

Abstract
Linear peptides containing the sequence WKTSRTSHY were used as lead compounds to synthesize a novel peptidomimetic antagonist of α2β1 integrin, with platelet aggregation-inhibiting activity, named Vipegitide. Vipegitide is a 13-amino acid, folded peptidomimetic molecule, containing two α-aminoisobutyric acid residues at positions 6 and 8 and not stable in human serum. Substitution of glycine and tryptophan residues at positions 1 and 2, respectively, with a unit of two polyethylene glycol (PEG) molecules yielded peptidomimetic Vipegitide-PEG2, stable in human serum for over 3 hours. Vipegitide and Vipegitide-PEG2 showed high potency (7×10−10 M and 1.5×10−10 M, respectively) and intermediate efficacy (40% and 35%, respectively) as well as selectivity toward α2 integrin in inhibition of adhesion of α1/α2 integrin overexpressing cells toward respective collagens. Interaction of both peptidomimetics with extracellular active domain of α2 integrin was confirmed in cell-free binding assay with recombinant α2 A-domain. Integrin α2β1 receptor is found on the platelet membrane and triggers collagen-induced platelet aggregation. Vipegitide and Vipegitide-PEG2 inhibited α2β1 integrin-mediated adhesion of human and murine platelets under the flow condition, by 50%. They efficiently blocked adenosine diphosphate- and collagen I-induced platelet aggregation in platelet rich plasma and whole human blood. Higher potency of Vipegitide than Vipegitide-PEG2 is consistent with results of computer modeling of the molecules in water. These peptidomimetic molecules were acutely tolerated in mice upon intravenous bolus injection of 50 mg/kg. These results underline the potency of Vipegitide and Vipegitide-PEG2 molecules as platelet aggregation-inhibiting drug lead compounds in antithrombotic therapy.
Keywords: adhesion, collagen I, platelets, integrin antagonist, peptidomimetic
Introduction
Platelets play a critical role in hemostasis and thrombosis. The initial step in thrombus formation requires the capturing of platelets from flowing blood to the exposed sub-endothelial collagen of an injured vessel wall. Arterial blood flow of high shear stress induces conformational changes within von Willebrand factor (vWF) previously bound to collagen and allows binding of the platelet glycoprotein (GP) Ib/V/IX receptor complex to vWF. This interaction mediates stable platelet binding via two major collagen receptors, GPVI and integrin α2β1.1 Recently, strong evidence has been provided to show that GPVI mainly functions as a signaling molecule to activate platelets by releasing chemical messengers such as adenosine diphosphate (ADP), thrombin, and thromboxane A2, whereas α2β1 is more involved in firm adhesion of platelets.2 This adhesion triggers platelet aggregation via platelet αΙΙbβ3 interactions with fibrinogen, which is essential in the interactions in central blood vessels under shear stress.3 Under pathological atherosclerotic conditions, this process of platelet adhesion to collagen may lead to thrombus formation and manifest as an acute coronary syndrome (ACS). Each component of this process is a potential target for antiplatelet activity. Drugs that affect platelet function are a fundamental approach in the management of ACS.4 Food and Drug Administration (FDA)-approved drugs such as ticlopidine, clopidogrel, prasugrel, and ticagrelor are antagonists of purinergic P2Y12 receptor, through which ADP elicits platelet aggregation.5,6 Fibrinogen receptor αΙΙbβ3 integrin plays a significant role in platelet adhesion and aggregation. The studies of this receptor resulted in development of three antagonists, which were approved by the FDA for the therapy of ACS. These drugs include abciximab, a humanized-murine monoclonal antibody;7 eptifibatide, a cyclic peptide;8 and tirofiban, a nonpeptide small molecule.9
α2β1 Integrin (platelet membrane glycoprotein [GPIa], VLA-2 subunit alpha, CD antigen 49b) belongs to a family of collagen receptors, which is expressed on the surface of a variety of cells. Integrins are heterodimeric receptors composed of two noncovalently linked α and β subunits.10 The α1β1 and α2β1 integrins have been recognized as the most selective receptors for various types of collagens. Integrin α2β1 is a receptor for laminin, collagen, collagen C-propeptides, fibronectin, and E-cadherin. It recognizes the proline-hydroxylated sequence GFPGER in collagen. This receptor mediates adhesion of platelets and other cells to collagens and also modulates collagen and collagenase gene expression, force generation, and organization of newly synthesized extracellular matrix. The I-domain of the integrin is responsible for collagen binding. It consists of approximately 200 amino acid residues and is localized near the amino terminus in the extracellular part of the α subunit. The studies with α1/α2 integrin I-domain chimeras confirmed that α1-I-domain-containing receptors have higher affinity toward collagen IV, whereas α2-I-domain-expressing cells better adhere to collagen I.11
Screening a large series of linear synthetic peptides characterized as α1β1/α2β1 dual antagonists,12 we identified an integrin recognition sequence: WKTSRTSHY. In this study, this sequence was modified by introducing two molecules of α-aminoisobutyric acid (AIB), which is an established alanine analog known to strongly favor helical folded conformation.13 AIB does not belong to the standard amino acids, and therefore peptides containing AIB are more protease resistant and more stable in vivo.13 Here we describe the synthesis and characterization of novel peptidomimetics Vipegitide and its pegylated analog (Vipegitide-PEG2). Moreover, we measure their ability to inhibit adhesion of integrin α2β1 overexpressing cells, human platelets, as well as antiaggregation platelet activity. These peptidomimetics inhibit platelet aggregation, show selective and high affinity binding to α2 integrin receptors, and were tolerable upon injection in mice. These unique peptidomimetic antagonists are lead structures, which might be further exploited for development of novel antiplatelet aggregation drugs.
Materials and methods
Materials
Collagen IV (from bovine placenta villi) was purchased from Chemicon (Temecula, CA, USA) and collagen I (from rat tail) from BD Biosciences (Bedford, MA, USA). The 96-well polystyrene plates were obtained from Nunc (Roskilde, Denmark). Bovine serum albumin (BSA), Hank’s balanced salt solution (HBSS) sulfate, alkaline phosphatase-conjugated anti-rabbit antibody, and p-nitrophenyl phosphate were purchased from Sigma-Aldrich (St Louis, MO, USA). CellTracker™ green 5-chloromethylfluorescein diacetate (CMFDA) and rabbit polyclonal antibodies against glutathione s-transferase (GST) were purchased from Molecular Probes (Eugene, OR, USA).
C-type lectin protein
Vixapatin was obtained from the venom of Vipera xanthina palestinae as previously described.14 Rhodocetin was purified from Calloselasma rhodostoma venom as reported.15
Cell lines
K562 cells transfected with α1 or α2 integrin subunits14 were used in this study.
Peptidomimetic synthesis reagents
All amino acids and Rink resin were purchased from GL Biochem Ltd. (Shanghai, People’s Republic of China). N,N-diisopropylethylamine (DIPEA), 2-(1H-benzotriazole-1yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), and 1-hydroxybenzo-triazole (HOBt) were purchased from BioLab Ltd (Jerusalem, Israel). All coupling reagents, chemicals, and solvents were purchased from Sigma-Aldrich.
Procedure for phthalimide-protected polyethylene glycol unit synthesis and characterization
The phthalimide (Pht)-protected polyethylene glycol (PEG) unit, Pht-PEG2, was synthesized as previously described.16,17 Briefly, 2-(2-aminoethoxy)ethanol (10.5 g, 0.1 mol) and phthalic anhydride (14.8 g, 0.1 mol) were dissolved in toluene (450 mL) and the resulting solution placed in a round bottomed flask fitted with a Dean–Stark apparatus. The solution was then heated under reflux for 4 hours, with the water evolved from the reaction periodically removed from the system. The reaction mixture was allowed to cool, dried over magnesium sulfate, filtered, and then the solvent removed in vacuo to give the pure, white Pht-protected amine in quantitative yield. The phthalimido derivative (5 mmol) was dissolved in acetone (60 mL) at 0°C. Chromium trioxide (12 mmol) dissolved in 35% sulfuric acid (25 mL) was added dropwise. The temperature of the reaction mixture was then allowed to reach room temperature, and stirring was continued for 3 hours. The reaction mixture was poured into water (80 mL) and the product extracted with ethyl acetate (4×50 mL). The combined organic phases were washed with brine (70 mL or until the organic phase was colorless), dried with sodium sulfate, and the solvent evaporated under reduced pressure.18 Pht-PEG2 was purified using analytical high-performance liquid chromatography (HPLC), molecular weight was confirmed using electrospray ionization (ESI) mass spectrometry (MS), and the structure was confirmed with proton nuclear resonance (1H-NMR) spectroscopy.
General procedure for peptidomimetics preparation and characterization
The peptidomimetics were synthesized on a solid phase by standard fluorenylmethyloxycarbonyl (Fmoc) chemistry.19 The synthesis was carried out manually on a Rink amide resin using Fmoc-protected amino acids. AIB was also Fmoc protected. Coupling was performed for 1 hour with four equivalents of HBTU and one equivalent of amino acid in the presence of four equivalents of HOBt and eight equivalents of DIPEA. Fmoc groups were removed with 20% piperidine in N-methyl-2-pyrrolidinone. For the synthesis of Vipegitide-PEG2, Pht-PEG2 previously synthesized was used.
Cleavage from the resin and full deprotection of peptidomimetics was carried out using a mixture of trifluoroacetic acid (TFA)/phenol/water/triisopropylsilane (88:5:5:2 v/v/v/v) for 3 hours at room temperature.20 The resin was filtrated, and the peptide was precipitated by addition of cold diethyl ether to the filtrate. The precipitate was separated by centrifugation at 4,000× g for 10 minutes, solubilized in water, and lyophilized. Cleavage of Vipegitide-PEG2 peptide from the resin first was done with hydrazine monohydrate, as described previously.21 Briefly, a solution of hydrazine monohydrate (41.2 mM, 2 mL) in 10 mL methanol/tetrahydrofuran (1:1) was added to the resin and reaction slurry was shaken for 6 hours. The resin was washed with methanol (5×) and with dichloromethane (3×). The second cleavage was done using TFA.
The synthesized peptidomimetics were purified by preparative reverse-phase HPLC (RP-HPLC) using a C18 column with an elution gradient of 0%–90% acetonitrile with 0.1% TFA in water. The compounds’ purity was verified by Thermo Scientific Dionex UltiMate 3000 analytical HPLC. Identities of both peptidomimetics were confirmed using high resolution ESI MS using a LTQ Orbitrap (Thermo Scientific, San Jose, CA, USA) in the positive ion mode.12
Stability of the peptidomimetics in human serum
One milliliter of Roswell Park Memorial Institute media supplemented with 25% (v/v) of human serum was introduced into a 1.5 μL Eppendorf tube and was temperature equilibrated at 37°C for 15 minutes before adding 5 mL peptidomimetic stock solution to make a final peptide concentration of 50 μg/mL. The initial time was recorded, and at known intervals, 100 μL of the reaction solution was removed and added to 200 μL of 96% ethanol to precipitate serum proteins. The sample was cooled to 4°C for 15 minutes and then centrifuged at 18,000× g for 2 minutes to precipitate serum proteins. The supernatant was then applied to a C18 column for separation by RP-HPLC. A linear gradient from 100% buffer A (0.1% TFA in water), to 50%–50% buffer A and buffer B (0.1% TFA in acetonitrile), was applied for over 30 minutes. The flow rate was 1 mL/minute and absorbance was detected at 220 nm.22
Modeling of the peptidomimetics in water
The starting points for the simulations of Vipegitide and Vipegitide-PEG2 were built in Discovery Studio as extended peptides. Prior to minimization, the structures were prepared using the prepare protein protocol as implemented in Discovery Studio to determine residues protonation states.23
Molecular dynamics simulations were performed using the Gromacs Molecular Dynamics package24,25 with the AMBER99SB-ILDN force field.26 Peptidomimetics were submerged in transferable intermolecular potential 3 points (TIP3P) water27 in a cubic box with an extra extension along each axis of the peptide of 10 Å. Ions were added to the solution to make the system electrically neutral. Structures were minimized, equilibrated (first under amount of substance, volume, and temperature conditions for 1 ns and then under amount of substance, volume, and temperature conditions for an additional 1 ns), and finally simulated under amount of substance, volume, and temperature conditions for 1 μs. The simulations were performed at 300°K with a time step of 2 fs using the leap-frog algorithm.28 Long-range electrostatic interactions were computed using Particle Mesh Ewald Summation.29,30 The cutoff for van der Waals and Coulomb interactions was set to 10 Å. Periodic boundary conditions were applied, and the LINCS algorithm31 was used to constrain bond lengths.
To examine the conformational ensembles of the peptidomimetics, we measured the distance between their termini and their radius of gyration along the simulations. For Vipegitide, the distance between the termini of the peptide chain was measured between the centers of mass of the backbone atoms (N, CA, C) of residues 1 and 13. For Vipegitide-PEG2, the distance was measured between the centers of mass of the heavy atoms in the N-terminus (atoms N, C1, C2) and the backbone atoms of residue 13. Distances were measured for the last 400 collected snapshots only (last 800 ns of the simulation) to remove the effect of the starting conditions. We defined peptidomimetics as folded when the distance between the two termini of the peptide chain was smaller than 5 Å. Radii of gyration (root mean square deviation [RMSD] of atoms from their center of gravity) were calculated using gyrate procedure as implemented in Gromacs.24,25
To investigate the conformational space of the peptidomimetics, we examined their secondary structures along the simulations. This analysis was performed only for residues 4–13, to allow for a valid comparison between the two peptidomimetics. For this purpose, we used the DSSP program32 (Gromacs’s do_dssp procedure which interfaces with DSSP), which assigns the most likely secondary structure to a given peptide chain. The do_dssp procedure provides a list of the secondary structures adopted by each residue for all the collected snapshots. From this list we calculated the fraction of conformations which adopt helical secondary structure and the fraction of conformations with high curvature (ie, adopting a bend or turn conformation) among the inner residues (6–11).
Finally, to assess the similarity of the conformational spaces of Vipegitide and Vipegitide-PEG2, we used a matrix termed “contained,” which was developed in an earlier study.12 This matrix calculates the fraction (in percentage) of the conformations of Vipegitide that are similar (ie, reside within a predefined RMSD cutoff) to at least one of the conformations of Vipegitide-PEG2 and vice versa. In this study, two conformations were deemed similar if the backbone RMSD between them (calculated over residues 4–13) was found to be smaller than 2 Å. Calculations performed with a 1 Å RMSD threshold resulted in no similarity between the ensembles.
Cell adhesion assay
The assay was carried out as described previously, with minor modifications.33 The day before the experiment, each well of a 96-well plate was coated with 10 μg/mL collagen I or 1 μg/mL collagen IV in 0.02 M acetic acid and incubated overnight at 4°C. Thereafter, nonspecific binding was blocked by incubating the wells with 1% (w/v) BSA in HBSS containing 5 mM magnesium chloride (MgCl2), at room temperature for 1 hour before use. The cells were labeled by incubation with 12.5 μM CMFDA in HBSS without 1% BSA at 37°C for 30 minutes. The labeled cells were then centrifuged at 1,000 rpm and washed twice with HBSS containing 1% BSA to remove excess CMFDA. Labeled cells (1×105 cells/well) were added to each well in the presence or absence of peptidomimetics and incubated at 37°C for 60 minutes. In the presence of peptide, cells were added to the well after prior incubation with peptide for 30 minutes at 37°C. Unbound cells were removed by washing the wells three times with 1% (w/v) BSA in HBSS, and bound cells were lysed by the addition of 0.5% Triton X-100 (diluted in double-distilled water). The fluorescence in each well was quantified with a SPECTRAFluor Plus plate reader (Tecan, San Jose, CA, USA) at excitation wavelength=485 nm and emission wavelength=530 nm. To determine the number of adhered cells from the fluorescence values, a standard curve was generated by serial dilution of known numbers of CMFDA-labeled cells.
GST-α1A/GST-α2A binding to type IV/I collagens binding assay
Inhibition enzyme-linked immunosorbent assay (ELISA) was performed as described previously,34 with the following modifications: CB3 (collagen IV fragment) or collagen I was immobilized overnight at 4°C on a microtiter plate at 10 μg/mL in Tris-buffered saline (TBS)/MgCl2 (50 mM Tris-HCl, 150 mM sodium chloride [NaCl], and 2 mM MgCl2, pH 7.4) and 0.1 M acetic acid, respectively. After blocking the plate with 1% BSA in TBS/MgCl2, the GST-tagged α1A/GST-tagged α2A domain was allowed to bind to type IV collagen/collagen I in the presence or absence of different peptidomimetics for 2 hours at room temperature. The bound GST-α1A/GST-α2A domain was fixed for 10 minutes with 2.5% glutaraldehyde in 2-(4-(2-Hydroxyethyl)-1-piperazinyl)-ethansulfonsäure (HEPES) buffer (50 mM HEPES, 150 mM NaCl, and 2 mM MgCl2, and 1 mM manganese chloride, pH 7.4). The bound GST-α1A/GST-α2A was quantified with rabbit polyclonal antibodies against GST, followed by alkaline phosphatase-conjugated anti-rabbit antibody, which served as the primary and secondary antibodies, respectively, each diluted in 1% BSA in TBS/MgCl2. The conversion of p-nitrophenyl phosphate was measured at 405 nm in an ELISA reader (BioTek, Bad Friedrichshall, Germany). Nonspecific binding was assessed by binding of GST-α1A/GST-α2A to BSA or of α1β1/α2β1 integrin to collagen IV/collagen I in the presence of 10 mM ethylenediaminetetraacetic acid (EDTA).
The cone and platelet analyzer (CPA) procedure
The study was approved by the Ethics Committee of the Hadassah Hospital, in accordance with the guidelines of the Declaration of Helsinki.35 For evaluation of the peptides on human and mouse platelet adhesion under flow conditions, the Impact-CPA (DiaMed, Cressier, Switzerland) technology was used.36 Control adhesion was obtained with blood previously incubated with only saline for 3 minutes. Platelets from whole blood were activated by incubation with 1 μg/mL of collagen I, with gentle mixing for 2 minutes. To test the inhibitory capacity of peptidomimetics, whole blood was preincubated with 35 μM of Vipegitide-PEG2 for 3 minutes and thereafter stimulated with collagen I. A volume of 130 μL of treated whole blood was placed in four-well polystyrene plates (Nunc, Roskilde, Denmark) and after 2 minutes subjected to a shear rate of 1,800 s−1 using a rotating Teflon cone. The wells were then thoroughly washed with tap water, stained with May–Grünwald stain (Sigma-Aldrich), and analyzed with an inverted light microscope connected to an image analysis system. Platelet adhesion was evaluated by percent of surface coverage, which is the percentage of total area covered by platelets (single platelets and clusters/aggregates). Each set of experiments was done with the blood of an individual donor. Tests with murine platelets were performed with blood samples pooled from ten mice. All CPA tests were performed in five replicates (different donors for humans and pool of ten animals) for each experiment.
Platelet aggregation assay in platelet rich plasma and whole blood
The freshly collected human blood from healthy donors, who were nonmedicated at least for the previous 15 days, was mixed with 3.8% trisodium citrate (citrate/blood 1:9) and centrifuged at 90× g for 15 minutes to obtain platelet rich plasma (PRP). Then, the remnant of blood was again centrifuged at 500× g for 15 minutes to obtain the platelet-poor plasma. The platelet number in the PRP was adjusted to 2.5×108 platelets/mL by diluting it with platelet-poor plasma and used within 2 hours. All the above preparations were carried out using plastic ware or siliconized glassware. The turbidimetric method of Born and Cross37 was followed, using the Platelet Aggregation Profiler®, model PAP-8E S/W, version 1.0.8. aggregometer (Bio/Data Corp, Horsham, PA, USA). Briefly, 225 μL of PRP suspension was maintained at 37°C in a siliconized glass cuvette and preincubated with different doses of peptidomimetics or double-distilled water (control) in 10 μL phosphate buffer saline for 3–30 minutes, and the aggregation was initiated by adding 2 μg/mL of collagen I or 10 μM ADP. In experiments where whole human blood was used, Vipegitide was added to whole blood, incubated for 10 minutes, and then PRP was isolated and used further in the experiments. The aggregation was then followed for 6–12 minutes with constant stirring at the speed of 900 rpm. In each case, aggregation induced by collagen I or ADP alone was considered as 100% aggregation. All aggregation tests were performed in quadruplicate (four donors).
Toxicity in vivo
Experiments involving animals and their care were approved by the Committee of Ethics of The Hebrew University and were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. A 50 mg/kg quantity of Vipegitide or Vipegitide-PEG2 in 0.2 mL was injected intravenously in C57BL/6 mice (n=6). They were monitored for 3 consecutive weeks. The animals were examined for autonomic symptoms by measuring salivation, urinary delivery, pupillary constriction, heart rate, blood pressure, and hair contraction. Neurotoxicity was evaluated by general locomotor activity of the animals in the cage and the ability of the peptide-injected mice to maintain balance and motor coordination by crossing 3 cm, 2 cm, and 1 cm wide balance beams. Occurrences of either flaccid or spastic paralysis of the legs were also measured. Blood samples were taken from control and peptide-injected mice after 10 hours from the time of injection and submitted for hematocrit and biochemical analysis.
Statistics
Student’s t-test and Analysis Of Variance were used to determine the significance of the differences between the various treatments compared with the control groups. The results were presented as mean ± standard deviation (SD); P≤0.05 was considered significant.
Results
Synthesis of Vipegitide-PEG2 unit
Pht-PEG2 unit (Figure 1A) was synthesized as described in the general procedure. ESI MS m/z calculated for C12H11NO5 249.22; found 249.98. The results of 1H-NMR spectroscopy are (CDCl3, 300 MHz) δ: 3.81 (2H, t, CH2N), 3.94 (2H, t, CH2CH2O), 4.12 (2H, s, CH2COOH), 7.73 (2H, m, ArH), 7.85 (2H, m, ArH), 11.6 (1H, s, COOH). These results confirm the chemical structures.
Figure 1.
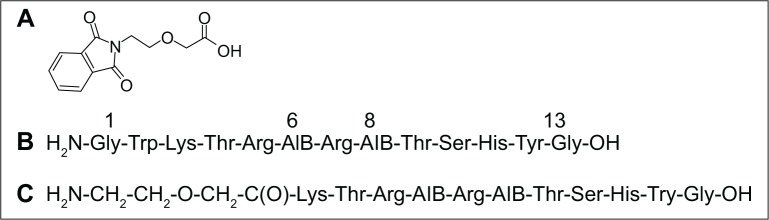
Synthesized peptides.
Notes: Chemical structure of (A) Pht-PEG2 unit, (B) Vipegitide, and (C) Vipegitide-PEG2.
Abbreviations: AIB, α-aminoisobutyric acid; PEG, polyethylene glycol; Pht, phthalimide.
Synthesis of the peptidomimetics
Peptidomimetics were synthesized as described in the general procedure. After their purification by analytical HPLC, their identities were confirmed with high resolution ESI MS. For Vipegitide (Figure 1B): m/z calculated for C67H104N23O18, 1518.7924; found 1518.7921. Vipegitde-PEG2 (Figure 1C): m/z calculated for C58H97N21O18, 1375.7315; found 1375.7508. These results establish the chemical structures.
Stability of the peptidomimetics in human serum
To investigate the effects of peptidomimetics pegylation on proteolytic susceptibility, the degradation of the intact peptidomimetics incubated in human serum at 37°C was followed by RP-HPLC. Incubations were done for different periods of time, and results are presented in Figure 2. Vipegitide was degraded with a half-life of 15 minutes. Pegylated peptidomimetic Vipegitide-PEG2 showed a more complex degradation behavior, and 50% of the starting peptidomimetic amount was preserved for 120 minutes in human serum.
Figure 2.
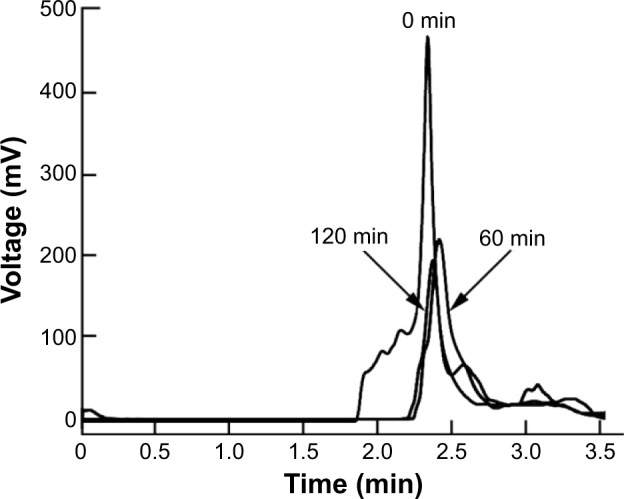
Stability of Vipegitide-PEG2 in human serum.
Notes: Chromatograms of Vipegitide-PEG2 after 0, 60, and 120 minutes of incubation in human serum.
Abbreviation: PEG, polyethylene glycol; min, minutes.
Modeling of the peptidomimetics in water
To evaluate the effect of replacing the first two residues of Vipegitide (glycine1 and tryptophan2) with two polyethylene glycol molecules, molecular dynamics simulations were performed for both peptidomimetics. A total of 500 snapshots were collected along the simulation of each peptidomimetic, but only the last 400 snapshots were used for the following analysis. This was done to remove the effect of the initial arbitrary conformations. Nine evenly spread conformations collected from the last 800 ns of the trajectory are presented for Vipegitide and for Vipegitide-PEG2 in Figure 3A and B, respectively. The conformations are superimposed over the backbone of residues 4–13, which is common for both peptidomimetics.
Figure 3.

Modeling of peptidomimetics in water.
Notes: Conformational space sampled by (A) Vipegitide and (B) Vipegitide-PEG2 peptidomimetics are represented by nine evenly spread snapshots taken from the last 400 snapshots collected during the simulation. Snapshots are shown as solid ribbons with residues colored according to secondary structure, ie, helices are red, beta sheets are cyan, turns are green, and coils are white. The two PEG molecules and lysine3 of Vipegitide-PEG2 are shown as lines. Atoms N, C1, C2 in the N-terminus of Vipegitide-PEG2 are shown in ball and stick representation; (C) Histogram of the radii of gyration of Vipegitide (black) and Vipegitide-PEG2 (white) along the simulation. Please note that Vipegitide adopts a larger range of values, indicating that it is somewhat more flexible than Vipegitide-PEG2.
Abbreviation: PEG, polyethylene glycol.
To examine and compare the conformational spaces of the two peptidomimetics, the distance between their termini was calculated. The percentage of conformations having this distance <5 Å is 9.5% for Vipegitide and 5.3% for Vipegitide-PEG2, suggesting the former has a larger preference for a folded conformation. In addition, we calculated the average radius of gyration of the peptidomimetics and found only a negligible difference between them, 6.9±0.6 Å and 7.0±0.4 Å for Vipegitide and Vipegitide-PEG2, respectively. Yet, the radius of gyration of Vipegitide spans a greater range of values (Figure 3C), indicating that Vipegitide is somewhat more flexible than Vipegitide-PEG2.
To investigate the conformational space of the two peptidomimetics, we examined their tendency to form secondary structure. We found that Vipegitide adopts a helical secondary structure (α-helix) in 10.3% of the collected conformations, with an average helix length of three residues, while Vipegitide-PEG2 adopts a helical secondary structure in 31.8% of the collected conformations, with an average helix length of 3.5 residues. Furthermore, 99.8% and 96.3% of Vipegitide and Vipegitide-PEG2 collected conformations, respectively, assume a bent conformation (ie, characterized by a turn or a bend among their inner residues).
Finally, we examined the similarity between the conformational spaces of Vipegitide and Vipegitide-PEG2 by using the "contained" matrix. We found that 63.3% of the conformational space of Vipegitide is contained within the conformational space of Vipegitide-PEG2, while 75.1% of the conformational space of Vipegitide-PEG2 is contained within the conformational space of Vipegitide. While both peptidomimetics sample the conformational space of each other rather extensively (ie, have similar conformational spaces), the higher percentage of Vipegitide-PEG2 conformational space which is “contained” in the Vipegitide conformational space may suggest that Vipegitide-PEG2 has more restricted conformational space, ie, it is somewhat less flexible.
Peptidomimetic effect on cell adhesion
In the cellular adhesion assay, the inhibitory effect of both peptidomimetics was first tested using α2-K562 overexpressor cells. To measure the potency and efficacy of the peptidomimetics, we performed dose-response experiments in a range of physiological concentrations between 1×10−10 M and 1×10−6 M, using collagen I-coated plates. Vipegitide blocked α2-mediated cell adhesion to collagen I with potency, characterized by an apparent half maximal inhibitory concentration (IC50) of 7×10−10 M and efficacy of 40% (Figure 4A). Vipegitide-PEG2 was characterized with IC50 of 1.5×10−10 M and very similar efficacy of 35% (Figure 4B). To verify differential selectivity of the peptidomimetics toward the α1β1 and α2β1 integrins we used K562 transfected with α1 integrin subunit and measured the potency and efficacy of the peptidomimetics in the same range of concentrations, using collagen IV-coated plates. Neither Vipegitide nor Vipegitide-PEG2 inhibited α1β1 integrin (Figure 4A and B). Apparently, Vipegitide is more selective for α2β1 integrin. In addition, pegylation of Vipegitide using PEG2 unit did not significantly affect the selectivity of Vipegitide and Vipegitide-PEG2 compared with nonpegylated peptidomimetic. However, both compounds inhibited adhesion of integrin α2 overexpressing transfectants with similar efficacy, but the potency of Vipegitide-PEG2 was 4.5 fold higher compared to Vipegitide.
Figure 4.

Inhibitory effect of the peptidomimetics on α2 overexpressing cells and GST-A domain adhesion.
Notes: Dose response curves of the inhibition of α2 (open symbols) and α1 (solid symbols) cell adhesion to respective collagens (A) Vipegitide and (B) Vipegitide-PEG2; (C) binding of the GST-α1A domain to CB3 (collagen IV fragment) (solid symbols); and the GST-α2A domain to collagen I (open symbols) in the presence of Rhodocetin (triangle), Vixapatin (square), Vipegitide (circle), and Vipegitide-PEG2 (rhombus). The mean number of adherent cells ± SD is derived from three independent experiments.
Abbreviations: GST, glutathione s-transferase; PEG, polyethylene glycol; SD, standard deviation.
Peptidomimetics compete for binding of GST-α2A to collagen I
A cell-free assay was performed to assess whether the synthesized peptidomimetics directly interact with the recombinant A domain of α1 or α2 integrin. For this purpose, Vixapatin and Rhodocetin, C-type lectin proteins selective for α2β1 integrin, as well as Vipegitide and Vipegitide-PEG2 were incubated together with the GST-linked A domains and allowed to bind to the immobilized collagen I or collagen IV. The amount of the bound recombinant A domain provided information on the inhibitory potential and biochemical recognition ability of the synthesized peptidomimetics (Figure 4C). As previously reported,34 Rhodocetin, in the range of concentrations between 1×10−10 M and 1×10−7 M, inhibited binding of the GST-α2A domain to collagen I in a dose-dependent manner. A similar inhibition was observed for Vixapatin (Figure 4C). Vipegitide and Vipegitide-PEG2 inhibited binding of GST-α2A to collagen I by 30%, in the range of concentration from 1×10−9 M to 1×10−7 M (Figure 4C). Higher concentrations of the peptidomimetics did not further increase inhibitory effect. These findings suggest that both peptides affect α2A-domain, most probably by conformational changes. Neither Vipegitide nor Vipegitide-PEG2 affected binding of GST-α1A to collagen IV (Figure 4C), indicating their selectivity toward α2β1 integrin.
Peptidomimetics inhibit adhesion of human platelets under flow conditions
In the previous experiments, we investigated the adhesion-inhibiting effect of synthesized peptidomimetics toward transfected cells overexpressing the α2 integrin subunit. Platelets express α2β1 integrin at physiological levels. Therefore, to investigate a potential antithrombotic activity of Vipegitide and Vipegitide-PEG2, first we analyzed their effect on adhesion of human and mouse platelets under shear stress conditions. Human whole blood incubated with only saline showed normal surface coverage of 9.5%, control adhesion (control in Figure 5A). Preactivation of the platelets with collagen I before applying shear stress decreased platelet adhesion, showing only 3.5% of surface coverage (collagen I in Figure 5A). When blood samples were incubated with Vipegitide or Vipegitide-PEG2 before activation with collagen, 7% and 6.5% of surface coverage, respectively, was obtained (Vipegitide and Vipegitide-PEG2 in Figure 5A). Therefore, both peptides reduced collagen-induced human platelet adhesion, as compared to adhesion of collagen-activated platelets.
Figure 5.
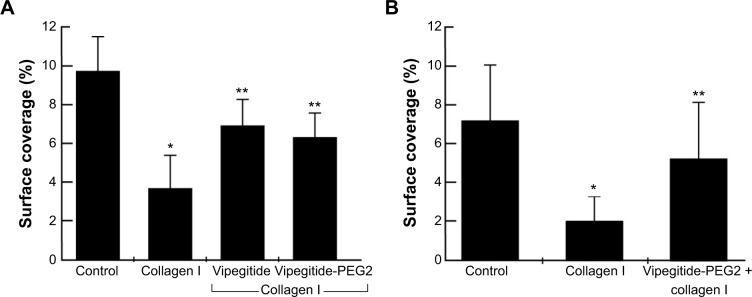
The effect of the peptidomimetics on human and mouse platelet adhesion under flow conditions.
Notes: Surface coverage of (A) human and (B) mouse blood samples incubated with saline (control), 1 μg/mL collagen I (collagen I), 35 μM Vipegitide, or 35 μM Vipegitide-PEG2 and collagen I (Vipegitide, Vipegitide-PEG2) before applying shear stress; *,**P<0.05.
Abbreviation: PEG, polyethylene glycol.
Similar results were obtained using a mouse platelet adhesion assay. Whole blood incubated with only saline showed normal surface coverage of 7%, control adhesion (control in Figure 5B). Previous activation of the platelets with collagen I before applying shear stress decreased platelet adhesion, indicating only 2% of surface coverage (collagen I in Figure 5B). When blood samples were incubated with Vipegitide-PEG2 before activation with collagen, 5% of surface coverage was obtained (Vipegitide-PEG2 + collagen I in Figure 5B).
Peptidomimetics inhibition of ADP-and collagen I-induced human platelets aggregation in PRP
To investigate the antiaggregation properties of the peptidomimetics in PRP, we determined the percentage of ADP- and collagen-induced aggregation of the platelets by aggregometry. Vipegitide and Vipegitide-PEG2 inhibit ADP-induced platelet aggregation in PRP by 40% at a concentration of 2.5 μM (Figure 6A and B). In addition, we investigated peptidomimetics inhibition of collagen-induced platelet aggregation in PRP. Saline, showed 85%±19% of platelet aggregation while 10 μM clopidogrel and prasugrel, the “gold standard” for inhibition of platelet aggregation, gave 38%±22% and 34%±18% of aggregation, respectively. At the same concentration (10 μM), Vipegitide reduced platelet aggregation to 60%±23%. Therefore, Vipegitide was half as potent as these compounds. In the range of concentrations from 4–10 μM for Vipegitide and 45–250 μM for Vipegitide-PEG2, collagen-induced platelet aggregation was inhibited by 25%–50% (Figure 7A and B). Vipegitide concentrations higher than 10 μM and Vipegitide-PEG2 concentrations higher than 200 μM did not further increase inhibition of platelet aggregation. Therefore, Vipegitide was found to be a ten to 20 fold more potent inhibitor of human platelet aggregation in PRP than Vipegitide-PEG2.
Figure 6.

Effect of the peptidomimetics on ADP-activated aggregation of human platelets in PRP.
Notes: *Marks the point in time when the agonist (ADP) was added to the reaction. Aggregation tracing for 0 μM (red), 1.5 μM (green), and 2.5 μM (blue) of (A) Vipegitide and (B) Vipegitide-PEG2.
Abbreviations: ADP, adenosine diphosphate; PEG, polyethylene glycol; PRP, platelet rich plasma.
Figure 7.
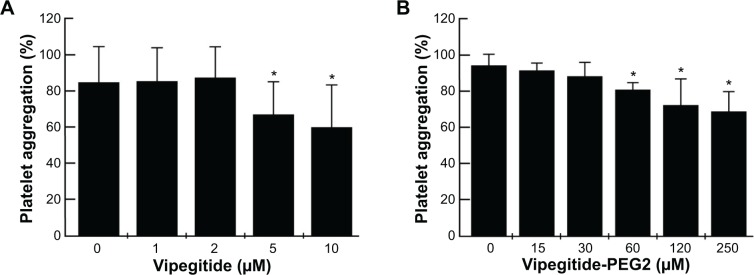
Effect of the peptidomimetics on collagen I-induced aggregation of platelets in PRP.
Notes: Platelet aggregation inhibited by (A) Vipegitide and (B) Vipegitide-PEG2; *P<0.05.
Abbreviations: PEG, polyethylene glycol; PRP, platelet rich plasma.
Peptidomimetics inhibition of collagen I-induced human platelets aggregation in whole blood
Vipegitide and Vipegitide-PEG2 were also evaluated for their effect on platelet aggregation in human whole blood. Both peptides inhibited platelet aggregation in whole blood in a dose-dependent manner (Figure 8A and B). The potency of Vipegitide was characterized with IC50 of 1 μM and Vipegitide-PEG2 with IC50 of 8 μM. The efficacy of 30 μM Vipegitide and 35 μM Vipegitide-PEG2 was 45% and 30%, respectively. It is clear that Vipegitide is eight fold more potent and 1.5 fold more efficient then Vipegitide-PEG2.
Figure 8.
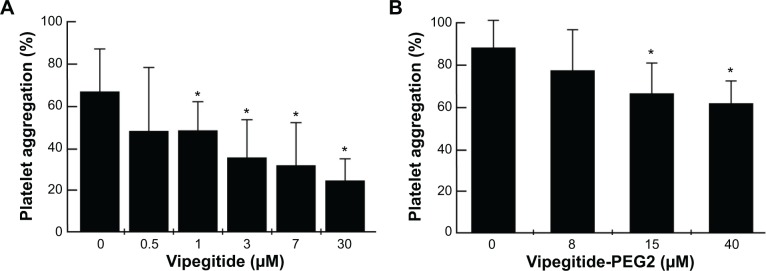
Effect of the peptidomimetics on collagen I-induced aggregation of human platelets in whole blood.
Notes: Platelet aggregation inhibited by (A) Vipegitide and (B) Vipegitide-PEG2; *P<0.05.
Abbreviation: PEG, polyethylene glycol.
Safety of the peptidomimetics in vivo
To investigate the safety of the peptidomimetics in mice, we intravenously injected for 3 consecutive weeks male mice with a dose of 50 mg/kg of Vipegitide or Vipegitide-PEG2. Acute tolerability was observed. At injection of this high dose of 50 mg/kg body weight, the mice did not suffer from visible weakness and/or exhaustion. No paralysis, altered motor activity, or irregular behavior were observed in mice treated with Vipegitide or Vipegitide-PEG2, suggesting lack of neurotoxicity. No cutaneous hematomas around the injection or at distant sites within 24 hours after injection were observed. Furthermore, no sudden deaths of mice occurred within 24 hours after the injection or during the 3 weeks of observation. The blood of mice injected with Vipegitide and Vipegitide-PEG2 after 10 hours was submitted for hematological and biochemical analysis. The values for white blood cells, red blood cells, and platelet counts were in the normal range of 6–15×103/μL, 7–12×106/μL, and 200–450×103/μL, respectively, for mice injected with Vipegitide and Vipegitide-PEG2, similar to the values obtained for control mice. Additional evidence of the lack of hemorrhage or anemic conditions was indicated by similar values for hematocrit, in the range of 35%–45%, and mean corpuscular hemoglobin, 11.1–12.7 pg/mouse, between Vipegitide- and Vipegitide-PEG2-injected mice compared to control mice. Lack of lymphopenia, monocytopenia, and granulocytopenia were indicated by similar values in the range of 20%–40%, 3%–5%, 7%–13%, respectively, between Vipegitide- and Vipegitide-PEG2-injected mice compared to control mice. Alkaline phosphatase and lactate dehydrogenase values were in the range of 100–214 U/L and 1,000–2,400 U/L, respectively, between Vipegitide- and Vipegitide-PEG2-injected mice compared to control mice, suggesting no toxic effects to liver and other tissues.
Discussion
We describe the synthesis, modeling, and antiplatelet characterization of selective α2β1 integrin, folded peptidomimetic antagonists with potential antithrombotic activity. Vipegitide and Vipegitide-PEG2 were characterized as bent peptidomimetics, having nanomolar potency, intermediate efficacy of binding, selectivity toward α2 integrin, different stability in human serum, safety in mice, and antiplatelet aggregation activity. The results clearly demonstrate improved stability of Vipegitide-PEG2 in human serum, underlining the stabilizing effect of the PEG2 unit to the peptidomimetic sequence. A similar effect has been reported for the stability of Integrilin, another antiplatelet drug.4 However, from a pharmacological point of view, the drop of eight fold in the potency of Vipegitide-PEG2 compared to Vipegitide in inhibition of platelet aggregation in whole blood apparently suggests that this chemical modification is not yet satisfactory. A possible structural explanation for the lower potency of the pegylated peptidomimetic compared to the nonpegylated one is therefore that the latter more readily adopts conformations important for α2β1 integrin binding, perhaps due to its higher flexibility, which allows it to more readily adapt to the requirements of the binding site. This hypothesis is in line with the induced fit mechanism.38 An alternative hypothesis is that Vipegitide a priori spends a larger fraction of time in α2β1 integrin-binding conformations, either due to its higher flexibility, which allows it to span a larger conformational space, or due to its lower preference for helical structures or larger preference for a folded conformation, if such conformations are required by the integrin binding site. This hypothesis is in line with the conformational selection mechanism.39 Choosing between these two hypotheses and identifying the α2β1 integrin-binding conformations of the two peptidomimetics will require additional experimental and computational work. Also, it is possible that other modifications, such as methylation or back-bone cyclization, could be more suitable to generate a stable drug and need to be considered. However, it is important to stress that Vipegitide-PEG2 can be used to investigate the in vivo antithrombotic effects caused by its platelet-targeting activity. Noteworthy, Vipegitide-PEG2 effective dose to inhibit platelet aggregation in whole blood is similar to the one of Integrilin.4
We measured inhibitory effect of the peptidomimetics not only in PRP but also in whole blood. Platelet aggregometry in whole blood more closely approximates physiological conditions than PRP, which contains only platelets but no erythrocytes and leukocytes, which both affect platelet aggregation. Both types of cells influence platelet activation: erythrocytes bind adenosine, which has antiaggregation effects, and thus enhances aggregation,40,41 whereas leukocytes produce prostacyclin and nitric oxide, which also impair platelet aggregation.42 In the present study, higher inhibition of 20%–30% aggregation in whole blood was measured compared to PRP with both peptidomimetics. Similarly, some antiplatelet drugs show greater activity in the presence of erythrocytes (eg, dipyridamole)43 or leukocytes (eg, aspirin).44 Efficacy of propofol inhibition of aggregation was higher in whole blood than in PRP, and also the presence of either erythrocytes or leukocytes enhanced antiaggregation effect of propofol in PRP than in whole blood.45
Our attempt in drug discovery of a novel molecular entity such as Vipegitide is in line with previously reported attempts to generate inhibitors of α2β1 integrin. The arylamide derivatives, a group of small molecules, allosteric inhibitors of α2β1 integrin, have been reported.46 One of these molecules is able to inhibit aggregation by blocking collagen I binding to integrin α2β1 on the platelet surface. Also, the same molecule blocked α2 I-domain function in vitro.46 Choi et al47 described small-molecule α2β1 selective inhibitors, derivatives of 2,3-diaminopropionic acid. However, those inhibitors do not inhibit the binding of the isolated α2 I-domain to immobilized type I collagen. By modification of the 2,3-diaminopropionic acid derivatives structure, Miller et al48 developed high-affinity, small-molecule inhibitors of integrin α2β1 that interact with β1 I-domain and prevent pathological thrombus formation via allosteric mechanism. Recently, small molecules acting as α2β1 integrin inhibitor, which recognize the collagen-binding, metal ion-dependent adhesion site in the human α2 I-domain, were presented. One of these molecules showed antiplatelet efficacy by selectively inhibiting α2β1 integrin-mediated collagen I binding.49 Ivaska et al50 reported the development of peptide inhibitor of platelet aggregation, the cyclic peptide interacting with platelet α2β1 integrin. This cyclic peptide inhibited collagen binding function of integrin α2 A-domain to collagen I, IV, and laminin-1, and also inhibited collagen-induced platelet aggregation.
Summarizing all experiments, we suggest that Vipegitide and Vipegitide-PEG2 may shed light on the role of α2β1 integrin in human platelet aggregation and may serve as lead structure for the development of novel α2β1 receptor antagonists, antiplatelet drugs.
Conclusion
This study describes the synthesis of selective α2 integrin receptor antagonist, bent peptidomimetic, Vipegitide and its pegylated analog, Vipegitide-PEG2, with antiadhesive and antiplatelet properties. We propose that these two peptidomimetics may represent a new group of antiplatelet drugs, which may be useful in secondary therapy of ACS.
Acknowledgments
We would like to thank Mrs Zehava Cohen for help with art work, and Dr Dinorah Barasch for extensive help in evaluation of the mass-spectra data. This work was supported by the Yissum Research & Development Company of the Hebrew University of Jerusalem (PL and JK), Intramural Research Funds (PL), the Israel Ministry of Industry and Commerce, Kamin (PL, JK, and DV), The German-Israeli Foundation (GIF-994-3.9/2008) (PL, JK, and JAE), and Deutsche Forschungsgemeinschaft through grant SFB/1009, project A09 (JAE), and the US National Institutes of Health and National Cancer Institute (1R01CA133262-01A2; 1R01CA100145-01A1) (CM). PL holds The Jacob Gitlin Chair in Physiology and is affiliated and partially supported by The Grass Center for Drug Design and Synthesis of Novel Therapeutics, The Hebrew University of Jerusalem, Israel.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Kumar RA, Dong JF, Thaggard JA, Cruz MA, López JA, McIntire LV. Kinetics of GPIbalpha-vWF-A1 tether bond under flow: effect of GPIbalpha mutations on the association and dissociation rates. Biophys J. 2003;85(6):4099–4109. doi: 10.1016/S0006-3495(03)74822-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pugh N, Simpson AM, Smethurst PA, de Groot PG, Raynal N, Farndale RW. Synergism between platelet collagen receptors defined using receptor-specific collagen-mimetic peptide substrata in flowing blood. Blood. 2010;115(24):5069–5079. doi: 10.1182/blood-2010-01-260778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28(3):403–412. doi: 10.1161/ATVBAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- 4.Sakhuja R, Yeh RW, Bhatt DL. Antiplatelet agents in acute coronary syndromes. Curr Prob Cardiology. 2010;35(3):123–170. doi: 10.1016/j.cpcardiol.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov. 2003;2(1):15–28. doi: 10.1038/nrd985. [DOI] [PubMed] [Google Scholar]
- 6.Yeung J, Holinstat M. Newer agents in antiplatelet therapy: a review. J Blood Med. 2012;3:33–42. doi: 10.2147/JBM.S25421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tcheng JE, Ellis SG, George BS, et al. Pharmacodynamics of chimeric glycoprotein IIb/IIIa integrin antiplatelet antibody Fab 7E3 in high-risk coronary angioplasty. Circulation. 1994;90(4):1757–1764. doi: 10.1161/01.cir.90.4.1757. [DOI] [PubMed] [Google Scholar]
- 8.Phillips DR, Scarborough RM. Clinical pharmacology of eptifibatide. Am J Cardiol. 1997;80(4A):11B–20B. doi: 10.1016/s0002-9149(97)00572-9. [DOI] [PubMed] [Google Scholar]
- 9.Hartman GD, Egbertson MS, Halczenko W, et al. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J Med Chem. 1992;35(24):4640–4642. doi: 10.1021/jm00102a020. [DOI] [PubMed] [Google Scholar]
- 10.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69(1):11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 11.Knight CG, Morton LF, Peachey AR, Tuckwell DS, Farndale RW, Barnes MJ. The collagen-binding A-domains of integrins alpha(1) beta(1) and alpha(2)beta(1) recognize the same specific amino acid sequence, GFOGER, in native (triple-helical) collagens. J Biol Chem. 2000;275(1):35–40. doi: 10.1074/jbc.275.1.35. [DOI] [PubMed] [Google Scholar]
- 12.Momic T, Katzehendler J, Benny O, et al. Vimocin and vidapin, cyclic KTS peptides, are dual antagonists of α1β1/α2β1 integrins with anti-angiogenic activity. J Pharmacol Exp Ther. 2014;350(3):506–519. doi: 10.1124/jpet.114.214643. [DOI] [PubMed] [Google Scholar]
- 13.Banerjee R, Basu G, Chène P, Roy S. Aib-based peptide backbone as scaffolds for helical peptide mimics. J Pept Res. 2002;60(2):88–94. doi: 10.1034/j.1399-3011.2002.201005.x. [DOI] [PubMed] [Google Scholar]
- 14.Staniszewska I, Walsh EM, Rothman VL, et al. Effect of VP12 and viperistatin on inhibition of collagen-receptor-dependent melanoma metastasis. Cancer Biol Ther. 2009;8(15):1507–1516. doi: 10.4161/cbt.8.15.8999. [DOI] [PubMed] [Google Scholar]
- 15.Eble JA, Beermann B, Hinz HJ, Schmidt-Hederich A. alpha 2beta 1 integrin is not recognized by rhodocytin but is the specific, high affinity target of rhodocetin, an RGD-independent disintegrin and potent inhibitor of cell adhesion to collagen. J Biol Chem. 2001;276(15):12274–12284. doi: 10.1074/jbc.M009338200. [DOI] [PubMed] [Google Scholar]
- 16.Lankshear MD, Cowley AR, Beer PD. Cooperative AND receptor for ion-pairs. Chem Commun (Camb) 2006;(6):612–614. doi: 10.1039/b515634c. [DOI] [PubMed] [Google Scholar]
- 17.Lang T, Guenet A, Graf E, Kyritsakas N, Hosseini MW. Porphyrin based molecular turnstiles. Chem Commun (Camb) 2010;46(20):3508–3510. doi: 10.1039/b927112k. [DOI] [PubMed] [Google Scholar]
- 18.Pajk S, Pečar S. Synthesis of novel amphiphilic spin probes with the paramagnetic doxyl group in the polar region. Tetrahedron. 2009;65(3):659–665. [Google Scholar]
- 19.Chan WC, White PD, editors. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford, UK: Oxford University Press; 2000. [Google Scholar]
- 20.Fields CG, Fields GB. Minimization of tryptophan alkylation following 9-fluorenylmethoxycarbonyl solid-phase peptide synthesis. Tetrahedron Lett. 1993;34(42):6661–6664. [Google Scholar]
- 21.Kocí J, Krchnák V. Solid-phase synthesis and chemical properties of 2-(2-amino/hydroxyethyl)-1-aryl-3,4-dihydropyrazino[1, 2-b]indazol-2-iums. J Comb Chem. 2010;12(1):168–175. doi: 10.1021/cc9001422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jenssen H, Aspmo SI. Serum stability of peptides. In: Otvos L, editor. Peptide-Based Drug Design. New York, NY: Humana Press; 2008. pp. 177–186. [DOI] [PubMed] [Google Scholar]
- 23.Accelrys Software Inc . Discovery Studio Modeling Environment. 2.5 ed. San Diego: Accelrys Software Inc; 2005–2009. [Google Scholar]
- 24.Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comp Phys Comm. 1995;91:43–56. [Google Scholar]
- 25.Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J Chem Theory Comput. 2008;4(1):116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 26.Lindorff-Larsen K, Piana S, Palmo K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78(8):1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- 28.Hockney RW, Goel SP, Eastwood JW. Quiet high-resolution computer models of a plasma. J Comput Phys. 1974;14(2):148–158. [Google Scholar]
- 29.Darden T, York D, Pedersen L. Particle Mesh Ewald – an N. Log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. [Google Scholar]
- 30.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577–8593. [Google Scholar]
- 31.Hess B, Bekker H, Berendsen HJC, Fraaije J. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472. [Google Scholar]
- 32.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 33.Bazan-Socha S, Kisiel DG, Young B, et al. Structural requirements of MLD-containing disintegrins for functional interaction with alpha 4 beta 1 and alpha 9 beta1 integrins. Biochemistry. 2004;43(6):1639–1647. doi: 10.1021/bi035853t. [DOI] [PubMed] [Google Scholar]
- 34.Eble JA, Tuckwell DS. The alpha2beta1 integrin inhibitor rhodocetin binds to the A-domain of the integrin alpha2 subunit proximal to the collagen-binding site. Biochem J. 2003;376(Pt 1):77–85. doi: 10.1042/BJ20030373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.WMA Declaration of Helsinki - Ethical Principles for Medical Research Involving Human Subjects. World Medical Association, Inc; 2013. [Accessed December 1, 2014]. Available from: http://www.wma.net/en/30publications/10policies/b3/index.html. [PubMed] [Google Scholar]
- 36.Goldschmidt N, Spectre G, Brill A, et al. Increased platelet adhesion under flow conditions is induced by both thalassemic platelets and red blood cells. Thromb Haemost. 2008;100(5):864–870. [PubMed] [Google Scholar]
- 37.Born GV, Cross MJ. Effect of adenosine diphosphate on the concentration of platelets in circulating blood. Nature. 1963;197:974–976. doi: 10.1038/197974a0. [DOI] [PubMed] [Google Scholar]
- 38.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci U S A. 1958;44(2):98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma B, Kumar S, Tsai CJ, Nussinov R. Folding funnels and binding mechanisms. Protein Eng. 1999;12(9):713–720. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 40.Valles J, Santos MT, Aznar J, et al. Erythrocytes metabolically enhance collagen-induced platelet responsiveness via increased thromboxane production, adenosine diphosphate release, and recruitment. Blood. 1991;78(1):154–162. [PubMed] [Google Scholar]
- 41.Rocca B, FitzGerald GA. Simply read: erythrocytes modulate platelet function. Should we rethink the way we give aspirin? Circulation. 1997;95(1):11–13. doi: 10.1161/01.cir.95.1.11. [DOI] [PubMed] [Google Scholar]
- 42.Schattner MA, Geffner JR, Isturiz MA, Lazzari MA. Inhibition of human platelet activation by polymorphonuclear leukocytes. Br J Pharmacol. 1990;101(2):253–256. doi: 10.1111/j.1476-5381.1990.tb12696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gresele P, Zoja C, Deckmyn H, Arnout J, Vermylen J, Verstraete M. Dipyridamole inhibits platelet aggregation in whole blood. Thromb Haemost. 1983;50(4):852–856. [PubMed] [Google Scholar]
- 44.De la Cruz JP, Camara S, Bellido I, Carrasco T, Sanchez de la Cuesta F. Platelet aggregation in human whole blood after chronic administration of aspirin. Thromb Res. 1987;46(1):133–140. doi: 10.1016/0049-3848(87)90213-1. [DOI] [PubMed] [Google Scholar]
- 45.De la Cruz JP, Carmona JA, Paez MV, Blanco E, Sanchez De La Cuesta F. Propofol inhibits in vitro platelet aggregation in human whole blood. Anesth Analg. 1997;84(4):919–921. doi: 10.1097/00000539-199704000-00040. [DOI] [PubMed] [Google Scholar]
- 46.Yin H, Gerlach LO, Miller MW, et al. Arylamide derivatives as allosteric inhibitors of the integrin alpha2beta1/type I collagen interaction. Bioorg Med Chem Lett. 2006;16(13):3380–3382. doi: 10.1016/j.bmcl.2006.04.037. [DOI] [PubMed] [Google Scholar]
- 47.Choi S, Vilaire G, Marcinkiewicz C, Winkler JD, Bennett JS, DeGrado WF. Small molecule inhibitors of integrin alpha2beta1. J Med Chem. 2007;50(22):5457–5462. doi: 10.1021/jm070252b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller MW, Basra S, Kulp DW, et al. Small-molecule inhibitors of integrin alpha2beta1 that prevent pathological thrombus formation via an allosteric mechanism. Proc Natl Acad Sci U S A. 2009;106(3):719–724. doi: 10.1073/pnas.0811622106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nissinen L, Pentikäinen OT, Jouppila A, et al. A small-molecule inhibitor of integrin alpha2 beta1 introduces a new strategy for antithrombotic therapy. Thromb Haemost. 2010;103(2):387–397. doi: 10.1160/TH09-06-0358. [DOI] [PubMed] [Google Scholar]
- 50.Ivaska J, Käpylä J, Pentikäinen O, et al. A peptide inhibiting the collagen binding function of integrin alpha2I domain. J Biol Chem. 1999;274(6):3513–3521. doi: 10.1074/jbc.274.6.3513. [DOI] [PubMed] [Google Scholar]