

Abstract
IL12RB1 is a human gene that is important for resistance to Mycobacterium tuberculosis infection. IL12RB1 is expressed by multiple leukocyte lineages, and encodes a type I transmembrane protein (IL12Rβ1) that associates with IL12p40 and promotes the development of host-protective TH1cells. Recently, we observed that il12rb1—the mouse homolog of IL12RB1—is alternatively spliced by leukocytes to produce a second isoform (IL12Rβ1ΔTM) that has biological properties distinct from IL12Rβ1. Although the expression of IL12Rβ1ΔTM is elicited by M. tuberculosis in vivo, and its overexpression enhances IL12p40 responsiveness in vitro, the contribution of IL12Rβ1ΔTM to controlling M. tuberculosis infection has not been tested. Here, we demonstrate that IL12Rβ1ΔTM represents a secreted product of il12rb1 that, when absent from mice, compromises their ability to control M. tuberculosis infection in extrapulmonary organs. Furthermore, elevated M. tuberculosis burdens in IL12Rβ1ΔTM-deficient animals are associated with decreased lymph node cellularity and a decline in TH1 development. Collectively, these data support a model wherein IL12Rβ1ΔTM is a secreted product of il12rb1 that promotes resistance to M. tuberculosis infection by potentiating TH cells response to IL-12.
INTRODUCTION
Tuberculosis (TB) is a communicable disease that affects a large portion of the global population (1). TB is caused by aerogenic transmission of the intracellular pathogen Mycobacterium tuberculosis, which primarily infects macrophages in the lung alveoli (2). In its active form, TB is associated with “consumption” of the lung tissue and dissemination of M. tuberculosis to other organs; in its latent form, TB is asymptomatic and not infectious (3). Improved public health practices and the use of effective drug treatment have reduced exposure and disease rates in many countries. However, the efforts to control TB in many other countries are not optimal, leading to mortality and morbidity rates that fall short of World Health Organization's goal of reversing TB incidence by 2015 (4). Complicating these efforts is the increasing inability to control TB with short-course chemotherapy, given the emergence of multidrug-resistant and extensively drug-resistant M. tuberculosis strains (5). For these reasons, continuing research into understanding the host responses that limit M. tuberculosis activity in vivo remains important.
It is now well established that the genes IL12B and IL12RB1 are important for host restriction of M. tuberculosis activity (6, 7). IL12B encodes the IL12p40-subunit of the cytokines interleukin-12 (IL-12), IL-23, and IL-12(p40)2, each of which serves a protective role during experimental TB (8). IL12p40 is expressed in the pleura of actively infected individuals (9, 10), where it sustains CD4+ T cell production of IFN-γ (11). IL12RB1 is also transcribed by pleural cells of patients with active TB (12, 13), and encodes the protein IL12Rβ1, a type I transmembrane receptor that binds the IL12p40-subunit of IL-12, IL-23, and IL-12(p40)2 (14–16). Reflecting the importance of IL12RB1 expression, IL12RB1null individuals are susceptible to disseminated forms of disease caused by the M. tuberculosis complex (7, 17, 18), as well as nontuberculous mycobacteria (19–21). Given the importance of IL12RB1 and IL-12-family members to limiting M. tuberculosis activity, several promising, experimental vaccine strategies that specifically target these cytokine pathways are being developed (22, 23).
Recently, we observed that il12rb1—the mouse homolog of IL12RB1—produces a second isoform referred to as IL12Rβ1ΔTM, which is expressed after experimental M. tuberculosis infection and enhances IL12p40-dependent responses in vitro (24). IL12Rβ1ΔTM is a product of il12rb1 alternative splicing that was first discovered by Chua et al. upon cloning of the IL-12 receptor (25). IL12Rβ1ΔTM is similar to IL12Rβ1 in its retention of a signal peptide, cytokine-binding region, fibronectin domains (25, 26), and expression by both innate and lymphocyte lineages (26). IL12Rβ1ΔTM is dissimilar to IL12Rβ1, however, in that it lacks a transmembrane domain, has an alternate C-terminal amino acid sequence, and localizes to an intracellular reticulum that resembles the endoplasmic reticulum (ER) (26). IL12Rβ1ΔTM's characteristics make it similar to other cytokine receptor splice variants that pass through the ER prior to secretion (27); however, it has not yet been demonstrated that IL12Rβ1ΔTM is a secreted protein. IL12Rβ1ΔTM expression in the lungs coincides with a period of active M. tuberculosis replication and dissemination to lymphoid organs (24), suggesting that it may have a role in limiting M. tuberculosis infection of tissues peripheral to the lung. However, whether IL12Rβ1ΔTM limits M. tuberculosis activity in vivo or influences IL12p40-dependent responses in vivo has also not yet been determined.
Here, we demonstrate that IL12Rβ1ΔTM represents a secreted product of il12rb1 that promotes the control of experimental TB in extrapulmonary organs. Relative to wild-type controls, mice that are unable to produce IL12Rβ1ΔTM have elevated numbers of M. tuberculosis in the spleen, mediastinal lymph node, and liver. These differences associate with a decline in the potential of TH cells to produce gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α), two cytokines that are positively regulated by IL12p40 in vivo (28). Collectively, these data support a model wherein IL12Rβ1ΔTM production during TB protects the host by promoting TH1 cell differentiation and limiting M. tuberculosis growth.
MATERIALS AND METHODS
Plasmids.
The cloning of mouse IL12Rβ1 and IL12Rβ1ΔTM into pEF-BOS expression plasmids has been described (25); these plasmids are referred to as pIL12Rβ1 and pIL12Rβ1ΔTM, respectively, and were generously provided by Uli Gubler (Hoffmann-La Roche, Nutley, NJ). Empty vector control plasmid was prepared by XbaI digestion of pIL12Rβ1ΔTM, gel purification, and religation of the parent pEF-BOS. To generate a tagged version of IL12Rβ1ΔTM, the pIL12Rβ1ΔTM cDNA insert was modified to contain a 6×His encoding sequence (CATCATCACCATCACCAC) at its C terminus. This recombinant cDNA was then cloned into expression vector pcDNA3.1 (Life Technologies, Grand Island, NY) to generate pIL12Rβ1ΔTM-6×His. For cell transfections, preparations of pIL12Rβ1, pIL12Rβ1ΔTM, pIL12Rβ1ΔTM-6×His, and pcDNA3.1 (empty vector) were made using a Pureyield plasmid midiprep system (Promega, Madison, WI).
Transfections and cell culture.
The NIH 3T3 mouse fibroblast line (American Type Culture Collection, Manassas, VA) was used for all transfection and subcellular fractionation studies. NIH 3T3 cells were maintained in complete Dulbecco modified Eagle medium (cDMEM; 10% bovine calf serum) supplemented with l-glutamine, minimal essential medium amino acids, sodium pyruvate, and penicillin-streptomycin. Transient transfections of NIH 3T3 cells were done using the Lipofectamine 2000 method (Life Technologies). For the generation of concanavalin A (ConA) blasts, spleens were dispersed through a 70-μm-pore-size nylon cell strainer; cell preparations were then centrifuged over Ficoll-Paque to enrich mononuclear lineages. Splenocytes were then washed and resuspended at 20 × 106 cells/ml in cDMEM; 1 ml of splenocytes was then cultured with ConA (Sigma-Aldrich; final concentration, 5 μg/ml) for 3 days before protein was collected for IL12Rβ1ΔTM localization. For assaying splenocyte responsiveness to IL-12, spleen mononuclear cells were cultured in the presence of increasing concentrations of IL-12 according to the methods of Wu et al. (29); 24 h later, culture supernatants were clarified and used to measure IFN-γ levels (BD Biosciences OptEIA mouse IFN-γ ELISA set).
IL12Rβ1ΔTM localization.
The supernatants and cell contents of pIL12Rβ1, pIL12Rβ1ΔTM, pIL12Rβ1ΔTM-6×His, and empty-vector transfectants were collected 24 h posttransfection. Supernatants were spun down to remove any contaminating cellular debris (10,000 × g for 30 min, 4°C); cell contents were fractionated into membrane and cytosol components using the Qproteome cell compartment method (Qiagen, Germantown, MD). Supernatant and subcellular fractions were buffer exchanged into phosphate-buffered saline (PBS)-Tween and then used for either immunoprecipitation or Western blotting to assay for the presence of proteins recognized by polyclonal anti-mouse IL12Rβ1 (R&D Systems; note that the antigen used to generate anti-IL12Rβ1 is a peptide sequence that is present in both IL12Rβ1 and IL12Rβ1ΔTM). For immunoprecipitation of proteins recognized by polyclonal anti-IL12Rβ1, we used the protein G/Dynabead method (Life Technologies); for immunoprecipitation of 6×His-containing proteins, PBS-equilibrated fractions were used in the nickel-Dynabead method (Life Technologies). For Western blotting, eluted fractions were first reduced and then separated by SDS-PAGE using the Mini-Protean system (Bio-Rad, Hercules CA); proteins were then transferred to nitrocellulose using a semidry system (Trans-Blot SD cell) and probed with anti-IL12Rβ1 and appropriate secondary reagents.
Mice.
Mice were bred at the Medical College of Wisconsin (MCW) in the MCW Biomedical Resource Center and were treated according to National Institutes of Health and MCW Institute Animal Care and Use Committee (IACUC) guidelines. The C57BL/6 and FVB/N-Tg(EIIa-cre)C5379Lmgd/J (i.e., EIIa-cre mice) strains were purchased from the Jackson Laboratory (Bar Harbor, ME). Andrea M. Cooper (Trudeau Institute, Saranac Lake, NY) kindly provided the B6.129S1-Il12rb1tm1jm/J strain (il12rb1−/− mice) (29), as well as the “il12rb1 KOKI” strain (described below) that we used to generate IL12Rβ1ΔTM knockout mice.
Generation of IL12Rβ1ΔTM knockout mice.
Depending on the nature of the inflammatory stimulus, mouse il12rb1 may be expressed as either IL12Rβ1 or IL12Rβ1ΔTM (see Fig. 2A). IL12Rβ1 is generated via inclusion of il12rb1 exons 1 to 16 (Fig. 2B), whereas IL12Rβ1ΔTM is generated by inclusion of exons 1 to 13, 15, and 16 (exon 14 is skipped; Fig. 2C). Therefore, in order to generate IL12Rβ1ΔTM knockout (ΔTM−/−) mice, the splice acceptor sites necessary for exon 14 skipping were removed from the wild-type il12rb1 locus. Specifically, a targeting vector was generated (Fig. 2D) to introduce, after recombination at the short arm (SA) and long arm (LA) of homology, a target allele (Fig. 2E) that differs from wild-type il12rb1 in two important ways. First, the target allele contains a 4.2-kb lox-P-flanked (FR) region comprising positive selection markers (NeoR and PuroR), intron 13, exon and intron 14, exon and intron 15, and exon 16; immediately downstream of the exon 16 3′ untranslated region (UTR) is a transcriptional STOP cassette (introduced to prevent transcriptional read-through [30]). NeoR and PuroR are flanked by FRT and F3 sites, respectively, and were inserted into intron 13 and downstream of the transcriptional STOP cassette, respectively. Second, the target allele contains, downstream of the distal loxP site, the 3′ portion of il12rb1 intron 13 containing the splice acceptor site and the cDNA sequence corresponding to il12rb1 exons 14 to 16. according to this strategy, a conditional knockin (KI) allele (Fig. 2F) is generated after Flp-mediated removal of selection markers. These mice, referred to as “il12rb1 KOKI mice,” were generously provided to our lab by Andrea Cooper (Trudeau Institute). Upon transfer to MCW, il12rb1 KOKI mice were crossed with Ella-cre mice to generate F1 progeny that were heterozygous for the recombined, constitutive allele (Fig. 2G); CRE recombination allows for production of a constitutive KI allele (Fig. 2G) that effectively removes those splice acceptor sites necessary for exon 14 skipping and ensures the exclusive expression of IL12Rβ1. After intercrossing F1 mice, F2 progeny were genotyped by PCR (Fig. 2H) to identify which pups were homozygous for the constitutive, or ΔTMnull, allele (here referred to as ΔTM−/− mice). To confirm that IL12Rβ1ΔTM production was absent in ΔTM−/− mice, the levels of IL12Rβ1ΔTM protein in ConA blasts were compared between C57BL/6, ΔTM−/− mice, and il12rb1−/− mice (which are deficient in both il12rb1 isoforms [29]). The results of this analysis are shown in Fig. 2I, with pIL12Rβ1ΔTM-transfected NIH 3T3 cells serving as a positive control for IL12Rβ1ΔTM expression.
FIG 2.

IL12Rβ1ΔTM-deficient mice retain the ability to produce IL12Rβ1 but are unable to produce IL12Rβ1ΔTM. (A) Wild-type il12rb1 is located on mouse chromosome 8, comprises a 5′ UTR, 16 exons, and a 3′ UTR, and is neighbored by the genes mast3 and arrdc2. The first 13 exons of il12rb1 encode the extracellular domains of IL12Rβ1, while exon 14 encodes the transmembrane (TM) domain, and exons 15 and 16 encode intracellular signaling domains. (B and C) The pre-mRNA produced by il12rb1 transcription is normally spliced to produce two distinct isoforms, IL12Rβ1 and IL12Rβ1ΔTM. (B) IL12Rβ1 is produced by inclusion of all il12rb1 exons, including exons 13 to 16; this requires joining of the splice donor “D” and acceptor “A” sites depicted by the dashed lines. (C) IL12Rβ1ΔTM is produced by inclusion of every il12rb1 exon but exon 14. This requires joining of the intron 13 splice D and intron 14 A sites, as depicted by dashed lines. Upon joining the intron 13 “D” and intron 14 “A” sites, exon 14 is removed from the il12rb1 pre-mRNA. This effectively results in “skipping” of exon 14. To generate mice that are unable to produce IL12Rβ1ΔTM but able to produce IL12Rβ1, the wild-type il12rb1 allele (A) was replaced with a targeted allele that, following Cre-recombination, is nonpermissive to il12rb1 pre-mRNA exon 14 skipping (E). Specifically, recombination between il12rb1 and the short arm (SA) and long arm (LA) of a targeting vector was used to introduce a loxP-flanked region (FR) containing the targeted allele, as well as two resistance cassettes (NeoR and PuroR) (D). (F) After FLP recombination, a conditional knockin (KI) allele was generated that, similar to the wild-type allele, was permissive to pre-mRNA exon 14 skipping (i.e., contained the necessary donor and acceptor splice sites). Transcriptional readthrough past the floxed region was prevented via a transcriptional STOP cassette. The mice containing this conditional KI allele are referred to as “il12rb1 KOKI mice.” This conditional allele was removed (G) after crossing mice onto the EIIA-Cre background, leaving a constitutive KI (i.e., ΔTMnull) allele that lacked the introns necessary for exon 14 skipping, rendering mice IL12Rβ1ΔTM deficient. (H) PCR genotyping with the primers mus15_F and mus16_R was used to discriminate mice harboring either a wild-type il12rb1 allele or conditional KI allele and those homozygous for the ΔTMnull allele (i.e., ΔTM−/− mice). Shown is a representative gel image demonstrating the results of genotyping il12rb1 KOKI mice (lane 6), C57BL/6 mice (lane 5), three F2 progeny (lanes 2 to 4), and a “no gDNA control” (lane 1). Indicated in panels A, F, and G are the relative positions of primers mus15_F and mus16_R along the wild-type (A), conditional KI (F), and ΔTMnull (G) alleles. On the sides of the gel in panel H are indicated the expected amplicons from either the wild-type or conditional KI allele amplification (1,556 bp), as well as the ΔTMnull allele amplification (805 bp). (I) To confirm that ΔTM−/− mice are unable to produce IL12Rβ1ΔTM, total cell lysates from C57BL/6 (lane 1), ΔTM−/− (lane 2), and il12rb1−/− ConA blasts were probed via Western blot with anti-IL12Rβ1. Cell lysates from NIH 3T3 cells transfected with pIL12Rβ1ΔTM served as a positive control for IL12Rβ1ΔTM expression (lane 4). All protein lysates were probed with anti-GAPDH as a loading control (bottom panel).
PCR genotyping.
Genomic DNA (gDNA) was prepared from mouse tail snips using the Wizard SV genomic DNA purification method (Promega). To distinguish between mice carrying a wild-type or conditional KI allele versus those homozygous for the ΔTMnull allele (i.e., ΔTM−/− mice), gDNA was amplified with the primers mus15_F and mus16_R. The relative positions of mus15_F and mus16_R along each allele are indicated in Fig. 2A, F, and G; the sequence of the forward primer mus15_F (5′-CCCACCCCTGCCTACACCCTGT-3′) is specific to exon 15, while the sequence of the reverse primer mus16_R (5′-GGCAAACTGAGCCTGTGACTGA-3′) is specific to exon 16. The reaction conditions were the following: 2 μl of gDNA preparation were added to a 50-μl reaction mixture comprised of 75 mM Tris-HCl (pH 9.0), 20 mM (NH4)2SO4, 0.01% Tween 20, 2 mM MgCl2, 0.1 mg/ml bovine serum albumin, 0.2 mM deoxynucleoside triphosphates, 0.2 μM mus15_F, 0.2 μM mus16_R, and 1 U of Taq polymerase (New England BioLabs, Ipswich, MA). The following cycling parameters were performed on a MyCycler PCR machine (Bio-Rad, Hercules, CA): incubation at 95°C for 5 min (1 cycle); denaturation at 95°C for 30 s, annealing at 58°C for 30 s, and extension at 72°C for 90 s (35 cycles); and a final extension at 72°C for 10 min (1 cycle). Products were visualized on a 1% agarose gel using standard electrophoresis methods. Since mus15_F and mus16_R span intron 15, PCR amplification of either the wild-type il12rb1 allele (Fig. 2A) or the conditional KI allele (Fig. 2F) with these primers results in a 1,556-bp product, while amplification of the ΔTMnull allele (Fig. 2G) results in an 805-bp product. Shown in Fig. 2H are representative results of our amplifying gDNA from a il12rb1 KOKI founder mouse (Fig. 2H, lane 6), C57BL/6 mouse (Fig. 2H lane 5), and three F2 progeny (M1-M3, Fig. 2H, lanes 2 to 4). A “no gDNA” control was used in each screening to discriminate primer dimers (Fig. 2H, lane 1). From the genotyping shown, M3 was judged to be a ΔTM−/− mouse (Fig. 2H, lane 4) and was used for subsequent M. tuberculosis infection.
Experimental M. tuberculosis infection.
The H37Rv strain of M. tuberculosis (Trudeau Institute) was grown in Proskauer Beck medium containing 0.05% Tween 80 to mid-log phase and frozen in 1-ml aliquots at −70°C. For aerosol delivery of ∼80 bacteria, animals were placed in a Glas-Col inhalation exposure system (Glas-Col, Terre Haute, IN) at a maximum 20 mice per sector. After loading the nebulizer (Glas-Col) with 10 ml of diluted H37Rv (5 × 106 CFU/ml in deionized water), mice were aerogenically infected using the following exposure settings: 900-s preheat; 3,600-s nebulizing, 1,800-s cloud decay, and 900-s UV decontamination (vacuum pressure, 50 cubic feet per hour (CFH); comp air pressure, 15 CFH). Immediately after infection, mice were placed in microisolator cages and, throughout the postinfection period, the animals were monitored for outward signs of distress per IACUC oversight. Lungs from a group of control mice were plated at day 1 postinfection to confirm the delivery of ∼80 CFU.
Bacterial load determination.
Infected mice were euthanized by CO2 asphyxiation; the indicated organs were aseptically removed and individually homogenized in sterile normal saline using the Gentle Macs, program RNA2.1 (Miltenyi, Bergisch Gladbach, Germany). Serial dilutions of the organ homogenate were plated on nutrient 7H11 agar. The number of mycobacterial CFU was determined after incubating plates for 12 to 14 days at 37°C in 7% CO2.
Cell preparations.
Mediastinal lymph node (MLN) cell suspensions were prepared by first removing these organs from M. tuberculosis-infected animals and then pressing them through a nylon tissue strainer (70-μm pore size). The resulting cell suspension was treated with red blood cell (RBC) lysis solution, washed, counted, and prepared for intracellular cytokine staining and fluorescence-activated cell sorting (FACS) analysis.
Intracellular cytokine staining and FACS analysis.
All antibodies used for FACS analysis were purchased from BD Pharmingen (San Diego, CA). For intracellular cytokine staining, cells were collected, washed, and placed in a V-bottom, 96-well plate in complete medium with 50 ng/ml PMA and 1 μg/ml ionomycin. Cells were placed in a 37°C incubator for 4 h in the presence of brefeldin A (Sigma-Aldrich; 5 μg/ml, final concentration). After washing, cells were stained with allophycocyanin-conjugated anti-CD4 and fluorescein isothiocyanate-conjugated anti-CD8, fixed with 4% formaldehyde in PBS, permeabilized in 0.1% saponin (Sigma-Aldrich) in PBS with 2% fetal calf serum, and stained with either phycoerythrin (PE)-conjugated anti-IFN-γ or PE-conjugated anti-TNF-α. After all staining, cells were washed twice and acquired on a biosafety cabinet-contained Guava 8HT flow cytometer (Millipore). Acquired data were analyzed with FlowJo software (Tree Star, Inc., Ashland, OR).
Histological analysis.
The lungs, MLNs, and livers of M. tuberculosis-infected C57BL/6, ΔTM−/−, and il12rb1−/− mice were removed on day 40 postinfection and placed in 10% neutral buffered formalin. Subsequent sections from each genotype were stained simultaneously with hematoxylin and eosin (H&E) or with Ziehl-Neelsen (acid-fast stain) to allow comparison of the staining intensities. Once the slides were generated, images of both H&E and acid-fast stains were taken with a Labophot-2 upright microscope (Nikon, Tokyo, Japan) using a Retiga 2000R camera (QImaging, Surrey, British Columbia, Canada) and analyzed using NIS Elements software (Nikon).
Statistical analysis.
Figures were prepared using GraphPad Prism v5.0a. Statistical analyses used the bundled software. Bars in the figures show means ± the standard deviations (SD). Numbers shown between data points represent P values for the comparisons indicated. Statistical comparisons involving more than two experimental groups were made using analysis of variance (ANOVA). All other statistical comparisons were conducted using the Student t test.
RESULTS
IL12Rβ1ΔTM is a secreted product of il12rb1.
Since several cytokine receptor splice variants pass through the ER prior to being secreted (27), we hypothesized that IL12Rβ1ΔTM represents a secreted product of il12rb1. To test this hypothesis, we transfected NIH 3T3 mouse fibroblasts with the mammalian expression vector pEF-BOS containing cDNAs for either IL12Rβ1ΔTM (pIL12Rβ1ΔTM) or IL12Rβ1 (pIL12Rβ1) (25). After 24 h, the supernatants of transfected cells were collected, clarified, and incubated with bead-conjugated anti-mouse IL12Rβ1 (a polyclonal that recognizes residues that are present in both IL12Rβ1 and IL12Rβ1ΔTM); bead-bound proteins were then examined by Western blotting with the same antibody. In a manner identical to that done for NIH 3T3 transfectants, the supernatants of il12rb1+/+ ConA blasts (which express both IL12Rβ1 and IL12Rβ1ΔTM [25]) and il12rb1−/− ConA blasts (which lack expression of any il12rb1 isoform [29]) were also incubated with bead-conjugated anti-IL12Rβ1 so as to immunoprecipitate any secreted proteins recognized by this antibody. As shown in Fig. 1A, supernatants from il12rb1+/+ ConA blasts contained a protein recognized by anti-IL12Rβ1 that was not present in the supernatants of il12rb1−/− ConA blasts (Fig. 1A, lanes 1 and 2). The size of this protein was ∼80 kDa, which is close to the predicted size of IL12Rβ1ΔTM (74.5 kDa) based on the amino acid sequence alone (25). The supernatants of pIL12Rβ1ΔTM transfectants also contained a protein recognized by anti-IL12Rβ1 (Fig. 1A, lane 5) that was similar in size to that secreted by il12rb1+/+ ConA blasts and was not present in the supernatants of pIL12Rβ1 or empty-vector-transfected controls (Fig. 1A, lanes 3 and 4). We conclude from this that expression of IL12Rβ1ΔTM, but not IL12Rβ1, results in secretion of a protein recognized by anti-IL12Rβ1.
FIG 1.
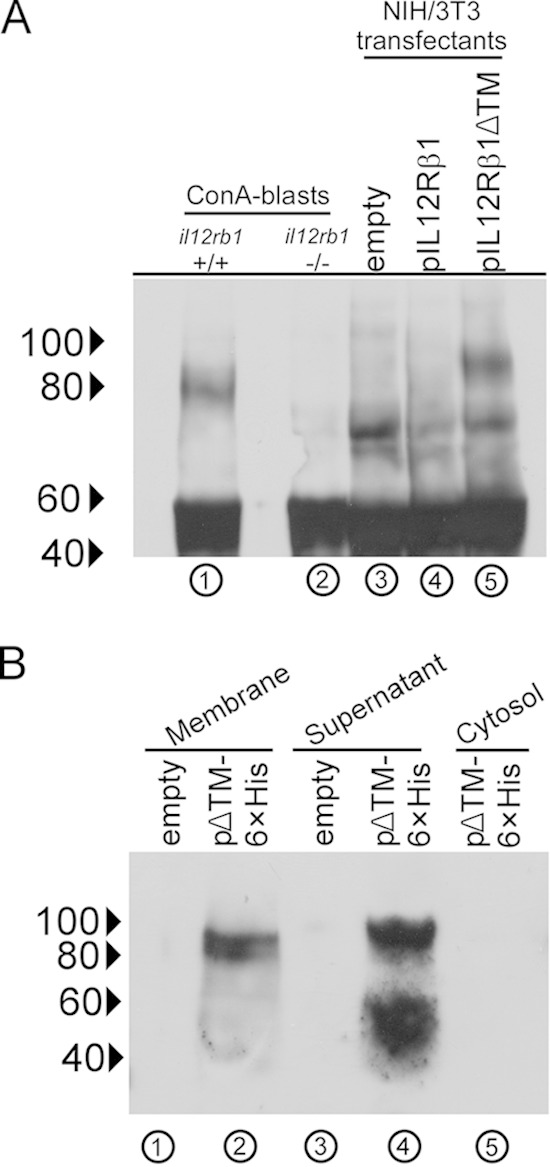
IL12Rβ1ΔTM is a secreted protein. (A) il12rb1+/+ and il12rb1−/− splenocytes were cultured in the presence of ConA for 3 days, after which supernatants were immunoprecipitated with anti-IL12Rβ1. In an identical manner, supernatants from empty-vector-, pIL12Rβ1-, and pIL12Rβ1ΔTM-transfected NIH 3T3 cells were also collected and immunoprecipitated. Eluted proteins were reduced, subjected to SDS-PAGE, blotted, and probed with anti-IL12Rβ1 after membrane transfer. Shown is an anti-IL12Rβ1 blot from each type of supernatant (indicated above the blot); lanes are also enumerated (below the blot) for clarity. (B) NIH 3T3 cells were transfected with either pIL12Rβ1ΔTM-6×His (abbreviated as pΔTM-6×His) or empty vector control. After 24 h, the supernatants of transfected cells were collected, along with the membrane and cytosol fractions of each group, and passed over nickel beads. Nickel-bound proteins were reduced and blotted with anti-IL12Rβ1 as performed for panel A. For panels A and B, the blots are representative of two separate transfection experiments; indicated along the side of each blot are the positions of 100, 80, 60, and 40-kDa markers.
As an additional means of testing whether IL12Rβ1ΔTM is secreted protein, we modified the cDNA present in pIL12Rβ1ΔTM to contain a C-terminal His tag (6×His). Tagged IL12Rβ1ΔTM was subsequently cloned into the pcDNA3.1 expression vector (pIL12Rβ1ΔTM-6×His) and transfected into NIH 3T3 cells as described above. In addition to collecting their supernatants, transfected cells were also fractionated into both cytosol and membrane-containing components (ER, Golgi body, and plasma membrane). Supernatants and cell fractions were then passed over nickel beads to bind tagged IL12Rβ1ΔTM; bound proteins were then examined by Western blotting for reactivity with anti-IL12Rβ1. Consistent with localization to the ER, the membrane fraction of pIL12Rβ1ΔTM-6×His transfectants contained a protein recognized by anti-IL12Rβ1 (Fig. 1B, lane 2). This protein was not present in the membrane fraction of cells transfected with empty vector (Fig. 1B, lane 1), nor was it observed in the cytosol fraction of pIL12Rβ1ΔTM-6×His transfectants (Fig. 1B, lane 5). The supernatant of pIL12Rβ1ΔTM-6×His transfectants also contained a protein recognized by anti-IL12Rβ1 (80- to 100-kDa range of Fig. 1B, lane 4) that, albeit slightly larger than its membrane fraction counterpart, was also not present in the supernatants of empty-vector transfectants (Fig. 1B, lane 3). Curiously, additional products were found in the supernatant of pIL12Rβ1ΔTM-6×His transfectants that, while smaller than the predicted size of IL12Rβ1ΔTM, were nevertheless recognized by anti-IL12Rβ1 and not present in the supernatants of empty-vector transfectants (40- to 60-kDa range of Fig. 1B, lane 4). Collectively, when combined with the results of Fig. 1A, these data demonstrate that IL12Rβ1ΔTM is a secreted product of il12rb1.
IL12Rβ1ΔTM-deficient mice retain the ability to express IL12Rβ1 but are unable to produce IL12Rβ1ΔTM.
IL12Rβ1ΔTM production is induced following M. tuberculosis infection (24). To test the significance of IL12Rβ1ΔTM production to in vivo M. tuberculosis infection, it was first necessary to generate mice that were unable to produce this isoform. However, since IL12Rβ1 and IL12Rβ1ΔTM mRNAs are derived from the same il12rb1 allele (Fig. 2A)—with IL12Rβ1 being produced via inclusion of all 16 il12rb1 pre-mRNA exons (Fig. 2B) and IL12Rβ1ΔTM being produced via deletion of exon 14 from the il12rb1 pre-mRNA (24) (Fig. 2C)—it was necessary to modify the wild-type il12rb1 allele in a way that knocked out the animals' ability to express IL12Rβ1ΔTM without affecting IL12Rβ1. To do this, we took advantage of the fact that for eukaryotic genes such as il12rb1, such deletions are caused by splicesome-mediated exon skipping (SMES) (31). Since SMES pertains to il12rb1 pre-mRNA splicing, rather than joining the intron 13 splicing donor site (at the 5′ end of intron 13) to the nearest splicing acceptor site (at the 3′ end of intron 13) (Fig. 2B), the splicesome instead joins the intron 13 donor site to the next closest splicing acceptor site at the 3′ end of intron 14 (Fig. 2C). Therefore, to disable animals' expression of IL12Rβ1ΔTM, a targeting vector (Fig. 2D) was introduced into mouse embryos to create a targeted allele (Fig. 2E), which upon FLP and CRE recombination (Fig. 2F and G), produced mice with a constitutive KI allele lacking introns 13 and 14 (i.e., the introns necessary for SMES of exon 14). Mice that were homozygous for this ΔTMnull allele (Fig. 2G) were discernible by PCR genotyping (Fig. 2H) and are here referred to as ΔTM−/− mice. Western analysis of spleen ConA blasts confirmed the absence of the IL12Rβ1ΔTM expression in ΔTM−/− mice (Fig. 2I); as is also true of il12rb1−/− mice (29), ΔTM−/− mice did not exhibit any growth defects, gross anatomical abnormalities, or breeding difficulties (data not shown). Collectively, these data support a model wherein IL12Rβ1ΔTM is produced by SMES and wherein ΔTM−/− mice are viable and retain the ability to express IL12Rβ1 but are unable to produce IL12Rβ1ΔTM.
ΔTM−/− mice are compromised in their ability to control M. tuberculosis in extrapulmonary organs.
Given the host-protective roles that IL-12 family members have during experimental TB (8), we hypothesized that IL12Rβ1ΔTM production is also host-protective after M. tuberculosis infection. To test this hypothesis, ΔTM−/− mice were aerogenically infected with a low dose (∼80 CFU) of virulent M. tuberculosis strain H37Rv. At select times postinfection, the M. tuberculosis burdens were determined for the lungs and extrapulmonary organs (i.e., MLNs, liver and spleen) and compared to those of C57BL/6 and il12rb1−/− controls. Consistent with previous observations (32), C57BL/6 mice were able to control M. tuberculosis growth in the lung by 40 days postinfection, while il12rb1−/− mice were unable to do the same (Fig. 3A). In addition to the lung, the importance of il12rb1 to controlling M. tuberculosis growth also extends to extrapulmonary organs, as evidenced by higher M. tuberculosis CFU numbers in the il12rb1−/− spleen (Fig. 3B), MLN (Fig. 3C), and liver (Fig. 3D) by 40 days postinfection. There is also an abundance of acid-fast bacilli in the il12rb1−/− MLN (Fig. 3E) and il12rb1−/− liver (Fig. 3F) at day 40 postinfection. Like C57BL/6 controls, ΔTM−/− mice were also able to contain M. tuberculosis growth in the lung (Fig. 3A); while at day 60 postinfection a modest increase in ΔTM−/− lung M. tuberculosis burdens was observed, this difference was not statistically different from C57BL/6 controls (P = 0.08). However, relative to C57BL/6 controls, ΔTM−/− mice had consistently higher M. tuberculosis CFU in extrapulmonary organs beginning at day 40 postinfection (Fig. 3B to D). Consistent with elevated M. tuberculosis CFU, ΔTM−/− mice also had noticeably more acid-fast bacilli in their MLNs (Fig. 3E) and livers (Fig. 3F) relative to C57BL/6 controls (compare the top and middle rows of Fig. 3E and F). Importantly, the M. tuberculosis burdens in ΔTM−/− mice were below those observed in il12rb1−/− mice, whether assessed by CFU burden (Fig. 3A to D) or acid-fast staining (compare the middle and bottom rows of Fig. 3E and F). Collectively, these data demonstrate that ΔTM−/− mice are compromised in their ability to control M. tuberculosis infection in extrapulmonary organs, albeit not to the same degree as il12rb1−/− mice.
FIG 3.
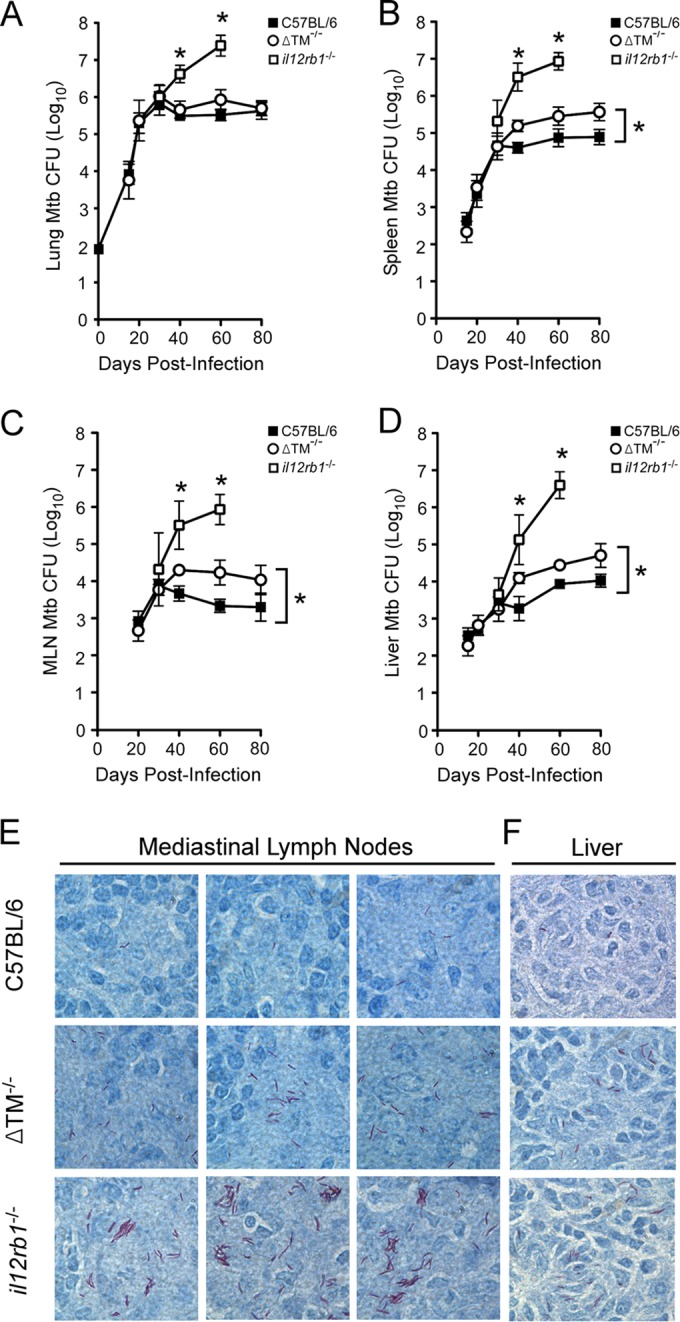
IL12Rβ1ΔTM-deficient mice are compromised in their ability to control M. tuberculosis in extrapulmonary organs. ΔTM−/− mice, along with C57BL/6 and il12rb1−/− controls, were aerogenically infected with ∼80 CFU of M. tuberculosis strain H37Rv. On days 15, 20, 30, and 40 postinfection, the lungs and spleens from the indicated groups were resected and plated on 7H11 agar. Shown for each group are the mean M. tuberculosis CFU counts (± the standard deviations) present in the lung (A), spleen (B), MLN (C), and liver (D) at each time point (three to four mice per group per time point). Significant differences between ΔTM−/− mice and either C57BL/6 or il12rb1−/− controls are indicated by asterisks (P ≤ 0.05, Student t test). The data are representative of two separate experiments. (E and F) From the same groups, Ziehl-Neelsen staining was used to determine the extent to which acid-fast bacilli were present in the MLNs (E) and liver (F) of each mouse. Shown are representative micrographs of each tissue (three MLN micrographs, one liver micrograph) from C57BL/6 (top row), ΔTM−/− (middle row), and il12rb1−/− mice (bottom row) on day 40 postinfection.
ΔTM−/− TH cells have a diminished capacity to produce IFN-γ and TNF-α.
M. tuberculosis-driven TH1 development is positively regulated by IL12p40 in vivo (28) and is an important determinant of host control (33). To determine whether IL12Rβ1ΔTM's ability to potentiate IL12p40-responsiveness extends in vivo, we compared the ability of ΔTM−/− mice to generate a TH1 response following M. tuberculosis infection to that of C57BL/6 and il12rb1−/− controls. Specifically, groups of C57BL/6, ΔTM−/−, and il12rb1−/− mice were infected with M. tuberculosis. On days 15 and 30 postinfection, the MLNs were resected, and the frequency and number of IFN-γ+ CD4+ and TNF-α+ CD4+ cells were assessed by intracellular cytokine staining.
Upon visual examination of the MLNs, we noted that ΔTM−/− MLNs were smaller than those of C57BL/6 mice, with il12rb1−/− MLNs being the smallest of all genotypes (insets, Fig. 4A to C). The smaller sizes of ΔTM−/− and il12rb1−/− MLNs (relative to C57BL/6 MLNs) associated with decreased cellularity, as observed by less-intense hematoxylin staining (Fig. 4A to C) and fewer total cell numbers (Fig. 4D). When we compared the ability of each genotype to generate TH1 cells, we noted that the frequency of CD4+ IFN-γ+ cells in ΔTM−/− MLNs was reduced compared to C57BL/6 MLNs on days 15 and 30 postinfection (Fig. 5A, B, and D). A similar decline in the frequency of CD4+ TNF-α+ cells was also observed in ΔTM−/− MLNs (Fig. 5E, F, and H). Notably, the frequency of CD4+ IFN-γ+ cells in ΔTM−/− MLNs, albeit lower than C57BL/6 controls, was higher than that observed in il12rb1−/− MLNs (Fig. 5B, C, and D). This intermediate pattern was also true of CD4+ TNF-α+ cells (Fig. 5F, G, and H). CD8+ production of IFN-γ and TNF-α was unaffected by the absence of IL12Rβ1ΔTM (data not shown). Consistent with a decline in TH1 development, granulomatous regions in M. tuberculosis-infected ΔTM−/− lungs (day 40 postinfection) appeared to be less lymphocytic than C57BL/6 controls at the same time (Fig. 5I and J). However, the relative ratio of lymphocytes to histiocytes in ΔTM−/− lungs appeared to be similar to that of C57BL/6 controls. This was not the case in the granulomatous regions of il12rb1−/− lungs (Fig. 5K), which relative to C57BL/6 controls (Fig. 5I) appeared to be less lymphocytic and have a lower lymphocyte/histiocyte ratio. Collectively, the data demonstrate that ΔTM−/− TH cells have a diminished capacity to produce IFN-γ and TNF-α, albeit not as extensive as that of il12rb1−/− TH cells.
FIG 4.
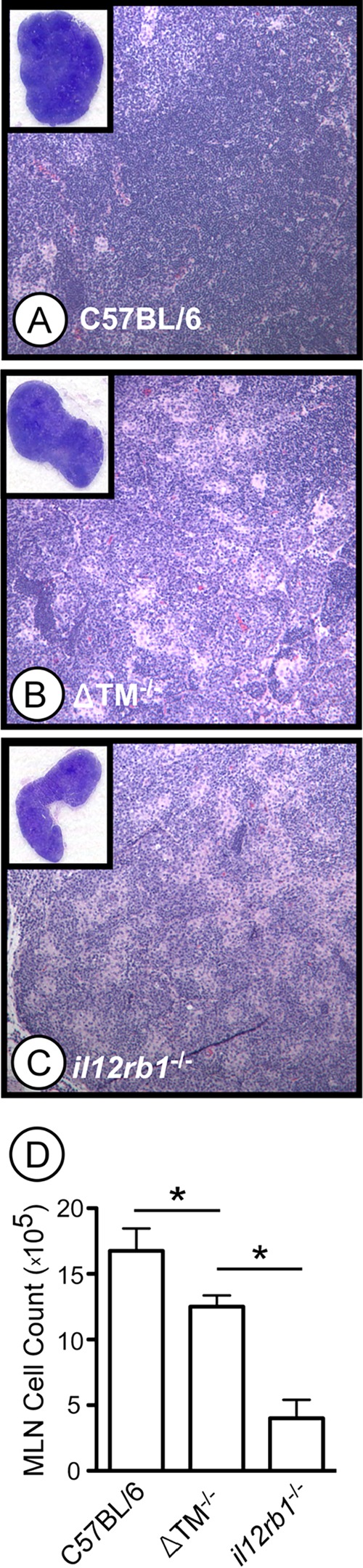
M. tuberculosis-infected ΔTM−/− mice have decreased MLN cellularity. MLNs from M. tuberculosis-infected C57BL/6 (A), ΔTM−/− (B), and il12rb1−/− (C) mice were collected on day 40 postinfection, fixed, and stained with H&E. Shown in the insets are magnified sections (×4) of the MLNs from mice of each genotype, along with ×100 magnifications of the lymph node cortex. (D) MLNs from the same genotypes were collected on day 30 postinfection and physically disrupted into to generate single cell suspensions. After RBC lysis, the cell preparations were counted, and the results were compared between each genotype. Bars represent the mean MLN cell count of three to four mice per genotype (± the standard deviations). Significant differences between counts of each genotype are indicated by asterisks (P ≤ 0.05, Student t test). The data are representative of two separate experiments.
FIG 5.

ΔTM−/− TH cells have a diminished capacity to produce IFN-γ and TNF-α. MLNs from M. tuberculosis-infected C57BL/6 (A and E), ΔTM−/− (B and F), and il12rb1−/− (C and G) mice were collected on days 15 and 30 postinfection. After RBC lysis, cell preparations were stimulated in vitro with PMA-ionomycin, and stained for either CD4 and intracellular IFN-γ (A to C) or CD4 and intracellular TNF-α (E to G). Panels A to C and E to G show dot plots representing the FACS data collected from each genotype on day 30 postinfection. The number in each inset is the percentage of CD4+ cells that were positive for the indicated cytokine. For both CD4 and intracellular cytokine staining, isotype controls were used to establish positive gates (data not shown). Panels D and H show the combined percent CD4+ IFN-γ+ (D) and percent CD4+ TNF-α+ (H) data for each genotype (three to four mice per genotype) on days 15 and 30 postinfection. The data points represent the means ± the standard deviations for each genotype at each time point. Significant differences between ΔTM−/− mice and either C57BL/6 or il12rb1−/− controls are indicated by asterisks (P ≤ 0.05, Student t test). (I to K) Micrographs of H&E-stained sections from M. tuberculosis-infected lungs of C57BL/6 (I), ΔTM−/− (J), and il12rb1−/− (K) mice. The lungs were taken at day 40 postinfection. The images shown are representative of sections of four mice per group. All data are representative of two separate experiments.
IL12Rβ1ΔTM potentiates IL-12-responsiveness in an environment where IL-12 is limiting.
The lower numbers of CD4+ IFN-γ+ cells in the lungs of M. tuberculosis-infected ΔTM−/− mice suggest that lymphocytes in these animals are less sensitive to IL-12. To test the hypothesis that IL12Rβ1ΔTM is a positive regulator of IL-12 responsiveness, we cultured spleen mononuclear preparations from C57BL/6, ΔTM−/−, and il12rb1−/− mice in the presence of ConA and increasing concentrations of IL-12. After 24 h, the IFN-γ-levels produced by each genotype at each IL-12-concentration were measured and compared. The results of this experiment are shown in Fig. 6. Consistent with the results of Wu et al. (29), C57BL/6 splenocytes responded to IL-12 by producing IFN-γ in an IL-12 concentration-dependent manner, while IFN-γ production by il12rb1−/− splenocytes was significantly lower at all IL-12-concentrations. Relative to the C57BL/6 controls, ΔTM−/− splenocytes produced significantly less IFN-γ at lower levels of IL-12 (0.005 to 0.05 ng/ml). At higher levels of IL-12 (0.5 to 50 ng/ml), no differences in IL-12 responsiveness existed between C57BL/6 and ΔTM−/− splenocytes. At all concentrations of IL-12 (0.005 to 50 ng/ml), ΔTM−/− splenocytes produced significantly more IFN-γ than il12rb1−/− controls. Collectively, these data demonstrate that IL12Rβ1ΔTM potentiates IL-12-elicited IFN-γ in an environment where the IL-12 concentration is limiting.
FIG 6.
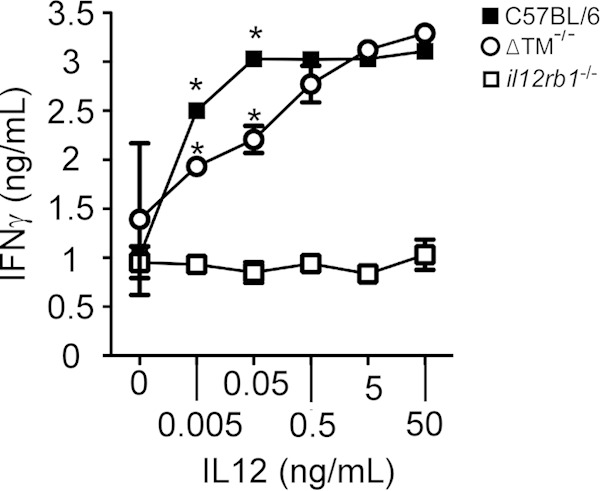
IL12Rβ1ΔTM potentiates IL-12 responsiveness in an environment where IL-12 is limiting. Mononuclear cells were purified from the spleens of C57BL/6, ΔTM−/−, and il12rb1−/− mice, normalized for cell number, and cultured in the presence of ConA (1 μg/ml) and increasing concentrations of IL-12 (i.e., 0, 0.005, 0.05, 0.5, 5, and 50 ng/ml). At 24 h later, the supernatants of each culture condition were clarified and used to measure IFN-γ levels by enzyme-linked immunosorbent assay. Shown for each IL-12 concentration are the means ± the standard deviations for the IFN-γ levels produced by C57BL/6 splenocytes (●), ΔTM−/− splenocytes (○), and il12rb1−/− splenocytes (■). Significant differences between ΔTM−/− mice and either C57BL/6 or il12rb1−/− controls are indicated by an asterisk (P ≤ 0.05, Student t test). The data are representative of four separate experiments.
DISCUSSION
Alternative splicing is a posttranscriptional process that can produce, from a single cytokine receptor gene, multiple mRNAs that translate into cytokine receptors with distinct, isoform-specific functions (27, 34). These alternate receptor isoforms can suppress or potentiate the activity of their cognate cytokine. Suppressive isoforms include splice variants of the IL-1 receptor accessory protein (IL-1RAcP) (35), IL-1 receptor type II (IL-1RII) (36), TNF receptor 2 (TNFR2) (37), IL-4 receptor α (IL4Rα) (38), leukemia inhibitory factor receptor α (LIFRα) (39), erythropoietin receptor (EPOR) (40), glycoprotein 130 (gp130) (41), IL-5 receptor α (IL5Rα) (42), oncostatin M receptor (OSMR) (43), and vascular endothelial growth-factor receptor 1 (VEGFR1) (44) and receptor 2 (VEGFR2) (45). Potentiating isoforms are fewer, and include splice variants of the IL-6 receptor α (IL6Rα) (46), IL-7 receptor α (IL7Rα) (47), IL-15 receptor α (IL15Rα) (48), and transforming growth factor β receptor 1 (TβR-I) (49). Rather than potentiating its cytokine, a splice variant of the interferon α receptor 2 (IFNAR2) can actually substitute for type I IFN in in vitro assays (50).
Given the number of cytokine receptor splice variants, it is no surprise that changing the levels of several receptor variants can have significant in vivo effects. The IL6Rα splice variant enhances IL-6's activities in the mouse central nervous system, leading to changes in body temperature and locomotor activity (51), while IL6Rα/IL-6 double transgenic mice display increases in hepatic hematopoiesis and hepatocellular hyperplasia (52, 53). Spliced IL7Rα exacerbates experimental autoimmune encephalomyelitis (47), while signal-mediated changes in VEGFR1 splicing alters vasculogenesis in tissue culture models systems (54). Increased serum concentrations of select cytokine receptor splice variants can also be indicative of an underlying pathology. For example, increased serum levels of spliced TNFR2 are found in both septic individuals and in rheumatoid arthritis patients (37). The levels of spliced EPOR in the serum of dialysis patients correlates with the amount of therapeutic recombinant erythropoietin required by the same individuals (40). Bronchoalveolar lavage levels of spliced IL6Rα are indicative of chronic lung disease in premature infants (55), while increased serum levels of spliced IL5Rα are indicative of systemic abundance of mast cells (56). Pertaining to cancer, multiple myeloma patients have increased serum levels of spliced OSMR relative to healthy individuals (43), while an increased level of IL15Rα in head and neck cancer patients is predictive of a poor clinical outcome (48). These studies demonstrate that, in addition to the levels of cytokines themselves, the relative levels of potentiating or suppressive receptor isoforms can also influence cytokine-driven phenomena.
Here we have shown that IL12Rβ1ΔTM, an alternative splice variant of the il12rb1 gene, is a secreted protein that is host-protective in the context of experimental TB—a model of human TB whose outcome is dependent on the expression of il12b and il12rb1 gene products (32, 57). IL12Rβ1ΔTM was first observed by Chua et al. (25) nearly 20 years ago upon cloning and characterization of the mouse IL-12 receptor (therein referred to as “clone 3”), as well as by Showe et al. (58) in the spleens of Mycobacterium bovis BCG/lipopolysaccharide-treated animals (referred to as “β1b” in that study). Our own interest in IL12Rβ1ΔTM stems from observing its expression in the lungs of M. tuberculosis-infected animals and that overexpression of IL12Rβ1ΔTM enhances IL12p40-dependent responses in vitro (24). That IL12Rβ1ΔTM-deficient mice have decreased numbers of M. tuberculosis-elicited TH1 cells, as well as increased M. tuberculosis burdens in peripheral organs, demonstrates that IL12Rβ1ΔTM's enhancement of IL12p40-dependent response extends in vivo. Our results also suggest that IL12Rβ1's ability to inhibit M. tuberculosis growth in extrapulmonary organs and promote TH1 development is greater than that of IL12Rβ1ΔTM, since mice that are deficient in both IL12Rβ1 and IL12Rβ1ΔTM (i.e., il12rb1−/− mice) exhibit higher bacterial burdens, and greater differences in TH1 numbers, than mice deficient in IL12Rβ1ΔTM alone (i.e., ΔTM−/− mice). The balance between IL12Rβ1 versus IL12Rβ1ΔTM is especially important in the lung, since il12rb1−/− mice displayed significantly higher M. tuberculosis burdens than ΔTM−/− mice.
Compared to wild-type controls, aerosol-infected ΔTM−/− mice develop higher M. tuberculosis burdens in organs peripheral to the lung, but not in the lung itself. The specificity of this phenotype to peripheral organs is reminiscent of mice deficient in other pathways that modulate delayed-type hypersensitivity (DTH), including p19-dependent pathways and tryptophan starvation (59, 60). p19 encodes a subunit of IL-23, a cytokine that compensates for the absence of IL-12 during experimental TB; aerosol-infected p19−/− mice have consistently higher M. tuberculosis burdens in the spleen after 120 days postinfection, with no differences observed in the lung until later chronic disease stages (60, 61). TH cells use tryptophan starvation in synergy with IFN-γ to limit M. tuberculosis (59); the significance of tryptophan starvation is also more apparent in the periphery, since treating aerosol-infected mice with an inhibitor of tryptophan biosynthesis (6-FABA) has a greater impact on M. tuberculosis numbers in the spleen than in the lungs (59). Why the significance of these mechanisms—whether they are driven by IL12Rβ1ΔTM, IL-23 or tryptophan starvation—becomes apparent only in the periphery (and not the lungs) is likely a reflection of the different manners by which M. tuberculosis arrives to the lung and periphery of aerosol-infected animals. For our studies of ΔTM−/− mice, and for others' studies of p19−/− or 6-FABA-treated mice (59, 60), ≤102 M. tuberculosis CFU arrive in the lung in aerosolized droplets of media (62); upon deposition in the alveoli, M. tuberculosis's thick cell wall is its only protection against the innate immune defenses that directly inhibit or kill M. tuberculosis, including surfactant protein D (63), antimicrobial peptides (AMPs) (64), and phagolysosomal fusion (65). M. tuberculosis adapts to this environment by inhibiting phagolysosome fusion (65), residing in phagocytes away from extracellular AMPs and surfactant proteins. Approximately 9 days postinfection, the lung burden has increased to ≥104 M. tuberculosis CFU, and a “spillover” occurs wherein M. tuberculosis disseminates from the lungs to the draining mediastinal lymph nodes (MLNs) and spleen (66). Since mouse lungs do not display overt signs of epithelial or vascular damage at this time (67), the appearance of M. tuberculosis in the MLNs and spleen is likely due to M. tuberculosis dissemination via an innate cell carrier. Supporting this model, Wolf et al. (68) demonstrated that after infecting animals with green fluorescent protein (GFP)-expressing M. tuberculosis, multiple innate lineages carry GFP-M. tuberculosis to the MLN; the majority of these GFP+ emigrants are CD11chi CD11bhi myeloid DCs. As M. tuberculosis arrives in the periphery using DCs as its vehicle (as opposed to media), effective DTH becomes all the more important for host protection since M. tuberculosis is now shielded from innate defenses in the extracellular environment (e.g., AMPs). Therefore, a tempered DTH response—whether due to a deficiency in IL12Rβ1ΔTM, IL-23, or tryptophan starvation—is manifested greater in these organs. Perhaps for this same reason, the functional inhibition of M. tuberculosis by mouse splenocytes is a useful surrogate for BCG-vaccine-mediated protection in mice (69).
As recently and extensively reviewed by van de Vosse et al. (70), multiple IL12RB1 polymorphisms confer susceptibility to mycobacterial infection. The importance of these IL12RB1 polymorphisms on IL-12 signaling and TH1 development has historically been interpreted in the context of human IL12Rβ1, which, relative to IL12Rβ1ΔTM, is better studied since its cloning by Chua et al. (71). However, given our observation that IL12Rβ1ΔTM is host protective in experimental TB, albeit less so than IL12Rβ1 itself, polymorphisms specifically affecting human IL12RB1 splicing should also be considered in the future when a relationship between IL12RB1 expression and mycobacterial susceptibility is suspected. Efforts are under way to identify the cis-elements in the human genome that direct preferential splicing of the various human IL12RB1 isoforms. Sequence variation in these elements may underlie why, between healthy individuals with otherwise identical IL12RB1 exon sequences, such wide variation in IL12RB1 splicing exists (26).
Finally, the mechanism by which IL12Rβ1ΔTM promotes IL12p40-dependent responses is likely distinct from that of IL12Rβ1, a type I transmembrane protein that associates with the IL12p40 domain of IL-12, and signals in concert with IL12Rβ2 (which binds the IL12p35 domain of IL-12). The extracellular portion of IL12Rβ1 contains the cytokine binding region necessary for physical association with IL-12, while the cytoplasmic portion acts in concert with IL12Rβ2 to transmit intracellular signals via preassociated TYK2 (72). IL12Rβ1ΔTM differs from IL12Rβ1 in its lacking a transmembrane-domain and having a distinct C terminus. These characteristics of IL12Rβ1ΔTM, when placed in the context of the classical IL-12 signaling pathway and how other potentiating cytokine receptor splice variants function (47, 73), have led us to a model wherein, after its secretion, IL12Rβ1ΔTM enhances IL-12's stability and/or bioavailability, prolonging its interaction with IL12Rβ1 and IL12Rβ2. IL12Rβ1ΔTM's potentiation of IL-12 responsiveness depends on the concentration of IL-12 in the extracellular environment: IL12Rβ1ΔTM's enhancement of IL-12 responsiveness is apparent in an environment where the concentration of IL-12 is limited but absent in an environment where the concentration of IL-12 is high (Fig. 6). These data are consistent with a model wherein IL12Rβ1ΔTM stabilizes the IL-12–IL12Rβ1's interaction when IL-12's concentration is nonsaturating. Although biochemical experiments are being initiated to test this model, additional data supporting this model include the fact that IL12Rβ1ΔTM's unique C terminus does not have any obvious signaling domains and that its enhancement of DC IL-12(p40)2 responsiveness is also IL12Rβ1 dependent (24). Given that IL12Rβ1ΔTM exists in both membrane-associated and secreted forms (Fig. 1), it is possible that it can function both in cis (i.e., on the cell expressing IL12Rβ1ΔTM) and in trans (i.e., on other cells) to potentiate of IL-12 responsiveness. Preliminary experiments testing this possibility have thus far been inconclusive (data not shown); however, should IL12Rβ1ΔTM function in trans, it raises the exciting possibility that recombinant IL12Rβ1ΔTM could be used to boost both IL-12 responsiveness as part of a vaccine strategy. Although IL12Rβ1ΔTM is expressed by both innate and adaptive lineages (24, 26), our recent demonstration that il12rb1 gene products must be expressed by rag1-dependent lineages for TB control to occur (32) supports a model wherein T cell expression of IL12Rβ1ΔTM, and not innate expression, is primarily responsible for IL12Rβ1ΔTM's protective effects in extrapulmonary sites. Therefore, it will be important to understand IL12Rβ1ΔTM's mechanism in the context of TH1 cell biology and how the balance between IL12Rβ1 and IL12Rβ1ΔTM fine-tunes the TH1 response, since this lineage is required for TB control (33).
ACKNOWLEDGMENTS
We thank Nicole Ford for developing our PCR genotyping protocol and Matthew Rodgers (MCW Medical Scientist Training Program) for assistance with IL12Rβ1ΔTM immunoprecipitation and Western analysis.
This study was supported by the Medical College of Wisconsin, the MCW Center for Infectious Disease Research, Advancing Healthier Wisconsin grant 5520189, a BD Biosciences Immunology Grant, and the National Institutes of Health (R01AI067723 to A.M.C. and R21AI099661 to R.T.R.).
REFERENCES
- 1.Lawn SD, Zumla AI. 2011. Tuberculosis. Lancet 378:57–72. doi: 10.1016/S0140-6736(10)62173-3. [DOI] [PubMed] [Google Scholar]
- 2.Russell DG. 2011. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol Rev 240:252–268. doi: 10.1111/j.1600-065X.2010.00984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ernst JD. 2012. The immunological life cycle of tuberculosis. Nat Rev Immunol 12:581–591. doi: 10.1038/nri3259. [DOI] [PubMed] [Google Scholar]
- 4.Raviglione M, Marais B, Floyd K, Lonnroth K, Getahun H, Migliori GB, Harries AD, Nunn P, Lienhardt C, Graham S, Chakaya J, Weyer K, Cole S, Kaufmann SH, Zumla A. 2012. Scaling up interventions to achieve global tuberculosis control: progress and new developments. Lancet 379:1902–1913. doi: 10.1016/S0140-6736(12)60727-2. [DOI] [PubMed] [Google Scholar]
- 5.Maartens G, Wilkinson RJ. 2007. Tuberculosis. Lancet 370:2030–2043. doi: 10.1016/S0140-6736(07)61262-8. [DOI] [PubMed] [Google Scholar]
- 6.Prando C, Samarina A, Bustamante J, Boisson-Dupuis S, Cobat A, Picard C, Al Sum Z, Al-Jumaah S, Al-Hajjar S, Frayha H, Alangari A, Al-Mousa H, Mobaireek KF, Ben-Mustapha I, Adimi P, Feinberg J, de Suremain M, Janniere L, Filipe-Santos O, Mansouri N, Stephan JL, Nallusamy R, Kumararatne DS, Bloorsaz MR, Ben-Ali M, Elloumi-Zghal H, Chemli J, Bouguila J, Bejaoui M, Alaki E, AlFawaz TS, Al Idrissi E, El Ghazali G, Pollard AJ, Murugasu B, Wah Lee B, Halwani R, Al-Zahrani M, Al Shehri MA, Bin-Hussain I, Mahdaviani SA, Parvaneh N, Abel L, Mansouri D, Barbouche R, Al-Muhsen S, Casanova JL. 2013. Inherited IL-12p40 deficiency: genetic, immunologic, and clinical features of 49 patients from 30 kindreds. Medicine 92:109–122. doi: 10.1097/MD.0b013e31828a01f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, Al-Muhsen S, Janniere L, Rose Y, de Suremain M, Kong XF, Filipe-Santos O, Chapgier A, Picard C, Fischer A, Dogu F, Ikinciogullari A, Tanir G, Al-Hajjar S, Al-Jumaah S, Frayha HH, Al Sum Z, Al-Ajaji S, Alangari A, Al-Ghonaium A, Adimi P, Mansouri D, Ben-Mustapha I, Yancoski J, Garty BZ, Rodriguez-Gallego C, Caragol I, Kutukculer N, Kumararatne DS, et al. . 2010. Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Medicine 89:381–402. doi: 10.1097/MD.0b013e3181fdd832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper AM, Khader SA. 2008. The role of cytokines in the initiation, expansion, and control of cellular immunity to tuberculosis. Immunol Rev 226:191–204. doi: 10.1111/j.1600-065X.2008.00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okamoto M, Kawabe T, Iwasaki Y, Hara T, Hashimoto N, Imaizumi K, Hasegawa Y, Shimokata K. 2005. Evaluation of interferon-gamma, interferon-gamma-inducing cytokines, and interferon-gamma-inducible chemokines in tuberculous pleural effusions. J Lab Clin Med 145:88–93. doi: 10.1016/j.lab.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 10.Zhang M, Gately MK, Wang E, Gong J, Wolf SF, Lu S, Modlin RL, Barnes PF. 1994. Interleukin 12 at the site of disease in tuberculosis. J Clin Invest 93:1733–1739. doi: 10.1172/JCI117157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng CG, Jankovic D, Kullberg M, Cheever A, Scanga CA, Hieny S, Caspar P, Yap GS, Sher A. 2005. Maintenance of pulmonary Th1 effector function in chronic tuberculosis requires persistent IL-12 production. J Immunol 174:4185–4192. doi: 10.4049/jimmunol.174.7.4185. [DOI] [PubMed] [Google Scholar]
- 12.Taha RA, Minshall EM, Olivenstein R, Ihaku D, Wallaert B, Tsicopoulos A, Tonnel AB, Damia R, Menzies D, Hamid QA. 1999. Increased expression of IL-12 receptor mRNA in active pulmonary tuberculosis and sarcoidosis. Am J Respir Crit Care Med 160:1119–1123. doi: 10.1164/ajrccm.160.4.9807120. [DOI] [PubMed] [Google Scholar]
- 13.Zhang M, Gong J, Presky DH, Xue W, Barnes PF. 1999. Expression of the IL-12 receptor beta 1 and beta 2 subunits in human tuberculosis. J Immunol 162:2441–2447. [PubMed] [Google Scholar]
- 14.Hoeve MA, de Boer T, Langenberg DM, Sanal O, Verreck FA, Ottenhoff TH. 2003. IL-12 receptor deficiency revisited: IL-23-mediated signaling is also impaired in human genetic IL-12 receptor β1 deficiency. Eur J Immunol 33:3393–3397. doi: 10.1002/eji.200324343. [DOI] [PubMed] [Google Scholar]
- 15.van de Vosse E, Lichtenauer-Kaligis EG, van Dissel JT, Ottenhoff TH. 2003. Genetic variations in the interleukin-12/interleukin-23 receptor (β1) chain, and implications for IL-12 and IL-23 receptor structure and function. Immunogenetics 54:817–829. doi: 10.1007/s00251-002-0534-9. [DOI] [PubMed] [Google Scholar]
- 16.Presky DH, Minetti LJ, Gillessen S, Wilkinson VL, Wu CY, Gubler U, Chizzonite R, Gately MK. 1998. Analysis of the multiple interactions between IL-12 and the high-affinity IL-12 receptor complex. J Immunol 160:2174–2179. [PubMed] [Google Scholar]
- 17.Aytekin C, Dogu F, Tuygun N, Tanir G, Guloglu D, Boisson-Dupuis S, Bustamante J, Feinberg J, Casanova JL, Ikinciogullari A. 2011. Bacille Calmette-Guerin lymphadenitis and recurrent oral candidiasis in an infant with a new mutation leading to interleukin-12 receptor β-1 deficiency. J Investig Allergol Clin Immunol 21:401–404. [PMC free article] [PubMed] [Google Scholar]
- 18.Lichtenauer-Kaligis EG, de Boer T, Verreck FA, van Voorden S, Hoeve MA, van de Vosse E, Ersoy F, Tezcan I, van Dissel JT, Sanal O, Ottenhoff TH. 2003. Severe Mycobacterium bovis BCG infections in a large series of novel IL-12 receptor β1 deficient patients and evidence for the existence of partial IL-12 receptor beta1 deficiency. Eur J Immunol 33:59–69. doi: 10.1002/immu.200390008. [DOI] [PubMed] [Google Scholar]
- 19.Sakai T, Matsuoka M, Aoki M, Nosaka K, Mitsuya H. 2001. Missense mutation of the interleukin-12 receptor beta1 chain-encoding gene is associated with impaired immunity against Mycobacterium avium complex infection. Blood 97:2688–2694. doi: 10.1182/blood.V97.9.2688. [DOI] [PubMed] [Google Scholar]
- 20.de Jong R, Altare F, Haagen IA, Elferink DG, Boer T, van Breda Vriesman PJ, Kabel PJ, Draaisma JM, van Dissel JT, Kroon FP, Casanova JL, Ottenhoff TH. 1998. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science 280:1435–1438. doi: 10.1126/science.280.5368.1435. [DOI] [PubMed] [Google Scholar]
- 21.Altare F, Durandy A, Lammas D, Emile JF, Lamhamedi S, Le Deist F, Drysdale P, Jouanguy E, Doffinger R, Bernaudin F, Jeppsson O, Gollob JA, Meinl E, Segal AW, Fischer A, Kumararatne D, Casanova JL. 1998. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science 280:1432–1435. doi: 10.1126/science.280.5368.1432. [DOI] [PubMed] [Google Scholar]
- 22.Okada M, Kita Y, Nakajima T, Kanamaru N, Hashimoto S, Nagasawa T, Kaneda Y, Yoshida S, Nishida Y, Nakatani H, Takao K, Kishigami C, Nishimatsu S, Sekine Y, Inoue Y, Matsumoto M, McMurray DN, de la Cruz EC, Tan EV, Abalos RM, Burgos JA, Saunderson P, Sakatani M. 2011. Novel therapeutic vaccine: granulysin and new DNA vaccine against tuberculosis. Hum Vaccin 7(Suppl):60–67. doi: 10.4161/hv.7.0.14563. [DOI] [PubMed] [Google Scholar]
- 23.Okada M, Kita Y, Nakajima T, Kanamaru N, Hashimoto S, Nagasawa T, Kaneda Y, Yoshida S, Nishida Y, Nakatani H, Takao K, Kishigami C, Nishimatsu S, Sekine Y, Inoue Y, McMurray DN, Sakatani M. 2011. Novel prophylactic vaccine using a prime-boost method and hemagglutinating virus of Japan-envelope against tuberculosis. Clin Dev Immunol 2011:549281. doi: 10.1155/2011/549281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson RT, Khader SA, Martino CA, Fountain JJ, Teixeira-Coelho M, Pearl JE, Smiley ST, Winslow GM, Woodland DL, Walter MJ, Conejo-Garcia JR, Gubler U, Cooper AM. 2010. Mycobacterium tuberculosis infection induces il12rb1 splicing to generate a novel IL-12Rβ1 isoform that enhances DC migration. J Exp Med 207:591–605. doi: 10.1084/jem.20091085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chua AO, Wilkinson VL, Presky DH, Gubler U. 1995. Cloning and characterization of a mouse IL-12 receptor-beta component. J Immunol 155:4286–4294. [PubMed] [Google Scholar]
- 26.Ford NR, Miller HE, Reeme AE, Waukau J, Bengtson C, Routes JM, Robinson RT. 2012. Inflammatory signals direct expression of human IL12RB1 into multiple distinct isoforms. J Immunol 189:4684–4694. doi: 10.4049/jimmunol.1200606. [DOI] [PubMed] [Google Scholar]
- 27.Levine SJ. 2008. Molecular mechanisms of soluble cytokine receptor generation. J Biol Chem 283:14177–14181. doi: 10.1074/jbc.R700052200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper AM, Magram J, Ferrante J, Orme IM. 1997. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with Mycobacterium tuberculosis. J Exp Med 186:39–45. doi: 10.1084/jem.186.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu C, Ferrante J, Gately MK, Magram J. 1997. Characterization of IL-12 receptor β1 chain (IL-12Rβ1)-deficient mice: IL-12Rβ1 is an essential component of the functional mouse IL-12 receptor. J Immunol 159:1658–1665. [PubMed] [Google Scholar]
- 30.Lakso M, Sauer B, Mosinger B Jr, Lee EJ, Manning RW, Yu SH, Mulder KL, Westphal H. 1992. Targeted oncogene activation by site-specific recombination in transgenic mice. Proc Natl Acad Sci U S A 89:6232–6236. doi: 10.1073/pnas.89.14.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Black DL. 2003. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 32.Miller HE, Robinson RT. 2012. Early control of Mycobacterium tuberculosis infection requires il12rb1 expression by rag1-dependent lineages. Infect Immun 80:3828–3841. doi: 10.1128/IAI.00426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Green AM, Difazio R, Flynn JL. 2013. IFN-gamma from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J Immunol 190:270–277. doi: 10.4049/jimmunol.1200061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine SJ. 2004. Mechanisms of soluble cytokine receptor generation. J Immunol 173:5343–5348. doi: 10.4049/jimmunol.173.9.5343. [DOI] [PubMed] [Google Scholar]
- 35.Jensen LE, Muzio M, Mantovani A, Whitehead AS. 2000. IL-1 signaling cascade in liver cells and the involvement of a soluble form of the IL-1 receptor accessory protein. J Immunol 164:5277–5286. doi: 10.4049/jimmunol.164.10.5277. [DOI] [PubMed] [Google Scholar]
- 36.Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, Giri JG, Dower SK, Sims JE, Mantovani A. 1993. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science 261:472–475. doi: 10.1126/science.8332913. [DOI] [PubMed] [Google Scholar]
- 37.Lainez B, Fernandez-Real JM, Romero X, Esplugues E, Canete JD, Ricart W, Engel P. 2004. Identification and characterization of a novel spliced variant that encodes human soluble tumor necrosis factor receptor 2. Int Immunol 16:169–177. doi: 10.1093/intimm/dxh014. [DOI] [PubMed] [Google Scholar]
- 38.Mosley B, Beckmann MP, March CJ, Idzerda RL, Gimpel SD, VandenBos T, Friend D, Alpert A, Anderson D, Jackson J, Wignall JM, Smith C, Gallis B, Sims JE, Urdal D, Widmer MB, Cosman D, Park LS. 1989. The murine interleukin-4 receptor: molecular cloning and characterization of secreted and membrane bound forms. Cell 59:335–348. doi: 10.1016/0092-8674(89)90295-X. [DOI] [PubMed] [Google Scholar]
- 39.Layton MJ, Cross BA, Metcalf D, Ward LD, Simpson RJ, Nicola NA. 1992. A major binding protein for leukemia inhibitory factor in normal mouse serum: identification as a soluble form of the cellular receptor. Proc Natl Acad Sci U S A 89:8616–8620. doi: 10.1073/pnas.89.18.8616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khankin EV, Mutter WP, Tamez H, Yuan HT, Karumanchi SA, Thadhani R. 2010. Soluble erythropoietin receptor contributes to erythropoietin resistance in end-stage renal disease. PLoS One 5:e9246. doi: 10.1371/journal.pone.0009246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, Fischer M, Neurath MF, Rose-John S. 2001. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem 268:160–167. doi: 10.1046/j.1432-1327.2001.01867.x. [DOI] [PubMed] [Google Scholar]
- 42.Tavernier J, Devos R, Cornelis S, Tuypens T, Van der Heyden J, Fiers W, Plaetinck G. 1991. A human high affinity interleukin-5 receptor (IL5R) is composed of an IL5-specific alpha chain and a beta chain shared with the receptor for GM-CSF. Cell 66:1175–1184. doi: 10.1016/0092-8674(91)90040-6. [DOI] [PubMed] [Google Scholar]
- 43.Diveu C, Venereau E, Froger J, Ravon E, Grimaud L, Rousseau F, Chevalier S, Gascan H. 2006. Molecular and functional characterization of a soluble form of oncostatin M/interleukin-31 shared receptor. J Biol Chem 281:36673–36682. doi: 10.1074/jbc.M607005200. [DOI] [PubMed] [Google Scholar]
- 44.Kendall RL, Wang G, Thomas KA. 1996. Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochem Biophys Res Commun 226:324–328. doi: 10.1006/bbrc.1996.1355. [DOI] [PubMed] [Google Scholar]
- 45.Pavlakovic H, Becker J, Albuquerque R, Wilting J, Ambati J. 2010. Soluble VEGFR-2: an antilymphangiogenic variant of VEGF receptors. Ann N Y Acad Sci 1207(Suppl 1):E7–E15. doi: 10.1111/j.1749-6632.2010.05714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM. 2001. The soluble interleukin 6 receptor: mechanisms of production and implications in disease. FASEB J 15:43–58. doi: 10.1096/fj.99-1003rev. [DOI] [PubMed] [Google Scholar]
- 47.Lundstrom W, Highfill S, Walsh ST, Beq S, Morse E, Kockum I, Alfredsson L, Olsson T, Hillert J, Mackall CL. 2013. Soluble IL7Rα potentiates IL-7 bioactivity and promotes autoimmunity. Proc Natl Acad Sci U S A 110:E1761–E1770. doi: 10.1073/pnas.1222303110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Badoual C, Bouchaud G, Agueznay Nel H, Mortier E, Hans S, Gey A, Fernani F, Peyrard S, Puig PL, Bruneval P, Sastre X, Plet A, Garrigue-Antar L, Quintin-Colonna F, Fridman WH, Brasnu D, Jacques Y, Tartour E. 2008. The soluble alpha chain of interleukin-15 receptor: a proinflammatory molecule associated with tumor progression in head and neck cancer. Cancer Res 68:3907–3914. doi: 10.1158/0008-5472.CAN-07-6842. [DOI] [PubMed] [Google Scholar]
- 49.Choi ME. 1999. Cloning and characterization of a naturally occurring soluble form of TGF-beta type I receptor. Am J Physiol 276:F88–F95. [DOI] [PubMed] [Google Scholar]
- 50.Han CS, Chen Y, Ezashi T, Roberts RM. 2001. Antiviral activities of the soluble extracellular domains of type I interferon receptors. Proc Natl Acad Sci U S A 98:6138–6143. doi: 10.1073/pnas.111139598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schobitz B, Pezeshki G, Pohl T, Hemmann U, Heinrich PC, Holsboer F, Reul JM. 1995. Soluble interleukin-6 (IL-6) receptor augments central effects of IL-6 in vivo. FASEB J 9:659–664. [DOI] [PubMed] [Google Scholar]
- 52.Schirmacher P, Peters M, Ciliberto G, Blessing M, Lotz J, Meyer zum Buschenfelde KH, Rose-John S. 1998. Hepatocellular hyperplasia, plasmacytoma formation, and extramedullary hematopoiesis in interleukin (IL)-6/soluble IL-6 receptor double-transgenic mice. Am J Pathol 153:639–648. doi: 10.1016/S0002-9440(10)65605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peters M, Schirmacher P, Goldschmitt J, Odenthal M, Peschel C, Fattori E, Ciliberto G, Dienes HP, Meyer zum Buschenfelde KH, Rose-John S. 1997. Extramedullary expansion of hematopoietic progenitor cells in interleukin (IL)-6-sIL-6R double transgenic mice. J Exp Med 185:755–766. doi: 10.1084/jem.185.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qi JW, Qin TT, Xu LX, Zhang K, Yang GL, Li J, Xiao HY, Zhang ZS, Li LY. 2013. TNFSF15 inhibits vasculogenesis by regulating relative levels of membrane-bound and soluble isoforms of VEGF receptor 1. Proc Natl Acad Sci U S A 110:13863–13868. doi: 10.1073/pnas.1304529110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chakraborty M, McGreal EP, Davies PL, Nowell MA, Jones S, Kotecha S. 2013. Role of interleukin-6, its receptor and soluble gp130 in chronic lung disease of prematurity. Neonatology 104:161–167. doi: 10.1159/000351015. [DOI] [PubMed] [Google Scholar]
- 56.Wilson TM, Maric I, Shukla J, Brown M, Santos C, Simakova O, Khoury P, Fay MP, Kozhich A, Kolbeck R, Metcalfe DD, Klion AD. 2011. IL-5 receptor alpha levels in patients with marked eosinophilia or mastocytosis. J Allergy Clin Immunol 128:1086–1092; e1081–e1083. doi: 10.1016/j.jaci.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cooper AM, Kipnis A, Turner J, Magram J, Ferrante J, Orme IM. 2002. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular responses to mycobacterial infection only if the IL-12 p40 subunit is present. J Immunol 168:1322–1327. doi: 10.4049/jimmunol.168.3.1322. [DOI] [PubMed] [Google Scholar]
- 58.Showe LC, Wysocka M, Wang B, Lineman-Williams D, Peritt D, Showe MK, Trinchieri G. 1996. Structure of the mouse IL-12R beta 1 chain and regulation of its expression in BCG/LPS-treated mice. Ann N Y Acad Sci 795:413–415. doi: 10.1111/j.1749-6632.1996.tb52708.x. [DOI] [PubMed] [Google Scholar]
- 59.Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, Trauner A, Wallis D, Galaviz S, Huttenhower C, Sacchettini JC, Behar SM, Rubin EJ. 2013. Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell 155:1296–1308. doi: 10.1016/j.cell.2013.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, Ghilardi N, de Sauvage F, Cooper AM. 2005. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol 175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 61.Khader SA, Guglani L, Rangel-Moreno J, Gopal R, Junecko BA, Fountain JJ, Martino C, Pearl JE, Tighe M, Lin YY, Slight S, Kolls JK, Reinhart TA, Randall TD, Cooper AM. 2011. IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J Immunol 187:5402–5407. doi: 10.4049/jimmunol.1101377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ordway DJ, Orme IM. 2011. Animal models of mycobacteria infection. Curr Protoc Immunol Chapter 19:Unit 19.5. doi: 10.1002/0471142735.im1905s94. [DOI] [PubMed] [Google Scholar]
- 63.Ferguson JS, Martin JL, Azad AK, McCarthy TR, Kang PB, Voelker DR, Crouch EC, Schlesinger LS. 2006. Surfactant protein D increases fusion of Mycobacterium tuberculosis-containing phagosomes with lysosomes in human macrophages. Infect Immun 74:7005–7009. doi: 10.1128/IAI.01402-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Padhi A, Sengupta M, Sengupta S, Roehm KH, Sonawane A. 2014. Antimicrobial peptides and proteins in mycobacterial therapy: current status and future prospects. Tuberculosis 94:363–373. doi: 10.1016/j.tube.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 65.Armstrong JA, Hart PD. 1971. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J Exp Med 134:713–740. doi: 10.1084/jem.134.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. 2002. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun 70:4501–4509. doi: 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rhoades ER, Frank AA, Orme IM. 1997. Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tuberc Lung Dis 78:57–66. doi: 10.1016/S0962-8479(97)90016-2. [DOI] [PubMed] [Google Scholar]
- 68.Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, Ernst JD. 2007. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol 179:2509–2519. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- 69.Marsay L, Matsumiya M, Tanner R, Poyntz H, Griffiths KL, Stylianou E, Marsh PD, Williams A, Sharpe S, Fletcher H, McShane H. 2013. Mycobacterial growth inhibition in murine splenocytes as a surrogate for protection against Mycobacterium tuberculosis. Tuberculosis 93:551–557. doi: 10.1016/j.tube.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 70.van de Vosse E, Haverkamp MH, Ramirez-Alejo N, Martinez-Gallo M, Blancas-Galicia L, Metin A, Garty BZ, Sun-Tan C, Broides A, de Paus RA, Keskin O, Cagdas D, Tezcan I, Lopez-Ruzafa E, Arostegui JI, Levy J, Espinosa-Rosales FJ, Sanal O, Santos-Argumedo L, Casanova JL, Boisson-Dupuis S, van Dissel JT, Bustamante J. 2013. IL-12Rβ1 deficiency: mutation update and description of the IL12RB1 variation database. Hum Mutat 34:1329–1339. doi: 10.1002/humu.22380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chua AO, Chizzonite R, Desai BB, Truitt TP, Nunes P, Minetti LJ, Warrier RR, Presky DH, Levine JF, Gately MK, Gubler U. 1994. Expression cloning of a human IL-12 receptor component: a new member of the cytokine receptor superfamily with strong homology to gp130. J Immunol 153:128–136. [PubMed] [Google Scholar]
- 72.Zou J, Presky DH, Wu CY, Gubler U. 1997. Differential associations between the cytoplasmic regions of the interleukin-12 receptor subunits β1 and β2 and JAK kinases. J Biol Chem 272:6073–6077. doi: 10.1074/jbc.272.9.6073. [DOI] [PubMed] [Google Scholar]
- 73.Peters M, Jacobs S, Ehlers M, Vollmer P, Mullberg J, Wolf E, Brem G, Meyer zum Buschenfelde KH, Rose-John S. 1996. The function of the soluble interleukin 6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice toward IL-6 and prolongation of the plasma half-life of IL-6. J Exp Med 183:1399–1406. doi: 10.1084/jem.183.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]