, Italy). Real-time PCR procedure has already been described in detail (Sacerdote et al., 2013). Taqman probes for mouse Prok2 (Mm 01182450_g1); prokineticin receptors (Prokr1: Mm 00517546_m1; Prokr2: Mm 00769571_m1); interleukins (IL-1β: Mm 00434228_m1; IL-10: Mm00439616_m1) and GAPDH (Mm99999915_g1) were purchased from Applied Biosystems (Monza, Italy). The reaction conditions were as follows: 95°C for 2 min (initial denaturation), followed by 45 cycles at 95°C for 15 s (cycled template denaturation) and at 60°C for 60 s (annealing and extension). The Ct value of the specific gene of interest was normalized to the Ct value of the endogenous control, GAPDH, and the comparative Ct method (2−ΔΔCt) was then applied using sham group as calibrator.
Cytokine protein measurement
The nerve and spinal cord samples were homogenized in ice-cold PBS containing a protease inhibitor cocktail (Roche Diagnostics, Monza, Italy). IL-1β and IL-10 protein contents were determined by ELISA using ultra-sensitive ELISA (IL-1β R&D Systems, Minneapolis, MN, USA; IL-10 eBioscience, San Diego, CA, USA).
Neutrophil content in the sciatic nerve
Neutrophil content in the sciatic nerve was evaluated measuring the myeloperoxidase activity (MPO) using the method of Bradley et al. (1982).
Immunofluorescence
L4–L5 spinal cord, DRG and sciatic nerve were dissected from transcardially perfused (PBS followed by 4% paraformaldehyde) mice, embedded in cryostat medium and frozen. Spinal cord sections (40 µm, free-floating) were incubated at 4°C for 48 h, whereas DRG and sciatic nerve sections (20 µm), mounted on slides, were incubated at 4°C overnight with the following primary antibodies diluted in PBS-0.3% Triton X-100: 1/200 rabbit polyclonal anti-PROK2 (AbCam, Cambridge, UK), 1/200 rabbit polyclonal anti-PKR1 and PKR2 (Alomone Labs, Jerusalem, Israel), 1/500 mouse monoclonal anti-neuronal nuclei (NeuN), 1/400 mouse polyclonal anti-glial fibrillary acidic protein (GFAP) (Immunological Sciences, Rome, Italy), 1/300 mouse polyclonal anti-Synaptophysin (Sigma-Aldrich, Milan, Italy), 1/100 rat monoclonal anti-CD11 (BD Pharmigen, Milan, Italy). The sections were then incubated for 2 h at room temperature in 1:200 anti-species IgG antibodies coupled to Alexa Fluor®-488 or 555 (Immunological Sciences). Nuclei were stained with DAPI 1/500. The stained sections were examined at confocal laser scanning microscope (Leica SP5, Leica Microsystems, Wetzlar, Germany). Immunofluorescence intensity or immunoreactive area was measured in five fields (300 µm2) for every section in at least 10 sections for every experimental group (http://imagej.nih.gov/ij/index.html, free software).
To assess the specificity of the anti-PROK2 antibody, we pre-adsorbed it with the protein PROK2 (500 ng) overnight at 4°C prior to incubation with tissue. To assess the specificity of the anti-PKR1 and anti-PKR2 antibody, we pre-adsorbed them with the respective blocking peptides (Alomone Labs) overnight. We also checked these antibodies on DRG and spinal cord sections from PKR1-KO to PKR2-KO mice.
Sciatic nerve immunohistochemistry
Paraffin-embedded sciatic nerve sections (5 µm), deparaffinized and rehydrated, were incubated with normal horse serum (3%, 1 h, 37°C) then with goat polyclonal anti-PROK2 (1:100, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA, overnight, 4°C), washed and incubated with biotinylated secondary antibody (Vector Laboratories, Burlingame, CA, USA) and avidin-biotin-horseradish peroxidase complex (Vectastain ABC kit; Vector Laboratories), stained with 3,3N-diaminobenzidine tetrahydrochloride (DAB, Sigma-Aldrich). All slides were counterstained with Mayer's haematoxylin, visualized and photographed with an Olympus DP12 microscope equipped with a digital camera. PROK2-staining intensity was computed as integrated optical density (IOD) for arbitrary areas and measured in six samples for each experimental group (Image Pro-Plus, 4.5.1, Milan, Italy).
Data analysis
Results are expressed as mean ± SEM. When appropriate, one-way anova with Tukey's test for multiple comparisons or two-way repeated measures anova with Bonferroni's post tests, was performed using GraphPad Prism 5 for Windows version 5.4. Differences were considered significant at P < 0.05.
Results
Pharmacological blockade of the prokineticin receptors alleviated hyperalgesia
Before CCI induction, the overall mean baseline PWL to noxious heat stimuli and PWT to mechanical stimuli were similar in all groups of mice and there were no significant differences between left and right PWL and PWT. CCI of the sciatic nerve caused a marked reduction of the ipsilateral PWL and PWT. Thermal hyperalgesia appeared from the first day after surgery and reached maximal values on day 3 after surgery. The PWL of the injured paw was 4.4 ± 0.2 s; 9.2 ± 0.2 s in the contralateral paw and 9.1 ± 0.19 s in sham-operated mice (P < 0.001). Thermal hypersensitivity of neuropathic mice returned to normal value after about 40 days. When tactile allodynia was assessed with von Frey filaments, it was clearly evident in the injured paw from day 12 after CCI, reached maximal levels on day 17 (P < 0.001) and was still significant (P < 0.01) on day 40. The tactile sensitivity of the contralateral paw remained near the basal level throughout the experiment (Figure 1A and B).
Figure 1.
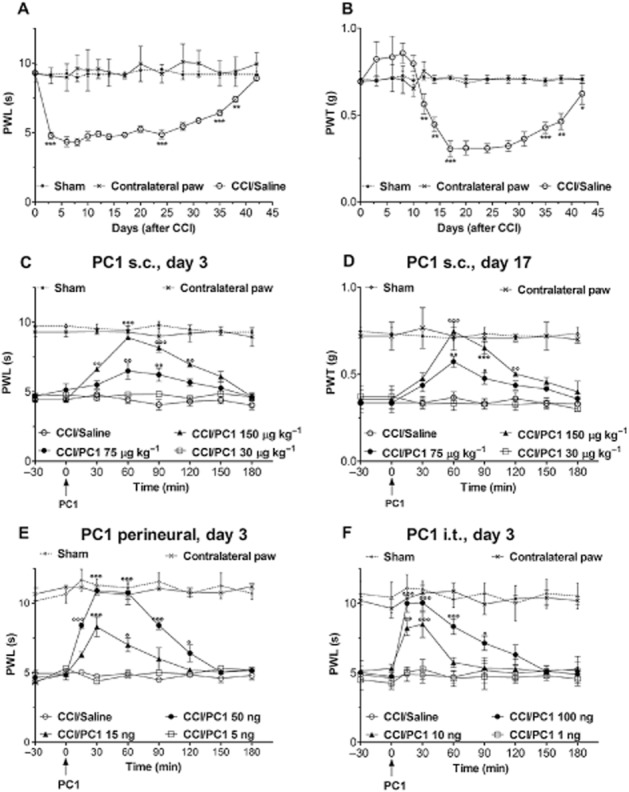
Antihyperalgesic effect of PC1 administration as single bolus. (A) CCI of the sciatic nerve caused a marked reduction of the ipsilateral PWL, assessed with the plantar test, from the first day after surgery. PWL reached minimal values on day 3 and returned to normal values 40 days after surgery. (B) PWT, assessed with the von Frey filament stimulation, was clearly reduced in the injured paw from day 12 after CCI reached minimal levels on day 17 and was still significantly lower than in contralateral paw on day 40. Thermal and tactile sensitivity of the contralateral paw remained near the basal level throughout the experiment. (C) Single s.c. administration of PC1 (30, 75 and 150 µg kg−1 s.c.) on day 3 after CCI, when thermal hyperalgesia was maximal, dose-dependently reduced the established CCI-induced thermal hyperalgesia. (D) Single s.c. administration of PC1 (30, 75 and 150 µg kg−1 s.c.) on day 17 after CCI, when allodynia have reached maximum level, dose-dependently reduced the established allodynia. The highest dose (150 µg kg−1 s.c) abolished thermal and tactile hyperalgesia for about 2 h. (E) A single bolus p.n. injection of PC1 (5, 15, 50 ng per mice) in the injured paw on day 3 after CCI dose-dependently reverted the established CCI-induced thermal hypersensitivity. The highest dose of PC1 (50 ng) abolished hyperalgesia for about 2 h. (F) A single bolus i.t. injection of PC1 (1, 10 and 100 ng per mice) induced a rapid, dose-dependent reduction of the established thermal hyperalgesia. The highest dose of PC1 (100 ng) abolished thermal hyperalgesia for about 2 h. Data represent means ± SEM of five mice. *P < 0.05, **P < 0.01, ***P < 0.001 CCI/saline versus sham; °P < 0.05; °°P < 0.01; °°°P < 0.001 CCI/PC1 versus CCI/saline mice; two-way anova, followed by Bonferroni's test.
A single bolus systemic injection of PC1 (30, 75 and 150 µg kg−1) on postoperative day 3, when thermal hyperalgesia was clearly evident (Figure 1C) or on postoperative day 17, when tactile allodynia reached full development (Figure 1D), dose-dependently reduced the established CCI-induced thermal hyperalgesia and tactile allodynia. The effect was maximum in about 20 min, suggesting a direct action on nociceptor PKRs, whose blockade hinders the transmission of pain stimuli. The antihyperalgesic effect of the highest dose (150 µg kg−1, s.c.) lasted for about 2 h.
To evaluate if this antihyperalgesic effect would depend on peripheral or spinal mechanisms or both, we injected PC1 p.n. or i.t. on day 3 after CCI, when thermal hyperalgesia was clearly evident. A significant dose-related antihyperalgesic effect was observed after p.n. injection of very low doses of PC1 (5, 15, 50 ng per·mouse). The onset of the antihyperalgesia was rapid suggesting that this action might be mediated through receptors expressed on DRG neurons (Figure 1E). PC1, dose-dependently (1, 10 and 100 ng per mouse), also reversed the established hyperalgesia when injected i.t. (Figure 1F). The effect was maximal in 10–15 min and lasted for 1 to 2 h, suggesting that nerve damage had induced activation of the prokineticin system at the level of the spinal cord. Intrathecal injection of PC1 did not induce any changes in the pain threshold of contralateral paw which maintained values similar to those measured in sham animals.
Then, we chose the highest, more effective s.c. dose of PC1 (150 µg kg−1) for chronic treatment of the established hyperalgesia (therapeutic schedules). Repeated administration of PC1 (150 µg kg−1, s.c. twice a day) from day 3 to 9 produced total recovery of the decreased PWL for at least 9 days (Figure 2A and B). This treatment was effective as soon as 2 days after the start of treatment (P < 0.05) and lasted for 4 days after cessation of treatment. Indeed, the thermal nociceptive threshold was maintained at the basal level (P < 0.001) up to day 13 after surgery. From day 14 onward, thermal hyperalgesia slowly reappeared but at a level significantly lower than that of CCI/saline mice (P < 0.05). Interestingly, such a treatment schedule prevented the later development of tactile allodynia. Indeed, the PWT of the CCI/PC1 mice remained at the same level as that of the sham group for the entire period of evaluation (40 days after CCI).
Figure 2.
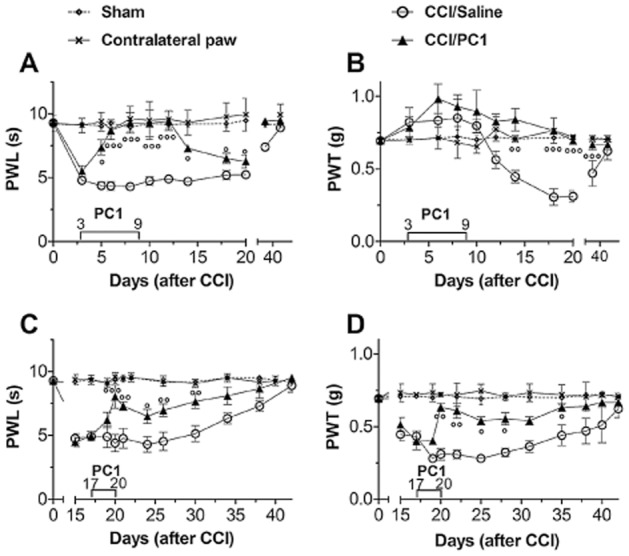
Antihyperalgesic effect of repeated administration of PC1. Repeated systemic injections of PC1 (150 µg kg−1, twice a day) from day 3 to 9 after CCI abolished the CCI-induced thermal hyperalgesia for about 2 weeks (A) and prevented the later development of tactile allodynia (B). Repeated administration of PC1 (150 µg kg−1 s.c. twice a day) from day 17 to 20 significantly reduced but did not abolish thermal (C) and tactile (D) hyperalgesia which slowly reappeared after treatment withdrawal. Data represent means ± SEM of six to nine mice. °P < 0.05; °°P < 0.01; °°°P < 0.001 CCI/PC1 versus CCI/saline mice; two-way anova, followed by Bonferroni's test.
Repeated administration of PC1 (150 µg kg−1, s.c. twice a day) from day 17 to 20 significantly reduced thermal hyperalgesia (Figure 2C). Tactile allodynia too was reduced but not abolished (Figure 2D). Nociceptive responses were significantly reduced after 2 days of treatment, for both tactile allodynia (P < 0.01) and thermal hyperalgesia (P < 0.01). After 4 days of treatment, thermal and tactile nociceptive thresholds of the CCI/PC1 group reached almost the levels of the sham group (P < 0.01, for both measures). After cessation of treatment, the nociceptive thresholds of CCI/PC1 mice were maintained significantly higher than those of CCI/saline mice for the entire period of observation.
As the hyperalgesia in our model was rapidly reversed by acute s.c., i.t. and p.n. administration of the PKR antagonist and its development was controlled by repeated systemic administration of the PKR antagonist, it was likely that the prokineticin system was involved. Hence, we decided to compare the expression of PROK2 and its receptors at peripheral and central sites of pain transmission in the experimental groups, on day 10 after surgery when the antihyperalgesic effect of PC1 was at its maximum.
PROK2-, PKR1- and PKR2-mRNA in the spinal cord, DRG and in the sciatic nerve
As illustrated in Figure 0, 10 days after CCI, a general activation of the prokineticin system was evident in the sciatic nerve, in L4–L5 DRGs and the dorsal spinal cord. In the injured sciatic nerve PROK2 (Figure 3A), PKR1 (Figure 3B) and PKR2 (Figure 3C) mRNA levels were significantly higher than in sham nerves. The up-regulation of PKR2 was most obvious, as the receptor expression was 150-fold higher than in control mice. In L4–L5 ipsilateral DRG and in the L4–L6 spinal cord, PROK2 (Figure 3D and G) and PKR2 (Figure 3F and I) mRNA levels were significantly higher in CCI than in sham nimals, whereas a non-significant increase was measured for PKR1 (Figure 3E and H). Therapeutic treatment with the receptor antagonist, PC1, blocked the PROK2 mRNA increase in all these tissues without significantly affecting PKR1- or PKR2-mRNA levels.
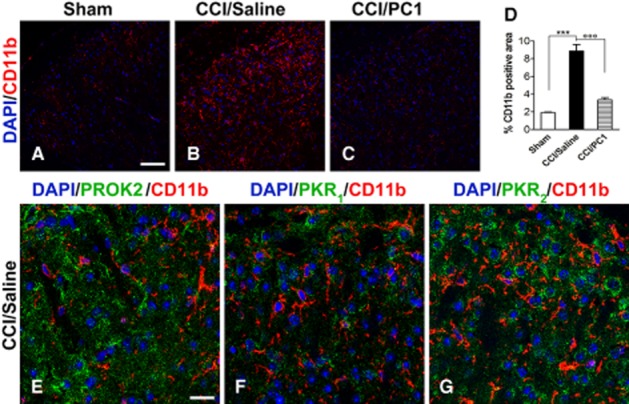
Immunofluorescence staining of CD11b (microglia marker, red) in the spinal cord ipsilateral dorsal horn of sham (A), CCI/saline (B) and CCI/PC1 (C) 10 days after injury demonstrated that PC1 treatment significantly reduced the CCI-induced microgliosis as demonstrated by quantification of immunoreactive area (D). Scale bar, 50 µm. We never detected PROK2 (E), PKR1 (F) or PKR2 (G) in microglia as demonstrated by lack of any signal associated with CD11b-positive cells, not even in CCI/saline mice. Scale bar, 20 µm. In (D), data are mean ± SEM. ***P < 0.001 CCI/saline versus sham mice; °°°P < 0.001 CCI/PC1 versus CCI/saline mice; one-way anova, followed by Tukey's test for multiple comparisons.
Figure 3.
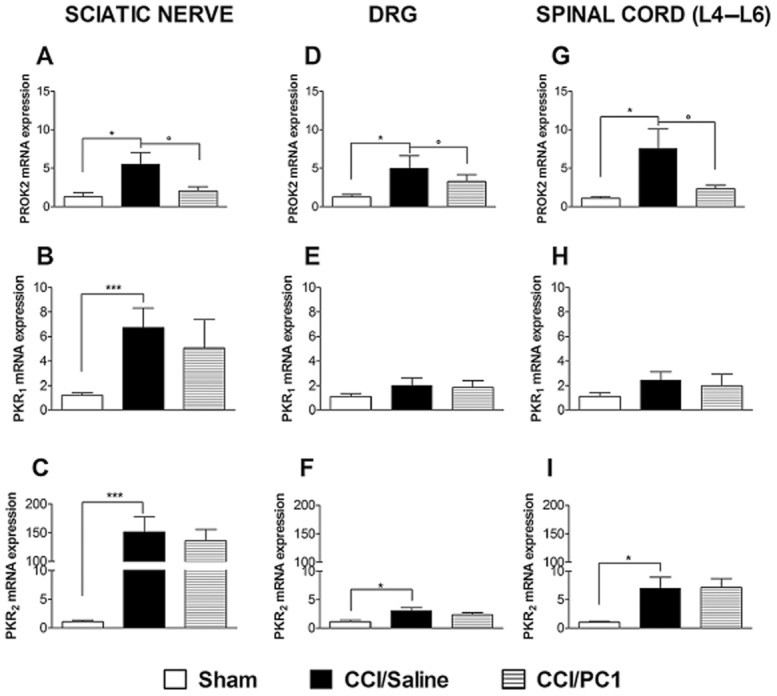
Expression of mRNA for PROK2 (A, D and G), PKR1 (B, E and H), PKR2 (C, F and I) in ipsilateral sciatic nerve, DRG and the spinal cord (L4–L6) 10 days after CCI. The mRNA expression levels, determined by RT-PCR, were expressed in relation to GAPDH and presented as fold of increase relative to sham animals. Data are means ± SEM of four to five animals. Overall analysis: panel A, F(2,25) = 4.923, P = 0.0166; panel B, F(2,29) = 8.73, P = 0.0012; panel C F(2,29) = 15.3, P < 0.0001; panel D, F(2,15) = 3.93, P = 0.0462; panel E, F(2,11) = 1.306, P = 0.3177; panel F, F(2,12) = 5.756, P = 0.0217; panel G, F(2,15) = 4.517, P = 0.0324; panel H F(2,15) = 0.679, P = 0.5241; panel I, F(2,15) = 5.645, P = 0.0172. Post hoc comparisons: *P < 0.05, ***P < 0.001 CCI/saline versus sham; °P < 0.05 CCI/PC1 versus CCI/saline; one-way anova, followed by Tukey's test for multiple comparisons, when necessary.
PROK2, PKR1 and PKR2 localization in sciatic nerve
Immunohistochemical staining using anti-PROK2 antibody failed to demonstrate any PROK2 immunoreactivity in the sciatic nerve of sham-operated mice whereas, 10 days after CCI, a heavy infiltration of PROK2-positive cells (brown colour, Figure 4A) was evident in the neuroma in the immediate proximity of the injury. PC1 treatment did not affect the number of neutrophils, evaluated by the MPO assay (Figure 4C) but significantly reduced PROK2 immunoreactivity (Figure 4A inset), evaluated as IOD (Figure 4B), with lower levels of PROK2 mRNA in the CCI/PC1 group, relative to the CCI/saline group. Immunofluorescence staining, such as immunohistochemical staining, did not reveal PROK2 signal in the sciatic nerve of sham mice (see Figure 5A), whereas immunofluorescence staining of the neuroma in CCI/saline mice demonstrated that PROK2 signal was associated with GFAP-positive Schwann cells (Figure 4D) and with CD11b-positive-activated neutrophils and macrophages (Figure 4E). PKR1 (Figure 4F) was mainly associated with CD11b-positive cells, suggesting that the PKR1 mRNA increase depends on the high number of infiltrating cells (Giannini et al., 2009), whereas PKR2 is mainly associated with GFAP-positive cells (Figure 4G).
Figure 4.
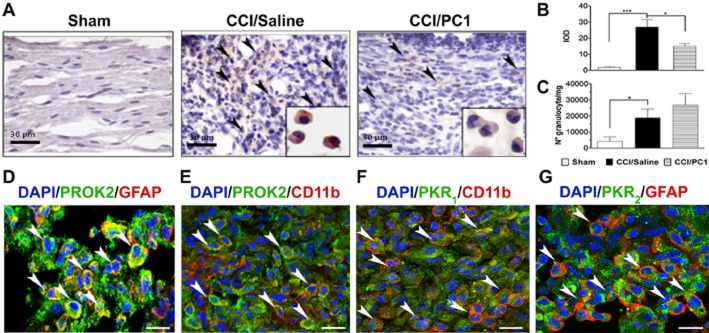
Representative images of sciatic nerve in the immediate proximity of the injury. (A) Immunohistochemical staining of ipsilateral sciatic nerve, on day 0 after CCI, from sham, CCI/saline and CCI/PC1 mice with anti-PROK2 antibody and haematoxylin. Scale bar = 30 µm. Arrowheads indicate the infiltrating cells expressing the PROK2 protein. A sustained infiltration of PROK2-positive cells was evident 10 days after CCI. PC1 treatment significantly reduced the PROK2 immunoreactivity (brown colour) in the cytoplasm of these cells (inset) as demonstrated in (B) by quantitative analysis of PROK2 signal computed as integrated optical density for arbitrary areas (six sections per animal, six animals). (C) Number of infiltrating neutrophils evaluated as MPO activity mg−1 tissue. Data are means ± SEM of four to six animals. *P < 0.05; ***P < 0.001 CCI/saline versus sham mice; °P < 0.05 CCI/PC1 versus CCI/saline mice; one-way anova, followed by Tukey's test for multiple comparisons. (D and E) Immunofluorescence double staining showing colocalization (yellow, arrowheads) of PROK2 (green) with GFAP (Schwann cell marker, red) and CD11b (macrophage marker, red) in the immediate proximity of the injury in the sciatic nerve of CCI/saline mice. (F and G) Representative images showing the localization (arrowheads) of the receptor PKR1 (green) in CD11b-positive macrophages (red) and of the receptor PKR2 (green) in GFAP-positive Schwann cells (red) in the immediate proximity of the injury of the sciatic nerve in CCI/saline mice. Cell nuclei were counterstained with DAPI (blue fluorescence). Scale bar, 20 µm.
Figure 5.
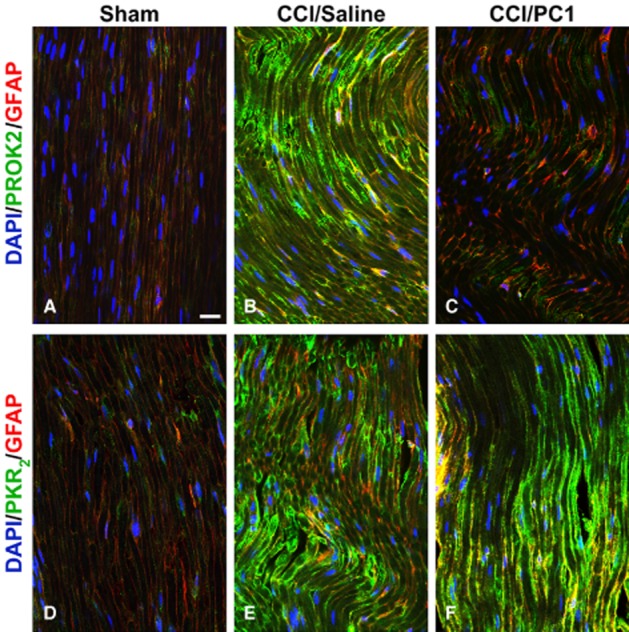
Representative images of CCI-induced up-regulation of PROK2 and PKR2 in the longitudinally sliced sciatic nerve proximal to the lesion. PROK2 immunofluorescence was never found in uninjured nerve (A). Only a very faint PKR2 signal was evident in the non-activated Schwann cells (GFAP-positive cells, red) (D). A dramatic increase of PROK2 and PKR2 signal (B and E, green) in fibres and in GFAP-positive structures was evident 10 days after nerve ligation. PC1 treatment prevented the injury-induced PROK2 up-regulation (C) but was ineffective against PKR2 up-regulation (F). Cell nuclei were counterstained with DAPI (blue fluorescence). Scale bar: 20 µm.
Immunofluorescence staining of sciatic nerve proximal to the lesion demonstrated a dramatic increase of PROK2 and PKR2 signal (Figure 5B and E, green) in fibres and in GFAP-positive structures, 10 days after CCI. PC1 treatment prevented the injury-induced PROK2 up-regulation but was ineffective against PKR2 up-regulation (Figures 5C and F). In the uninjured nerve, only a very faint PKR2 signal was evident in Schwann cells (Figure 5D).
PROK2, PKR1 and PKR2 localization in DRG
In lumbar DRG of sham-operated mice, the PROK2 signal was very faint (Figure 6A). PKR1 and PKR2 immunoreactivity was mainly localized on cell membrane of some neurons (Figure 6D and G). Ten days after CCI, in the ipsilateral DRG, the number of PROK2-positive neurons was clearly increased relative to sham mice, with immunofluorescence distributed in the whole cell body (Figure 6B). PKR1 immunoreactivity was unchanged compared with sham, whereas a strong increase of PKR2 immunoreactivity, staining the whole cell body, was evident in many neurons (Figure 6E and H). PKR1, PKR2 and PROK2 showed a cytoplasmic vesicular pattern, characteristic of proteins that are packaged, transported and released. PROK2 and PKR2 fluorescence was also increased in some GFAP-positive satellite cells (Figure 6B and H). In PC1-treated mice, the PROK2 signal was significantly reduced compared with the CCI/saline group (Figure 6B and C). PKR2 immunoreactivity (Figure 6I), as well as PKR2 mRNA, showed a non-significant tendency to decrease; a bright PKR2 signal appeared redistributed on the neuronal cell membranes (Figure 6I).
Figure 6.

Representative cross sections of mouse L4–L5 ipsilateral DRG from 10 days sham (A, D, G), CCI/saline (B, E, H) and CCI/PC1 (C, F, I) mice. Immunofluorescence double staining of PROK2 (A, B, C), PKR1 (D, E, F) and PKR2 (G, H, I) (green) with GFAP (marker for satellite cells, red). Cell nuclei were counterstained with DAPI (blue fluorescence). Scale bar, 30 µm. Evaluation of PROK2 (J), PKR1 (K) and PKR2 (L) fluorescence intensity (green). In neurons, PROK2, PKR1 and PKR2 immunofluorescence shows a vesicular cytoplasmic pattern which is dense in proximity of the neuronal membrane (arrowheads). PROK2, PKR1 and PKR2 immunofluorescence is also present in satellite cells as assessed by their colocalization with GFAP-positive cells (arrows). PROK2 immunoreactivity significantly increased in the DRG of CCI/saline mice (B) and was reduced by PC1 treatment (C and J). In CCI/saline group, PKR1 immunoreactivity was unchanged in respect to sham (D, E), whereas a sustained increase of PKR2 immunoreactivity, staining the whole cell body (H), was evident. PC1 treatment left the PKR1 and PKR2 fluorescence intensity unchanged (K, L). However, the PKR2 signal appeared redistributed on the neuronal cell membranes (I). Data of fluorescence intensity are means ± SEM of four to six animals. **P < 0.01; ***P < 0.001 CCI/saline versus sham mice; °°°P < 0.001 CCI/PC1 versus CCI/saline mice; one-way anova, followed by Tukey's test for multiple comparisons.
PROK2, PKR1 and PKR2 localization in the spinal cord
PROK2 immunofluorescence, localized in superficial layers (I and II) of the spinal cord in naive animals, was strongly increased in the ipsilateral dorsal horn 10 days after CCI, staining also the deeper layers. Therapeutic PC1 treatment prevented this increase in immunofluorescence so that the PROK2 signal in CCI/PC1 mice resembled that observed in sham animals (Figure 7A, panels a, d, g). Diffuse punctuate pattern PROK2 immunoreactivity partially colocalized with synaptophysin, a presynaptic marker, mainly in superficial laminae of dorsal horn as illustrated in Figure 7C. The lower PROK2 immunofluorescence in the ipsilateral dorsal horn of CCI/PC1 mice (Figure 7A, panel g) in respect to that in CCI/saline mice (Figure 7A, panel d) was related to the strong reduction of PROK2 in the presynaptic terminals. PROK2 immunoreactivity in ipsilateral spinal cord of the CCI/saline mice was also associated with GFAP-positive proliferating and activated astrocytes (Figure 7A panel f and Figure 7B panel c). In the contralateral horn, the number of PROK2/GFAP immunoreactive cells was slightly but not significantly higher than that in sham mice (not shown). Seven-day PC1 treatment strongly reduced the PROK2 immunofluorescence in the activated astrocytes as illustrated by Figure 7B, panel f. Moreover, it reduced the number of the large sized GFAP-positive cells (Figure 7A, panel h in respect to Figure 7A, panel e), as indicated by measuring the percent GFAP-positive area (Figure 8).
Figure 7.
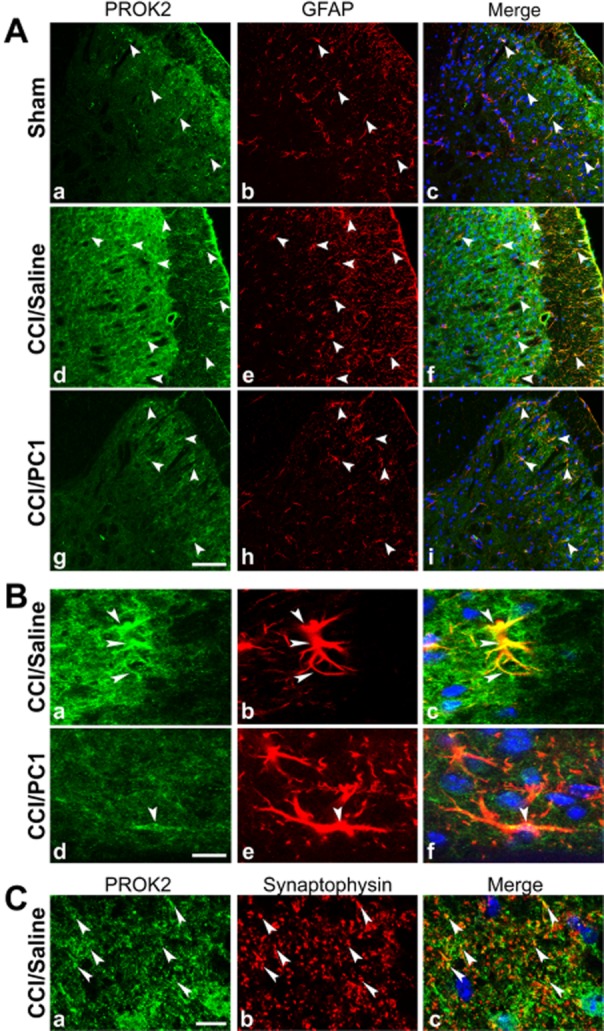
Representative images showing PROK2 localization in the mouse L4–L5 spinal cord dorsal horn. (A) PROK2-positive profiles (green) in sham (a), CCI/saline (d) and CCI/PC1 (g) mice. GFAP (astrocyte marker) positive profiles (red) in sham (b), CCI/saline (e) and CCI/PC1 (h) mice. Sciatic nerve ligation induced a substantial increase in PROK2 and in GFAP signal 10 days after ligation. Double staining reveals a colocalization of PROK2 with the astrocyte marker GFAP (f). Scale bar: 50 µm. (B) High-magnification image (scale bar: 10 µm): double immunofluorescence labelling for PROK2 (green) and GFAP (red) shown in single channels (a, d and b, e) and as a merged image (c, f) showing that activated astrocytes (GFAP-positive enlarged cell bodies and thick processes) contain PROK2 (yellow, c). PC1 treatment (150 µg kg−1 s.c. twice a day for 7 days) reduced PROK2 immunofluorescence in the astrocytes (f). (C) Representative images showing colocalization (c) of PROK2 (green, a) with synaptophysin (red, b). Scale bar: 10 µm. Cell nuclei were counterstained with DAPI (blue).
Figure 8.
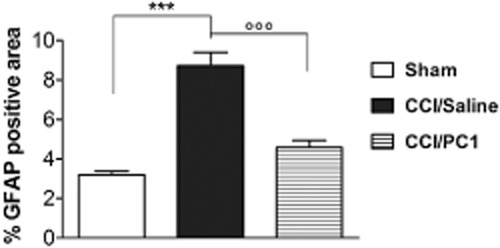
Quantification of the area occupied by the astrocyte marker GFAP in 300 µm2 area, intersecting laminae I and II of cross-sectional spinal cord from sham, CCI/saline and CCI/PC1 mice (five sections per animal, three animals per group). PC1 treatment significantly reduced the CCI-induced astrocytosis and microgliosis. Data are mean ± SEM of four to six animals. ***P < 0.001 CCI/saline versus sham mice; °°°P < 0.001 CCI/PC1 versus CCI/saline mice; one-way anova, followed by Tukey's test for multiple comparisons.
In sham animals, PKR2 immunoreactivity showed a faint diffuse punctuate pattern partially colocalized with synaptophysin (not shown). PKR2 positive neuronal cell bodies were clearly evident in superficial laminae of the dorsal horn (Figure 9A arrow, inset), the same localization shown by EGFP epifluorescence in Gensat database (http://www.gensat.org/imagenavigator). Ten days after CCI, PKR2 positive neuronal cell bodies were more evident also in deeper layers of the dorsal horn (Figure 9B arrows) as illustrated in Figure 9G, showing PKR2 immunofluorescence in NeuN positive cells. The PKR2 signal was clearly increased in the activated astrocytes (Figure 9B head arrows and insert) and also in the diffuse punctuate pattern. PC1 treatment did not induce any variations in PKR2 immunoreactivity (Figure 9C).
Figure 9.
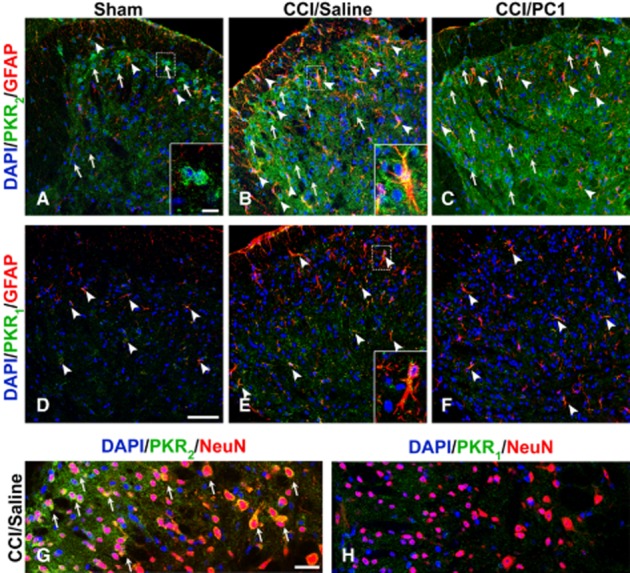
Representative images showing PKR2 and PKR1 localization in the mouse L4–L5 spinal cord dorsal horns from 10 days sham (A, D), CCI/saline (B, E) and CCI/PC1 (C, F) mice. PKR2 immunofluorescence (green) is clearly evident in sham animals, localized in some neuronal cells (a, arrows and inset), and in some astrocytes (A, arrowheads). Ten days after CCI (B), PKR2 positive neuronal cell bodies were more evident also in deeper layers of the dorsal horn (B, arrow) as demonstrated by colocalization with the neuronal marker NeuN (G). The localization of PKR2 in activated astrocytes is demonstrated by the double staining of PKR2 (green) with the astrocytes marker GFAP (B, arrowheads and inset). The diffuse punctuate pattern PKR2 signal appeared clearly increased. PC1 treatment did not modify the PKR2 immunofluorescence intensity. In the spinal cord, the PKR1 signal was very faint and was not affected by nerve injury nor by PC1 treatment. PKR1 immunoreactivity was clearly evident in GFAP-positive resting and activated astrocytes (D, E) and was not modified by PC1 treatment. We never detected PKR1 signal in NeuN-positive cells. Cell nuclei were counterstained with DAPI (blue fluorescence). Scale bar, 50 µm in A to F; 30 µm in G, H and 10 µm insets.
The PKR1 signal in the spinal cord sections was very faint and was unaffected by CCI or by PC1 treatment, mirroring the results of PKR1 mRNA evaluation. It clearly colocalized only with GFAP-positive astrocytes (Figure 9D–F) whereas we never detected PKR1 signals in NeuN-positive cells (Figure 9H). We also did not observe co-staining of PROK2, PKR1 or PKR2 with CD11b (Figure 0E–G), suggesting that the spinal cord microglial cells, even when activated, do not express any elements of the prokineticin system. Nevertheless, PC1 treatment significantly reduced the nerve injury-induced microgliosis (Figure 0A–C) as demonstrated by evaluating the proportion of CD11b-positive area (Figure 0D).
Effect of PC1 treatment on cytokine levels in the sciatic nerve and the spinal cord
As cytokines are regulated at several post-transcriptional and post-translational levels, we measured both mRNA and protein levels. mRNA and protein levels of IL-1β were both increased in the injured sciatic nerve, and in the spinal cord. PC1 treatment prevented the IL-1β increase, significantly reducing it in the sciatic nerve (Figure 1A and B) and restoring IL-1β levels to basal values in the spinal cord (Figure 11E and F).
Figure 11.
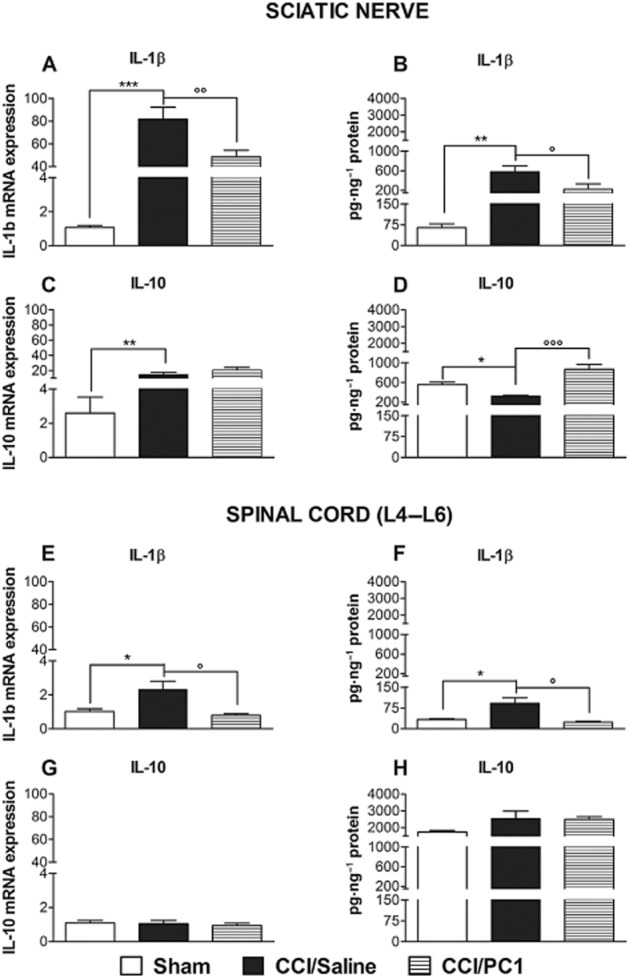
IL-1β (A, B, E and F) and IL-10 (C, D, G and H) mRNA expression and protein content in ipsilateral sciatic nerve and spinal cord 10 days after CCI. The cytokines mRNA levels, determined by RT-PCR, were expressed in relation to GAPDH and are presented as fold of increase relative to sham animals (A, C, E, G). Cytokine protein content is normalized to sample total protein (B, D, F, H). Data are mean ± SEM of four to five animals. *P < 0.05; **P < 0.01; ***P < 0.001 CCI/saline versus sham; °P < 0.05; °°P < 0.01; °°°P < 0.001 CCI/PC1 versus CCI/saline; one-way anova, followed by Tukey's test for multiple comparisons.
The concentration of IL-10 protein decreased in injured sciatic nerve (Figure 1D), whereas its mRNA expression increased (Figure 1C) probably because of activation of the synthetic machinery in order to counteract the pro-inflammatory cascade induced by the lesion (Sacerdote et al., 2013). PC1 treatment led to an increased expression of IL-10 protein. In our experimental setting, we did not find any modulation of IL-10 in the spinal cord, either after CCI or PC1 treatment (Figure 1G and H).
Discussion
Here, for the first time, we provide evidence that the prokineticin system plays a role in neuroinflammation and in the evolution of the neuropathic pain model. Peripheral nerve damage induced up-regulation of PROK2 and of its receptor PKR2 both in the periphery and in the spinal cord.
One-week treatment with the PKR antagonist PC1 alleviated the established neuropathic hyperalgesia and prevented the activation of glia and the increased production of inflammatory cytokines. Moreover, it reduced the neuropathy-induced overexpression of PROK2 itself – a potent pro-nociceptive and pro-inflammatory agent – in sciatic nerve, DRG and spinal cord.
Inflammatory responses occurring in injured nerves play an important role in the genesis of neuropathic pain. In fact 10 days after CCI, we detected a strong increase of PROK2 mRNA in the sciatic nerve: PROK2 immunoreactivity was increased in the neuroma, associated with activated Schwann cells and invading activated neutrophils/macrophages (Figure 4), and was also dramatically increased in the sciatic nerve fibres proximal to ligation (Figure 5). As previously demonstrated in inflammatory pain models (Giannini et al., 2009), the release of PROK2 in the nerve contributes to the fall in the nociceptor activation thresholds and to the recruitment of neutrophils and macrophages, which PROK2 drives towards a pro-inflammatory phenotype, increasing the release of IL-1β and decreasing the release of the anti-inflammatory cytokine IL-10. In our CCI model, we showed that PC1 treatment was highly effective against the peripheral inflammatory component in the injured sciatic nerve. Acute perineural injection of very low doses of PC1 blocked the neuronal PKR1 and PKR2 and prevented the PROK2-induced nociceptor activation. Also, repeated treatment reduced the availability of PROK2 in the nerve and in the infiltrating cells. Moreover, blocking the macrophage PKR1 and the PKR2 highly expressed on activated Schwann cells restored the pro-inflammatory and anti-inflammatory cytokines to normal, physiological levels, as we have already demonstrated in macrophages from PKR1-KO mice (Martucci et al., 2006).
Ten days after the ligation, PROK2 mRNA and protein were significantly increased also in the DRG and in the spinal cord. In myeloid cells, PROK2 up-regulation is induced through activation of STAT3 that binds the enhancer site of its promoter (Shojaei et al., 2007; Qu et al., 2012; Yan et al., 2013). STAT3 activation by G-CSF, IL-6 and IL-1β signalling was recently demonstrated in DRG neurons and astrocytes, but not in microglia (Schweizerhof et al., 2009; Tsuda et al., 2011). Accordingly, we found a significant increase of PROK2 in DRG neurons and in GFAP-positive activated astrocytes in the spinal cord but not in CD11b-positive microglial cells.
The increased PROK2 immunofluorescence in presynaptic terminals in the spinal cord suggests that PROK2 may be transported to the central endings where it induces central sensitization through synthesis and release of the neuropeptides CGRP and SP (De Felice et al., 2012), activation of glutamate interneurons (Yuill et al., 2007) and reduction of GABAA receptor functions (Ren et al., 2011). These PROK2-induced effects may contribute to microgliosis, astrocytosis and production of proinflammatory cytokines, such as IL-1β and IL-6, which in turn stimulate astrocytes to induce further PROK2 expression.
Astrocyte reaction after nerve injury is more persistent than microglial reaction and displays a better correlation with chronic pain-related behaviours (Gao and Ji, 2010b). The fact that PROK2 is associated with astrogliosis confirms the involvement of the prokineticin system at the spinal cord level. As previously demonstrated also in vitro (Koyama et al., 2006), PROK2 induces proliferation of astrocytes expressing both PKR1 and PKR2 so functioning as an astrocytic autocrine growth factor. In mice with CCI and treated with PC1, the astrocyte immunoreactive area was significantly lower than in CCI/saline mice (Figures 7E, H and 8) and their PROK2 content was clearly reduced (Figure 7B) indicating that repeated PC1 treatment directly controls astrogliosis. Further demonstration that the prokineticin system involved in neuropathic pain at the spinal cord level comes from the observations that intrathecal injection of very low doses of PC1 dose-dependently reduced/abolished hyperalgesia in CCI/saline mice (Figure 1F).
Both PROK2 and PKR2 are present also in brain regions associated with pain as medial preoptic area, periaqueductal grey, amygdala, thalamus and hypothalamus (Cheng et al., 2006; de Novellis et al., 2007), where the prokineticin system has a role in modulating the descending inhibitory control of pain (de Novellis et al., 2007; Lattanzi et al., 2012). At the moment, we have no data regarding a potential role of brain PROK2 in neuropathic pain models, but interestingly a PROK2 increase has been demonstrated also in brain astrocytes and neurons after brain ischaemia (Cheng et al., 2012).
The PROK2 released in the spinal cord by astrocytes and primary sensory neurons may activate the PKR2 constitutively localized in the spinal cord neurons (Gensat data base) and up-regulated after nerve injury (Figure 9A and B). There is already evidence suggesting that PKR2 is the inducible receptor (Kisliouk et al., 2005). Our RT-PCR and immunohistochemical data indicate that, 10 days after nerve damage, PKR2 is overexpressed in all the examined tissues: PKR2-mRNA and immunoreactive protein were dramatically increased in the injured sciatic nerve and in the activated Schwann cells as well as being significantly increased in DRG and the spinal cord. Data from our laboratory has demonstrated that Bv8-induced or inflammation-induced tactile allodynia was deficient in PKR2-KO mice (R. Lattanzi, unpublished). As PKR2 is constitutively expressed in some medium to large DRG neurons which also contain the ion channel TRPA1, which is involved in tactile allodynia (Nassini et al., 2011; Negri and Lattanzi, 2011), and also in the spinal cord neurons, we suggest that the increased expression of PKR2 in nociceptors and in the spinal cord neurons together with the increased expression of its agonist might have a crucial role in induction and maintenance of allodynia. Hence, it is possible that prolonged pharmacological blocking of these receptors together with the reduced availability of the agonist prevented and/or reduced sensitization of the pathway responsible for allodynia.
Interestingly, after cessation of PC1 treatment – performed from day 3 to 10 after CCI – thermal hyperalgesia reappeared whereas mechanical allodynia did not develop. It is possible that when PC1 administration started, on day 3 after CCI, the unique plasticity of the CNS that underlies allodynia (Woolf, 2011; Baron et al., 2013) was not fully completed. Thus, by blocking neuronal PKR2 and inhibiting astrocyte activation and the PROK2 synthesis in nociceptors and in astrocytes, allodynia can be prevented. However, when PC1 was administered to mice later, at day 17, when allodynia had already fully developed, the antagonist was able to significantly reduce it but, on cessation of the treatment, the allodynia reappeared.
Last but not least, a peculiar effect of PC1 treatment was to prevent the CCI-induced PROK2 up-regulation, reducing the availability of a potent pro-algesic and pro-inflammatory agent, both in periphery and at the spinal cord level. Indeed, in all the examined tissues, the PROK2 mRNA levels and immunofluorescence were significantly lower in the CCI/PC1 mice than in CCI/saline group.
In conclusion, the prokineticin system plays a critical role in the peripheral nervous system and the CNS in the development and maintenance of neuropathic pain, regulating neuronal–glial interactions. By acting on the peripheral PKRs, the antagonist PC1 controlled the injury-induced abnormal activity of sensory neurons (Xie et al., 2009; Woolf, 2011), so preventing the CCI-induced microglia activation and abolishing the neuropathy-induced IL-1β increase, an effect that correlates well with the ability of PC1 to switch off microglia activation (Watkins and Maier, 2002; Martucci et al., 2008). In the spinal cord, astrocytes appeared to be the preferred target for PC1 and this antagonist reduced astrogliosis and astrocyte-PROK2 production simultaneously. Moreover, its anti-inflammatory and immunomodulatory action had an important effect in the injured sciatic nerve where its reduction of the concentration of IL-1β and increase of the expression of the anti-inflammatory cytokine IL-10 are likely to contribute to the therapeutic effect observed (Sacerdote et al., 2013). In addition, we demonstrated that the final effect of PC1 was also due to a direct modulation of the spinal prokineticin system as shown by the ability of i.t. administration of PC1 to abolish the nerve injury-induced hyperalgesia. Because the daily dose of PC1 peripherally injected in a mouse is about a hundred times higher than equi-effective i.t. doses, it is most likely that that the systemic doses of PC1 act also at the spinal cord.
Compounds such as PC1, which in addition to directly targeting the receptors, also controls PROK2 synthesis and release, would provide more effective treatment of neuroinflammation, by suppressing both the pain symptoms and the factors underlying disease progression.
Acknowledgments
This work was supported by grants from the Italian Ministry of University and Scientific Research (grant number 20099F3XPM_003) and from Sapienza University of Rome.
Glossary
- CFA
complete Freund's adjuvant
- PC1
prokineticin receptor antagonist
- PKR1
prokineticin receptor 1
- PKR2
prokineticin receptor 2
- PROK2
prokineticin 2
Author contributions
D. M. with F. F. performed all the immunofluorescence experiments and coordinated the behavioural experiments performed by V. M. and L. A. G. E. B. and L. F. R. first characterized the PROK2 and PKR antibodies especially in the sciatic nerve. P. S. supervised and evaluated the immunomodulatory role of PC1 coordinating the work of M. C., S. M. and V. M. who performed RT-PCR and ELISA experiments to evaluate the levels of PROK2 and cytokines in sham and CCI mice and in CCI mice pretreated with PC1. S. M. and L. L. evaluated microglia activation in the spinal cord. G. B. synthesized and provided PC1. R. L. supervised all the behavioural, biochemical and immunohistochemical experiments performed by the Roman group. L. N. coordinated the work of the various groups and wrote the paper.
Conflict of interest
None.
References
- Abbadie C, Bhangoo S, De Koninck Y, Malcangio M, Melik-Parsadaniantz S, White FA. Chemokines and pain mechanisms. Brain Res Rev. 2009;60:125–134. doi: 10.1016/j.brainresrev.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to Pharmacology 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balboni G, Lazzari I, Trapella C, Negri L, Lattanzi R, Giannini E, et al. Triazine compounds as antagonists at Bv8-prokineticin receptors. J Med Chem. 2008;51:7635–7639. doi: 10.1021/jm800854e. [DOI] [PubMed] [Google Scholar]
- Baron R, Hans G, Dickenson AH. Peripheral input and its importance for central sensitization. Ann Neurol. 2013;74:634–636. doi: 10.1002/ana.24017. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cheng MY, Leslie FM, Zhou QY. Expression of prokineticins and their receptors in the adult mouse brain. J Comp Neurol. 2006;498:796–809. doi: 10.1002/cne.21087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MY, Lee AG, Culbertson C, Sun G, Talati RK, Manley NC, et al. Prokineticin 2 is an endangering mediator of cerebral ischemic injury. Proc Natl Acad Sci U S A. 2012;109:5475–5480. doi: 10.1073/pnas.1113363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice M, Melchiorri P, Ossipov MH, Vanderah TW, Porreca F, Negri L. Mechanisms of Bv8-induced biphasic hyperalgesia: increased excitatory transmitter release and expression. Neurosci Lett. 2012;521:40–45. doi: 10.1016/j.neulet.2012.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsch M, Qiu Y, Soler D, Frank N, Duong T, Goodearl A, et al. PK1/EG-VEGF induces monocyte differentiation and activation. J Leukoc Biol. 2005;78:426–434. doi: 10.1189/jlb.0205061. [DOI] [PubMed] [Google Scholar]
- Franchi S, Giannini E, Lattuada D, Lattanzi R, Tian H, Melchiorri P, et al. The prokineticin receptor agonist Bv8 decreases IL-10 and IL-4 production in mice splenocytes by activating prokineticin receptor-1. BMC Immunol. 2008;9:60. doi: 10.1186/1471-2172-9-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Chemokines, neuronal–glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010a;126:56–68. doi: 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurother. 2010b;7:482–493. doi: 10.1016/j.nurt.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini E, Lattanzi R, Nicotra A, Campese AF, Grazioli P, Screpanti I, et al. The chemokine Bv8/prokineticin 2 is up-regulated in inflammatory granulocytes and modulates inflammatory pain. Proc Natl Acad Sci U S A. 2009;106:14646–14651. doi: 10.1073/pnas.0903720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WP, Zhang C, Li JD, Luo ZD, Amadesi S, Bunnett N, et al. Impaired pain sensation in mice lacking prokineticin 2. Mol Pain. 2006;2:35–44. doi: 10.1186/1744-8069-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Kiguchi N, Maeda T, Kobayashi Y, Fukazawa Y, Kishioka S. Macrophage inflammatory protein-1 alpha mediates the development of neuropathic pain following peripheral nerve injury through interleukin-1 beta up-regulation. Pain. 2010;149:305–315. doi: 10.1016/j.pain.2010.02.025. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisliouk T, Podlovni H, Spanel-Borowski K, Ovadia O, Zhou QY, Meidan R. Prokineticins (endocrine gland-derived vascular endothelial growth factor and BV8) in the bovine ovary: expression and role as mitogens and survival factors for corpus luteum-derived endothelial cells. Endocrinology. 2005;146:3950–3958. doi: 10.1210/en.2005-0297. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Kiyo-oka M, Osakada M, Horiguchi N, Shintani N, Ago Y, et al. Expression of prokineticin receptors in mouse cultured astrocytes and involvement in cell proliferation. Brain Res. 2006;1112:65–69. doi: 10.1016/j.brainres.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Lattanzi R, Sacerdote P, Franchi S, Canestrelli M, Miele R, Barra D, et al. Pharmacological activity of a Bv8 analogue modified in position 24. Br J Pharmacol. 2012;166:950–963. doi: 10.1111/j.1476-5381.2011.01797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCouter J, Zlot C, Tejada M, Peale F, Ferrara N. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc Natl Acad Sci U S A. 2004;101:16813–16818. doi: 10.1073/pnas.0407697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martucci C, Franchi S, Giannini E, Tian H, Melchiorri P, Negri L, et al. Bv8, the amphibian homologue of the mammalian prokineticins, induces a proinflammatory phenotype of mouse macrophages. Br J Pharmacol. 2006;147:225–234. doi: 10.1038/sj.bjp.0706467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martucci C, Trovato AE, Costa B, Borsani E, Franchi S, Magnaghi V, et al. The purinergic antagonist PPADS reduces pain related behaviours and interleukin-1 beta, interleukin-6, iNOS and nNOS overproduction in central and peripheral nervous system after peripheral neuropathy in mice. Pain. 2008;137:81–95. doi: 10.1016/j.pain.2007.08.017. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele R, Lattanzi R, Bonaccorsi di Patti MC, Paiardini A, Negri L, Barra D. Expression of Bv8 in Pichia pastoris to identify structural features for receptor binding. Protein Expr Purif. 2010;73:10–14. doi: 10.1016/j.pep.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Nassini R, Gees M, Harrison S, De Siena G, Materazzi S, Moretto N, et al. Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain. 2011;152:1621–1631. doi: 10.1016/j.pain.2011.02.051. [DOI] [PubMed] [Google Scholar]
- Negri L, Lattanzi R. Bv8-prokineticins and their receptors: modulators of pain. Curr Pharm Biotechnol. 2011;12:1720–1727. doi: 10.2174/138920111798357410. [DOI] [PubMed] [Google Scholar]
- Negri L, Lattanzi R. Bv8/PROK2 and prokineticin receptors: a druggable pronociceptive system. Curr Opin Pharmacol. 2012;12:62–66. doi: 10.1016/j.coph.2011.10.023. [DOI] [PubMed] [Google Scholar]
- Negri L, Lattanzi R, Giannini E, Melchiorri P. Modulators of pain: Bv8 and prokineticins. Curr Neuropharmacol. 2006a;4:207–215. doi: 10.2174/157015906778019518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri L, Lattanzi R, Giannini E, Colucci M, Margheriti F, Melchiorri P. Impaired nociception and inflammatory pain sensation in mice lacking the prokineticin receptor PKR1: focus on interaction between PKR1 and the capsaicin receptor TRPV1 in pain behavior. J Neurosci. 2006b;26:6716–6727. doi: 10.1523/JNEUROSCI.5403-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri L, Lattanzi R, Giannini E, Melchiorri P. Bv8/prokineticin proteins and their receptors. Life Sci. 2007;81:1103–1116. doi: 10.1016/j.lfs.2007.08.011. [DOI] [PubMed] [Google Scholar]
- de Novellis V, Negri L, Lattanzi R, Rossi F, Palazzo E, Marabeze I, et al. The prokineticin receptor agonist Bv8 increases GABA release in the periaqueductal grey and modifies RVM cell activities and thermoceptive reflexes in the rat. Eur J Neurosci. 2007;26:3068–3078. doi: 10.1111/j.1460-9568.2007.05910.x. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucleic Acids Res. 2014;42(D1):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Zhuang G, Yu L, Meng G, Ferrara N. Induction of Bv8 expression by granulocyte colony-stimulating factor in CD11b+ Gr1+ cells: key role of Stat3 signaling. J Biol Chem. 2012;287:19574–19584. doi: 10.1074/jbc.M111.326801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren P, Zhang H, Qiu F, Liu YQ, Gu H, O'Dowd DK, et al. Prokineticin 2 regulates the electrical activity of rat suprachiasmatic nuclei neurons. PLoS ONE. 2011;6:20263. doi: 10.1371/journal.pone.0020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacerdote P, Franchi S, Moretti S, Castelli M, Procacci P, Magnaghi V, et al. Cytokine modulation is necessary for efficacious treatment of experimental neuropathic pain. J Neuroimmune Pharmacol. 2013;8:202–211. doi: 10.1007/s11481-012-9428-2. [DOI] [PubMed] [Google Scholar]
- Schweizerhof M, Stösser S, Kurejova M, Njoo C, Gangadharan V, Agarwal N, et al. Hematopoietic colony-stimulating factors mediate tumor-nerve interactions and bone cancer pain. Nat Med. 2009;15:802–807. doi: 10.1038/nm.1976. [DOI] [PubMed] [Google Scholar]
- Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–831. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Kohro Y, Yano T, Tsujikawa T, Kitano J, Tozaki-Saitoh H, et al. JAK-STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain. 2011;134:1127–1139. doi: 10.1093/brain/awr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellani V, Colucci M, Lattanzi R, Giannini E, Negri L, Melchiorri P. Sensitization of transient receptor potential vanilloid 1 by the prokineticin receptor agonist Bv8. J Neurosci. 2006;26:5109–5116. doi: 10.1523/JNEUROSCI.3870-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev. 2002;82:981–1011. doi: 10.1152/physrev.00011.2002. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152:S2–S15. doi: 10.1016/j.pain.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Strong JA, Zhang JM. Early blockade of injured primary sensory afferents reduces glial cell activation in two rat neuropathic pain models. Neuroscience. 2009;160:847–857. doi: 10.1016/j.neuroscience.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B, Wei JJ, Yuan Y, Sun R, Li D, Luo J, et al. IL-6 cooperates with G-CSF to induce protumor function of neutrophils in bone marrow by enhancing STAT3 activation. J Immunol. 2013;190:5882–5893. doi: 10.4049/jimmunol.1201881. [DOI] [PubMed] [Google Scholar]
- Yuill EA, Hoyda TD, Ferri CC, Zhou QY, Ferguson AV. Prokineticin 2 depolarizes paraventricular nucleus magnocellular and parvocellular neurons. Eur J Neurosci. 2007;25:425–434. doi: 10.1111/j.1460-9568.2006.05293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]