

Abstract
Background and Purpose
Sepsis is a clinical condition characterized by overwhelming systemic inflammation with high mortality rate and high prevalence, but effective treatment is still lacking. Toll-like receptor 3 (TLR3) is an endogenous sensor, thought to regulate the amplification of immune response during sepsis. Modulators of TLR3 have an advantage in the treatment of sepsis. Here, we aimed to explore the mechanism of a monosubstituted 1,2-benzenediamine derivative FC-99 {N1-[(4-methoxy)methyl]-4-methyl-1,2-benzenediamine}on modulating TLR3 expression and its therapeutic potential on mouse model of sepsis.
Experimental Approach
Cells were pretreated with FC-99 followed by poly(I:C) or IFN-α stimulation; TLR3 and other indicators were assayed. Female C57BL/6 mice were subjected to sham or caecal ligation puncture (CLP) surgery after i.p. injection of vehicle or FC-99; serum and tissues were collected for further experiments.
Key Results
FC-99 suppressed inflammatory response induced by poly(I:C) with no effect on cell viability or uptake of poly(I:C). FC-99 also inhibited TLR3 expression induced by not only poly(I:C) but also by exogenous IFN-α. This inhibition of FC-99 was related to the poly(I:C)-evoked IRF3/IFN-α/JAK/STAT1 signalling pathway. In CLP-induced model of sepsis, FC-99 administration decreased mice mortality and serum levels of inflammatory factors, attenuated multiple organ dysfunction and enhanced bacterial clearance. Accordingly, systemic and local expression of TLR3 was reduced by FC-99 in vivo.
Conclusion and Implications
FC-99 reversed TLR3 expression and ameliorate CLP-induced sepsis in mice. Thus, FC-99 will be a potential therapeutic candidate for sepsis.
Table of Links
| TARGETS | LIGANDS | |
| IFNAR1, interferon α/β receptor 1 | CCL5 | IL-6 |
| TLR3, Toll-like receptor 3 | Eritoran | Poly I:C |
| JAK, Janus kinase | IFN-α | TNF-α |
This Table lists the protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Sepsis is a serious human health problem that is associated with a high mortality rate of 30–50% despite appropriate therapy and advances in supportive care (Boyd, 2012). It often occurs following a whole-body inflammatory state caused by the immune system's response to serious infection or sterile infection and causes death by severe complications such as acute lung injury (ALI) and multiple organ failure (MOF) (Russell, 2006; Yasuda et al., 2008; Chen and Nunez, 2010; Mei et al., 2010). Since Xygris (recombinant activated protein C) and Eritoran (an inhibitor of TLR4) were withdrawn for their lack of clinical efficacy (Angus, 2011), there is no FDA-approved drug for use in sepsis (Ward, 2012). Hence, effective agents for the treatment of sepsis remain an unmet clinical need.
The pathogenetic mechanism of sepsis is highly complex as both infection and non-infection can trigger sepsis (Akira et al., 2006). It is clear that microbial components from infectious agents such as bacteria, fungal and viral can initiate innate response through interacting with Toll-like receptors (TLRs) (Ward, 2012). These TLR agonists from exogenous pathogen components are called pathogen-associated molecular patterns (Bianchi, 2007). However, in the case of ‘sterile infection’, endogenous factors (such as nucleic acid, histones and heat shock protein) released from damaged tissues or necrotic cells, serving as danger-associated molecular patterns (DAMPs), could also target TLRs and activate the innate immune response (Chen and Nunez, 2010; Boyd, 2012). When the innate immunity is dys-regulated, the inflammatory mediator cascade can amplify the inflammatory response and develop it into severe pathophysiological conditions, such as sepsis (Cavassani et al., 2008).
TLR3 is a member of the TLR family and plays a fundamental role in innate immune response. TLR3 recognizes double-stranded RNA (dsRNA) from virus, degraded bacteria, damaged tissues and necrotic cells (Alexopoulou et al., 2001; Yu et al., 2010), and transmits signals via the adaptor Toll-IL-1 receptor (TIR) domain-containing adaptor molecule-1 (TICAM-1, also named TRIF) (Savva et al., 2013). Recent studies indicated that TLR3 could serve as an endogenous sensor of necrosis and amplify inflammatory response (Doughty et al., 2006). Excessive or persistent TLR3 activation was involved in the pathogenic mechanisms of sepsis and aggravated pre-existing systemic inflammation. Meanwhile, TLR3-deficient mice showed an increased survival rate in caecal ligation puncture (CLP)-induced model of sepsis (Cavassani et al., 2008).
FC-99 (C15N2OH18, whose chemical structure is shown in Figure 1A) is a novel 1,2-benzenediamine derivative. In the original studies, the biological activity of FC-99 was assayed by inhibition of LPS-induced NO production in RAW264.7 cells. However, the effect of FC-99 on poly(I:C)-induced TLR3 signal was not tested. Here, we have shown that FC-99 exhibited an inhibitory effect on the inflammatory response induced by poly(I:C), a synthetic dsRNA,, and on TLR3 expression. Thus, we assumed that FC-99 could serve as a modulator of TLR3 and be used in the treatment of sepsis. To elucidate the mechanism of action of FC-99 in sepsis, we used the model of sepsis induced by CLP in mice Our data showed that FC-99 reduced the serum level of inflammatory factors, improved multiple organ dysfunction, enhanced bacterial clearance and improved survival. Of note, FC-99 suppressed TLR3 expression both in vitro and in vivo. This inhibition by FC-99 was related to the poly(I:C)-evoked IRF3/IFN-α/JAK/STAT1 signalling pathway and contributed to the amelioration of CLP-induced sepsis. Our findings suggest that FC-99 may be a potential therapeutic candidate for sepsis.
Figure 1.
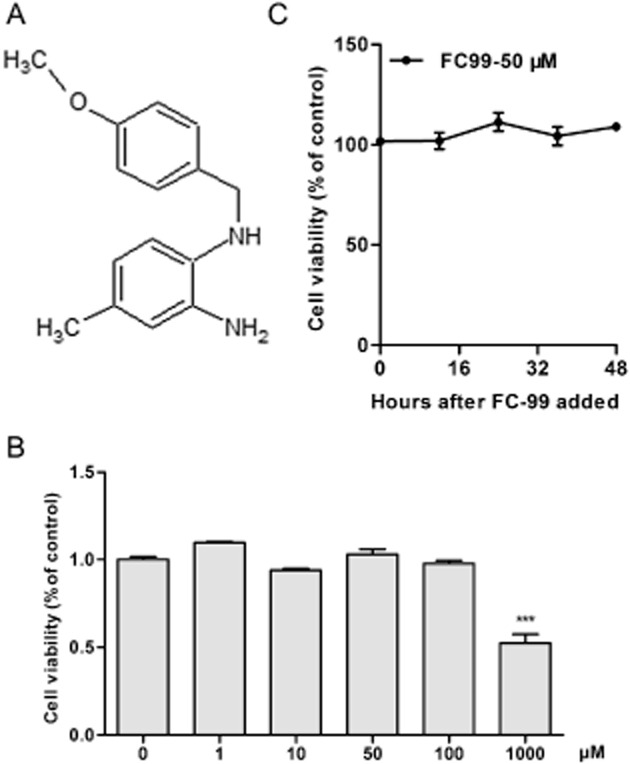
The chemical structure of FC-99 and its effect on peritoneal macrophage viability. (A), the chemical structure of FC-99. In (B), peritoneal macrophages were treated with various concentrations of FC-99 for 24 h or (C) with FC-99 at 50 μM for the indicated time periods. Cell viability was measured by the CCK-8 kit assay. Data are means ± SEM. n = 5 in each condition. ***P < 0.001 versus control.
Methods
Mice and CLP model
All animal welfare and experimental procedures were in strict accordance with the Research Ethics Committee of Nanjing University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 180 animals were used in the experiments described here.
Specific pathogen-free, female C57BL/6 mice (aged 8–10 weeks) were purchased from Model Animal Genetics Research Center of Nanjing University (Nanjing, China). Vehicle (DMSO-containing saline) or FC-99 was administered (100 mg·kg−1) i.p. to mice 2 h before CLP surgery. For polymicrobial sepsis, mice were anaesthetized with 200 mg·kg−1 ketamine and 10 mg·kg−1 xylazine by i.p. injection. Under aseptic conditions, mice were subjected to sham or CLP surgery as previously described (Zhao et al., 2013). After 24 h, all mice were anaesthetized, bled and killed, and the lung, liver and kidney were collected and fixed for haematoxylin and eosin (H&E) staining or stored at −80°C for further analysis. Peritoneal lavage fluids were harvested and serially diluted, and the bacterial colonies in these fluids were determined by plating on agar plates and incubated at 37°C overnight. In a separate experiment, groups in the same conditions were monitored for survival over 6 days. Animals exhibiting extreme morbidity were killed.
Cell cultures
The mouse macrophage cell line RAW264.7 purchased from ATCC (American Type Culture Collection, Manassas, VA, USA) were cultured in DMEM containing 10% heat-inactivated FBS. Thioglycolate-elicited mouse peritoneal macrophages, mouse bone marrow-derived dendritic cells (BM-DCs) and splenocytes were prepared as described before (Liu et al., 2008; Xie et al., 2011; Coquery et al., 2012) and maintained in RPMI 1640 supplemented with 10% FBS for further culture.
Cell viability assay
The cytotoxicity of FC-99 was determined using the CCK-8 kit, according to the manufacturer's protocol. The average optical density formed in control was taken as 100% viability, and the results were expressed as a percentage of the control.
Flow cytometry
Peritoneal macrophages from normal mice were treated with FC-99 for 2 h before poly(I:C) stimulation. After treatment, cells were collected and blocked by CD16/32 and then stained with PE-F4/80. After fixation and permeabilization, cells were incubated with APC-conjugated anti-TLR3. For in vivo, splenic cells from different treated groups were collected. After being blocked with CD16/32, cells were stained with PE-CD11b. Then cells were fixed, permeabilized and stained with APC-TLR3. All stained cells were analysed using a BD FACS Calibur flow cytometer (Bedford, MA, USA).
Small interfering RNA (siRNA) knockdown
For silencing of IFN-α/β receptor 1 (IFNAR1, NM_010508.2), siRNA knockdown was performed in RAW264.7 cells using siRNA duplexes purchased from Ribobio (Guangzhou, China). The negative control siRNA (scrambled siRNA) was provided by Ribobio. The three independent oligonucleotides designed for the IFNAR1 were as follows: for 01, 5′-GGAAUGAGGUUGAUCCGUU dTdT-3′; for 02, 5′-CCAAUACCAACCUGUGCAA dTdT-3′; and for 03, 5′-GCACAUGUGAUGGACUCAA dTdT-3′. RAW264.7 cells were transfected by electroporation using Cell Line Nucleofector Kit V (Lonza, Basel, Switzerland) and program D-32 of Amaxa Nucleofector (Amaxa, Cologne, Germany) according to the manufacturer's protocol. Cells were then recovered for 48 h and the silencing effect of siRNA was detected by real-time quantitative PCR (Q-PCR).
Reverse transcription and Q-PCR
Total RNA was isolated following the manufacturer's instructions. Then, 1 μg of RNA was reverse-transcribed in a 20 μL system using a Revert Aid First-Strand cDNA Synthesis kit (Fermentas, Vilnius, Lithuania). Subsequently, 1 μL complementary DNA was used as the template for the Q-PCR, which was performed using SybrGreen PCR Master Mix (with Rox) (Invitrogen) and 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The relative expression levels of the target genes against that of the GAPDH was calculated as 2-ΔΔCt according to the manufacture's specification. The sequences of primers were as follows: TLR3 forward 5′-TGCCAAATACTCCCTTTGTTGAA-3′ and reverse 5′-CCCGTTCCCAACTTTGTAGATG-3′. IFN-α forward 5′-TGTGACCTGCCTCAGACTCATAAC-3′ and reverse 5′-AATCCAAAGTCCTTCCTGTCCTTCA-3′. TNF-α forward 5′-CCCTCACACTCAGATCATCTTCT-3′ and reverse 5′-GCTACGACGTGGGCTACAG-3′. IL-6 forward 5′-TAGTCCTTCCTACCCCAATTTCC-3′ and reverse 5′-TTGGTCCTTAGCCACTCCTTC-3′. CCL-5 forward 5′-GCTGCTTTGCCTACCTCTCC-3′ and reverse 5′-TCGAGTGACAAACACGACTGC-3′. IP-10 forward 5′-GTCATTTTCTGCCTCATCCT-3′ and reverse 5′-GAGCCCTTTTAGACCTTTT-3′. IFNAR1 forward 5′-GACAACTACACCCTAAAGTGGAG-3′ and reverse 5′-GCTCTGACACGAAACTGTGTTTT-3′. GAPDH forward 5′-GGTGAAGGTCGGTGTGAACG-3′ and reverse 5′-CTCGCTCCTGGAAGATGGTG-3.
Western blot analysis
Western blot analysis was performed as previously described (Gong et al., 2012). Antibodies for phospho-JNK, JNK, phospho-ERK, ERK, phospho-P38, P38, phosphor-IκB, IκB, phospho-IRF3, IRF3 (interferon regulatory factor 3), phospho-JAK1, JAK1, phospho-STAT1, STAT1, TLR3, GAPDH, HRP-conjugated goat anti-mouse IgG and goat anti-rabbit IgG for Western blot were ordered from Cell Signaling Technology (Danvers, MA, USA). Protein bands were visualized using ECL Plus Western blotting detection reagents (Millipore, Bedford, MA, USA). GAPDH was used as an internal control. Each blot was a representative of three independent experiments. Band intensity was measured using Image J software (NIH, Bethesda, MD, USA) and the data were shown as fold value relative to the untreated control.
Respiratory syncytial virus (RSV) infection assay
RSV was provided by Professor Erguang Li. Human epithelial cells A549 were pretreated with FC-99 for 2 h before infected with RSV (MOI = 1 PFU·per cell). Fusion proteins F0 and F1 of RSV in host cells were detected by special antibody (Santa Cruz, CA, USA) using Western blot assay.
Cytokine/chemokine assays
Mouse peritoneal macrophage was pretreated with FC-99 (0.5, 5 and 50 μM) for 2 h before poly(I:C) stimulation for another 24 h. Levels of TNF-α and IL-6 in the culture supernatants were measured using elisa kit (Biolegend, San Diego, CA, USA) according to the manufacturer's instructions.
For in vivo experiments, IL-6, TNF-α, CCL-5 and KC (murine homologue of human IL-8) amounts in the serum were measured by the Bio-Plex system (Bio-Rad, Hercules, CA, USA) following the manufacturer's instructions.
Measurement of biochemical markers
Serum levels of creatinine, urea nitrogen, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured at the biochemistry laboratory of Zhongda Hospital (Nanjing, China).
Data analysis
Results are expressed as mean ± SEM and analysed by one-way anova followed by Bonferroni's comparison test. Statistical comparison between survival curves were performed using log-rank (Mantel-Cox) test. The level of significance was set to P < 0.05. All analyses were performed with GraphPad Prism (GraphPad Software, San Diego, CA, USA). Each experiment was repeated at least three times.
Materials
FC-99 {N1-[(4-methoxy)methyl]-4-methyl-1,2-benzenediamine, purity ≥ 98%} was synthesized following the method in the Supporting Information. It was dissolved in DMSO and diluted with saline before using. DMEM, RPMI 1640 and FBS were purchased from Gibco Inc. (Grand Island, NY, USA). DMSO and thioglycolate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Poly(I:C) and rhodamine-poly(I:C) were obtained from Invitrogen (San Diego, CA, USA). Anti-mouse CD16/32, PE-F4/80, PE-CD11b and APC-TLR3 were obtained from eBioscience (San Diego, CA, USA). The Cell Counting Kit-8 (CCK-8) was purchased from DojinDo Molecular Technologies (Kyushu, Japan).
Results
FC-99 inhibited inflammatory response induced by poly(I:C) in macrophage
To identify the small compound with anti-inflammatory activity in vitro, we assayed the inhibitory ability on the inflammatory cytokine production induced by poly(I:C) in peritoneal macrophage. FC-99 clearly suppressed TNF-α and IL-6 mRNA levels at the indicated time periods (Figure 2A and B). Furthermore, this inhibitory ability acted in a dose-dependent manner as exhibited in protein levels (Figure 2C and D). Similar results were also observed in poly(I:C)-induced chemokines (Supporting Information Fig. S1). However, this inhibition was not due to the cytotoxicity, as the concentration and time of FC-99 used in experiments had no effect on cell viability (Figure 1B and C), or related to the ligand uptake by macrophage, as FC-99 did not affect the uptake of poly(I:C) in the macrophage (Supporting Information Fig. S2).
Figure 2.
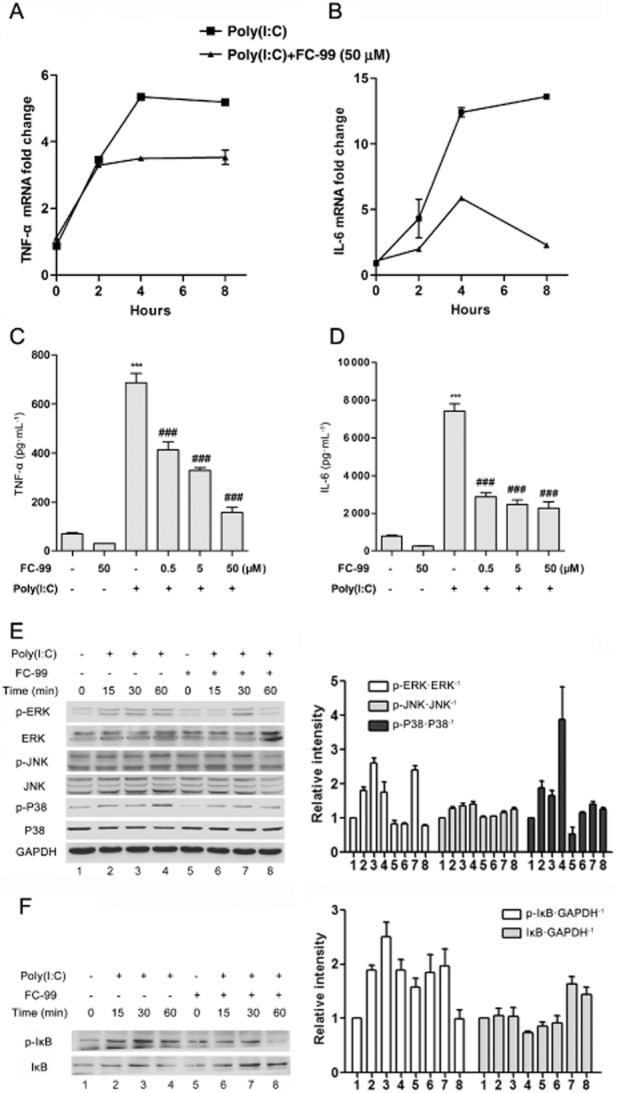
FC-99 inhibited the inflammatory response induced by poly(I:C). Peritoneal macrophages were pretreated with FC-99 for 2 h before 25 μg·mL−1 poly(I:C) stimulation for the indicated time periods; TNF-α and IL-6 levels were measured by Q-PCR (A, B, n = 4) or elisa (C,D, n = 4). (E, F) Protein levels of phospho-ERK, ERK, phospho-JNK, JNK, phospho-P38, P38, phospho-IκB, IκB and GAPDH were measured by Western blot, and a representative of three independent experiments is given. Histograms showed the relative band intensity of Western blot from three independent experiments. The data are means ± SEM. ***P < 0.001 versus control; ###P < 0.001 versus poly(I:C) treated alone.
To clarify the underlying mechanism of FC-99 on the macrophage, activations of ERK, JNK, P38 MAPK and IκB induced by poly(I:C) were examined. FC-99 suppressed the phosphorylation levels of ERK, JNK, and P38 in varying degrees without altering the total protein (Figure 2E). Similar results were observed in IκB phosphorylation, and accordingly, poly(I:C)-induced IκB degradation was abolished by FC-99 (Figure 2F).
FC-99 inhibited TLR3 expression induced by poly(I:C)
FC-99 pretreatment significantly inhibited TLR3 expression induced by poly(I:C) at the indicated time periods (Figure 3A and B), and this inhibitory effect showed positive correlation with the concentration of FC-99 (Figure 3C and D). Besides, this inhibitory effect was non-specific on cell type, as evidenced by the results from parallel studies in RAW264.7, BM-DCs and splenocytes (Figure 3E).
Figure 3.
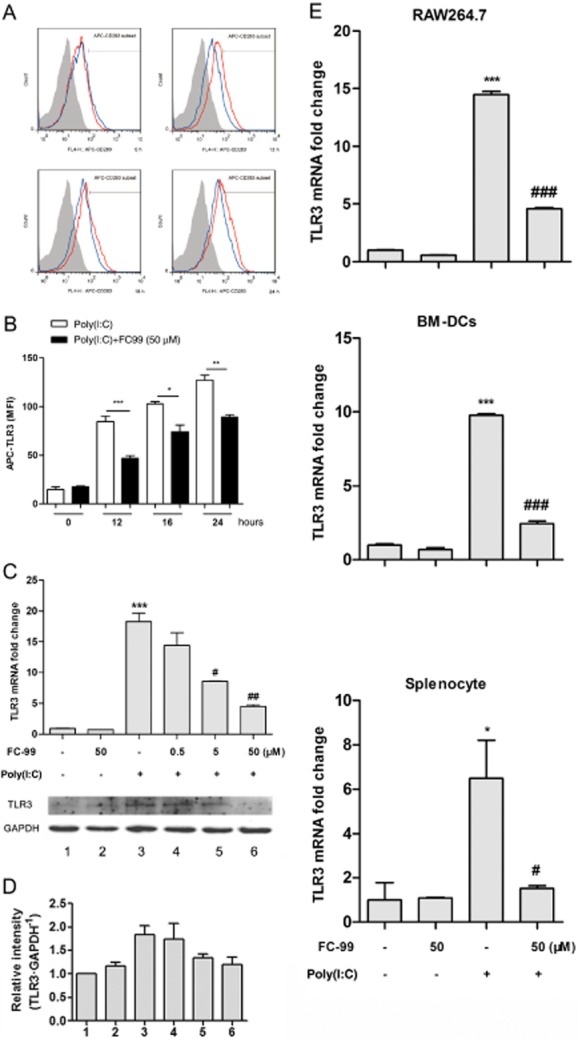
FC-99 suppressed TLR3 expression induced by poly(I:C) in vitro. (A) Peritoneal macrophages were pretreated with FC-99 for 2 h and exposed to 25 μg·mL−1 poly(I:C) for the indicated time periods. TLR3 expression was measured by flow cytometry. Shaded grey, isotype control; red line, treated with poly(I:C); blue line, treated with FC-99 and poly(I:C). In (B), the MFI calculated from the histogram. n = 5 in each condition. *P < 0.05, **P < 0.01, ***P < 0.001. (C) Peritoneal macrophages were pretreated with FC-99 for 2 h and exposed to 25 μg·mL−1 poly(I:C) for another 6 h (up, mRNA level) or 24 h (down, protein level) and TLR3 expression in different levels were measured by Q-PCR (n = 4) or Western blot. (D) Histogram shows the relative band intensity of Western blot from three independent experiments. (E) RAW264.7, BM-DCs and splenocytes were pretreated with FC-99 for 2 h and exposed to 25 μg·mL−1 poly(I:C) for another 6 h. TLR3 mRNA levels were measured by Q-PCR (n = 4). Data are means ± SEM. *P < 0.05, ***P < 0.001 versus control; #P < 0.05, ##P < 0.01, ###P < 0.001 versus poly(I:C) treated alone.
FC-99 inhibited the production of IFN-α, which was an inducer of TLR3 expression
We assayed the effect of IFN-α on TLR3 expression in poly(I:C)-induced mouse macrophage line RAW264.7 cells. Induction of TLR3 by poly(I:C) occurred at 4 h (Figure 4B), which was later than IFN-α-evoked time point (Figure 4A). siRNA for IFNAR1 was used in RAW264.7 cells. Si-IFNAR1-01 was chosen for further test because of its significant reduction in the level of IFNAR1 mRNA (Figure 4C). TLR3 was induced after poly(I:C) stimulation in the presence of negative control (NC-control), but this was suppressed when siIFNAR1-01 was present (Figure 4D). These data indicated that poly(I:C)-induced TLR3 expression relied upon the IFN-α. However, the IFN-α production induced by poly(I:C) was significantly inhibited by FC-99 (Figure 4E), and accordingly, the IRF3 phosphorylation was suppressed (Figure 4F).
Figure 4.

TLR3 expression was induced by IFN-α via IFNAR1, whereas FC-99 inhibited the poly(I:C)-induced IFN-α via IRF3. RAW264.7 cells were treated with 25 μg·mL−1 poly(I:C) for the indicated time periods, mRNA levels of IFN-α (A) and TLR3 (B) were measured by Q-PCR (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001 versus control. (C) RAW264.7 cells were transfected with control siRNA (si-NC) or siRNA targeting IFNAR1 (siIFNAR1) for a total of 48 h and IFNAR1 mRNA levels were determined by Q-PCR (n = 4). (D) After transfected for 48 h, cells were treated with 25 μg·mL−1 poly(I:C) for 6 h and TLR3 mRNA were measured by Q-PCR (n = 4). RAW264.7 were pretreated with FC-99 for 2 h before 25 μg·mL−1 poly(I:C) stimulation for the indicated time periods. (E) IFN-α mRNA levels were tested by Q-PCR (n = 4). (F) Protein levels of phospho-IRF3, IRF3 and GAPDH were tested by Western blot. Histogram showed the relative band intensity from three independent experiments. The data are means ± SEM. **P < 0.01, ***P < 0.001.
FC-99 decreased TLR3 expression induced by exogenous IFN-α
Exogenous IFN-α was used to identify whether the inhibitory effect of FC-99 was unique on the poly(I:C)-induced signal. FC-99 inhibited TLR3 expression induced by exogenous IFN-α (Figure 5A and B). Accordingly, FC-99 suppressed the phosphorylation of IFN-α-stimulated STAT1 at 1 h (the appropriate time for phosphorylation of STAT1) without alteration in STAT1 total protein (Figure 5C). Similar results were observed in the phosphorylation of JAK1 (Figure 5D). Above all, our data suggest that FC-99 inhibited TLR3 through a poly(I:C)-evoked IRF3/IFN-α/JAK/STAT1 signalling pathway.
Figure 5.
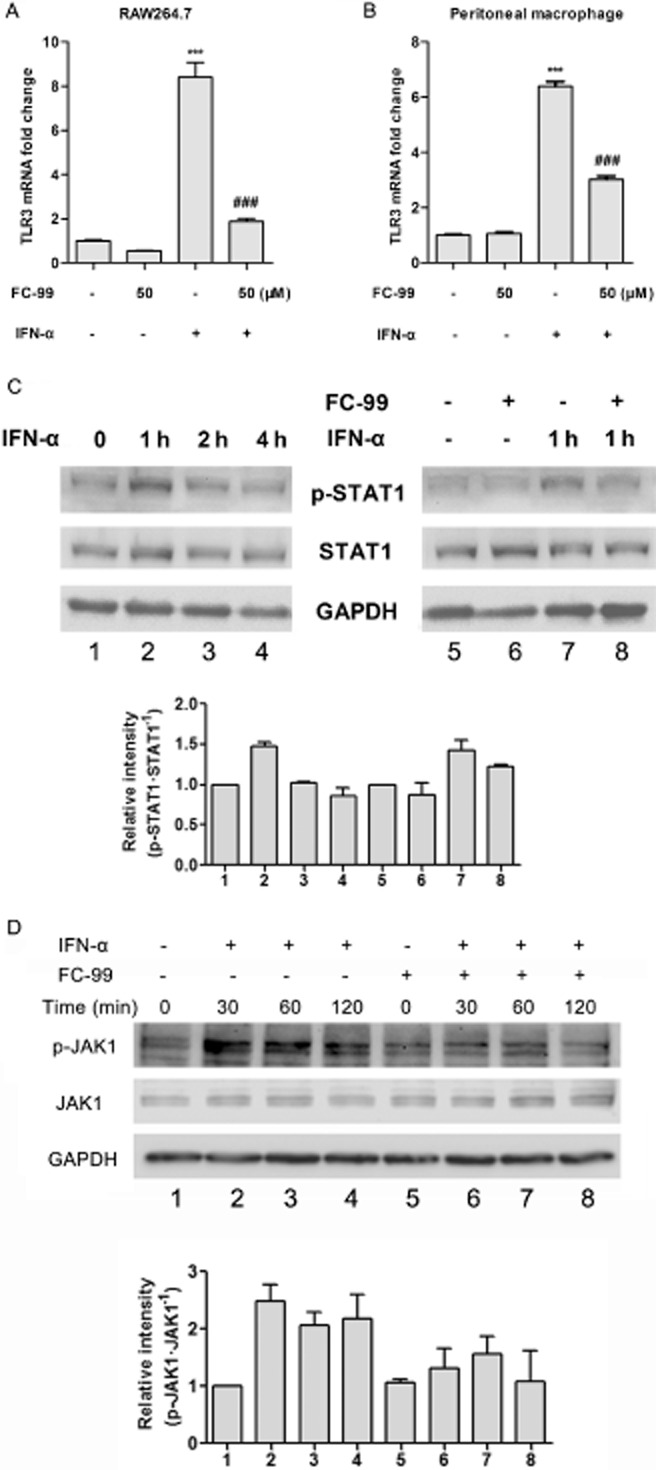
FC-99 reduced the TLR3 expression induced by exogenous IFN-α via STAT1. RAW264.7 cells (A) or peritoneal macrophage (B) was pretreated with FC-99 for 2 h before IFN-α (100 U·mL−1) stimulation for another 3 h and TLR3 mRNA levels were tested by Q-PCR (n = 4). (C, D) RAW264.7 cells were treated with IFN-α (100 U·mL−1) for the indicated time with or without pretreatment with FC-99 for 2 h; protein levels of phospho-STAT1, STAT1 phospho-JAK1, JAK1 and GAPDH were tested by Western blot. Histograms show the relative band intensity from three independent experiments. The data are means ± SEM. ***P < 0.001 versus control; ###P < 0.001 versus IFN-α treated alone.
FC-99 inhibited RSV replication in host cells
We used RSV to infect human epithelial cell line A549 to examine the effect of FC-99 on virus replication. A reduction expression in RSV-F0/F1 protein (subtypes of the RSV-fusion protein) at the indicated time periods suggested that FC-99 reduced the replication of RSV in A549 cell line (Figure 6).
Figure 6.
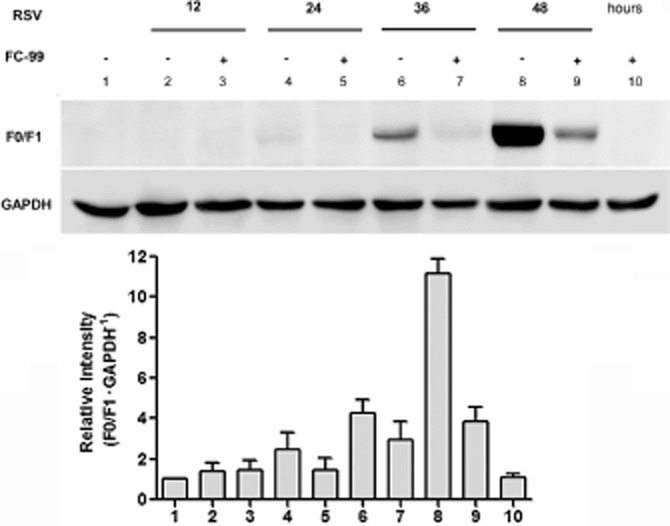
FC-99 inhibited RSV replication in host cells. A549 cells were pretreated with 50 μM FC-99 for 2 h and then infected with RSV (MOI = 1 PFU·per cell) for the indicated time periods. Cells were harvested and fusion proteins F0 and F1 of RSV in host cells were detected by Western blot. Histogram shows the relative band intensity from three independent experiments.
FC-99 attenuated CLP-induced sepsis
Now that the data showed that FC-99 reversed TLR3 expression in vitro, we used a mouse model of sepsis induced by CLP, to elucidate its potential therapeutic effect in vivo. Biochemical indicators in serum were measured to reveal organ function. FC-99 administration significantly reduced the serum levels of blood creatinine, urea nitrogen (indicators of renal dysfunction), and ALT and AST (indicators of hepatic dysfunction) (Figure 7A). Histological assessment of representative lung, liver and kidney sections also revealed evidence for multiple organ damage. Organ specimens from CLP/vehicle group displayed significant histological abnormalities, including (i) inflammatory infiltration, alveolar congestion and marked thickening of alveolar walls in lung; (ii) disarrangement of hepatocytes and morphological alterations of the nuclei in liver; and (iii) vacuolar degeneration and inflammatory infiltration in kidney. These histological alterations were dramatically attenuated in the CLP/FC-99 group (Figure 7B). Besides, FC-99 reduced serum inflammatory parameters, including IL-6, TNF-α, CCL-5 and KC (Figure 7C). Finally, FC-99 reduced mortality (Figure 7D) and the bacterial burden in peritoneal fluid (Figure 7E).
Figure 7.
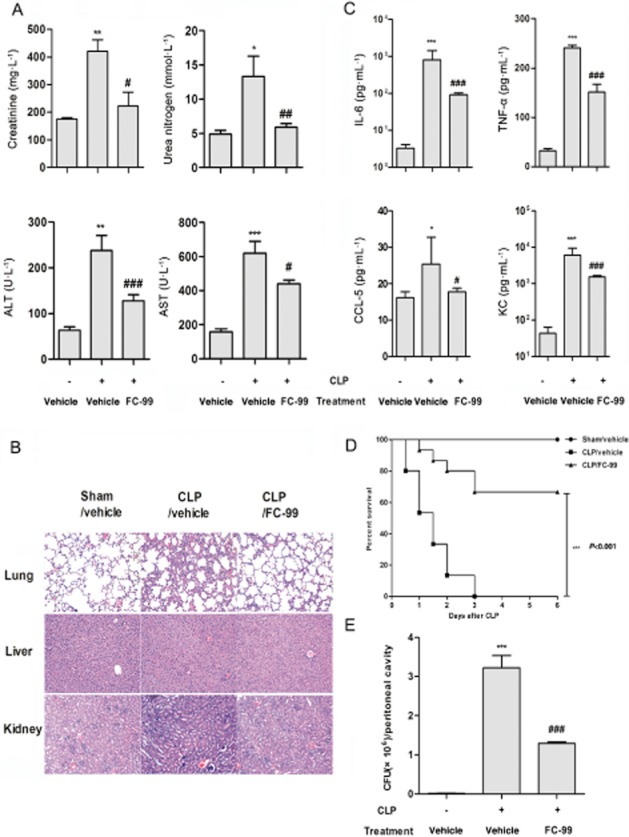
FC-99 improved survival and decreased symptoms of CLP-induced sepsis. Mice were challenged with CLP surgery after i.p. injection of FC-99 (100 mg·kg−1) or vehicle for 2 h. At 24 h after surgery, mice (n = 5 per group) were killed and the serum from each group was collected for biochemical indexes (A) including creatinine, urea nitrogen, ALT and AST, or cytokine/chemokine (C) including IL-6, TNF-α, CCL5 and KC analysis. (B) Lung, liver and kidney sections were subjected to H&E staining. (E) The numbers of bacterial colony forming units (CFU) in peritoneal lavage fluids were quantified. (D) In separate experiments (n = 15 per group), the survival rate was monitored for 6 days. The data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham/vehicle group; #P < 0.05, ##P < 0.01, ###P < 0.001 versus CLP/vehicle group.
FC-99 inhibited TLR3 expression in the model of sepsis
TLR3 expression was increased in splenic CD11b+ cells from CLP mice (CLP/vehicle), whereas FC-99 markedly diminished TLR3 expression in CLP/FC-99 group (Figure 8A and B). The same tendency could be observed in the change of mRNA levels in lung, liver and kidney tissue (Figure 8C–E). All data showed that FC-99 treatment ameliorated CLP-induced sepsis and reduced systemic and local TLR3 expression.
Figure 8.

FC-99 suppressed systemic and local expression of TLR3 in CLP-induced sepsis. Mice were challenged with CLP surgery after i.p. injection of FC-99 (100 mg·kg−1) or vehicle for 2 h and killed at 24 h. (A) Splenic cells were obtained and TLR3 expression was measured by flow cytometry. (B) Histogram shows TLR3 expression in splenic CD11b+ cells. (C–E) TLR3 mRNA expression in lung, liver and kidney tissue was quantified by Q-PCR. (F) Schematic representation showing the mechanism of action of FC-99. The data are means ± SEM (n = 5 per group). *P < 0.05, ***P < 0.001 versus sham/vehicle group; #P < 0.05, ###P < 0.001 versus CLP/vehicle group.
Discussion
Sepsis is a deadly medical condition with a whole-body inflammatory state (Levy et al., 2003). Since Xygris and Eritoran were withdrawn in 2011, there was no FDA-approved drug for sepsis (Angus, 2011; Ward, 2012). Therefore, new drug development and more effective therapies are highly desirable. During injury events, TLR exerts a prominent role in response to endogenous signals provided by the host (Marshak-Rothstein, 2006). Factors released from stressed or damaged tissues could serve as the DAMPs, which triggered the inflammatory response via TLRs (Cavassani et al., 2008; Ward, 2012). Exaggerated TLR signals mediated the inflammation and TLR antagonists had been developed as therapeutics for infectious and inflammatory diseases in clinic (Kanzler et al., 2007; O'Neill et al., 2009). More recently, TLR3, a TLR recognized for not only dsRNA but also endogenous nucleic acids from necrotic cells, had served as an endogenous sensor of tissue necrosis independent of viral activation (Cavassani et al., 2008). This deleterious role of TLR3 has been shown in inflammatory and autoimmune diseases including septic shock (Cavassani et al., 2008; Gao et al., 2012), ALI (Murray et al., 2008) and rheumatoid arthritis (Brentano et al., 2005; Kim et al., 2009). TLR3 deficiency or specific neutralizing antibodies were explored as therapeutic options and decreased the survival in septic shock (Cavassani et al., 2008; Gao et al., 2012). However, gene knockout is not feasible after a disease process has been initiated; neutralizing antibodies are high MW substances (about 150 kDa) and strong immunogenicity, all of which are disadvantageous to the evaluation of therapeutic TLR blockade. Here, we discovered a synthetic compound FC-99 with low MW, simple structure, stable nature and low toxicity, which significantly decreased TLR3 expression. Furthermore, FC-99 reduced mortality and moderated MOF in a clinically relevant model of sepsis induced by CLP. This improvement was in accompanied by enhanced bacterial clearance and an overall reduction in inflammatory factors. Accordingly, local and systemic TLR3 in vivo were suppressed by FC-99. Therefore, down-modulation of TLR3 might represent a novel anti-inflammatory and immune modulatory approach with potential therapeutic applications.
Poly(I:C) is a synthetic analogue of dsRNA often used in studies to investigate TLR3-mediated signals (Okahira et al., 2005). After uptake by cells, poly(I:C) bound with TLR3 and triggered TRIF recruitment. This progress resulted in varying outcomes through different branches: one was the activation of IRF3, leading to the production of type I IFN (Matsumoto and Seya, 2008); another one was the activation of MAPKs and NF-κB pathways, which were important for inflammatory responses in TLR3-modulated signals (Kawai and Akira, 2006) and further caused the generation of inflammatory cytokines. As one of the IFN-stimulated genes (ISGs), TLR3 could be induced by type I IFN through JAK/STAT1 (Heinz et al., 2003). Our data also demonstrated that poly(I:C)-induced TLR3 expression relied upon the type I IFN, as TLR3 expression was observed later than IFN-α, and RNA interference for IFNAR1 (a subunit of IFN-α/β receptor) suppressed TLR3 induced by poly(I:C). Thus, during TLR3-involved inflammation, dsRNA from exogenous pathogen or endogenous injury tissue could rapidly induce expression of inflammatory factors and type I IFN via binding with TLR3. Subsequently, type I IFN. released early in the process, interacted with IFNAR and promoted the activation of JAK/STAT pathway, which further enhanced TLR3 expression. Up-regulation of TLR3 formed a key factor in the positive feedback loop and amplified related responses. Based on this progress, the effect of FC-99 could be deduced from three findings. Firstly, FC-99 inhibited the expression of inflammatory factors induced by poly(I:C) and this effect was related to the suppression of phosphorylation in MAPKs and NF-κB. Secondly, FC-99 reduced the expression of IFN-α, which served as a stimulus for TLR3 expression. Thirdly, FC-99 inhibited IFN-α-induced TLR3 expression through suppressing the phosphorylation of JAK/STAT1, and for this FC-99 suppressed TLR3-modulated positive feedback loop. The combination of these actions allowed FC-99 to exhibit a marked anti-inflammatory ability (Figure 8F).
Previous studies defined TLR3 as a major sensor against virus in the antiviral response (Perales-Linares and Navas-Martin, 2013). TLR3 recognizes not only dsRNA from the dsRNA virus genomes but also dsRNA replication intermediates from some ssRNA viruses such as RSV (Zhang et al., 2007). Recent studies suggested a detrimental contribution of TLR3 during virus infection (Le Goffic et al., 2006). TLR3 deficiency could positively affect the load of viral infection. For example, TLR3−/− mice infected with vaccinia virus displayed less viral load, along with decreased morbidity compared with wild-type mice (Hutchens et al., 2008). Similarly, TLR3−/− mice infected with West Nile virus also exhibited less viral load in the brain (Wang et al., 2004). Given that infection of RSV could up-regulate TLR3 expression (Groskreutz et al., 2006), we used RSV to infect human epithelial cells in order to indirectly confirm the effect of FC-99 on reversing TLR3 expression. The result indicated that after infection, RSV-F0/F1 protein was highly expressed in host cells, whereas FC-99 treatment reduced the RSV-F0/F1 expression, suggesting that replication of RSV in host cells was suppressed. The point is that the exact role of TLR3 in viral infection was still controversial with varying susceptibility to infection and disease outcomes reported in TLR3-deficient mice. Our data indicated that the effect of FC-99 on reversing TLR3 might be involved in the inhibition of RSV replication, but we could not exclude a direct role of FC-99 on viral replication.
TLRs are a family of innate immune receptors that play an important role during sepsis. However, the relationship between TLRs and sepsis is still debatable. In both animal models and septic patients, TLRs are highly expressed on monocytes/macrophages, such as TLR2 (Tsujimoto et al., 2005), TLR3 (Cavassani et al., 2008), TLR4 (Williams et al., 2003) and TLR9 (Tsujimoto et al., 2006). It is more likely that up-regulation of TLRs might be associated with the positive feedback by continually recognizing harmful agonists released from pathogens or host (Tsujimoto et al., 2008). Therapeutic strategies aimed at TLRs have been implemented even though there is no approved clinical therapy (Boyd, 2012). For example, TLR3-deficient mice were protected from the lethal effects of sustained inflammation (Cavassani et al., 2008). TLR9 deficiency improved CLP sepsis-induced mortality and acute kidney injury, with improvement in renal function and histology (Yasuda et al., 2008). However, knockout of MyD88, but not TLR4, could attenuate CLP-induced acute kidney injury (Dear et al., 2006). In the present study, our data showed that FC-99 suppressed TLR3-modulated inflammatory response and TLR3 expression both in vitro and in vivo. In fact, FC-99 also exhibited inhibitory effects on CpG-induced cytokines in DCs with no effect on TLR9 expression (data were not shown). As more than one TLR are involved in the development of sepsis, we could not exclude possible actions of FC-99 on other TLRs, but at least, aggravation of sepsis required a functional TLR3 signal, and inhibitory effect of FC-99 on TLR3 provided a potential mechanism for therapeutic actions on CLP-induced sepsis model. Of course, FC-99 might perform other actions, in addition to inhibiting TLR3, for treatment of sepsis and further studies would be needed to elucidate the full biological effects of FC-99.
In summary, these data help us understand the mechanismsunderlying the biological activities and pharmacological use of FC-99. The ability of FC-99 to reverse TLR3 expression may account for its marked effect on the model of sepsis. FC-99 can serve as a therapeutic clinical option for sepsis.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31370899) and the Scientific Research Foundation of Graduate School of Nanjing University (2012CL03).
Glossary
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BM-DCs
marrow-derived dendritic cells
- CCK-8
cell counting kit-8
- CLP
cecal ligation puncture
- DAMPs
danger-associated molecular patterns
- FC-99
N1-[(4-methoxy)methyl]-4-methyl-1,2-benzenediamine
- IFNAR1
interferon-alpha/beta receptor 1
- IRF3
interferon regulatory factor 3
- Q-PCR
real-time quantitative PCR
- RSV
respiratory syncytial virus
- siRNA
small interfering RNA
- TLR
Toll-like receptor
Author contributions
W. G. and Y. H. conceived and designed the experiments. W. G., E. H., X. S., L. Y. and J. J. performed the experiments. W. G., E. H. and H. D. analysed and interpreted the data. E. L. and R. T. provided the regents and/or materials. W. G. and Y. H. drafted the article and/or gave the final version. All authors read and approved the manuscript.
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12797
Figure S1 Inhibitory effect of FC-99 on poly(I:C)-induced chemokines. Peritoneal macrophages were pretreated with FC-99 for 2 h before poly(I:C)-25 μg·mL−1 stimulation for the indicated time points (8 h in C and 4 h in D), the amounts of CCL5 and IP-10 were measured by Q-PCR. The data are means ± SEM (n = 4). ***P < 0.001 compared with control; #P < 0.05, ##P < 0.01, ###P < 0.001 compared with poly(I:C) alone.
Figure S2 FC-99 had no effect on uptake of poly(I:C) by macrophage. Peritoneal macrophages were incubated with poly(I:C) conjugated with rhodamine [rho-poly(I:C)] for the indicated time periods (A), or cells were pretreated with FC-99 (50 μM) for 2 h before incubated with rho-poly(I:C) for another 16 h (B), and then detected by flow cytometry for the mean fluorescent intensity (MFI) of intracellular rhodamine. The data are means ± SEM (n = 4). ***P < 0.001 compared with the indicated group.
References
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Angus DC. The search for effective therapy for sepsis: back to the drawing board? JAMA. 2011;306:2614–2615. doi: 10.1001/jama.2011.1853. [DOI] [PubMed] [Google Scholar]
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- Boyd JH. Toll-like receptors and opportunities for new sepsis therapeutics. Curr Infect Dis Rep. 2012;14:455–461. doi: 10.1007/s11908-012-0273-5. [DOI] [PubMed] [Google Scholar]
- Brentano F, Kyburz D, Schorr O, Gay R, Gay S. The role of Toll-like receptor signalling in the pathogenesis of arthritis. Cell Immunol. 2005;233:90–96. doi: 10.1016/j.cellimm.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquery CM, Loo W, Buszko M, Lannigan J, Erickson LD. Optimized protocol for the isolation of spleen-resident murine neutrophils. Cytometry A. 2012;81:806–814. doi: 10.1002/cyto.a.22096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dear JW, Yasuda H, Hu X, Hieny S, Yuen PS, Hewitt SM, et al. Sepsis-induced organ failure is mediated by different pathways in the kidney and liver: acute renal failure is dependent on MyD88 but not renal cell apoptosis. Kidney Int. 2006;69:832–836. doi: 10.1038/sj.ki.5000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughty LA, Carlton S, Galen B, Cooma-Ramberan I, Chung CS, Ayala A. Activation of common antiviral pathways can potentiate inflammatory responses to septic shock. Shock. 2006;26:187–194. doi: 10.1097/01.shk.0000223129.79759.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Ha T, Zhang X, Liu L, Wang X, Kelley J, et al. Toll-like receptor 3 plays a central role in cardiac dysfunction during polymicrobial sepsis. Crit Care Med. 2012;40:2390–2399. doi: 10.1097/CCM.0b013e3182535aeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W, Dou H, Liu XQ, Sun LY, Hou YY. Technetium-99 conjugated with methylene diphosphonate inhibits receptor activator of nuclear factor-kappa B ligand-induced osteoclastogenesis. Clin Exp Pharmacol Physiol. 2012;39:886–893. doi: 10.1111/j.1440-1681.2012.12006.x. [DOI] [PubMed] [Google Scholar]
- Groskreutz DJ, Monick MM, Powers LS, Yarovinsky TO, Look DC, Hunninghake GW. Respiratory syncytial virus induces TLR3 protein and protein kinase R, leading to increased double-stranded RNA responsiveness in airway epithelial cells. J Immunol. 2006;176:1733–1740. doi: 10.4049/jimmunol.176.3.1733. [DOI] [PubMed] [Google Scholar]
- Heinz S, Haehnel V, Karaghiosoff M, Schwarzfischer L, Muller M, Krause SW, et al. Species-specific regulation of Toll-like receptor 3 genes in men and mice. J Biol Chem. 2003;278:21502–21509. doi: 10.1074/jbc.M301476200. [DOI] [PubMed] [Google Scholar]
- Hutchens M, Luker KE, Sottile P, Sonstein J, Lukacs NW, Nunez G, et al. TLR3 increases disease morbidity and mortality from vaccinia infection. J Immunol. 2008;180:483–491. doi: 10.4049/jimmunol.180.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Cho ML, Oh HJ, Kim HR, Kang CM, Heo YM, et al. TLR-3 enhances osteoclastogenesis through upregulation of RANKL expression from fibroblast-like synoviocytes in patients with rheumatoid arthritis. Immunol Lett. 2009;124:9–17. doi: 10.1016/j.imlet.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006;2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–1256. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- Liu X, Yao M, Li N, Wang C, Zheng Y, Cao X. CaMKII promotes TLR-triggered proinflammatory cytokine and type I interferon production by directly binding and activating TAK1 and IRF3 in macrophages. Blood. 2008;112:4961–4970. doi: 10.1182/blood-2008-03-144022. [DOI] [PubMed] [Google Scholar]
- Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly(I:C) Adv Drug Deliv Rev. 2008;60:805–812. doi: 10.1016/j.addr.2007.11.005. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei SH, Haitsma JJ, Dos Santos CC, Deng Y, Lai PF, Slutsky AS, et al. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. Am J Respir Crit Care Med. 2010;182:1047–1057. doi: 10.1164/rccm.201001-0010OC. [DOI] [PubMed] [Google Scholar]
- Murray LA, Knight DA, McAlonan L, Argentieri R, Joshi A, Shaheen F, et al. Deleterious role of TLR3 during hyperoxia-induced acute lung injury. Am J Respir Crit Care Med. 2008;178:1227–1237. doi: 10.1164/rccm.200807-1020OC. [DOI] [PubMed] [Google Scholar]
- Okahira S, Nishikawa F, Nishikawa S, Akazawa T, Seya T, Matsumoto M. Interferon-beta induction through toll-like receptor 3 depends on double-stranded RNA structure. DNA Cell Biol. 2005;24:614–623. doi: 10.1089/dna.2005.24.614. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Bryant CE, Doyle SL. Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol Rev. 2009;61:177–197. doi: 10.1124/pr.109.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucleic Acids Research. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales-Linares R, Navas-Martin S. Toll-like receptor 3 in viral pathogenesis: friend or foe? Immunology. 2013;140:153–167. doi: 10.1111/imm.12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JA. Management of sepsis. N Engl J Med. 2006;355:1699–1713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- Savva A, Roger T. Targeting toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front Immunol. 2013;4:387. doi: 10.3389/fimmu.2013.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto H, Ono S, Majima T, Kawarabayashi N, Takayama E, Kinoshita M, et al. Neutrophil elastase, MIP-2, and TLR-4 expression during human and experimental sepsis. Shock. 2005;23:39–44. doi: 10.1097/01.shk.0000145936.31967.d7. [DOI] [PubMed] [Google Scholar]
- Tsujimoto H, Ono S, Matsumoto A, Kawabata T, Kinoshita M, Majima T, et al. A critical role of CpG motifs in a murine peritonitis model by their binding to highly expressed toll-like receptor-9 on liver NKT cells. J Hepatol. 2006;45:836–843. doi: 10.1016/j.jhep.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tsujimoto H, Ono S, Efron PA, Scumpia PO, Moldawer LL, Mochizuki H. Role of Toll-like receptors in the development of sepsis. Shock. 2008;29:315–321. doi: 10.1097/SHK.0b013e318157ee55. [DOI] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- Ward PA. New approaches to the study of sepsis. EMBO Mol Med. 2012;4:1234–1243. doi: 10.1002/emmm.201201375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DL, Ha T, Li C, Kalbfleisch JH, Schweitzer J, Vogt W, et al. Modulation of tissue Toll-like receptor 2 and 4 during the early phases of polymicrobial sepsis correlates with mortality. Crit Care Med. 2003;31:1808–1818. doi: 10.1097/01.CCM.0000069343.27691.F3. [DOI] [PubMed] [Google Scholar]
- Xie H, Hua C, Sun L, Zhao X, Fan H, Dou H, et al. 17beta-estradiol induces CD40 expression in dendritic cells via MAPK signaling pathways in a minichromosome maintenance protein 6-dependent manner. Arthritis Rheum. 2011;63:2425–2435. doi: 10.1002/art.30420. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Leelahavanichkul A, Tsunoda S, Dear JW, Takahashi Y, Ito S, et al. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am J Physiol Renal Physiol. 2008;294:F1050–F1058. doi: 10.1152/ajprenal.00461.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. 2010;14:2592–2603. doi: 10.1111/j.1582-4934.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- Zhao X, Liu D, Gong W, Zhao G, Liu L, Yang L, et al. Poly(I:C) Improves MSCs immune function: the toll-like receptor 3 ligand, poly(I:C), improves immunosuppressive function and therapeutic effect of mesenchymal stem cells on sepsis via inhibiting miR-143. Stem Cells. 2013;32:521–533. doi: 10.1002/stem.1543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Inhibitory effect of FC-99 on poly(I:C)-induced chemokines. Peritoneal macrophages were pretreated with FC-99 for 2 h before poly(I:C)-25 μg·mL−1 stimulation for the indicated time points (8 h in C and 4 h in D), the amounts of CCL5 and IP-10 were measured by Q-PCR. The data are means ± SEM (n = 4). ***P < 0.001 compared with control; #P < 0.05, ##P < 0.01, ###P < 0.001 compared with poly(I:C) alone.
Figure S2 FC-99 had no effect on uptake of poly(I:C) by macrophage. Peritoneal macrophages were incubated with poly(I:C) conjugated with rhodamine [rho-poly(I:C)] for the indicated time periods (A), or cells were pretreated with FC-99 (50 μM) for 2 h before incubated with rho-poly(I:C) for another 16 h (B), and then detected by flow cytometry for the mean fluorescent intensity (MFI) of intracellular rhodamine. The data are means ± SEM (n = 4). ***P < 0.001 compared with the indicated group.