

Abstract
The human fungal pathogen Candida albicans causes invasive candidiasis, characterized by fatal organ failure due to disseminated fungal growth and inflammatory damage. The suppressor of TCR signaling 1 (Sts-1) and Sts-2 are two homologous phosphatases that negatively regulate signaling pathways in a number of hematopoietic cell lineages, including T lymphocytes, mast cells, and platelets. Functional inactivation of both Sts enzymes leads to profound resistance to systemic infection by C. albicans, such that greater than 80% of mice lacking Sts-1 and -2 survive a dose of C. albicans (2.5 × 105 CFU/mouse) that is uniformly lethal to wild-type mice within 10 days. Restriction of fungal growth within the kidney occurs by 24 h postinfection in the mutant mice. This occurs without induction of a hyperinflammatory response, as evidenced by the decreased presence of leukocytes and inflammatory cytokines that normally accompany the antifungal immune response. Instead, the absence of the Sts phosphatases leads to the rapid induction of a unique immunological environment within the kidney, as indicated by the early induction of a proinflammatory cytokine (CXL10). Mice lacking either Sts enzyme individually display an intermediate lethality phenotype. These observations identify an opportunity to optimize host immune responses toward a deadly fungal pathogen.
INTRODUCTION
Changes in medical care are creating increasing opportunities for lethal systemic infections by the human fungal pathogen Candida albicans (1, 2). In recent years, C. albicans has become the fourth leading cause of hospital-acquired bloodstream infections, with close to 50,000 cases being reported yearly in the United States alone, and global mortality rates have not decreased in the last 20 years, in spite of advances in antifungal therapy (2–4). One underlying reason is that C. albicans ordinarily inhabits the skin and mucosa as a benign commensal. New medical procedures, such as cancer chemotherapy or organ transplantation, that reduce host immunity allow C. albicans to escape normal physiological barriers and disseminate to deeper tissues. In addition, factors that permit the overgrowth of C. albicans, including the use of broad-spectrum antibacterial antibiotics or biofilm formation on catheters and indwelling medical devices, allow this fungus to infect immunocompetent individuals (5). Better antifungal therapies are needed to address this escalating problem (6).
During the course of infection, kidneys are highly favorable niches for both fungal proliferation and the morphogenetic switch of C. albicans to a hyphal state (7, 8). Recent analysis has drawn attention to the intensity of the immune response following systemic infection. On the one hand, the innate immune system that is critical for host protection is rapidly activated. In particular, fungal cell wall constituents are recognized by Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) on the surface of phagocytes within the kidney, promoting the release of cytokines and chemokines that lead to the infiltration of additional waves of monocytes and neutrophils into the kidney (9, 10). On the other hand, it has become appreciated that host inflammatory responses targeting C. albicans lead to detrimental and often fatal collateral tissue damage (11). Indeed, in a well-characterized mouse model of systemic candidiasis that mimics the clinical progression of disseminated candidiasis in humans, progressive sepsis accompanied by renal failure has been identified as the cause of death (12). Thus, destructive inflammatory responses leading to pathological tissue destruction is a significant clinical problem.
Current antifungal medications suffer from a number of drawbacks, including high cost, toxicity, and a narrow spectrum of activity (13). These limitations are compounded by difficulties in making a rapid and accurate disease diagnosis (14). The emergence of strains resistant both to fluconazole and to newer members of the echinocandin class of compounds is now considered a major threat by the CDC (15). Because no effective C. albicans vaccine is currently available, developing strategies for enhancing host immune responses that synergize with current medications holds great promise (16). Indeed, attempts to develop combination therapies, such as the coadministration of antifungal monoclonal antibodies or adjuvant immunotherapy with recombinant cytokines to boost a patient's immune response, are ongoing (17–19). The development of combination therapies that are most effective will require a thorough understanding of the host antifungal immune response.
To gain new insight into the mechanisms that control the host response to fungal pathogens, we examined the role of the suppressor of TCR signaling 1 (Sts-1) and Sts-2, two homologous proteins that negatively regulate signaling pathways in a number of hematopoietic cell lineages, including T lymphocytes, mast cells, and platelets (20–23). These evolutionarily conserved proteins share 50% amino acid identity and appear to carry out overlapping functions. However, their expression patterns differ markedly, in that Sts-1 has been found to have a widespread pattern of expression in numerous tissues, while Sts-2 has a more limited pattern of expression, mainly in cells of hematopoietic origin. Critical to Sts function is a distinctive type of phosphatase domain in which the nucleophilic activity is mediated by a conserved histidine (24, 25). Other conserved features include a ubiquitin association (UBA) domain that can bind mono- and polyubiquitin and an SH3 domain that interacts with proline-rich segments of other polypeptides. Current models of Sts function suggest that these protein interaction domains play key roles in either localizing Sts enzymatic functions to specific intracellular regions or targeting the phosphatase domain to specific intracellular substrates (23). Of the two, Sts-1 is the more active enzyme in vitro, although the bona fide in vivo substrate(s) of both enzymes has not been established definitively (24, 25). The important role of Sts-1 and Sts-2 in controlling cellular responses is underscored by the hypersensitivity to TCR stimulation of T cells that lack both proteins. However, Sts-1/2−/− mice do not display an overt autoimmune phenotype, suggesting that other mechanisms of lymphocyte regulation can compensate for the absence of Sts function (20). We report here that functional inactivation of Sts activity has a profound effect on the ability to resist invasive candidiasis and ameliorate the destructive immunopathophysiology that accompanies the disease.
MATERIALS AND METHODS
Mice.
The generation of mice containing the Sts mutations, backcrossed 10 generations onto the C57/B6 background, has been described previously (26). Mice were housed and bred in the Stony Brook University Animal Facility under specific-pathogen-free conditions. Mice used for experiments were 8 weeks old. All mice were maintained in accordance with Stony Brook University Division of Laboratory Animal Resources (DLAR) guidelines. All animal experiments were approved by the Stony Brook University Institutional Animal Care and Use Committee (IACUC).
Infection assays.
Mouse infections were carried out as described previously (27). C. albicans wild-type (WT) strain SC5314 was grown overnight at 30°C in yeast extract-peptone-dextrose (YPD) medium with 80 μg/ml uridine, reinoculated into fresh medium, and incubated again overnight. Cells were harvested by centrifugation, washed twice in phosphate-buffered saline (PBS), counted, and diluted to the appropriate density with PBS. The cell counts were confirmed by plating dilutions onto solid YPD medium. Female mice were inoculated via the lateral tail vein and monitored for 28 days. If a mouse became moribund, humane euthanasia was performed. Graphing and statistical analysis of survival after infection were carried out using a log-rank test (Mantel-Haenszel test) with Prism (version 6) software (GraphPad Software, Inc., La Jolla, CA).
Analysis of numbers of CFU.
Organs were excised from the mice, placed in 5 ml PBS, and homogenized with a tissue homogenizer (Pro Scientific, Inc., Oxford, CT). The number of CFU per gram of tissue was determined by plating serial dilutions of the homogenates onto YPD medium plates and incubating at 30°C. Time course analysis of the numbers of CFU was performed using groups of mice preassigned to specific time points.
Calcofluor white staining.
Kidneys were homogenized in PBS, to which KOH was added to 20%. Following incubation at 37°C for 3 h, lysates were spun at 13,000 × g for 10 min. The pellet was washed 3 times with PBS, resuspended in a small volume of PBS, and stained with calcofluor white. Hyphae were visualized by fluorescence microscopy, and total hyphal clusters present in each homogenate were counted.
Histology.
Tissues were fixed in neutral buffered formalin, paraffin embedded, and sectioned at 5 μm. Sections were counterstained with either hematoxylin and eosin (H&E) or periodic acid-Schiff (PAS), the former at the Stony Brook Histology Core and the latter at McClain Laboratories (Smithtown, NY).
Generation of single-cell organ suspensions.
Kidney leukocyte suspensions were prepared as described previously (28). Briefly, kidneys were disassociated in PBS with 2% fetal calf serum and treated with 50 U/ml DNase (Roche) and 0.2 mg/ml Liberase TL enzyme (Roche) for 30 min at 37°C. Following erythrocyte lysis by addition of ACK buffer (150 mM NH4Cl, 1 mM KHCO3, 0.1 mM EDTA), the debris was removed by straining and centrifuged, and the resulting pellet was suspended in PBS with 2% fetal bovine serum and centrifuged at 1,000 × g at room temperature for 30 min over a 40% Ficoll cushion. The cell pellet was washed, filtered, and suspended in buffer for flow cytometric analysis.
Flow cytometry.
Cell suspensions were incubated for 15 min with FC block prior to staining with fluorochrome-conjugated antibodies to the following for 20 to 30 min: T cell receptor β (TCRβ; clone H57-597; BioLegend), CD19 (clone 6D5; BioLegend), NK1.1 (clone PK136; BioLegend), CD45 (clone 30-F11; BD Biosciences), Ly6C (clone AL-21; BioLegend), CD11c (clone N418; BD Biosciences), Ly6G (clone 1A8; BioLegend), F4/80 (clone BM8; BioLegend), CD11b (clone M1/70; BioLegend), CD4 (clone RM4-5; BD Biosciences), CD8 (clone 53-6-7; BD Biosciences), and I-A/I-E (clone M5/114.15.2; BioLegend). The total numbers of kidney-infiltrating leukocytes were quantified as the number of CD45+ cells in the live-cell gate. Flow cytometric analysis was conducted with a BD LSR Fortessa flow cytometer.
Cytokine analysis.
Kidneys were homogenized using a PRO250 homogenizer (ProScientific) in buffer containing 10 mM Tris, pH 7.8, 200 mM NaCl, 5 mM EDTA, 10% glycerol, and 1× Roche protease inhibitor cocktail. Clarified homogenates were analyzed by enzyme-linked immunosorbent assay (ELISA; for interleukin-6 [IL-6] and tumor necrosis factor alpha [TNF-α], R&D Systems; for alpha interferon [IFN-α], eBioscience, Inc.) or multiplex analysis (Millipore, Inc.). Data were analyzed with a Molecular Devices Spectramax M5 (ELISAs) or a Bio-Plex 200 (Bio-Rad) system.
RESULTS
Sts-1/2−/− mice are highly resistant to systemic C. albicans infection.
The role of Sts-1 and Sts-2 in regulating the host response to a fungal pathogen was examined by testing the susceptibility of Sts-1/2−/− mice to systemic infection by C. albicans. Infections were initiated by injecting wild-type strain SC5314 via the tail vein, and then the mice were monitored over a 28-day period. As expected, nearly 100% of wild-type mice became moribund at the three doses evaluated, with the median time to death being delayed by approximately 5 days with the lowest dose (105 CFU/mouse) relative to that with the highest dose (5 × 105 CFU/mouse) (Fig. 1A). In contrast, mice lacking the Sts proteins exhibited strikingly enhanced survival following induction of systemic candidiasis. Specifically, Sts-1/2−/− mice were almost uniformly resistant to an inoculum of 105 CFU of C. albicans, and at the higher doses they displayed a significantly delayed onset of disease and less than half became moribund (Fig. 1A).
FIG 1.

Resistance of Sts-1/2−/− mice to systemic infection by C. albicans. (A) Mice were infected with the indicated doses of C. albicans (number of CFU per mouse) and monitored for 28 days. Sts-1/2−/− mice demonstrated significantly enhanced survival (P < 0.0001 by the log-rank Mantel-Cox test) at all doses tested. For each dose, results are for a total of 16 to 20 mice analyzed in three independent experiments. (B) Representative histological analysis of wild-type and Sts-1/2−/− kidneys 6 days after infection with a dose of 2.5 × 106 CFU. Staining was with H&E. Arrows, large misshapen tubules indicative of pathological damage. Magnifications, ×100.
Because lethality is caused in part by inflammation-mediated damage to the kidneys, we examined histological sections of kidneys (8, 11). Kidneys harvested from uninfected WT and Sts-1/2−/− mice were similar in appearance (see Fig. S1 in the supplemental material). Following intravenous C. albicans administration (2.5 × 105 CFU/mouse), the kidneys from wild-type mice infected for 6 days showed the expected pathogenic effects resulting from the invasive growth of C. albicans combined with an inflammatory immune response. In particular, many misshapen tubules with large, open lumens were evident throughout the cortex region (Fig. 1B, left; see also Fig. S1 in the supplemental material) (11). In some cases, the lumenal regions contained large numbers of mononuclear leukocytes. In contrast, kidneys isolated from Sts-1/2−/− mice 6 days after infection appeared undamaged (Fig. 1B, right; see also Fig. S1 in the supplemental material). Thus, the widespread damage to the kidneys that is a hallmark of C. albicans systemic infection did not occur in the absence of the Sts proteins.
Individual contributions of Sts homologues to the host response to C. albicans.
To dissect the individual contributions of Sts-1 and Sts-2 to the regulation of the host response to systemic C. albicans infection, we compared the corresponding single mutant mice to the Sts-1/2−/− mice. When infected with 1 × 105 CFU/mouse, both Sts-1−/− and Sts-2−/− mice displayed resistance to C. albicans similar to that of Sts-1/2−/− mice (Fig. 2, top). However, when infected at a dose of 2.5 × 105 CFU/mouse, mice with an individual deletion of either Sts-1 or Sts-2 demonstrated an intermediate susceptibility to infection relative to that of wild-type and Sts-1/2−/− mice (Fig. 2, bottom). Thus, despite interesting functional differences between the Sts proteins, including their in vitro phosphatase activities and their patterns of tissue expression, they appear to carry out redundant functions in regulating the host response to C. albicans infection.
FIG 2.
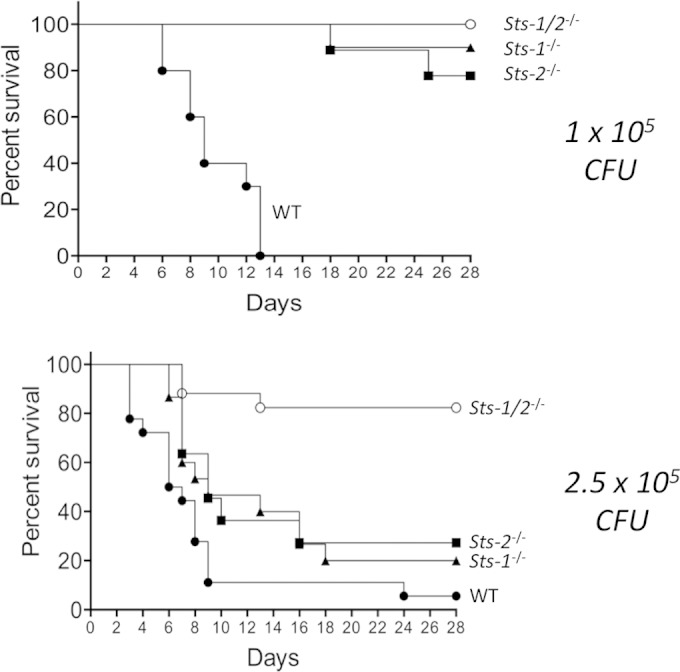
Resistance of Sts-1−/− and Sts-2−/− mice to systemic infection by C. albicans. Mice with an individual deletion of either Sts-1 or Sts-2 were infected intravenously and monitored for 28 days. Each result is for a total of 14 to 15 mice from two independent experiments. (Top) For a dose of 1 × 105 CFU/mouse, P was <0.01 (by log-rank Mantel-Cox analysis) for Sts-1−/− versus WT mice and Sts-2−/− versus WT mice. (Bottom) For a dose of 2.5 × 105 CFU/mouse, P was <0.01 (by log-rank Mantel-Cox analysis) for Sts-1−/− versus Sts-1/2−/− mice and Sts-2−/− versus Sts-1/2−/− mice.
Reduced fungal burden in mice lacking Sts proteins.
The enhanced ability of Sts-1/2−/− mice to survive a lethal systemic challenge of C. albicans suggested that they could be more effective than wild-type mice at either tolerating a higher kidney fungal burden or disabling pathogen virulence mechanisms and clearing the infection, or a combination of both. Following the introduction of C. albicans into the mouse bloodstream, the fungus spreads rapidly to a wide range of tissues, where it is brought under control by organ-specific resident and/or infiltrating leukocytes (8, 28). However, for reasons that are not completely understood, within the kidney there is rapid fungal growth accompanied by a morphogenic switch to the hyphal state. This extensive fungal proliferation appears to overwhelm the host immune response, eventually leading to kidney destruction and mortality. To determine whether the mutant mice were better at tolerating or clearing the pathogen, we infected mice and assessed the kidney fungal load when mice became moribund or at the end of 28 days. Uniformly, the numbers of CFU in wild-type mouse kidneys were significantly greater than those in Sts-1/2−/− mouse kidneys (see Fig. S2 in the supplemental material). Indeed, most of the Sts-1/2−/− mice survived to the end of the 28-day period, and most appeared to clear the infection, as there were no detectable CFU. Importantly, the few Sts-1/2−/− mice that did not survive also displayed a significant reduction in kidney fungal burden (see Fig. S2 in the supplemental material). Thus, the absence of the Sts proteins appeared to promote elimination of the pathogen from the kidney.
This conclusion is supported by several additional observations. First, the number and size of C. albicans hyphal clusters observed in Sts-1/2−/− kidney homogenates 24 h after infection were significantly diminished relative to those observed in wild-type kidney homogenates (Fig. 3A). Second, histological analysis indicated that while concentrated foci of filamentous C. albicans were readily apparent within the cortex of wild-type kidneys 2 days after infection, only small clusters of C. albicans extending hyphae that were not easily discernible could be found in the cortex of Sts-1/2−/− kidneys (Fig. 3B). Finally, in contrast to wild-type kidneys, which were moderately swollen and displayed a large number of infectious foci on their surface 3 days following inoculation, Sts-1/2−/− kidneys appeared normal, with the surface being devoid of similar infectious foci (Fig. 3C).
FIG 3.

Attenuated fungal growth in Sts-1/2−/− kidneys relative to wild-type kidneys. (A) Hyphae in kidney homogenates were visualized with calcofluor white stain at 24 h after infection. (Left) Representative hyphal clusters; (right) total number of hyphal cells per kidney homogenate observed microscopically from two independent experiments (3 mice per group). P was <0.01 (by Mann-Whitney analysis) for Sts-1/2−/− versus WT mice. (B) At 48 h postinfection, representative kidney histological sections were stained with PAS to visualize C. albicans hyphae. Black arrows, hyphae. Magnifications, ×400. (C) Multiple inflammatory foci appear on the surface of wild-type kidneys but not on Sts-1/2−/− kidneys. Kidneys were harvested 3 days after infection with 2.5 × 105 CFU.
Sts-1/2−/− mice suppress C. albicans growth within the kidney more readily than wild-type mice.
To more precisely define the kinetic parameters underlying pathogen clearance from Sts-1/2−/− kidneys, we infected mice and assessed the numbers of kidney CFU along a time course beginning at 4 h after infection. Importantly, we noted equivalent seeding in both wild-type and mutant mouse strains, indicating that a lack of the Sts proteins does not affect dissemination to the kidney. As expected, fungal growth in both the right and left wild-type kidneys was robust, achieving a 100-fold increase in numbers of CFU within 48 h (Fig. 4A). After 48 h, the numbers of CFU in the wild-type kidney were sustained at similarly high levels for the next 4 days. In contrast, by 24 h the numbers of CFU in both the right and left Sts-1/2−/− kidneys were significantly lower than those in wild-type organs, and they remained at levels significantly lower than those in wild-type kidneys for the duration of the time course of analysis. Interestingly, the kidney was the only organ for which the number of fungal CFU differed dramatically between wild-type and mutant mice. No differences in fungal burdens or rates of clearance were observed in the spleen, liver, lungs, or brain (Fig. 4B). Thus, rather than causing a global effect on C. albicans survival, these data indicate that inactivation of the Sts proteins transforms the mammalian kidney from an environment in which C. albicans can readily thrive to one that is hostile to the growth of the pathogen.
FIG 4.

Organ-specific reduction in fungal burden within Sts-1/2−/− mice. A time course analysis of fungal burden in kidneys (A) or additional peripheral organs (B) from mice infected with 2.5 × 105 CFU. Results represent the average of two independent experiments each carried out with three mice per group. **, P < 0.01 by Mann-Whitney analysis; *, P < 0.05 by Mann-Whitney analysis.
Altered cellular immune response induced by C. albicans in Sts-1/2−/− mice.
The survival of Sts-1/2−/− mice after inoculation with lethal doses of C. albicans suggested that their immune response to the fungal pathogen within the kidney was altered relative to the response of wild-type mice. Because immunopathology and organ failure resulting from lethal systemic C. albicans infection are associated with a large influx of inflammatory cells into the kidneys during the initial stage of the disease, we examined hematopoietic cell infiltrates into the kidney at regular time intervals after infection (8, 9). During the first 24 h, we observed an equivalent increase in the total numbers of CD45+ cells within the kidneys of both wild-type and Sts-1/2−/− mice (Fig. 5A). During the next 48 h, however, the responses of the two strains differed dramatically. Within wild-type kidneys, the leukocyte population continued to expand markedly. In striking contrast, within Sts-1/2−/− kidneys the number of leukocytes began to decline after 24 h following infection. The number, size, and cellularity of the inflammatory foci in the cortex of infected kidneys further demonstrated the attenuated response of Sts-1/2−/− mice. Histological analysis revealed that whereas a large number of dense, highly concentrated H&E-positive leukocyte abscesses were clearly evident throughout the wild-type kidneys 2 days after Candida infection, the few foci that were visible in Sts-1/2−/− kidneys were thinly populated and diffuse (Fig. 5B).
FIG 5.

Attenuated leukocyte response in the absence of Sts-1 and -2. (A) Comparison of total kidney-infiltrating leukocytes (CD45+). Hematopoietic cells were isolated from the kidneys of mice infected with 2.5 × 105 CFU for the indicated times. Results are from two independent experiments with 3 mice for each time point. *, P < 0.05 by Mann-Whitney analysis. (B) Inflammatory foci (black arrows) formed within the kidney cortex, visualized by H&E staining of histological sections of kidneys harvested 2 days after infection with 2.5 × 105 CFU. Foci within Sts-1/2−/− kidneys were reduced in size and cellularity. Magnifications, ×20 (top) and ×100 (bottom). Images with a higher magnification are presented in Fig. S3 in the supplemental material.
We then evaluated specific subsets of responding cells. We detected similar steady-state levels of kidney-resident leukocyte subpopulations prior to infection, with both mouse strains having low levels of CD45+ CD11b+ Ly6Chi monocytes and CD45+ CD11b+ Ly6Cmed neutrophils (Fig. 6) (8). In wild-type kidneys, the percentages of monocytes peaked at 24 h and began to decline thereafter, while the number of neutrophils continued to grow rapidly. However, within Sts-1/2−/− kidneys, the numbers and ratios of both monocytes and neutrophils exhibited a substantial decline after 24 h following infection (see representative plots and plot summaries in Fig. 6). Thus, while the initial stage of innate immune effector cell recruitment into the kidney appeared to be identical in wild-type and Sts-1/2−/− mice, the sustained influx of immune cells into the kidney that normally occurs was significantly attenuated in mice lacking the Sts proteins.
FIG 6.

Reduced kidney infiltration of Ly6C+ monocytes and Ly6Cmed neutrophils in the absence of Sts-1 and -2. (Top) Representative fluorescence-activated cell sorter plots illustrating the levels of CD11b+ Ly6Chi cells (monocytes) and CD11b+ Ly6Cmed cells (neutrophils) in the kidney at time points following fungal infection. (Bottom) Summary of fluorescence-activated cell sorter plots with total flow cytometric data from two independent experiments (3 mice per group) analyzed. **, P < 0.01 by Mann-Whitney analysis; *, P < 0.05 by Mann-Whitney analysis.
Altered cytokine profile induced by C. albicans in Sts-1/2−/− mice.
To gain more insight into the attenuated cellular immune response observed in Sts-1/2−/− mouse kidneys following systemic infection with C. albicans, we monitored the levels of inflammatory cytokines during the early stages of the infection (29–31). As expected, within wild-type kidneys the levels of TNF-α and IL-6 increased significantly during the first 48 h (Fig. 7A). Sts-1/2−/− kidney TNF-α and IL-6 levels were also increased at 24 h, but then the levels declined, such that by 48 h they were significantly lower than the corresponding wild-type kidney cytokine levels (Fig. 7A). The kidney levels of four chemokines that play an important role in recruiting monocytes and neutrophils to sites of infection, CCL2 (monocyte chemotactic protein 1), CCL3, CXCL1 (keratinocyte-derived chemokine), and CXCL2, also trended lower in Sts-1/2−/− mice than in wild-type mice by 24 h (Fig. 7B). In contrast, the levels of a variety of other cytokines were unchanged between the two strains (see Fig. S4 in the supplemental material). Thus, we observed reduced levels of specific inflammatory factors in Sts-1/2−/− kidneys relative to wild-type kidneys during the time frame that corresponded to both a reduction in the numbers of fungal CFU and reduced cellular immune responses.
FIG 7.

Altered inflammatory cytokine environment in the absence of Sts-1 and -2. Mice were infected with 2.5 × 105 CFU, and kidneys were harvested at the indicated time points and analyzed. (A) Levels of inflammatory cytokines TNF-α and IL-6 within the kidney. The results of one of two independent experiments with similar results (6 mice per group) are displayed. *, P < 0.05 by Mann-Whitney analysis. (B) Levels of kidney chemokines were determined by multiplex analysis. Scatter plots display the results of one of two independent experiments (each with 6 mice per group) with similar results. *, P < 0.05 by Mann-Whitney analysis. (C) Levels of kidney CXCL10 at 12 h postinfection. The results of one of two independent experiments (each with 6 mice per group) with similar results are displayed. **, P < 0.01 by Mann-Whitney analysis. Horizontal line represents the average of values.
The sharply reduced fungal burden in Sts-1/2−/− kidneys within 24 h after infection suggested that mice lacking the Sts proteins could mount a rapid immune response able to neutralize early fungal growth. To identify evidence of a more potent early inflammatory response in Sts-1/2−/− mice, we assessed the levels of cytokines and chemokines within the first 12 h following infection. Of 15 common cytokines and chemokines examined, including TNF-α, IL-6, CCL2, and CXCL1, only the level of CXCL10 was increased significantly at 12 h (Fig. 7C). However, antibody-mediated neutralization of either CXCL10 or its receptor, CXCR3, failed to abrogate the protective phenotype evident in Sts-1/2−/− mice (data not shown), suggesting that CXCL10 is not solely responsible for the resistance phenotype. Collectively, these observations suggest that a potent and directed innate inflammatory response that favors the elimination of freshly seeded fungal cells is rapidly generated within Sts-1/2−/− kidneys. By allowing the host-pathogen balance to be tipped quickly in favor of the host, functional inactivation of the negative regulatory Sts proteins could thereby lead to reduced inflammation and host survival in the face of an overwhelming C. albicans infection.
DISCUSSION
Mice lacking Sts-1 and -2 are significantly protected from death due to systemic infection by Candida albicans. In this disease model, the kidney is the organ most acutely affected, leading wild-type mice to succumb to pyelonephritis within days of infection (7). The reduction in kidney fungal burden and the maintenance of organ homeostasis that take place in Sts-1/2−/− mice are associated with sharply decreased levels of many inflammatory molecules beginning at 24 h postinfection. This phenotype is accompanied by a concomitant reduction in kidney leukocyte infiltrates after 24 h and an absence of inflammatory lesions. It is possible that the reduced inflammation evident in Sts-1/2−/− mice follows directly from the reduction in the numbers of fungal CFU that occurs within 24 h. Importantly, mice deficient in either Sts-1 or Sts-2 individually display an intermediate phenotype, suggesting a redundant effect.
Previous studies have demonstrated a functional role for the Sts proteins as negative regulators of TCR signaling in T cells, FcεRI receptor signaling in mast cells, and GPVI signaling in platelets (23). However, since the predominant cell types associated with the early innate immune response to C. albicans infection within the kidney are phagocytes, including monocytes and neutrophils (8), our results suggest that the Sts proteins have important negative regulatory functions in a range of cell types broader than that previously identified. Alternatively, the enhanced functions of Sts-1/2−/− T cells could directly or indirectly stimulate the candidacidal behavior of responding phagocytes within Sts-1/2−/− mice, for example, through increased production of specific cytokines. In this regard, however, it is important to point out that we saw no significant differences in the levels of several important T cell cytokines within wild-type and mutant kidneys, with IFN-γ and IL-17 being among them (see Fig. S4 in the supplemental material). Interestingly, previous studies have demonstrated that the absence of T cells has an overall beneficial effect on the host response to systemic C. albicans infection, suggesting that T cell-inhibitory effects predominate (32). Studies are under way to address the in vivo role of Sts-1/2−/− hyperresponsive T cells on the protective phenotype.
This study yields important insight into the physiological functions of the Sts proteins, stemming from the observation that antifungal effects were the strongest in the kidney. It is currently unclear why this is the case, although we speculate that the internal architecture of the organ and the functional properties of resident phagocytes could be important determinants, along with the relative ratio of resident and/or incoming phagocytes to freshly seeded fungal cells. Although the number of kidney CFU increased through 48 h in both strains of mice, the fungal burden remained consistently lower in Sts-1/2−/− mice than wild-type mice. Simultaneously, the levels of kidney cytokines and chemokines were significantly lower, consistent with a reduced fungal burden. These data suggest a model in which the loss of Sts inhibitory functions leads to activation of innate immune responses important for fungal clearance in the kidney. Analysis of cytokine expression indicates that the absence of the Sts proteins creates a novel type of immunological environment in the kidney. For example, although there was no detectable increase in IFN-γ, TNF-α, or IL-6 expression in the Sts-1/2−/− kidney relative to that in the wild-type kidney at either 12 or 24 h postinfection, the level of the proinflammatory chemokine CXCL10 was significantly increased in Sts-1/2−/− kidneys at 12 h postinfection. CXCL10 is a T lymphocyte recruitment factor, and increased circulating levels of CXCL10 have been correlated with different inflammatory conditions (33, 34). CXCL10 is also known to be upregulated by a number of different cell types in response to various cytokines, and in many physiological situations it is thought to promote an immune response (35, 36). Because antibody neutralization of CXCL10 and its receptor, CXCR3, has no effect on the resistance phenotype of Sts-1/2−/− mice, it is likely that additional inflammatory mediators are also involved in protection. Altogether, these results demonstrate that a lack of Sts proteins results in a distinct inflammatory environment in the kidney that is more capable of restricting the growth of C. albicans.
The remarkable ability of Sts-1/2−/− mice to withstand lethal doses of C. albicans is all the more striking because fungal clearance does not occur in the context of an overall hyperinflammatory response. This contrasts with previous reports of increased resistance to C. albicans in mice lacking a functional receptor for type I interferons (Ifnar1−/− mice) or lacking the important neutrophil chemokine receptor CCR1 (11, 31). While both of these mutant mouse strains survived longer after they received low inocula because of decreased inflammatory damage, their kidney fungal burden remained very high during the entire course of infection. In both cases, decreased lethality was primarily associated with the absence of an important component of the inflammatory response rather than a decreased fungal burden, underscoring the deleterious nature of collateral tissue damage that can often occur during an overwhelming immune response. In contrast to these mouse models, mice lacking the CD37 tetraspanin were more resistant to C. albicans because they displayed more efficient fungal clearance from the kidney (37). However, their ability to resist infection occurred in the context of significantly increased levels of serum IgA and proinflammatory IL-6 by day 3 postinfection, both of which were concluded to aid in the clearance of C. albicans. CD37 associates with Dectin 1, a pattern recognition receptor that recognizes fungal cell wall β-glucan, and inhibits IL-6 production (38). Tellingly, another study demonstrated that elevated serum IgA levels in the absence of CD37 led to glomerular IgA deposition and increased levels of inflammatory phagocytes within the kidney, making CD37−/− mice prone to spontaneous nephropathy (39). This stands in marked contrast to the findings for Sts-1/2−/− mice, which displayed no spontaneous autoimmunity.
In sum, our results underscore the important role of the Sts proteins in regulating host responses to a deadly fungal pathogen and have the potential to contribute to the development of much needed new antifungal therapies. Importantly, the antifungal protection evident in Sts-1/2−/− mice is not accompanied by an overt hyperinflammatory response that would cause tissue damage. Indeed, mice tolerate Sts deficiency well, as it does not lead to deleterious side effects. Therefore, our studies identify the Sts proteins to be potential molecular targets for inhibitory molecules that could be used prophylactically in high-risk patients. We have also demonstrated a novel inflammatory environment within Sts-1/2−/− kidneys that promotes the early clearance of C. albicans. Further understanding of this environment could aid in the development of tailored immunological approaches that could be combined with current antifungal medications to achieve synergistic effects.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Stony Brook University and funds from the National Institutes of Health: R01AI080892 (to N.C.) and R01AI047837 (to J.B.K.). D.F. was partially supported by NIGMS grant T32GM007518-36.
We thank Laurie Levine and Jean Rooney for help with animal studies, Todd Rueb and Rebecca Connor for help with fluorescence-activated cell sorter analysis, Mallory Korman for histological sample preparation, and Dimitri Gnatenko for help with cytokine multiplex analysis. We also thank Jorge Benach, Jim Bliska, and all members of the N. Carpino and J. B. Konopka laboratories for insightful discussions.
We declare no financial conflict of interest.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02789-14.
REFERENCES
- 1.Pfaller MA, Diekema DJ. 2010. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol 36:1–53. doi: 10.3109/10408410903241444. [DOI] [PubMed] [Google Scholar]
- 2.Lionakis MS. 2014. New insights into innate immune control of systemic candidiasis. Med Mycol 52:555–564. doi: 10.1093/mmy/myu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayer FL, Wilson D, Hube B. 2013. Candida albicans pathogenicity mechanisms. Virulence 4:119–128. doi: 10.4161/viru.22913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 5.van de Veerdonk FL, Kullberg BJ, Netea MG. 2010. Pathogenesis of invasive candidiasis. Curr Opin Crit Care 16:453–459. doi: 10.1097/MCC.0b013e32833e046e. [DOI] [PubMed] [Google Scholar]
- 6.Brown GD, Denning DW, Levitz SM. 2012. Tackling human fungal infections. Science 336:647. doi: 10.1126/science.1222236. [DOI] [PubMed] [Google Scholar]
- 7.Ashman RB. 1997. Genetic determination of susceptibility and resistance in the pathogenesis of Candida albicans infection. FEMS Immunol Med Microbiol 19:183–189. doi: 10.1111/j.1574-695X.1997.tb01087.x. [DOI] [PubMed] [Google Scholar]
- 8.Lionakis MS, Lim JK, Lee CC, Murphy PM. 2011. Organ-specific innate immune responses in a mouse model of invasive candidiasis. J Innate Immun 3:180–199. doi: 10.1159/000321157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown GD. 2011. Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol 29:1–21. doi: 10.1146/annurev-immunol-030409-101229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng SC, Joosten LA, Kullberg BJ, Netea MG. 2012. Interplay between Candida albicans and the mammalian innate host defense. Infect Immun 80:1304–1313. doi: 10.1128/IAI.06146-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majer O, Bourgeois C, Zwolanek F, Lassnig C, Kerjaschki D, Mack M, Muller M, Kuchler K. 2012. Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog 8:e1002811. doi: 10.1371/journal.ppat.1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spellberg B, Ibrahim AS, Edwards JE Jr, Filler SG. 2005. Mice with disseminated candidiasis die of progressive sepsis. J Infect Dis 192:336–343. doi: 10.1086/430952. [DOI] [PubMed] [Google Scholar]
- 13.Perlin DS. 2009. Antifungal drug resistance: do molecular methods provide a way forward? Curr Opin Infect Dis 22:568–573. doi: 10.1097/QCO.0b013e3283321ce5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kullberg BJ, Verweij PE, Akova M, Arendrup MC, Bille J, Calandra T, Cuenca-Estrella M, Herbrecht R, Jacobs F, Kalin M, Kibbler CC, Lortholary O, Martino P, Meis JF, Munoz P, Odds FC, De Pauw BE, Rex JH, Roilides E, Rogers TR, Ruhnke M, Ullmann AJ, Uzun O, Vandewoude K, Vincent JL, Donnelly JP. 2011. European expert opinion on the management of invasive candidiasis in adults. Clin Microbiol Infect 17(Suppl 5):S1–S12. doi: 10.1111/j.1469-0691.2011.03557.x. [DOI] [PubMed] [Google Scholar]
- 15.CDC. 2013. Antibiotic resistance threats in the United States, 2013. CDC, Atlanta, GA: http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. [Google Scholar]
- 16.Armstrong-James D, Harrison TS. 2012. Immunotherapy for fungal infections. Curr Opin Microbiol 15:434–439. doi: 10.1016/j.mib.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 17.van de Veerdonk FL, Kullberg BJ, Netea MG. 2012. Adjunctive immunotherapy with recombinant cytokines for the treatment of disseminated candidiasis. Clin Microbiol Infect 18:112–119. doi: 10.1111/j.1469-0691.2011.03676.x. [DOI] [PubMed] [Google Scholar]
- 18.Spellberg B. 2011. Vaccines for invasive fungal infections. F1000 Med Rep 3:13. doi: 10.3410/M3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filler SG. 2013. Can host receptors for fungi be targeted for treatment of fungal infections? Trends Microbiol 21:389–396. doi: 10.1016/j.tim.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carpino N, Turner S, Mekala D, Takahashi Y, Zang H, Geiger TL, Doherty P, Ihle JN. 2004. Regulation of ZAP-70 activation and TCR signaling by two related proteins, Sts-1 and Sts-2. Immunity 20:37–46. doi: 10.1016/S1074-7613(03)00351-0. [DOI] [PubMed] [Google Scholar]
- 21.Thomas DH, Getz TM, Newman TN, Dangelmaier CA, Carpino N, Kunapuli SP, Tsygankov AY, Daniel JL. 2010. A novel histidine tyrosine phosphatase, TULA-2, associates with Syk and negatively regulates GPVI signaling in platelets. Blood 116:2570–2578. doi: 10.1182/blood-2010-02-268136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Castro RO, Zhang J, Groves JR, Barbu EA, Siraganian RP. 2012. Once phosphorylated, tyrosines in carboxyl terminus of protein-tyrosine kinase Syk interact with signaling proteins, including TULA-2, a negative regulator of mast cell degranulation. J Biol Chem 287:8194–8204. doi: 10.1074/jbc.M111.326850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsygankov AY. 2013. TULA-family proteins: a new class of cellular regulators. J Cell Physiol 228:43–49. doi: 10.1002/jcp.24128. [DOI] [PubMed] [Google Scholar]
- 24.Mikhailik A, Ford B, Keller J, Chen Y, Nassar N, Carpino N. 2007. A phosphatase activity of Sts-1 contributes to the suppression of TCR signaling. Mol Cell 27:486–497. doi: 10.1016/j.molcel.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.San Luis B, Sondgeroth B, Nassar N, Carpino N. 2011. Sts-2 is a phosphatase that negatively regulates zeta-associated protein (ZAP)-70 and T cell receptor signaling pathways. J Biol Chem 286:15943–15954. doi: 10.1074/jbc.M110.177634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cieniewicz B, Carpino N, Krug LT. 2014. Enhanced response of T cells from murine gammaherpesvirus 68-infected mice lacking the suppressor of T cell receptor signaling molecules Sts-1 and Sts-2. PLoS One 9:e90196. doi: 10.1371/journal.pone.0090196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Douglas LM, Wang HX, Keppler-Ross S, Dean N, Konopka JB. 2012. Sur7 promotes plasma membrane organization and is needed for resistance to stressful conditions and to the invasive growth and virulence of Candida albicans. mBio 3(1):e00254–11. doi: 10.1128/mBio.00254-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ngo LY, Kasahara S, Kumasaka DK, Knoblaugh SE, Jhingran A, Hohl TM. 2014. Inflammatory monocytes mediate early and organ-specific innate defense during systemic candidiasis. J Infect Dis 209:109–119. doi: 10.1093/infdis/jit413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashman RB, Papadimitriou JM. 1995. Production and function of cytokines in natural and acquired immunity to Candida albicans infection. Microbiol Rev 59:646–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacCallum DM, Castillo L, Brown AJ, Gow NA, Odds FC. 2009. Early-expressed chemokines predict kidney immunopathology in experimental disseminated Candida albicans infections. PLoS One 4:e6420. doi: 10.1371/journal.pone.0006420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lionakis MS, Fischer BG, Lim JK, Swamydas M, Wan W, Richard Lee CC, Cohen JI, Scheinberg P, Gao JL, Murphy PM. 2012. Chemokine receptor Ccr1 drives neutrophil-mediated kidney immunopathology and mortality in invasive candidiasis. PLoS Pathog 8:e1002865. doi: 10.1371/journal.ppat.1002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahanty S, Greenfield RA, Joyce WA, Kincade PW. 1988. Inoculation candidiasis in a murine model of severe combined immunodeficiency syndrome. Infect Immun 56:3162–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lacotte S, Brun S, Muller S, Dumortier H. 2009. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci 1173:310–317. doi: 10.1111/j.1749-6632.2009.04813.x. [DOI] [PubMed] [Google Scholar]
- 34.Antonelli A, Ferrari SM, Giuggioli D, Ferrannini E, Ferri C, Fallahi P. 2014. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun Rev 13:272–280. doi: 10.1016/j.autrev.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Groom JR, Luster AD. 2011. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 89:207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu M, Guo S, Hibbert JM, Jain V, Singh N, Wilson NO, Stiles JK. 2011. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev 22:121–130. doi: 10.1016/j.cytogfr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Spriel AB, Sofi M, Gartlan KH, van der Schaaf A, Verschueren I, Torensma R, Raymakers RA, Loveland BE, Netea MG, Adema GJ, Wright MD, Figdor CG. 2009. The tetraspanin protein CD37 regulates IgA responses and anti-fungal immunity. PLoS Pathog 5:e1000338. doi: 10.1371/journal.ppat.1000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer-Wentrup F, Figdor CG, Ansems M, Brossart P, Wright MD, Adema GJ, van Spriel AB. 2007. Dectin-1 interaction with tetraspanin CD37 inhibits IL-6 production. J Immunol 178:154–162. doi: 10.4049/jimmunol.178.1.154. [DOI] [PubMed] [Google Scholar]
- 39.Rops AL, Figdor CG, van der Schaaf A, Tamboer WP, Bakker MA, Berden JH, Dijkman HB, Steenbergen EJ, van der Vlag J, van Spriel AB. 2010. The tetraspanin CD37 protects against glomerular IgA deposition and renal pathology. Am J Pathol 176:2188–2197. doi: 10.2353/ajpath.2010.090770. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.