

Abstract
The complement system is a key component of the host immune response for the recognition and clearance of Streptococcus pneumoniae. In this study, we demonstrate that the amidase LytA, the main pneumococcal autolysin, inhibits complement-mediated immunity independently of effects on pneumolysin by a complex process of impaired complement activation, increased binding of complement regulators, and direct degradation of complement C3. The use of human sera depleted of either C1q or factor B confirmed that LytA prevented activation of both the classical and alternative pathways, whereas pneumolysin inhibited only the classical pathway. LytA prevented binding of C1q and the acute-phase protein C-reactive protein to S. pneumoniae, thereby reducing activation of the classical pathway on the bacterial surface. In addition, LytA increased recruitment of the complement downregulators C4BP and factor H to the pneumococcal cell wall and directly cleaved C3b and iC3b to generate degradation products. As a consequence, C3b deposition and phagocytosis increased in the absence of LytA and were markedly enhanced for the lytA ply double mutant, confirming that a combination of LytA and Ply is essential for the establishment of pneumococcal pneumonia and sepsis in a murine model of infection. These data demonstrate that LytA has pleiotropic effects on complement activation, a finding which, in combination with the effects of pneumolysin on complement to assist with pneumococcal complement evasion, confirms a major role of both proteins for the full virulence of the microorganism during septicemia.
INTRODUCTION
Streptococcus pneumoniae (also termed the pneumococcus) colonizes the human nasopharynx in a high percentage of the population and can be carried asymptomatically from the first days of life (1). S. pneumoniae is the most common etiologic agent of acute otitis media and community-acquired pneumonia and a major cause of bacterial sepsis and meningitis, resulting in significant rates of morbidity and mortality worldwide (2). Prevention of pneumococcal disease requires efficient recognition and clearance of the invading pathogen by the complement system and professional phagocytes (3, 4). Activation of the three complement cascades—termed the classical pathway (CP), the alternative pathway (AP), and the lectin pathway—leads to the formation of the key complement component C3b, which plays a pivotal role in the host immune response, such as opsonization and clearance of invading pathogens (5–7). The CP is important for complement recognition of pneumococci and is generally activated by the recognition of antigen-antibody complexes on the bacterial surface (6, 8) as part of the adaptive immune response and by natural IgM, the lectin SIGN-R1, and acute-phase proteins as part of the innate immune response (6, 9, 10). In addition, the AP is activated by the spontaneous hydrolysis of the C3 component, triggering the amplification of C3 deposition (11, 12), and mannose binding lectin pathway activation has also recently been reported for S. pneumoniae (7). A finely controlled set of specific surface-bound and fluid-phase regulators, such as C4b-binding protein (C4BP) and factor H (FH), protects host cells from complement activation and complement-mediated damage (13–18).
Although the expression of the S. pneumoniae capsule is essential for the virulence of the microorganism, numerous pneumococcal proteins also contribute to pathogenesis, including by promoting complement evasion (3, 19). For example, the S. pneumoniae cell wall protein PspC can recruit the complement downregulators C4BP and FH to the bacterial cell surface, thereby inhibiting activation of the CP and AP, respectively (13–18). In addition, the cholesterol-dependent cytolysin pneumolysin (Ply) (20) prevents CP-mediated complement recognition of pneumococci through interactions with the CP component C1q (3, 21). However, export of Ply into extracellular fluid or for attachment to the cell wall seems to require lysis of the bacteria (22). The pneumococcal protein involved in lysis is the major autolytic enzyme of the bacterium and is termed LytA, an amidase that cleaves the N-acetylmuramoyl-l-alanine bonds of pneumococcal peptidoglycan (23). Previously, LytA was thought to contribute toward pneumococcal pathogenesis due to its importance for the release of Ply and inflammatory mediators, such as teichoic acids and peptidoglycan fragments from S. pneumoniae (23, 24), rather than direct effects on immune evasion independent of Ply.
In this study, we have investigated the contribution of Ply and LytA to the establishment of invasive pneumococcal disease (IPD), exploring their role in essential aspects of the pathogenesis process, including evasion of different components of the host immune response.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The S. pneumoniae clinical isolates used were D39 (NCTC 07466, serotype 2 [ST2]); strain S3 lytA (ST23F) and its complemented mutant, S3C (lytA+) (25); and strain 1515/97 (ST6B) and its lytA-deficient strain (26). Isogenic D39 mutants with mutations in lytA, ply, pspC, or lytB were constructed by transformation, using standard protocols (18, 21, 27, 28), with DNA prepared from mutants previously characterized. Pneumococcal strains expressing the green fluorescent protein (GFP) were constructed by genetic transformation with pMV158GFP (tetracycline resistant) as previously described (28). Kanamycin (250 μg/ml), erythromycin (0.2 μg/ml), and tetracycline (0.5 μg/ml) were added to blood agar plates for isolation of bacterial transformants. S. pneumoniae strains were cultured on blood agar plates at 37°C in a CO2 atmosphere or in Todd-Hewitt broth supplemented with 0.5% yeast extract to an optical density at 550 nm (OD550) of 0.5 and stored at −70°C in 10% glycerol as single-use aliquots.
Binding of complement factors to S. pneumoniae.
A pool of sera from five healthy male volunteers unvaccinated against S. pneumoniae (median age, 40 years) was obtained with informed consent according to institutional guidelines (LIB 14/2007, 3 July) and stored as single-use aliquots at −70°C as a source of complement and serum components. C1q, C3b, FH, C4BP, and C-reactive protein (CRP) were assessed using flow cytometry assays as previously described (10, 18, 28). Human sera depleted of C1q and factor B were purchased from Calbiochem. C3b deposition was detected by incubating 5 × 106 CFU of the bacteria opsonized with 20% serum using a fluorescein isothiocyanate (FITC)-conjugated polyclonal goat anti-human C3b antibody (ICN-Cappel) diluted 1/300 in phosphate-buffered saline (PBS)–0.1% Tween 20. After incubation, the bacteria were washed with PBS–Tween 20 (0.02%) to remove unbound components, fixed in 3% paraformaldehyde, and analyzed on a FACSCalibur flow cytometer (BD Biosciences) or a Beckman Coulter Cytomics FC500 flow cytometer using forward and side scatter parameters to gate on at least 25,000 bacteria. The results were expressed as a relative percent fluorescence index (FI) that measures not only the proportion of fluorescent bacteria positive for the host serum component investigated but also the intensity of fluorescence, which quantifies the immune component bound (8). This assay was adapted to assess the binding to C1q, CRP, FH, and C4BP using a conjugated polyclonal sheep anti-human C1q antibody (Serotec), a polyclonal rabbit anti-human CRP antibody (Calbiochem), a polyclonal sheep anti-human FH antibody (Serotec), and a polyclonal sheep anti-human C4BP antibody (Serotec). To detect CRP, FH, and C4BP, secondary staining in PBS–0.1% Tween 20 containing FITC-conjugated polyclonal goat anti-rabbit or FITC/DyLight 649 antisheep antibodies (Serotec) was performed. A direct interaction between purified LytA and purified C4BP or FH was determined by enzyme-linked immunosorbent assay (ELISA) as previously described (10). Briefly, Nunc MaxiSorp 96-well plates were coated with 10 μg/ml of purified LytA for 2 h at 37°C and blocked with a PBS–2% bovine serum albumin (BSA) solution before 50 μl of different concentrations of purified human C4BP or FH was added to each well. After 2 h of incubation at 37°C, the plates were incubated with 50 μl of sheep anti-human C4BP or FH (Serotec) diluted 1/2,000 in PBS. Finally, the plates were incubated with 50 μl of rabbit antisheep HRP antibody (Santa Cruz) for 30 min and developed using o-phenylenediamine (Sigma-Aldrich) before determining the OD492 using a microtiter plate reader (Anthos 2020).
Quantification of Pcho and PspC.
The level of phosphorylcholine (Pcho) and PspC on the bacterial surface was detected by flow cytometry as previously described (28). The conditions of the assays were the same as those described above for complement components, except that the bacterial strains were incubated for 1 h at 37°C with TEPC-15 antibody (a monoclonal antibody specific for Pcho [Sigma-Aldrich]) diluted 1/25 or rabbit polyclonal antibody to PspC (a kind gift from Sven Hammerschmidt, University of Greifswald, Greifswald, Germany) diluted 1/300. The secondary antibodies used for the detection of Pcho and PspC were rabbit antimouse FITC (Serotec) and goat antirabbit FITC (Serotec), respectively.
C3b/iC3b degradation by LytA.
Purified LytA (displaying amidase activity), the carboxy-terminal moiety or choline-binding domain of LytA (C-LytA), and the enzymatically inactive LytAH133A protein (a mutated LytA amidase containing a His133 → Ala substitution [LytAH133A] that inactivates the enzyme [29]) were obtained by overexpression of previously described plasmids in Escherichia coli (30). A lytA-deficient strain was opsonized for 20 min with human serum, and after two washes with PBS–Tween 20, proteins were added and the C3b level was explored by flow cytometry. To detect C3 fragments by Western blotting, a lytA-null strain was opsonized with 50% human serum as a source of C3b in the absence or in the presence of different pneumococcal proteins (LytA, C-LytA, LytAH133A, or the LytC lysozyme) for 2 h at 37°C. A sample of each supernatant was analyzed by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the proteins were transferred to a membrane, and fragments were revealed by immunoblotting using a goat anti-human C3b antibody. As controls, purified C3b proteins treated and not treated with FH and factor I were included. Additionally, purified C3b and iC3b proteins (3 μg) in sodium phosphate buffer (20 mM, pH 6.9) were treated with 3 ng of LytA or LytAH133A and incubated for 2 h at 37°C. Samples were analyzed by SDS-PAGE using Tricine instead of Tris-glycine (31, 32).
Interaction of S. pneumoniae with phagocytes.
Experiments investigating the recognition and phagocytosis by alveolar macrophages (AMs) were performed as previously described (27, 33). Briefly, murine MH-S cells (CRL-2019; American Type Culture Collection [ATCC]) were used as AMs and were grown in RPMI tissue culture medium supplemented with 10% heat-inactivated fetal calf serum and HEPES (10 mM). To test the recognition of the wild type and the different mutants by AMs, cells (seeded in 24-well plates containing 7 × 105 cells per well) were infected in triplicate with 50 μl of a suspension of the pneumococcal strains at a ratio of 50 bacteria to 1 cell and incubated at 37°C. For adhesion assays, cells were infected for 1 h, washed five times with PBS, and lysed with 300 μl of a solution containing 0.025% saponin-PBS for 10 min. For phagocytosis assays, cells that had previously been infected for 1 h were washed five times with PBS and incubated for an additional hour in tissue culture medium containing penicillin (10 μg/ml) and gentamicin (200 μg/ml) to kill extracellular bacteria. Viable bacteria recovered from infected cells were obtained by plating serial dilutions on blood agar plates.
Phagocytosis by neutrophils was evaluated using HL-60 cells (CCL-240; ATCC) differentiated to granulocytes, and the general conditions of the assay were based on those described previously (10, 27, 34). Briefly, S. pneumoniae strains were fluorescently labeled by incubation with 6-carboxyfluorescein, succinimidyl ester (FAM-SE; Molecular Probes), solution (10 mg/ml in dimethyl sulfoxide; Sigma-Aldrich) in 0.1 M sodium bicarbonate buffer for 1 h at 37°C and then washed six times with Hanks balanced salt solution (HBSS)–0.2% BSA and stored in aliquots at −70°C in 10% glycerol for further assays. Infection assays were performed with bacteria at a ratio of 10 bacteria per cell. A minimum of 6,000 cells were analyzed using a Cytomics flow cytometer. Results were expressed as a relative percent phagocytosis index, defined as the proportion of cells positive for fluorescent bacteria multiplied by the geometric mean of the fluorescence intensity, which correlates with the amount of bacteria phagocytosed per cell (8, 10, 27).
Confocal microscopy.
S. pneumoniae strains expressing GFP were obtained by transformation with pMV158GFP and were used for immunofluorescence microscopy. MH-S cells and HL-60 cells that had previously been infected as described above were seeded on 12-mm circular coverslips for immunofluorescence staining. As the HL-60 cells were in suspension, the cells were centrifuged at 70 × g for 2 min using a Cytospin centrifuge (Thermo Electron, Pittsburgh, PA). For the detection of late endosomal markers in AMs, we stained late antigen membrane protein 1 (LAMP1) or LAMP2. Coverslips containing the infected cells were washed twice in PBS containing 0.1% saponin and once in PBS and incubated for 30 min with primary antibodies. Staining was performed in PBS containing 10% horse serum, 0.1% saponin, and the primary antibodies using rat anti-mouse LAMP1 or LAMP2 (Southern Biotech) diluted 1/200, and the DNA was stained with Hoechst (Invitrogen) diluted 1/2,500. After 30 min incubation with primary antibodies at room temperature, the coverslips were washed twice with PBS–0.1% saponin and once with PBS (pH 7.0) before incubation for 30 min at room temperature with a 1/200 dilution of the secondary antibody goat antirat tetramethylrhodamine isocyanate (Serotec). The actin cytoskeleton was stained with rhodamine-phalloidin (Invitrogen) diluted 1:200. Finally, the coverslips were washed twice in PBS containing 0.1% saponin, once in PBS, and once in H2O; mounted with Aqua Poly/Mount (Polysciences); and analyzed with a Leica spectral SP5 confocal microscope using Leica software (LAS-AF).
Experimental models of infection.
C57BL/6 mice were bred by the CIB-CSIC and ISCIII animal facilities. All mice used were 8 to 16 weeks old, and within each experiment, groups of mice were matched for age and sex. In studies investigating the role of Ply and LytA in the establishment of pneumococcal sepsis and pneumonia, groups of 5 mice each were infected as previously described (10). Briefly, for the sepsis model of infection, mice were challenged with 1 × 106 CFU/ml of each strain (in a volume of 200 μl) by the intraperitoneal route, whereas for pneumonia, mice, which were under anesthesia with isoflurane, were inoculated intranasally with 50 μl containing 107 CFU/mouse. At 24 h after challenge, a lethal dose of pentobarbital was administered and bacterial counts were determined from samples recovered from bronchoalveolar lavage (BAL) fluid, lung, and blood. Experiments were repeated twice using 5 mice in each group, and results are expressed as the log number of CFU of bacteria/ml recovered from the different sites. All animal procedures were approved by the Animal Care and Use Committees of CIB-CSIC and ISCIII (approval reference numbers CIB-FJD 06010017 and CBBA-PA 52_2011-v2, respectively).
Quantification of capsular polysaccharide.
Serotype 2 pneumococcal capsular polysaccharide (CPS) was either purchased from ATCC or prepared as previously described (35). Glucuronic acid was determined with m-hydroxydiphenyl, as previously described (36), using type 2 CPS as a standard and measuring the OD520 using a microtiter plate reader (Anthos 2020). Recognition of the CPS of serotype 2 strains by specific antibodies was studied by the flow cytometry assay explained above using the wild-type D39 strain and the isogenic lytA mutant strain. The antibodies used for detection were rabbit anti-serotype 2 (Statens Serum Institut) diluted 1/200 and a secondary goat antirabbit FITC-conjugated antibody (Santa Cruz) diluted 1/300. Results are expressed as a fluorescence index, as explained above.
Statistical analysis.
Data are representative of the results obtained from repeated independent experiments, and each point represents the mean and standard deviation (SD) from 3 to 5 replicates. Statistical analysis was performed by using a two-tailed Student's t test (for two groups), whereas analysis of variance (ANOVA) followed by Dunnett's post hoc test was chosen for use for multiple comparisons. GraphPad InStat (version 5.0) software (GraphPad Software, San Diego, CA) was used for statistical analysis. Differences were considered statistically significant when P was <0.05 and highly significant when P was <0.01 and <0.001.
RESULTS
Ply and LytA divert C3b deposition following a cooperative strategy.
The complement system is an efficient immune surveillance system detecting foreign intruders and is one of the first lines of the host immune defense against S. pneumoniae (11). To identify the role of Ply and LytA in the subversion of pneumococcal recognition by the key complement component C3b, strains defective in Ply and LytA were constructed. Strains lacking either LytA or Ply had increased C3b deposition on the bacterial surface compared to the wild-type strain. Higher levels of C3b were found on the lytA mutant than on the ply-defective strain, suggesting that LytA might avoid recognition by C3b using a Ply-independent mechanism (Fig. 1A and B). Binding to C3b was even more pronounced for the lytA ply double mutant, indicating that both proteins might act in concert to avoid complement-mediated immunity (Fig. 1A and B). Incubation with human serum depleted of complement component C1q or factor B (in which CP or AP activity, respectively, was abolished) confirmed that the presence of Ply reduced the activation of only the CP, whereas the presence of LytA inhibited the activation of both complement pathways, providing additional evidence that LytA impairs complement activation independently of Ply (Fig. 1C). A lack of enhanced C3b deposition when the lytA ply double mutant was incubated in C1q- or factor B-depleted sera confirmed that the activity of both cascades was essential for the improved effect in complement evasion mediated by both proteins (Fig. 1).
FIG 1.

LytA of S. pneumoniae avoids complement activation by a Ply-independent mechanism. (A) C3b deposition on the surface of the wild-type and isogenic defective strains using NHS, as measured by a flow cytometry assay. (B) Example of a flow cytometry histogram for C3b deposition using NHS. (C) Deposition of C3b via AP (white bars) or CP (gray bars) activity in human serum in which C1q (white bars) or factor B (gray bars) was depleted, respectively. (D) Binding to C3b on the surface of the wild-type D39 strain and the lytB strain. (E) Phase-contrast microscopy images of pneumococcal wild-type D39 and isogenic lytA and lytB strains. Error bars represent SDs, and asterisks indicate statistically significant differences compared to the results for the wild-type strain: *, P < 0.05; **, P < 0.01; ***, P < 0.001. P was <0.01 for the comparison of the C3b results for the lytA ply double mutant versus the single mutants using NHS. For the results for all defective strains compared to those for the wild type, P was <0.001 (one-way ANOVA with Dunnett's post hoc test). FI, fluorescence intensity.
To exclude the possibility that cellular morphology (i.e., chain formation) might affect complement interaction, C3b deposition was analyzed on an isogenic lytB mutant strain, which forms long chains of bacteria (Fig. 1D and E). There was no increase in complement deposition on the lytB mutant, suggesting that, at least under our experimental conditions, the increased C3b levels on the lytA-null strain were not due to differences in cell separation (Fig. 1). The capsule is known to inhibit complement activity against the pneumococcus (19), but the content of glucuronic acid (a component of the CPS of serotype 2) was actually higher for the lytA mutant strain (0.185 for the wild-type strain versus 0.361 for the lytA mutant). In addition, the recognition of S. pneumoniae by antibodies specific to serotype 2 increased in the absence of LytA (100 ± 17 for the wild-type strain versus 201 ± 121 for the lytA mutant; P < 0. 05). These results suggest that the increased C3b observed in the lytA-null strain cannot be due to reduced levels of CPS expression by the lytA mutant.
Ply and LytA prevent activation of the CP on S. pneumoniae.
Activation of the CP by binding of C1q or acute-phase proteins, such as CRP or serum amyloid P component (SAP), to the bacterium is essential for complement-mediated immunity against S. pneumoniae, whereas bacterial components impairing activation are factors critical for immune evasion (6, 10, 37). Hence, we investigated the effect of deposition of C1q or CRP on the bacterial surface using pneumococcal strains lacking Ply, LytA, or both proteins simultaneously. An S. pneumoniae strain defective in either Ply or LytA showed higher levels of C1q binding, confirming that both proteins allow S. pneumoniae to impair the activation of the CP (Fig. 2A and B). This effect was more pronounced in the absence of LytA, suggesting that this amidase assists with pneumococcal evasion of CP activation using a Ply-independent strategy (Fig. 2A and B). CRP deposition was also increased in the absence of LytA but not in the absence of Ply (Fig. 2C and D). These results together demonstrated that LytA is more effective than Ply at impairing CP activation. As the increased levels of CRP on the lytA mutant might be due to differences in the amount of Pcho residues exposed on the bacterial cell wall, the level of Pcho was measured using pneumococcal strains of two different serotypes and the corresponding isogenic lytA mutants. A lack of LytA was associated with increased detection of Pcho, indicating that LytA impaired the recognition of pneumococci by the CP through effects on the availability of Pcho for binding to CRP (Fig. 2E to G).
FIG 2.

Ply and LytA divert classical pathway activation. (A) Deposition of C1q on the surface of the different mutants compared to that on the surface of the wild-type strain. (B) Example of a flow cytometry histogram for C1q deposition. (C) Recognition of the wild-type strain and the different mutants by CRP. (D) Example of a flow cytometry histogram for CRP deposition. (E) Pcho levels on the surface of wild-type strains D39 and 1515 of ST2 and ST6B, respectively, and LytA-deficient mutants. (F) Example of a flow cytometry histogram for the Pcho level on the ST2 strain. (G) Example of a flow cytometry histogram for the Pcho level on the ST6B strain. Error bars represent SDs, and asterisks indicate a statistically significant difference compared to the results for the wild-type strain: **, P < 0.01; ***, P < 0.001. For the results for all defective strains compared to the result for the wild type, P was <0.001 (one-way ANOVA with Dunnett's post hoc test). CT, control (bacteria incubated in PBS).
LytA avoids complement immunity by recruiting fluid-phase downregulators.
To prevent damage of host cells by a constant low level of complement activation, a finely controlled set of soluble and membrane-bound regulators allows any complement activation on host cells to be either avoided or strongly inhibited (11). Trapping of fluid-phase downregulators, such as C4BP and FH, by certain pathogens is a successful strategy for avoiding the complement response (13). Binding of the CP inhibitor protein C4BP was greatly reduced in the lytA-null strain (Fig. 3A and B). As PspC has recently been reported to be a ligand for C4BP and FH, an isogenic pspC mutant strain was investigated (15, 17, 18). The loss of PspC resulted in a reduced proportion of C4BP binding, with the levels of binding being similar to those found in the absence of LytA, whereas the loss of both PspC and LytA resulted in a greater reduction in C4BP binding, demonstrating that both proteins play a key role in the recruitment of C4BP (Fig. 3A and B). Fluorescence intensity data also confirmed that a lack of either LytA or PspC was significantly associated with impaired recruitment of C4BP (23 ± 5 for the wild-type strain, 17 ± 2 for the ply strain, 8 ± 2 for the lytA strain, 9 ± 1 for the pspC strain, and 6 ± 1 for the pspC lytA strain). To confirm the requirement of LytA for binding to C4BP, the C4BP binding assays were performed using a serotype 23F lytA mutant strain (termed S3, the first described clinical isolate of S. pneumoniae deficient in LytA activity) and the corresponding lytA+ transformant (termed S3C) (25) (Fig. 3C and D). The loss of LytA in strain S3 was again associated with reduced C4BP binding, which was restored by transformation with the lytA+ allele (strain S3C), confirming that LytA is a novel ligand of S. pneumoniae for C4BP (Fig. 3C and D). Direct binding of purified LytA with different concentrations of purified human C4BP was observed, supporting a role for LytA in recruiting C4BP to the bacterial cell surface (Fig. 3F).
FIG 3.

LytA recruits C4BP to reduce classical pathway activation. (A) Proportion of bacteria positive for C4BP for the D39 wild-type strain and the different mutants. (B) Example of a flow cytometry histogram for C4BP binding for strains with the D39 genetic background. (C) Proportion of bacteria positive for C4BP for the S3 lytA mutant strain and the complemented S3C strain (lytA+) belonging to ST23F. (D) Example of a flow cytometry histogram for C4BP binding for the ST23F strains. (E) Direct binding of 10 μg/ml of LytA to different concentrations of C4BP by ELISA. Error bars represent SDs, and asterisks indicate a statistically significant difference between the results for single mutants and the wild-type strain or between the results for different concentrations of C4BP and those in the absence of protein: **, P < 0.01; ***, P < 0.001. P was <0.05 for the comparison of the C4BP results between the pspC lytA double mutant and the single mutants.
Interaction with FH, the downregulator of the AP, was also evaluated for the different strains. As expected, a lack of Ply did not affect FH binding, whereas a loss of LytA or PspC produced significantly lower levels of FH bound than those found with the wild-type strain (Fig. 4A and B). In addition, binding of FH to the pspC lytA double mutant was markedly impaired, indicating that both proteins are important pneumococcal ligands for FH binding (Fig. 4A and B). Fluorescence intensity values confirmed that a lack of either LytA or PspC on the D39 strain was significantly associated with impaired binding to FH (72 ± 30 for the wild-type strain, 58 ± 10 for the ply strain, 23 ± 7 for the lytA strain, 5 ± 1 for the pspC strain, and 5 ± 1 for the pspC lytA strain). Recruitment of FH was also evaluated for strains S3 and S3C of serotype 23F (Fig. 4C to E). The proportion of bacteria binding to FH was similar between both strains (Fig. 4C), but the intensity of the FH bound was significantly lower in the absence of LytA (Fig. 4D and E), further supporting a role for LytA in pneumococcal recruitment of high levels of FH. Furthermore, a direct interaction between purified LytA autolysin and human FH proteins was observed (Fig. 4F). There were no differences in the level of PspC measured between the wild-type and the lytA-deficient strains (Fig. 5), confirming that the reduced levels of both C4BP and FH on the lytA-deficient strain were not caused by the effects of the lytA mutation on PspC expression (28).
FIG 4.

LytA binds the downregulator FH to impair the activation of the alternative pathway. (A) Proportion of bacteria positive for FH for the D39 wild-type strain and the different mutants. (B) Example of a flow cytometry histogram for FH binding for strains with the D39 genetic background. WT, wild type. (C) Proportion of bacteria positive for FH for the S3 lytA mutant strain and the complemented S3C strain (lytA+) belonging to ST23F. (D) Mean fluorescence intensity (MFI) of FH binding on the surface of the S3 lytA strain and the complemented S3C (lytA+) strain. (E) Example of a flow cytometry histogram for FH binding of the ST23F strains. (F) Direct binding of 10 μg/ml of LytA to different concentrations of FH by ELISA. Error bars represent the SDs, and asterisks indicate statistically significant differences compared to the results for the wild-type strain or between the results for different concentrations of FH compared to those in the absence of protein: *, P < 0.05; **, P < 0.01; ***, P < 0.001. P was <0.01 for the comparison of the FH results between the pspC lytA double mutant and the single mutants.
FIG 5.

PspC levels are similar in the wild-type and lytA strains. (A) PspC levels on the surface of the D39 wild-type strain and the LytA-deficient mutant. (B) Example of a flow cytometry histogram for the PspC level. Error bars represent SDs.
LytA impairs opsonization by degradation of C3b and iC3b.
Proteolytic enzymes can counteract the effects of complement activation by bacterial pathogens (13). Hence, the ability of LytA to cleave the C3b deposited on the bacterial surface and purified C3b/iC3b components was investigated (Fig. 6). For these experiments, we used a fully active LytA protein (LytA), its C-terminal domain (C-LytA), and the enzymatically inactive protein LytAH133A (see above). The degradation of the C3b deposited was initially investigated by adding the different LytA proteins to a previously opsonized lytA-deficient strain (Fig. 6A). The results demonstrated full restoration of the wild-type phenotype only when the fully active LytA amidase was used (Fig. 6A). This result was confirmed by Western blotting using an anti-C3 antibody. A small C3 fragment was observed after addition of the LytA protein but not after addition of LytAH133A, C-LytA, or the LytC lysozyme (Fig. 6B). C3b degradation products were also found when LytA was incubated with either C3b (Fig. 6C) or iC3b (Fig. 6D), confirming the direct cleavage of C3b and iC3b components by LytA. Degradation of the C3b/iC3b deposited on the surface of the nonencapsulated R6 strain was also observed, indicating that LytA is involved in C3 degradation independently of capsule expression (Fig. 6E). These results demonstrated that the pneumococcal LytA autolysin can cause degradation of the complement components C3b and iC3b, partially explaining why the lytA-deficient strain had higher levels of opsonization with C3b/iC3b.
FIG 6.

LytA degrades C3b and iC3b to impair complement activation. (A) Flow cytometry assay showing degradation of the C3b deposited on a lytA strain that had previously been opsonized with serum in the presence of 0.3 μg of either LytA amidase, the choline-binding domain of LytA (C-LytA), or a mutated LytA protein without amidase activity (LytAH133A). Error bars represent SDs, and asterisks indicate a statistically significant difference compared to the results for the wild-type strain: ***, P < 0.001. (B) Coomassie-stained polyacrylamide gel showing C3b degradation by FH-factor I (FI) and Western blotting to detect C3 fragments using a lytA mutant opsonized with serum (lanes 1 to 5) and exposed to 3 ng of LytA having amidase activity (lane 2), LytC lysozyme (lane 3), C-LytA (lane 4), and LytAH133A (lane 5). (C and D) Western blotting to detect fragments of C3b and iC3b degradation, respectively, after exposure of purified C3b (C) and iC3b (D) to LytA or LytAH133A. (B and D) Arrowheads, typical bands of C3b degradation by FH-FI (α65 and α43); arrows, C3b fragments obtained after digestion with LytA. (E) Degradation of the C3b deposited on the R6 strain in the presence or absence of 0.3 μg of LytA.
LytA and Ply divert phagocytosis by professional phagocytes.
Neutrophils control pneumococcal dissemination by phagocytosis, a process that requires the opsonization of bacteria by the complement system (38, 39). Opsonization with HBSS or heat-killed human serum (HKS) did not support the phagocytosis of S. pneumoniae by human neutrophils, whereas opsonization with normal human serum (NHS) did, confirming the importance of complement-mediated immunity for this process (Fig. 7A to C). There was increased phagocytosis of the lytA and ply mutants compared to that of the wild-type strain, showing that both proteins are important bacterial factors involved in evasion of phagocytosis. The lytA mutant exhibited higher phagocytosis levels than the ply mutant, suggesting that LytA participates in resistance to phagocytosis by a mechanism that is independent of the release of Ply (Fig. 7A to C). Moreover, synergistic increases in phagocytosis were found for the lytA ply double mutant in comparison to the single mutants and the wild-type strain, demonstrating that both proteins contribute to evasion of phagocytosis by neutrophils (Fig. 7A to C).
FIG 7.

Evasion of the phagocytosis process mediated by pneumolysin and LytA. (A) Phagocytosis of the FAM-SE-labeled wild-type strain and the different mutant strains incubated in 20% normal human serum using a flow cytometry assay. Results are expressed as the fluorescent index (in percent) relative to the results for the wild-type D39 strain. (B) Example of a flow cytometry histogram for phagocytosis by neutrophils. (C) Opsonophagocytosis of the different strains expressing GFP by human neutrophils detected by confocal microscopy. DNA was stained by Hoechst, and the actin cytoskeleton was visualized with rhodamine-phalloidin (Phall-RRX) staining. (D) Attachment to AMs of the different mutant strains compared to that of the wild-type strain at 1 h postinfection. (E and F) Phagocytosis of the different strains by AMs at 1 h (E) and 4 h (F) postinfection. (G) Phagolysosomal maturation of AMs during infection with S. pneumoniae lytA ply expressing GFP. DNA was stained with Hoechst, whereas late endosomal markers were visualized using specific antibodies to recognize LAMP1 (top) and LAMP2 (bottom). Error bars represent the SDs, and asterisks indicate statistically significant differences compared to the results for the wild-type strain: *, P < 0.05; **, P < 0.01; ***, P < 0.001. P was < 0.01 for the comparison of the phagocytosis results for the lytA ply double mutant and the single mutants for all times except 4 h of phagocytosis by AMs between the lytA ply double mutant and the lytA mutant, for which P was equal to 0.21. For the results for all defective strains compared to those for the wild type, P was <0.001 (one-way ANOVA with Dunnett's post hoc test). CT, control (bacteria incubated in PBS).
Adhesion to a murine cell line of AMs was slightly increased in the absence of Ply or LytA and markedly increased in the double mutant, suggesting that these two proteins enable S. pneumoniae to divert the recognition by AMs (Fig. 7D). Time course experiments were performed to evaluate the phagocytosis process within the macrophage. At an early phase, phagocytosis of the ply and lytA single mutants was increased (Fig. 7A). Phagocytosis of the ply lytA double mutant strain was more efficient than that of the single mutants, confirming the additive effect of the loss of these two proteins on pneumococcal phagocytosis by AMs (Fig. 7E). Compared with the bacterial load at 1 h, the bacterial load was significantly reduced over time, with a more than 100-fold reduction being found at 4 h, suggesting that once the macrophage has recognized and phagocytosed pneumococcal strains lacking Ply and LytA, the machinery of the macrophage efficiently destroys the engulfed bacteria (Fig. 7F). To confirm this possibility, we investigated maturation of the phagosome containing the lytA ply-null strain using immunofluorescence microscopy to analyze the colocalization of phagocytosed GFP-expressing bacteria with early and late endosomal markers. The double mutant was observed in LAMP1- and LAMP2-positive compartments (Fig. 7G), suggesting that in the absence of Ply and LytA, AMs efficiently process S. pneumoniae by the conventional phagolysosomal pathway.
LytA and Ply enhance the establishment of pneumococcal pneumonia and invasive disease.
Mouse models of pneumonia and sepsis were used to characterize the contribution of Ply and LytA to the pathogenesis of S. pneumoniae. A lack of either Ply or LytA was associated with a significant attenuation of virulence in the sepsis model in comparison to the virulence of the wild-type strain (Fig. 8). In addition, the virulence of the lytA ply double mutant was greatly reduced compared to that of the single mutants and the wild-type strain, confirming that both proteins contribute separately to the establishment of pneumococcal sepsis (Fig. 8A).
FIG 8.
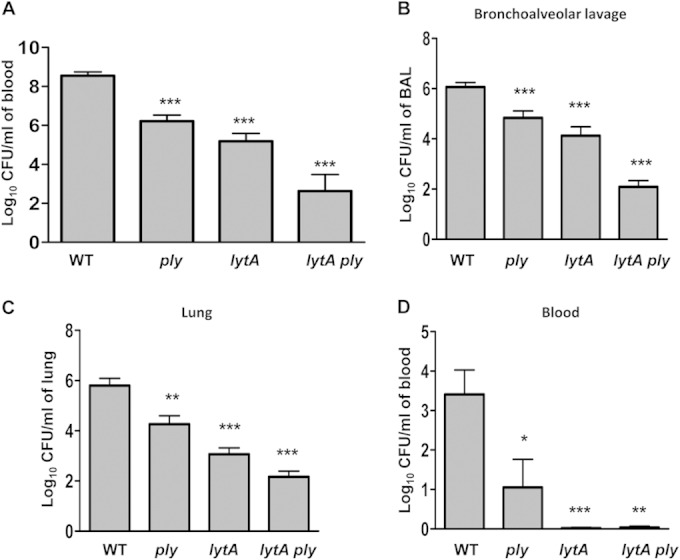
Role of Ply and LytA in the establishment of sepsis (intraperitoneal inoculation) and pneumococcal pneumonia (intranasal inoculation). (A) Bacterial levels recovered from blood at 24 h after pneumococcal sepsis produced with the wild-type and mutant strains. (B to D) Bacterial levels recovered at 24 h from BAL fluid (B), lung homogenate (C), and blood (D) after pneumonia caused by the wild-type and defective strains. Error bars represent SDs, and asterisks indicate statistically significant differences in the levels of the different mutant bacterial strains compared to the levels of the wild-type strain: *, P < 0.05; **, P < 0.01; ***, P < 0.001. P was <0.05 for the comparison of the bacterial levels for the lytA ply double mutant and those for the single mutants. For the results for all defective strains compared to the wild type, P was <0.01 (one-way ANOVA with Dunnett's post hoc test).
In the pneumonia model, the levels of the ply or lytA single mutants recovered from BAL fluid, lung, and blood samples were significantly lower than those obtained with the parental strain, indicating that both proteins are involved in the pathogenesis of pneumococcal pneumonia (Fig. 8B to D). Moreover, the loss of both Ply and LytA caused further falls in the number of CFU recovered from BAL fluid, lung, and blood samples, confirming that the activity of both proteins is required for the full virulence of the bacterium in the respiratory tract and for spread from the lung to the blood (Fig. 8B to D). These results are compatible with the complement interaction data and confirm that inhibition of complement deposition on S. pneumoniae by the combination of LytA and Ply is essential for full virulence during systemic infection. Collectively, these data suggest that the enhanced effect of LytA and Ply on virulence is mainly due to their combined inhibition of complement-dependent immunity and phagocytosis.
DISCUSSION
S. pneumoniae is the leading cause of community-acquired pneumonia and a major cause of sepsis and meningitis, which are associated with high morbidity and mortality rates worldwide (2, 40). As one of the most devastating human pathogens, S. pneumoniae has developed a wide arsenal of virulence factors to escape the well-balanced machinery of the immune system (3). Several proteins are involved in the establishment of IPD, which occurs when S. pneumoniae invades typically sterile sites, causing bacteremic pneumonia and sepsis, or when it crosses the blood-brain barrier, causing meningitis (3). Ply is a cytolytic protein with a significant role in pneumonia, sepsis, and meningitis but not, apparently, in carriage (3, 21, 24, 41–43). In contrast, the contribution of LytA to pneumococcal pathogenesis is poorly understood. The use of strains deficient in LytA has demonstrated attenuation of the virulence of these mutants in different models of infection, suggesting that LytA is important for virulence (39, 44, 45). Activation of complement-mediated immunity is an essential and critical component of the host immune response against S. pneumoniae (5–7, 9), and Ply has previously been reported to reduce the CP opsonic activity against S. pneumoniae. The effects of LytA on virulence have traditionally been linked to the release of Ply and not to a direct effect of LytA (3, 46, 47). However, our results demonstrate that LytA plays a critical role in complement evasion that is independent of the release of Ply. The lytA mutant had higher levels of CRP, C1q, and C3b binding to its surface than the ply mutant, and the ply lytA double mutant had increased levels of C3b deposition compared to those for the single mutant strains, confirming that both proteins confer complement resistance on S. pneumoniae. Our findings confirm previous data suggesting that Ply impairs the activation of the CP through C1q (21, 37, 46, 47), and this might be a possible explanation for the increased recognition by C1q in the absence of both Ply and LytA. Using a lytA mutant strain, other authors have reported increased sensitivity to complement-dependent clearance and attributed this attenuation to its increased bacterial chain length, suggesting that chain length is an important factor that increases the ability to fix complement C3b (39). Although a certain deficit in promoting the efficient separation of daughter cells in the absence of LytA may increase the level of recognition by C3b, our results suggest that this is not the full explanation for the increased complement activity against the lytA mutant, as the formation of longer chains by the lytB mutant strain does not affect complement deposition (27). Instead, the lytA mutation affects complement activity by a variety of mechanisms which lead to increased CP and AP activity.
One mechanism in the lytA mutant was an increased amount or accessibility to the cell surface of Pcho, the target for CRP, natural IgM, and SAP binding to S. pneumoniae, and therefore of innate CP activation. Modification of Pcho levels did not affect the expression of PspC, another important choline binding protein involved in complement evasion. Despite this, recruitment of the fluid-phase downregulators C4BP and FH (both of which are known to bind to PspC) was decreased in the lytA mutant, and assays using purified proteins demonstrated direct binding between LytA or C4BP and FH. Hence, our findings demonstrate that LytA is an additional pneumococcal protein that reduces complement-mediated immunity by recruiting C4BP and FH. Finally, we have shown evidence that LytA mediates the direct degradation of C3 by S. pneumoniae, which has previously been described (48).
Our data suggest that LytA can inhibit complement activation against S. pneumoniae by multiple mechanisms. The pneumococcal capsule also alters different aspects of complement- and phagocyte-mediated immunity, resulting in a profound inhibition of opsonophagocytosis (19), and effects on the thickness of the capsule layer could potentially explain the pleiotropic effects of LytA on complement activity. However, the content of glucuronic acid (a component of the serotype 2 CPS) and the recognition by specific antibodies to CPS were actually slightly increased for the lytA mutant. Hence, the effects of LytA on the capsule are unlikely to explain the increased complement deposition seen on the lytA mutant; in addition, the direct interactions of purified LytA with FH, C4BP, and C3 cannot be explained by the effects of the loss of LytA on other aspects of S. pneumoniae biology. The location of bound C3b is important because the accessibility of this component affects the recognition by phagocytic cells. In this sense, the opsonic activity of C3b deposited on the bacterial cell wall may be less efficient for induction of phagocytosis. The presence of antibodies to CPS and the cell wall can also influence complement deposition (49–51). Human serum contains antibodies to multiple S. pneumoniae antigens, but these would not affect our results, unless there was a marked difference in the expression of target antigens between the strains investigated. Instead, our data indicate that increased complement activity against the lytA mutant was mediated by several separate mechanisms independent of antibodies, including increased Pcho availability (which could be a nonphysiological effect of the reduced occupation of choline residues in the lytA mutant), direct binding of LytA to C4BP and FH (causing the negative regulation of CP and AP activity, respectively), and enzymatic activity against C3.
AMs are one of the first barriers of the host immune defense system against pathogens invading the lungs, and neutrophils are key players controlling the dissemination of relevant microorganisms through the systemic circulation (52, 53). A loss of Ply and LytA was associated with the enhanced uptake of S. pneumoniae by AMs and neutrophils in vitro, confirming that the effects of these two proteins on complement are important for avoidance of the recognition and engulfment of S. pneumoniae by phagocytic cells. This is in agreement with previous evidence showing synergistic inhibition of complement-dependent immunity and phagocytosis for S. pneumoniae proteins (21, 27, 54, 55). The efficiency of AM phagolysosomal processing and bacterial killing within the macrophage was increased for the ply and lytA mutants, demonstrating that a lack of Ply and LytA increases the efficiency of AM clearance of the bacteria through the phagolysosomal route (56).
Data from the mouse models confirmed that Ply and LytA are critical proteins that cooperate in the establishment of IPD and pneumonia. Chain length formation has been identified to be a factor that might affect bacterial virulence (39). However, pneumococci growing as chains due to a lytB mutation did not show impaired virulence in our models, suggesting that the attenuation of the virulence of our lytA mutant was not significantly related to chain formation (27). The strain with the simultaneous loss of LytA and Ply showed a marked attenuation in virulence, indicating that both proteins cooperate in the replication of the bacterium in the respiratory tract and systemic circulation. The impaired levels of the mutants in the blood, as previously reported (21, 41, 43, 47), suggest that pneumococcal strains lacking LytA and Ply have a reduced ability to breach the epithelial barrier. This is in agreement with the findings of a previous study showing that a lack of both LytA and Ply had an additive effect on the median survival time in a murine sepsis model of infection (43), and this phenotype has previously been shown to be related to complement sensitivity for the ply pspA double mutant (21). Overall, our results confirm that LytA plays an important role in bacteremic pneumonia and sepsis by a mechanism that is independent of Ply release and is likely to reflect the additive effects seen in vitro of Ply and LytA on complement inhibition and phagocytosis.
ACKNOWLEDGMENTS
We thank Eloisa Cano for skillful technical assistance.
This work was supported by grant SAF2012-39444-C01/02 from MINECO. The Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES) and Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) are initiatives of ISCIII. E.R.-S. was supported by an FPU fellowship from MINECO.
REFERENCES
- 1.Bogaert D, De Groot R, Hermans PWM. 2004. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis 4:144–154. doi: 10.1016/S1473-3099(04)00938-7. [DOI] [PubMed] [Google Scholar]
- 2.O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T, for the Hib and Pneumococcal Global Burden of Disease Study Team . 2009. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374:893–902. doi: 10.1016/S0140-6736(09)61204-6. [DOI] [PubMed] [Google Scholar]
- 3.Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol 6:288–301. doi: 10.1038/nrmicro1871. [DOI] [PubMed] [Google Scholar]
- 4.van der Poll T, Opal SM. 2009. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374:1543–1556. doi: 10.1016/S0140-6736(09)61114-4. [DOI] [PubMed] [Google Scholar]
- 5.Xu Y, Ma M, Ippolito GC, Schroeder HW Jr, Carroll MC, Volanakis JE. 2001. Complement activation in factor D-deficient mice. Proc Natl Acad Sci U S A 98:14577–14582. doi: 10.1073/pnas.261428398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown JS, Hussell T, Gilliland SM, Holden DW, Paton JC, Ehrenstein MR, Walport MJ, Botto M. 2002. The classical pathway is the dominant complement pathway required for innate immunity to Streptococcus pneumoniae infection in mice. Proc Natl Acad Sci U S A 99:16969–16974. doi: 10.1073/pnas.012669199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ali YM, Lynch NJ, Haleem KS, Fujita T, Endo Y, Hansen S, Holmskov U, Takahashi K, Stahl GL, Dudler T, Girija UV, Wallis R, Kadioglu A, Stover CM, Andrew PW, Schwaeble WJ. 2012. The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PLoS Pathog 8:e1002793. doi: 10.1371/journal.ppat.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuste J, Sen A, Truedsson L, Jönsson G, Tay LS, Hyams C, Baxendale HE, Goldblatt F, Botto M, Brown JS. 2008. Impaired opsonization with C3b and phagocytosis of Streptococcus pneumoniae in sera from subjects with defects in the classical complement pathway. Infect Immun 76:3761–3770. doi: 10.1128/IAI.00291-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang YS, Do Y, Lee HK, Park SH, Cheong C, Lynch RM, Loeffler JM, Steinman RM, Park CG. 2006. A dominant complement fixation pathway for pneumococcal polysaccharides initiated by SIGN-R1 interacting with C1q. Cell 125:47–58. doi: 10.1016/j.cell.2006.01.046. [DOI] [PubMed] [Google Scholar]
- 10.Yuste J, Botto M, Bottoms SE, Brown JS. 2007. Serum amyloid P aids complement-mediated immunity to Streptococcus pneumoniae. PLoS Pathog 3:1208–1219. doi: 10.1371/journal.ppat.0030120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walport MJ. 2001. Complement. First of two parts. N Engl J Med 344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 12.Walport MJ. 2001. Complement. Second of two parts. N Engl J Med 344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 13.Lambris JD, Ricklin D, Geisbrecht BV. 2008. Complement evasion by human pathogens. Nat Rev Microbiol 6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal V, Hammerschmidt S, Malm S, Bergmann S, Riesbeck K, Blom AM. 2012. Enolase of Streptococcus pneumoniae binds human complement inhibitor C4b-binding protein and contributes to complement evasion. J Immunol 189:3575–3584. doi: 10.4049/jimmunol.1102934. [DOI] [PubMed] [Google Scholar]
- 15.Dieudonné-Vatran A, Krentz S, Blom AM, Meri S, Henriques-Normark B, Riesbeck K, Albiger B. 2009. Clinical isolates of Streptococcus pneumoniae bind the complement inhibitor C4b-binding protein in a PspC allele-dependent fashion. J Immunol 182:7865–7877. doi: 10.4049/jimmunol.0802376. [DOI] [PubMed] [Google Scholar]
- 16.Ogunniyi AD, Grabowicz M, Mahdi LK, Cook J, Gordon DL, Sadlon TA, Paton JC. 2009. Pneumococcal histidine triad proteins are regulated by the Zn2+-dependent repressor AdcR and inhibit complement deposition through the recruitment of complement factor H. FASEB J 23:731–738. doi: 10.1096/fj.08-119537. [DOI] [PubMed] [Google Scholar]
- 17.Dave S, Brooks-Walter A, Pangburn MK, McDaniel LS. 2001. PspC, a pneumococcal surface protein, binds human factor H. Infect Immun 69:3435–3437. doi: 10.1128/IAI.69.5.3435-3437.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuste J, Khandavilli S, Ansari N, Muttardi K, Ismail L, Hyams C, Weiser J, Mitchell T, Brown JS. 2010. The effects of PspC on complement-mediated immunity to Streptococcus pneumoniae vary with strain background and capsular serotype. Infect Immun 78:283–292. doi: 10.1128/IAI.00541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyams C, Camberlein E, Cohen JM, Bax K, Brown JS. 2010. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect Immun 78:704–715. doi: 10.1128/IAI.00881-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tilley SJ, Orlova EV, Gilbert RJC, Andrew PW, Saibil HR. 2005. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell 121:247–256. doi: 10.1016/j.cell.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 21.Yuste J, Botto M, Paton JC, Holden DW, Brown JS. 2005. Additive inhibition of complement deposition by pneumolysin and PspA facilitates Streptococcus pneumoniae septicemia. J Immunol 175:1813–1819. doi: 10.4049/jimmunol.175.3.1813. [DOI] [PubMed] [Google Scholar]
- 22.Price KE, Camilli A. 2009. Pneumolysin localizes to the cell wall of Streptococcus pneumoniae. J Bacteriol 191:2163–2168. doi: 10.1128/JB.01489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernández-Tornero C, López R, García E, Giménez-Gallego G, Romero A. 2001. A novel solenoid fold in the cell wall anchoring domain of the pneumococcal virulence factor LytA. Nat Struct Biol 8:1020–1024. doi: 10.1038/nsb724. [DOI] [PubMed] [Google Scholar]
- 24.Canvin JR, Marvin AP, Sivakumaran M, Paton JC, Boulnois GJ, Andrew PW, Mitchell TJ. 1995. The role of pneumolysin and autolysin in the pathology of pneumonia and septicemia in mice infected with a type 2 pneumococcus. J Infect Dis 172:119–123. doi: 10.1093/infdis/172.1.119. [DOI] [PubMed] [Google Scholar]
- 25.Moscoso M, Domenech M, García E. 2010. Vancomycin tolerance in clinical and laboratory Streptococcus pneumoniae isolates depends on reduced enzyme activity of the major LytA autolysin or cooperation between CiaH histidine kinase and capsular polysaccharide. Mol Microbiol 77:1052–1064. doi: 10.1111/j.1365-2958.2010.07271.x. [DOI] [PubMed] [Google Scholar]
- 26.Ramos-Sevillano E, Rodríguez-Sosa C, Díez-Martínez R, Giménez MJ, Olmedillas E, García P, García E, Aguilar L, Yuste J. 2012. Macrolides and β-lactam antibiotics enhance C3b deposition on the surface of multidrug-resistant Streptococcus pneumoniae strains by a LytA autolysin-dependent mechanism. Antimicrob Agents Chemother 56:5534–5540. doi: 10.1128/AAC.01470-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramos-Sevillano E, Moscoso M, García P, Garciía E, Yuste J. 2011. Nasopharyngeal colonization and invasive disease are enhanced by the cell wall hydrolases LytB and LytC of Streptococcus pneumoniae. PLoS One 6:e23626. doi: 10.1371/journal.pone.0023626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Domenech M, Ramos-Sevillano E, García E, Moscoso M, Yuste J. 2013. Biofilm formation avoids complement immunity and phagocytosis of Streptococcus pneumoniae. Infect Immun 81:2606–2615. doi: 10.1128/IAI.00491-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mellroth P, Sandalova T, Kikhney A, Vilaplana F, Hesek D, Lee M, Mobashery S, Normark S, Svergun D, Henriques-Normark B, Achour A. 2014. Structural and functional insights into peptidoglycan access for the lytic amidase LytA of Streptococcus pneumoniae. mBio 5(1):e01120-13. doi: 10.1128/mBio.01120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.García JL, García E, López R. 1987. Overproduction and rapid purification of the amidase of Streptococcus pneumoniae. Arch Microbiol 149:52–56. doi: 10.1007/BF00423136. [DOI] [PubMed] [Google Scholar]
- 31.Alcorlo M, Tortajada A, Rodríguez de Córdoba S, Llorca O. 2013. Structural basis for the stabilization of the complement alternative pathway C3 convertase by properdin. Proc Natl Acad Sci U S A 110:13504–13509. doi: 10.1073/pnas.1309618110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alcorlo M, Martínez-Barricarte R, Fernández FJ, Rodríguez-Gallego C, Round A, Vega MC, Harris CL, de Córdoba SR, Llorca O. 2011. Unique structure of iC3b resolved at a resolution of 24 Å by 3D-electron microscopy. Proc Natl Acad Sci U S A 108:13236–13240. doi: 10.1073/pnas.1106746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morey P, Cano V, Martí-Lliteras P, López-Goómez A, Regueiro V, Saus C, Bengoechea JA, Garmendia J. 2011. Evidence for a non-replicative intracellular stage of nontypable Haemophilus influenzae in epithelial cells. Microbiology 157:234–250. doi: 10.1099/mic.0.040451-0. [DOI] [PubMed] [Google Scholar]
- 34.Romero-Steiner S, Libutti D, Pais LB, Dykes J, Anderson P, Whitin JC, Keyserling HL, Carlone GM. 1997. Standardization of an opsonophagocytic assay for the measurement of functional antibody activity against Streptococcus pneumoniae using differentiated HL-60 cells. Clin Diagn Lab Immunol 4:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Domenech M, García E, Moscoso M. 2009. Versatility of the capsular genes during biofilm formation by Streptococcus pneumoniae. Environ Microbiol 11:2542–2555. doi: 10.1111/j.1462-2920.2009.01979.x. [DOI] [PubMed] [Google Scholar]
- 36.Blumenkrantz N, Asboe-Hansen G. 1973. New method for quantitative determination of uronic acids. Anal Biochem 54:484–489. doi: 10.1016/0003-2697(73)90377-1. [DOI] [PubMed] [Google Scholar]
- 37.Agarwal V, Sroka M, Fulde M, Bergmann S, Riesbeck K, Blom AM. 2014. Binding of Streptococcus pneumoniae endopeptidase O (PepO) to complement component C1q modulates the complement attack and promotes host cell adherence. J Biol Chem 289:15833–15844. doi: 10.1074/jbc.M113.530212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Underhill DM, Goodridge HS. 2012. Information processing during phagocytosis. Nat Rev Immunol 12:492–502. doi: 10.1038/nri3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalia AB, Weiser JN. 2011. Minimization of bacterial size allows for complement evasion and is overcome by the agglutinating effect of antibody. Cell Host Microbe 10:486–496. doi: 10.1016/j.chom.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koedel U, Scheld WM, Pfister HW. 2002. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis 2:721–736. doi: 10.1016/S1473-3099(02)00450-4. [DOI] [PubMed] [Google Scholar]
- 41.Orihuela CJ, Gao G, Francis KP, Yu J, Tuomanen EI. 2004. Tissue-specific contributions of pneumococcal virulence factors to pathogenesis. J Infect Dis 190:1661–1669. doi: 10.1086/424596. [DOI] [PubMed] [Google Scholar]
- 42.Hirst RA, Gosai B, Rutman A, Guerin CJ, Nicotera P, Andrew PW, O'Callaghan C. 2008. Streptococcus pneumoniae deficient in pneumolysin or autolysin has reduced virulence in meningitis. J Infect Dis 197:744–751. doi: 10.1086/527322. [DOI] [PubMed] [Google Scholar]
- 43.Berry AM, Paton JC. 2000. Additive attenuation of virulence of Streptococcus pneumoniae by mutation of the genes encoding pneumolysin and other putative pneumococcal virulence proteins. Infect Immun 68:133–140. doi: 10.1128/IAI.68.1.133-140.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berry AM, Paton JC, Hansman D. 1992. Effect of insertional inactivation of the genes encoding pneumolysin and autolysin on the virulence of Streptococcus pneumoniae type 3. Microb Pathog 12:87–93. doi: 10.1016/0882-4010(92)90111-Z. [DOI] [PubMed] [Google Scholar]
- 45.Kharat AS, Tomasz A. 2006. Drastic reduction in the virulence of Streptococcus pneumoniae expressing type 2 capsular polysaccharide but lacking choline residues in the cell wall. Mol Microbiol 60:93–107. doi: 10.1111/j.1365-2958.2006.05082.x. [DOI] [PubMed] [Google Scholar]
- 46.Paton JC, Rowan-Kelly B, Ferrante A. 1984. Activation of human complement by the pneumococcal toxin pneumolysin. Infect Immun 43:1085–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alcantara RB, Preheim LC, Gentry-Nielsen MJ. 2001. Pneumolysin-induced complement depletion during experimental pneumococcal bacteremia. Infect Immun 69:3569–3575. doi: 10.1128/IAI.69.6.3569-3575.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Angel CS, Ruzek M, Hostetter MK. 1994. Degradation of C3 by Streptococcus pneumoniae. J Infect Dis 170:600–608. doi: 10.1093/infdis/170.3.600. [DOI] [PubMed] [Google Scholar]
- 49.Briles DE, Claflin JL, Schroer K, Forman C. 1981. Mouse Igg3 antibodies are highly protective against infection with Streptococcus pneumoniae. Nature 294:88–90. doi: 10.1038/294088a0. [DOI] [PubMed] [Google Scholar]
- 50.Briles DE, Forman C, Horowitz JC, Volanakis JE, Benjamin WH Jr, McDaniel LS, Eldridge J, Brooks J. 1989. Antipneumococcal effects of C-reactive protein and monoclonal antibodies to pneumococcal cell wall and capsular antigens. Infect Immun 57:1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li J, Szalai AJ, Hollingshead SK, Nahm MH, Briles DE. 2009. Antibody to the type 3 capsule facilitates immune adherence of pneumococci to erythrocytes and augments their transfer to macrophages. Infect Immun 77:464–471. doi: 10.1128/IAI.00892-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Standish AJ, Weiser JN. 2009. Human neutrophils kill Streptococcus pneumoniae via serine proteases. J Immunol 183:2602–2609. doi: 10.4049/jimmunol.0900688. [DOI] [PubMed] [Google Scholar]
- 53.Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. 2010. Neutrophil kinetics in health and disease. Trends Immunol 31:318–324. doi: 10.1016/j.it.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dalia AB, Standish AJ, Weiser JN. 2010. Three surface exoglycosidases from Streptococcus pneumoniae, NanA, BgaA, and StrH, promote resistance to opsonophagocytic killing by human neutrophils. Infect Immun 78:2108–2116. doi: 10.1128/IAI.01125-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martner A, Skovbjerg S, Paton JC, Wold AE. 2009. Streptococcus pneumoniae autolysis prevents phagocytosis and production of phagocyte-activating cytokines. Infect Immun 77:3826–3837. doi: 10.1128/IAI.00290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stuart LM, Ezekowitz RA. 2005. Phagocytosis: elegant complexity. Immunity 22:539–550. doi: 10.1016/j.immuni.2005.05.002. [DOI] [PubMed] [Google Scholar]