

SUMMARY
Despite the tremendous efficacy of trastuzumab against HER2-overexpressing metastatic breast cancers, a significant fraction of women demonstrate progressive disease during treatment. Multiple mechanisms have been proposed to mediate trastuzumab resistance. In this mini-review, we discuss the evidence supporting FOXM1 as a mediator of resistance and potential new therapeutic target in trastuzumab-refractory breast cancer. FOXM1 expression is significantly elevated in multiple breast cancer data sets. Some studies suggest a direct correlation between FOXM1 and HER2 expression levels. In addition, overexpression of FOXM1 reduces the sensitivity of HER2-positive breast cancer cells to trastuzumab or lapatinib. Conversely, knockdown or pharmacological inhibition of FOXM1 rescues resistance to HER2-targeted therapies. Current pre-clinical information supports further investigation of the role of FOXM1 in trastuzumab-resistant breast cancer.
Keywords: breast cancer, foxm1, her2, lapatinib, resistance, trastuzumab
Introduction
Breast cancer remains one of the most common cancers worldwide and is a major cause of cancer-related deaths among adult females in the United States. Molecular techniques, such as immunohistochemistry (IHC) and fluorescence in situ hybridization, are routinely performed to characterize breast tumor biopsies [1]. Gene expression profiling has identified at least five major subtypes of breast cancer- luminal A (ER+ and/or PR+, HER2−, low Ki67), luminal B (ER+ and/or PR+, HER2+ or HER2− with high Ki67), ER− and HER2+, triple negative (TNBC) and basal-like (ER−, PR−, HER2−) [2–7]. Histopathological and molecular classifications improve the abilities to predict outcomes and direct appropriate targeted treatment options to patients.
Among newly diagnosed breast cancer patients, an estimated 15–20% of breast tumors demonstrate overexpression of the human epidermal growth factor receptor 2 (HER2) receptor tyrosine kinase [8–10]. HER2 amplification is associated with a more aggressive tumor biology [11] and an increased incidence of metastasis [12] due to the constitutive activation of numerous downstream signaling networks involved in migration, cell-cycle regulation, proliferation, inhibition of apoptosis, and angiogenesis [13, 14]. The increased expression of this cell-surface molecule specifically in tumor cells and its association with unfavorable outcomes in patients with breast cancer provide rationale for selectively inhibiting this molecular target. The first anti-HER2 antibody to be translated to clinical use was trastuzumab [15], which is currently the main first-line therapy for patients with HER2-overexpressing breast cancer. Trastuzumab binds to domain IV of the HER2 extracellular domain and disrupts downstream PI3K signaling [16] and Ras-MAPK signaling [17]. Trastuzumab-mediated tumor regression appears to be partially dependent on the abilities to block angiogenesis [18, 19], induce antibody-dependent cellular cytotoxicity [20, 21], and suppress invasion and metastasis [22, 23], which may be related to the ability to target a HER2-positive stem cell population [24, 25].
Despite the tremendous efficacy of trastuzumab against HER2-overexpressing metastatic breast cancers, a significant fraction of women demonstrate progressive disease during treatment. There are many proposed mechanisms of resistance. One potential mechanism is masking of the HER2 epitope to which trastuzumab binds, which has been described as a result of overexpression of the mucin cell-surface protein MUC4 [26]. Compensatory signaling and receptor cross-talk have also been proposed as mechanisms through which HER2 signaling is sustained in resistant cells; for example, the insulin-like growth factor-I receptor [27, 28] and the hepatocyte growth factor receptor MET [29] have been shown to cluster and crosstalk with HER2. Increased signaling through the PI3K pathway is recognized as one of the most clinically relevant mechanisms of resistance and may occur due to down-regulation of PTEN [30], hyperactivating mutations in the catalytic subunit of PI3K [31], or subsequent to increased upstream growth factor receptor signaling. Further downstream, reduced expression or cellular relocalization of the p27 protein [16, 32–35] or increased expression of anti-apoptotic regulators, including Bcl-2 [36], have been described in models of trastuzumab resistance. Another potential mechanism is up-regulation of ligands that increase phosphorylation of HER2, such as the EGFR ligand TGF-alpha [37], HER3 ligand heregulin [37], and the cytokine growth differentiation factor 15 [38]. There are additional mechanisms of trastuzumab resistance that have been proposed, many of which have been comprehensively discussed in a number of excellent, recent reviews [39–42].
Attempts to overcome trastuzumab resistance have resulted in new therapeutic strategies targeted against HER2, including the small-molecule dual EGFR/HER2 kinase inhibitor lapatinib [43] [44]. Single-agent lapatinib reduces tyrosine phosphorylation of HER2 [45] and inhibits downstream signaling through PI3K and MAPK in trastuzumab-resistant cells [46, 47]. In addition, lapatinib monotherapy induces apoptosis and increases sensitivity to radiation in trastuzumab-resistant cells [48]. Clinical studies investigating the combinatorial effects of trastuzumab and lapatinib in HER2-overexpressing breast cancers demonstrated synergistic enhancement of trastuzumab-mediated antibody-dependent cellular cytotoxicity [49]. Lapatinib is currently approved as a second-line therapy in combination with chemotherapy for trastuzumab-refractory metastatic breast cancer [50]. However, a majority of patients who received prior trastuzumab therapy demonstrate resistance to lapatinib. Thus, improved understanding of the molecular mechanisms contributing to resistance to both trastuzumab and lapatinib is critical for developing new therapies and for identifying those who are most likely to respond to currently available agents.
FOXM1 and breast cancer
Forkhead box MI (FOXM1) is a member of the forkhead family of transcription factors [51]. There are more than 100 proteins in the forkhead family, which represents a subgroup of the helix-turn-helix class of transcription factors; this name refers to the winged nature of the DNA-binding domain, which is flanked by two side loops [51]. There are three known isoforms of human FOXM1, which are referred to as FOXM1a, FOXM1b, and FOXM1c; these isoforms result from alternative splicing of the transcript of the 25-kb foxm1 gene, which contains 10 exons and is found at chromosomal location 12p13.33 [51]. The FOXM1 isoforms are characterized by a highly conserved DNA-binding domain, an N-terminal repressor domain, and a strong transactivation domain. FOXM1b and FOXM1c recognize and activate transcription from consensus sequence 5′-A-C/T-AAA-C/T-AA-3′ [51]. Both are regulated by MEK signaling, but FOXM1c has two Erk1/2 phosphorylation sites, S330 and S703 [51]; thus, FOXM1c may be more dependent upon MEK signaling. The functions of FOXM1 are related to the functions of its target genes, of which there are more than 200; these targets regulate the majority of cancer-related processes, including proliferation, invasion, angiogenesis, senescence, stem cell function, and DNA repair (Figure 1) [51]. Normal physiological functions include regulation of replication, mitosis, and repair. Readers are guided to a recent, outstanding review article [51] for in-depth details regarding the normal and cancer-related functions and structure of FOXM1.
Figure 1. The role of FOXM1 in normal cells and the effects of its deregulation in tumor cell chemoresistance.
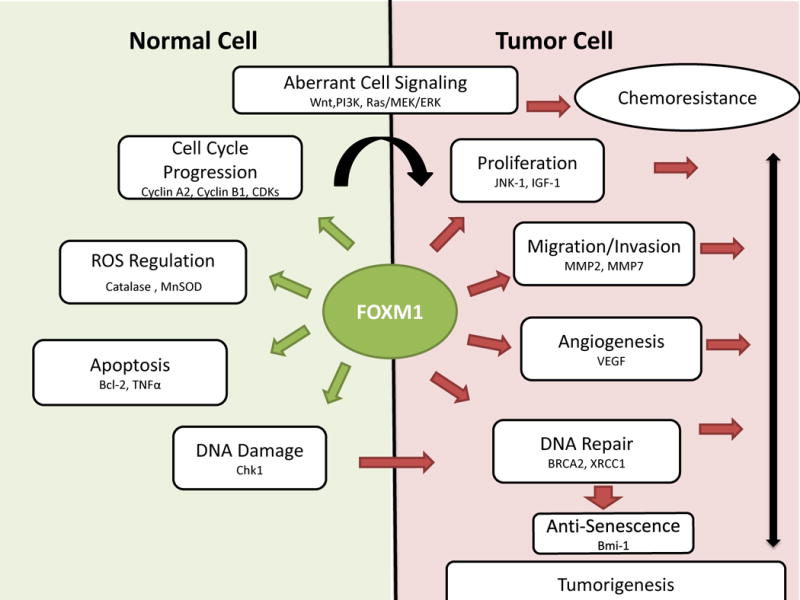
FOXM1 has several mechanisms of activation. Under normal conditions FOXM1 transcriptional activity and expression are tightly regulated. FOXM1 controls a variety biological process, by driving the transcription of target genes that regulate cell cycle progression/arrest, cellular responses to oxidative stress, DNA damage and cell death. In tumor cells, FOXM1 homeostatic regulation is compromised due to dysregulation of cell signaling pathways. For example, deregulation of Wnt, PI3K, and/or ras/MEK/ERK signaling has been shown to increase FOXM1 expression and activation. Sustained FOXM1 signaling promotes increased expression of FOXM1 target genes and evasion of cell death in tumor cells resulting in chemoresistance and tumorigenesis. Thus, FOXM1 antagonizes the effects of chemotherapy by upregulating DNA repair, self-renewal, proliferation, and migration.
Abbreviations:_Bcl-2, B cell lymphoma 2; Bmi-1, B lymphoma Mo-MLV (Moloney-murine leukemia virus) insertion region-1; BRAC2, Breast cancer 2, early onset; Chk1, Check point kinase; Cdk, Cyclin-dependent kinase; ERK, extracellular signal regulating kinase; IGF1, insulin-like growth factor-1; JNK, c-jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; MEK, MAPK kinase; MMP2, MMP9, matrix metalloproteinase; MnSOD, manganese superoxide dismutase; TNFα, tumor necrosis factor α; VEGF, vascular endothelial growth factor; WNT, wingless-type; XRCC, X-ray repair cross-complementing [68, 75, 76].
FOXM1 is associated with poor prognosis in breast cancer [52]. FOXM1 transcript levels are significantly elevated in multiple breast cancer tissue data sets (Figure 1). Studies indicate that FOXM1 plays a role in most subtypes of breast cancer, not just HER2-overexpressing forms. Intriple negative breast cancer (TNBC), FOXM1 overexpression protects cancer cells from DNA double-strand breaks, by interacting with NFκB to promote doxorubicin chemoresistance [53]. Accordingly, it has been shown that FOXM1 inhibition decreases transcription of DNA repair genes and restores Doxorubicin sensitivity in TNBC [53]. Constitutive overexpression of FOXM1 in MCF-7 breast cancer cells promotes acquired cisplatin resistance, by enhancing the expression of the DNA damage response genes; breast cancer-associated gene 2 (BRCA2) and X-ray cross complementing group 1 (XRCC1) [54]. Furthermore, in ERα positive and negative breast cancer cells, FOXM1 has been shown to interact with the coactivator CARM1 to regulate ERα transcription [55]. Increased FOXM1 levels amplify estrogen-mediated mitogenic actions and promote endocrine therapy resistance in ERα positive breast cancer [56]. FOXM1 inhibition has been demonstrated to decrease expression of ERα-regulated genes, suppress estrogen-induced breast cancer cell proliferation, and restore tamoxifen sensitivity [55, 56].
Studies investigating FOXM1 as a downstream target of HER2 signaling have demonstrated a direct correlation between HER2 and FOXM1 expression levels in vivo and in vitro [57, 58]. Stable overexpression of FOXM1 in HER2-overexpressing cell lines effectively diminished trastuzumab sensitivity, increased colony formation, and inhibited lapatinib-induced cytotoxicity [57, 59]. Interestingly, inhibition of EGFR/HER2 with lapatinib had no observable effect on FOXM1 protein levels in lapatinib-resistant lines. In contrast, combined inhibition of MEK signaling plus lapatinib diminished nuclear FOXM1 levels [58]. Consistent with these findings, inhibition of Raf/MEK/ERK signaling delays G2/M transition and inhibits expression of FOXM1 target genes [60]. Additionally, treatment of sensitive and resistant breast carcinoma lines with the anti-EGFR tyrosine kinase inhibitor gefitinib reduces FOXM1 and HER2 phosphorylation only in sensitive cell lines [61]. Moreover, FOXM1 blocks paclitaxel-induced apoptosis due to reduced levels of the microtubule-destabilizing protein stathmin in HER2-positive breast cancer cells [57]. Aberrant FOXM1 signaling can promote a drug-resistant phenotype, characterized by activation of anti-apoptotic proteins Bcl-2, up-regulation of genes important for homologous recombination, and promotion of epithelial to mesenchymal transition (EMT) [62–64]. Furthermore, FOXM1 has been shown to sustain TGFβ-induced formation of a SMAD3/SMAD4 nuclear transcription complex that up-regulates the downstream EMT target SLUG to promote breast cancer metastasis [65, 66]. This process may also be mediated by growth differentiation factor 15 (GDF15), a divergent member of the TGFβ superfamily, which promotes invasion, EMT, and is increased in the setting of acquired trastuzumab resistance [38]. Collectively, these studies provide validity for further investigation into the mechanisms of FOXM1-mediated chemoresistance in HER2-positive breast cancer.
Targeting FOXM1 in trastuzumab-resistant breast cancer
FOXM1 deregulation is a potential diagnostic and prognostic biomarker of oncogenic potential in several malignancies [67]. Aberrant HER2 signaling constitutively activates multiple downstream signaling pathways, including PI3K/Akt, and ERK, which enhance FOXM1 signaling [68]. FOXM1 regulates proliferation, mitosis, metastasis, tumor development, and progression in breast cancer [14–20]. Thus, FOXM1-mediated trastuzumab resistance may occur through a variety of molecular mechanisms. Treatment of HER2-positive breast cancer cells with thiostrepton, a selective inhibitor of FOXM1 mRNA, causes increased sensitivity to lapatinib. Furthermore, thiostrepton diminishes proliferation, invasiveness, and transformation, and induces apoptosis in breast cancer cells that express FOXM1, regardless of HER2 overexpression status, indicating that FOXM1-targeting is a relevant approach for multiple subtypes of breast cancer [54, 59]. Knockdown of FOXM1 with thiostrepton in micelle-nanoparticles administered to MDA-MB-231 breast cancer xenografts reduced tumor growth rates and increased apoptosis [69]. Additionally, co-administration of thiostrepton and lapatinib reduces the survival of HER2-positive breast cancer cells. The natural nontoxic agent 3,3′-diidolylmethane (DIM) combined with trastuzumab causes down-regulation of Akt, NFκB, and FOXM1 in breast cancer cells [70]. DIM enhances trastuzumab efficacy by selectively reducing FOXM1 expression and inhibiting tumor growth without toxicity [71]. Injection of FOXM1-specific siRNA into tumor xenografts suppresses tumor growth and reduces expression of FOXM1 transcriptional targets [72]. Similarly, treatment of resistant and sensitive breast cancer cells with the ARF-derived peptide, which is a FOXM1 inhibitor, decreases proliferation and restores sensitization to trastuzumab [57]. Knockdown of FOXM1 with shRNA diminishes proliferation, anchorage independence, and tumorigenesis of breast cancer cells in vitro and in vivo [73]. These studies demonstrate the therapeutic potential of co-targeting FOXM1 in drug-refractory breast cancer.
Future perspective
Battling the clinical challenge of drug resistance requires an understanding of the molecular mechanisms that facilitate escape from targeted therapies. FOXM1 is overexpressed in many breast cancers, including the HER2-overexpressing and triple-negative subtypes. However, the extent to which FOXM1 contributes to the development or progression of individual subtypes of breast cancer remains unknown. Specific inhibition of FOXM1 may have substantial clinical impact in these subtypes, including trastuzumab-refractory metastatic breast cancers. However, further translational and clinical investigations into the mechanisms through which HER2 regulates FOXM1 are needed to determine the true suitability of FOXM1 as a therapeutic target. Further, the mechanisms employed by FOXM1 to promote progression of HER2-positive cancers are not completely defined; knowledge of these mechanisms is needed to develop more effective targeted therapies or combination treatments. Finally, pharmacological approaches to target FOXM1 are lacking. Although the thiazole antibiotic and proteasome inhibitor, thiostrepton, effectively knocks down FOXM1 expression, its clinical utility is limited by the fact that it is insoluble in aqueous solution. Attempts to encapsulate thiostrepton in micelles achieved enhanced apoptosis and reduced tumor growth of FOXM1-expressing TNBC cells in culture and as xenografts [74]. Future efforts should focus on the development of FOXM1-targeted therapies, including nanoencapsulation of FOXM1-targeted siRNA, DNAzymes, or proteasome inhibitors. Targeting FOXM1 for degradation should ultimately improve responses to existing cancer therapies, such as trastuzumab, and should delay progression of breast cancer.
Figure 2. Increased FOXM1 transcript levels in breast cancer tissues.



(A) FoxM1 transcript levels in breast cancer tissues versus normal breast tissues are shown for the Curtis breast data set retrieved from Oncomine; this data set included 2,136 samples examined on the Illumina Human HT-12 V3.0 R2 Array consisting of 19,273 measured genes. Fold change = 2.21, **p<0.005 (B) FoxM1 transcript levels in breast cancer tissues versus normal breast tissues are shown for the TCGA breast data set retrieved from Oncomine; This data set included 593 samples; the array measured 20,423 genes, but the name of the array platform was not provided in the Oncomine database. Fold change = 5.213, **p<0.005 (C) FoxM1 transcript levels in breast cancer tissues versus normal breast tissues are shown for the Richardson breast 2 data set retrieved from Oncomine; This data set included 47 samples examined on the Human Genome U133 Plus 2.0 Array consisting of 19,574 measured genes. Fold change = 17.629, **p<0.005; [Citation: www.oncomine.org, April 2014, Compendia Bioscience, Ann Arbor, MI]
KEY POINTS.
HER2 is overexpressed in 15–20% of metastatic breast cancers.
Trastuzumab resistance eventually develops in a majority of patients.
FOXM1 transcript levels are significantly elevated in multiple breast cancer tissue data sets.
There is a direct correlation between HER2 and FOXM1 expression levels.
FOXM1 overexpression reduces sensitivity to HER2-targeted treatments.
FOXM1 knockdown increases sensitivity to HER2-targeted therapies.
Acknowledgments
Bridgette Peake acknowledges funding from the Molecular Systems and Pharmacology Training Grant at Emory University (5T32GM0080602-17). Rita Nahta acknowledges funding from NIH R01CA157754 and is a Glenn Breast Cancer Research Scholar at the Winship Cancer Institute of Emory University. We acknowledge Winship Cancer Institute P30 CA138292.
Footnotes
Disclosure
Bridgette Peake and Rita Nahta do not have any conflicts related to this manuscript.
References
- 1.Connolly CK, Van Schil PE, Milroy R, Braendli O, Prescott RJ. The approach to the surgical management of cancer in four European countries. The European respiratory journal. 2003;22(5):838–844. doi: 10.1183/09031936.03.00052703. [DOI] [PubMed] [Google Scholar]
- 2**.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. The initial study to identify distinct molecular subtypes of breast cancer based on genetic profiles. [DOI] [PubMed] [Google Scholar]
- 3.Nielsen TO, Hsu FD, Jensen K, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10(16):5367–5374. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 4.Neven P, Pochet N, Drijkoningen M, et al. Progesterone receptor in estrogen receptor-positive breast cancer: the association between HER-2 and lymph node involvement is age related. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24(16):2595. doi: 10.1200/JCO.2005.05.1334. author reply 2595–2597. [DOI] [PubMed] [Google Scholar]
- 5.Shipitsin M, Campbell LL, Argani P, et al. Molecular definition of breast tumor heterogeneity. Cancer cell. 2007;11(3):259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 6.Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Molecular oncology. 2011;5(1):5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida T, Ozawa Y, Kimura T, et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. British journal of cancer. 2014 doi: 10.1038/bjc.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, Mcguire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 9.Yaziji H, Gown AM. Accuracy and precision in HER2/neu testing in breast cancer: are we there yet? Human pathology. 2004;35(2):143–146. doi: 10.1016/j.humpath.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 10.Owens MA, Horten BC, Da Silva MM. HER2 amplification ratios by fluorescence in situ hybridization and correlation with immunohistochemistry in a cohort of 6556 breast cancer tissues. Clinical breast cancer. 2004;5(1):63–69. doi: 10.3816/cbc.2004.n.011. [DOI] [PubMed] [Google Scholar]
- 11.Tan GH, Choo WY, Taib NA, Yip CH. Factors associated with HER2 overexpression in breast cancer: Experience in an Asian developing country. Asian Pacific journal of cancer prevention : APJCP. 2009;10(5):837–840. [PubMed] [Google Scholar]
- 12.Carter WB, Niu G, Ward MD, Small G, Hahn JE, Muffly BJ. Mechanisms of HER2-induced endothelial cell retraction. Annals of surgical oncology. 2007;14(10):2971–2978. doi: 10.1245/s10434-007-9442-4. [DOI] [PubMed] [Google Scholar]
- 13.Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25(1):118–145. doi: 10.1200/JCO.2006.09.2775. [DOI] [PubMed] [Google Scholar]
- 14.Wang SC, Hung MC. HER2 overexpression and cancer targeting. Seminars in oncology. 2001;28(5 Suppl 16):115–124. doi: 10.1016/s0093-7754(01)90289-1. [DOI] [PubMed] [Google Scholar]
- 15.Carter P, Presta L, Gorman CM, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89(10):4285–4289. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002;62(14):4132–4141. [PubMed] [Google Scholar]
- 17.Albanell J, Codony J, Rovira A, Mellado B, Gascon P. Mechanism of action of anti-HER2 monoclonal antibodies: scientific update on trastuzumab and 2C4. Advances in experimental medicine and biology. 2003;532:253–268. doi: 10.1007/978-1-4615-0081-0_21. [DOI] [PubMed] [Google Scholar]
- 18.Du Manoir JM, Francia G, Man S, et al. Strategies for delaying or treating in vivo acquired resistance to trastuzumab in human breast cancer xenografts. Clin Cancer Res. 2006;12(3 Pt 1):904–916. doi: 10.1158/1078-0432.CCR-05-1109. [DOI] [PubMed] [Google Scholar]
- 19.Izumi Y, Xu L, Di Tomaso E, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416(6878):279–280. doi: 10.1038/416279b. [DOI] [PubMed] [Google Scholar]
- 20.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 21.Petricevic B, Laengle J, Singer J, et al. Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. Journal of translational medicine. 2013;11:307. doi: 10.1186/1479-5876-11-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplan MA, Ertugrul H, Firat U, et al. Brain metastases in HER2-positive metastatic breast cancer patients who received chemotherapy with or without trastuzumab. Breast cancer. 2014 doi: 10.1007/s12282-013-0513-z. [DOI] [PubMed] [Google Scholar]
- 23.Lazaro G, Smith C, Goddard L, et al. Targeting focal adhesion kinase in ER+/HER2+ breast cancer improves trastuzumab response. Endocrine-related cancer. 2013;20(5):691–704. doi: 10.1530/ERC-13-0019. [DOI] [PubMed] [Google Scholar]
- 24.Korkaya H, Kim GI, Davis A, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012;47(4):570–584. doi: 10.1016/j.molcel.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25**.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27(47):6120–6130. doi: 10.1038/onc.2008.207. This study demonstrates that HER2 signals sustain a stem cell-like breast cancer cell population. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price-Schiavi SA, Jepson S, Li P, et al. Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int J Cancer. 2002;99(6):783–791. doi: 10.1002/ijc.10410. [DOI] [PubMed] [Google Scholar]
- 27.Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65(23):11118–11128. doi: 10.1158/0008-5472.CAN-04-3841. [DOI] [PubMed] [Google Scholar]
- 28.Huang X, Gao L, Wang S, et al. Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-i receptor in breast cancer cells resistant to herceptin. Cancer Res. 2010;70(3):1204–1214. doi: 10.1158/0008-5472.CAN-09-3321. [DOI] [PubMed] [Google Scholar]
- 29.Shattuck DL, Miller JK, Carraway KL, 3rd, Sweeney C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008;68(5):1471–1477. doi: 10.1158/0008-5472.CAN-07-5962. [DOI] [PubMed] [Google Scholar]
- 30.Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 31.Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 32.Kute T, Lack CM, Willingham M, et al. Development of Herceptin resistance in breast cancer cells. Cytometry A. 2004;57(2):86–93. doi: 10.1002/cyto.a.10095. [DOI] [PubMed] [Google Scholar]
- 33.Lee-Hoeflich ST, Pham TQ, Dowbenko D, et al. PPM1H is a p27 phosphatase implicated in trastuzumab resistance. Cancer discovery. 2011;1(4):326–337. doi: 10.1158/2159-8290.CD-11-0062. [DOI] [PubMed] [Google Scholar]
- 34.Lu Y, Zi X, Pollak M. Molecular mechanisms underlying IGF-I-induced attenuation of the growth-inhibitory activity of trastuzumab (Herceptin) on SKBR3 breast cancer cells. Int J Cancer. 2004;108(3):334–341. doi: 10.1002/ijc.11445. [DOI] [PubMed] [Google Scholar]
- 35.Nahta R, Takahashi T, Ueno NT, Hung MC, Esteva FJ. P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004;64(11):3981–3986. doi: 10.1158/0008-5472.CAN-03-3900. [DOI] [PubMed] [Google Scholar]
- 36.Crawford A, Nahta R. Targeting Bcl-2 in Herceptin-Resistant Breast Cancer Cell Lines. Current pharmacogenomics and personalized medicine. 2011;9(3):184–190. doi: 10.2174/187569211796957584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ritter CA, Perez-Torres M, Rinehart C, et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13(16):4909–4919. doi: 10.1158/1078-0432.CCR-07-0701. [DOI] [PubMed] [Google Scholar]
- 38.Joshi JP, Brown NE, Griner SE, Nahta R. Growth differentiation factor 15 (GDF15)-mediated HER2 phosphorylation reduces trastuzumab sensitivity of HER2-overexpressing breast cancer cells. Biochem Pharmacol. 2011;82(9):1090–1099. doi: 10.1016/j.bcp.2011.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11(2):263–275. doi: 10.1586/era.10.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nahta R. Pharmacological Strategies to Overcome HER2 Cross-Talk and Trastuzumab Resistance. Curr Med Chem. 2012 doi: 10.2174/092986712799320691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nahta R. Deciphering the role of insulin-like growth factor-I receptor in trastuzumab resistance. Chemotherapy research and practice. 2012;2012:648965. doi: 10.1155/2012/648965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Critical reviews in oncogenesis. 2012;17(1):1–16. doi: 10.1615/critrevoncog.v17.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moy B, Goss PE. Lapatinib: current status and future directions in breast cancer. The oncologist. 2006;11(10):1047–1057. doi: 10.1634/theoncologist.11-10-1047. [DOI] [PubMed] [Google Scholar]
- 44.Johnston SR, Leary A. Lapatinib: a novel EGFR/HER2 tyrosine kinase inhibitor for cancer. Drugs of today. 2006;42(7):441–453. doi: 10.1358/dot.2006.42.7.985637. [DOI] [PubMed] [Google Scholar]
- 45.Lin NU, Dieras V, Paul D, et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res. 2009;15(4):1452–1459. doi: 10.1158/1078-0432.CCR-08-1080. [DOI] [PubMed] [Google Scholar]
- 46.Konecny GE, Pegram MD, Venkatesan N, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66(3):1630–1639. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- 47.Xia W, Gerard CM, Liu L, Baudson NM, Ory TL, Spector NL. Combining lapatinib (GW572016), a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases, with therapeutic anti-ErbB2 antibodies enhances apoptosis of ErbB2-overexpressing breast cancer cells. Oncogene. 2005;24(41):6213–6221. doi: 10.1038/sj.onc.1208774. [DOI] [PubMed] [Google Scholar]
- 48.Nahta R, Yuan LX, Du Y, Esteva FJ. Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol Cancer Ther. 2007;6(2):667–674. doi: 10.1158/1535-7163.MCT-06-0423. [DOI] [PubMed] [Google Scholar]
- 49.Scaltriti M, Verma C, Guzman M, et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene. 2009;28(6):803–814. doi: 10.1038/onc.2008.432. [DOI] [PubMed] [Google Scholar]
- 50.Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 51.Wierstra I. The transcription factor FOXM1 (Forkhead box M1): proliferation-specific expression, transcription factor function, target genes, mouse models, and normal biological roles. Advances in cancer research. 2013;118:97–398. doi: 10.1016/B978-0-12-407173-5.00004-2. [DOI] [PubMed] [Google Scholar]
- 52*.Bektas N, Haaf A, Veeck J, et al. Tight correlation between expression of the Forkhead transcription factor FOXM1 and HER2 in human breast cancer. BMC Cancer. 2008;8:42. doi: 10.1186/1471-2407-8-42. This study provides evidence of a correlation between HER2 and FOXM1 expression levels in breast cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park YY, Jung SY, Jennings NB, et al. FOXM1 mediates Dox resistance in breast cancer by enhancing DNA repair. Carcinogenesis. 2012;33(10):1843–1853. doi: 10.1093/carcin/bgs167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwok JM, Myatt SS, Marson CM, Coombes RC, Constantinidou D, Lam EW. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol Cancer Ther. 2008;7(7):2022–2032. doi: 10.1158/1535-7163.MCT-08-0188. [DOI] [PubMed] [Google Scholar]
- 55.Sanders DA, Ross-Innes CS, Beraldi D, Carroll JS, Balasubramanian S. Genome-wide mapping of FOXM1 binding reveals co-binding with estrogen receptor alpha in breast cancer cells. Genome biology. 2013;14(1):R6. doi: 10.1186/gb-2013-14-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Millour J, Constantinidou D, Stavropoulou AV, et al. FOXM1 is a transcriptional target of ERalpha and has a critical role in breast cancer endocrine sensitivity and resistance. Oncogene. 2010;29(20):2983–2995. doi: 10.1038/onc.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carr JR, Park HJ, Wang Z, Kiefer MM, Raychaudhuri P. FoxM1 mediates resistance to herceptin and paclitaxel. Cancer Res. 2010;70(12):5054–5063. doi: 10.1158/0008-5472.CAN-10-0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58**.Francis RE, Myatt SS, Krol J, et al. FoxM1 is a downstream target and marker of HER2 overexpression in breast cancer. Int J Oncol. 2009;35(1):57–68. doi: 10.3892/ijo_00000313. This study indicates that FOXM1 reduces sensitivity to trastuzumab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59**.Gayle SS, Castellino RC, Buss MC, Nahta R. MEK inhibition increases lapatinib sensitivity via modulation of FOXM1. Curr Med Chem. 2013;20(19):2486–2499. doi: 10.2174/0929867311320190008. This study indicates that reduced MEK signaling alters FOXM1 function and restores lapatinib sensitivity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baselga J, Gelmon KA, Verma S, et al. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol. 2010;28(7):1138–1144. doi: 10.1200/JCO.2009.24.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mcgovern UB, Francis RE, Peck B, et al. Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in breast cancer. Mol Cancer Ther. 2009;8(3):582–591. doi: 10.1158/1535-7163.MCT-08-0805. [DOI] [PubMed] [Google Scholar]
- 62.Zhao F, Lam EW. Role of the forkhead transcription factor FOXO-FOXM1 axis in cancer and drug resistance. Frontiers of medicine. 2012;6(4):376–380. doi: 10.1007/s11684-012-0228-0. [DOI] [PubMed] [Google Scholar]
- 63.Millour J, De Olano N, Horimoto Y, et al. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Molecular cancer therapeutics. 2011;10(6):1046–1058. doi: 10.1158/1535-7163.MCT-11-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Halasi M, Gartel AL. Suppression of FOXM1 sensitizes human cancer cells to cell death induced by DNA-damage. PloS one. 2012;7(2):e31761. doi: 10.1371/journal.pone.0031761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xue J, Lin X, Chiu WT, et al. Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-beta-dependent cancer metastasis. J Clin Invest. 2014;124(2):564–579. doi: 10.1172/JCI71104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang C, Chen H, Tan G, et al. FOXM1 promotes the epithelial to mesenchymal transition by stimulating the transcription of Slug in human breast cancer. Cancer Lett. 2013;340(1):104–112. doi: 10.1016/j.canlet.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 67.Raychaudhuri P, Park HJ. FoxM1: a master regulator of tumor metastasis. Cancer Res. 2011;71(13):4329–4333. doi: 10.1158/0008-5472.CAN-11-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koo CY, Muir KW, Lam EW. FOXM1: From cancer initiation to progression and treatment. Biochimica et biophysica acta. 2012;1819(1):28–37. doi: 10.1016/j.bbagrm.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 69.Liu GY, Liu XS, Wang SS, Chen CJ, Ji J. Biomimetic polymersomes as carriers for hydrophilic quantum dots. Langmuir : the ACS journal of surfaces and colloids. 2012;28(1):557–562. doi: 10.1021/la2033669. [DOI] [PubMed] [Google Scholar]
- 70.Ahmad A, Ali S, Ahmed A, et al. 3, 3′-Diindolylmethane enhances the effectiveness of herceptin against HER-2/neu-expressing breast cancer cells. PloS one. 2013;8(1):e54657. doi: 10.1371/journal.pone.0054657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ahmad A, Sakr WA, Rahman KM. Novel targets for detection of cancer and their modulation by chemopreventive natural compounds. Frontiers in bioscience. 2012;4:410–425. doi: 10.2741/e388. [DOI] [PubMed] [Google Scholar]
- 72*.Wang M, Gartel AL. The suppression of FOXM1 and its targets in breast cancer xenograft tumors by siRNA. Oncotarget. 2011;2(12):1218–1226. doi: 10.18632/oncotarget.359. This manuscript provides proof-of-concept that FOXM1 is an important molecular target in breast cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang C, Chen H, Yu L, et al. Inhibition of FOXM1 transcription factor suppresses cell proliferation and tumor growth of breast cancer. Cancer gene therapy. 2013;20(2):117–124. doi: 10.1038/cgt.2012.94. [DOI] [PubMed] [Google Scholar]
- 74.Wang M, Gartel AL. Micelle-encapsulated thiostrepton as an effective nanomedicine for inhibiting tumor growth and for suppressing FOXM1 in human xenografts. Mol Cancer Ther. 2011;10(12):2287–2297. doi: 10.1158/1535-7163.MCT-11-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang IC, Chen YJ, Hughes DE, et al. FoxM1 regulates transcription of JNK1 to promote the G1/S transition and tumor cell invasiveness. The Journal of biological chemistry. 2008;283(30):20770–20778. doi: 10.1074/jbc.M709892200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wierstra I. FOXM1 (Forkhead box M1) in tumorigenesis: overexpression in human cancer, implication in tumorigenesis, oncogenic functions, tumor-suppressive properties, and target of anticancer therapy. Advances in cancer research. 2013;119:191–419. doi: 10.1016/B978-0-12-407190-2.00016-2. [DOI] [PubMed] [Google Scholar]