

Abstract
An undetermined feature of Staphylococcus aureus pathogenesis is its persistence and then relapse of disease. This has been explained by its switch to alternative lifestyles, mainly as biofilm or small-colony variants (SCVs). Studying the native characteristics of SCVs has been problematic due to their reversion to the parental lifestyle. We have observed that for a number of S. aureus strains as they switch to an SCV lifestyle, there is the formation of an extracellular matrix. We focused our analysis on one strain, WCH-SK2. For bacterial survival in the host, the combination of low nutrients and the prolonged time frame forms a stress that selects for a specific cell type from the population. In this context, we used steady-state growth conditions with low nutrients and a controlled low growth rate for a prolonged time and with methylglyoxal. These conditions induced S. aureus WCH-SK2 into a stable SCV cell type; the cells did not revert after subculturing. Analysis revealed these cells possessed a metabolic and surface profile that was different from those of previously described SCVs or biofilm cells. The extracellular matrix was protein and extracellular DNA but not polysaccharide. The SCV cells induced expression of certain surface proteins (such as Ebh) and synthesis of lantibiotics while downregulating factors that stimulate the immune response (leucocidin, capsule, and carotenoid). Our data reveal cell heterogeneity within an S. aureus population and under conditions that resemble long-term survival in the host have identified a previously unnoticed S. aureus cell type with a distinctive metabolic and molecular profile.
INTRODUCTION
Staphylococcus aureus has an incredible ability to survive, either by adapting to environmental conditions or defending against exogenous stress. In part, this ability is provided by the breadth of lifestyles or modes of growth S. aureus can adopt. Key to an understanding of chronic, persistent, and relapsing S. aureus infections is determining the basis for their switch to quasi-dormant lifestyles. Across different bacterial species, these alternative lifestyles form a population known as persister cells (1). It has been proposed that while within their host, a subpopulation of S. aureus survives host-generated and therapeutic antimicrobial stresses by inducing biofilm growth on host tissue or by growing as small-colony variants (SCVs). It is likely this is not an on-off switch, from planktonically growing cells to a biofilm or likewise to SCVs, but a continuum of cell types; the bacterial population will have the potential for a diverse range of lifestyles defined by different metabolic pathways and surface structures. In a multicellular biofilm, the metabolically quiescent bacterial community produces a highly protective extracellular polymeric substance (EPS). The EPS is variously composed of polysaccharides (mainly the ica operon-encoded polysaccharide intercellular adhesin [PIA]), extracellular DNA (eDNA), and protein, and its protection results in persistent bacterial infections (2). S. aureus forms biofilms in different human tissues, and to some degree its associated EPS has been studied (3, 4). In clinical settings, SCVs of S. aureus have been observed for many years (5). When cultured, these cells form nonpigmented colonies that are ca. 10 times smaller than their counterparts on agar plates. They have long been associated with persistent infections, with intrinsic resistance to antibiotics, and with relapsing infections by S. aureus (6, 7). Largely it is difficult to clearly study these variants or to define their native properties because they readily revert to their parental cell type. Mutations in hemin and menadione biosynthesis (hemB and menA) have produced laboratory-generated SCVs, and these and other mutations have been studied (8–11). There have now been enough studies to implicate a breadth of potential pathways involved at some level in the development of SCVs as a response to harsh conditions, ribosomal proteins, RNA processing, stringent response, aerobic/anaerobic growth pathways, reduced virulence factors, and an intracellular life cycle. (These ideas are excellently reviewed in references 12 and 13.) The presence of gentamicin also impedes S. aureus metabolism and results in SCVs. Both methods do result in stable forms of SCVs, but these are artificially generated. Various genotypic factors (single nucleotide polymorphisms, mutations, and gene deletions) have now been identified to attempt to characterize S. aureus SCVs (10, 14–16). S. aureus SCVs have now been clearly associated with different diseases, and importantly these include chronic infections, such as those in patients with cystic fibrosis, chronic rhinosinusitis, endocarditis, and chronic osteomyelitis (17–19). In the context of bacterial survival, what has been shown to be a key part of these chronic infections is the long-term nature of infection and time-dependent genetic changes (20). Also important are the physical and chemical natures of conditions of the niche and how the bacteria are able to grow under these conditions. For S. aureus and their SCVs, in the context of the lung and during colonization of the nasal passage, it is known that nutrients will be limiting (hypoxia and iron limitation, for instance) but also there will be antimicrobial agents and interspecies competition (21, 22). For nasal carriage, for example, there are no common genetic or phenotypic traits that define a successful colonizing S. aureus strain from one that is unable to colonize. In addition to this, there are host factors and the microbiome of the nasal passage (23, 24). Certainly bacterial growth is inhibited by this combination of factors in the nasal passage, which means bacterial growth is slow. The chemical components of host tissue have been analyzed, and the exact parameters of bacterial growth in the host have to some extent been studied (21, 25–27). In the lung, the ability of an infecting bacterial species to grow (and its consequent growth rate) will be determined by host factors, such as the immune response (28) and other bacteria present (29). For Streptococcus pneumoniae, there have been different calculations of its growth rate in the lung (27, 30), but a doubling time between 2 and 3 h seems likely. From studies with S. aureus in the host, there is large variation in the growth rates. In the lung, it has been suggested that while S. pneumoniae does colonize, it has a very low growth rate and may only have a doubling time close to or even more than 24 h; in osteomyelitis, the generation time can be between 8 and 24 h (25, 26). In studies on the lungs of cystic fibrosis patients (where S. aureus SCVs are present), there are strain-specific traits that allow adaptation to the lung over a prolonged time frame (these have reinforced this time factor as being important), and with sites around the anatomically heterologous lung tissue, bacteria will encounter various levels (starting from no response) of the immune response and interspecies competition, antibiotics, as well as hypoxia and nutrient starvation (31, 32). In the nasal passage and the lung, there will be low nutrients, and the bacterial growth will be low. When bacteria are within a host niche, either during colonization or when existing in a state of persistence, there are factors such as the nutrient level and subsequent bacterial growth rate and the impact of the milieu of chemicals that are vastly different from those when bacteria are in batch culture growth. S. aureus, with or without an immune response, will encounter these factors as an environmental stress and therefore be under the selective pressure of limiting nutrients and low growth rate, and then the prolonged time frame will be important. Continuous culture has been recognized as providing these conditions; independent of growth phase (33, 34). While tissue culture, animal models, and bacterial culture conditions will permit the survival and study of certain types of S. aureus cells, they will omit others. The use of a chemostat to allow prolonged growth analysis of S. aureus with the stresses of low nutrient and low growth rate will permit other cell types to exist in the population. Based on the literature on S. aureus growth in host infections discussed above, the combination of a prolonged time component with limiting nutrients and a low growth rate would be relevant to a study of S. aureus SCV development.
MATERIALS AND METHODS
Bacterial strains and batch growth conditions.
Clinical isolates were obtained from patients' samples at the Women and Children's Hospital (Adelaide, Australia). Staphylococcus aureus WCH-SK2 is a blood isolate. For routine growth, bacterial cultures were incubated overnight at 37°C in 5% CO2 on tryptone soya broth (TSB) (Oxoid, Melbourne, Australia) with 1.2% agar. Cells were inoculated into 10 ml TSB broth in a 100-ml flask and incubated at 37°C with shaking (200 rpm) overnight.
Analysis in batch culture of SCV colony type and growth kinetics.
To screen clinical isolates for small-colony variants (SCVs), bacterial strains were grown in 10 ml TSB for 8 h, and 20, 50, and 100 μl were plated onto chemically defined medium (CDM) (35), Columbia blood agar (Oxoid Melbourne, Australia), or TSB and incubated overnight at 37°C in 5% CO2. Colonies were observed at 24 h and then left to incubate for a further 24 h to allow the development of SCVs. Colonies were measured at these fixed time points and classified into non-SCVs or SCVs. The batch response to methylglyoxal was determined by growing the bacteria in preculture for 1 to 2 h and inoculating them into a 96-well plate with various concentrations of the specific chemical stress (such as methylglyoxal). These were incubated overnight in an incubating plate reader with shaking at 37°C (BioTek ES260), and the optical density at 630 nm (OD630) was assessed every 0.5 h. Each assay was performed in triplicate. Cells were then plated as described above, and colony type was assessed.
For analysis of the growth requirements and subsequent growth kinetics of S. aureus WCH-SK2, we established a highly chemically defined medium (CDM) that was based on the published HHW medium for slime production by coagulase-negative staphylococci (35). We placed the CDM constituents into defined groups (see Table S1 in the supplemental material), and bacteria were inoculated from an overnight preculture into CDM containing various amounts of certain amino acids or glucose. The culture was grown at 37°C (200 rpm) for 24 h, and growth was monitored by measuring the OD600 at 1-h time points. It is established that the amino acids arginine, proline, and glutamic acid are required for S. aureus growth, so these were added to CDM at increasing concentrations. Accordingly the concentrations of the three amino acids and glucose were set so that growth rate (μ) was proportional to the dilution rate (D) of CDM into the chemostat culture vessel (Fig. 1) (36). We could then change the growth rate relative (μrel) to the organism's maximum growth rate (μmax). The μmax was determined by growing WCH-SK2 under batch culture in the chemostat using the modified CDM. The μmax or maximum generation time (Tg) in the log phase was 1.03 h. Using the relationship Tg = ln2/D, we initially maintained WCH-SK2 at a growth rate of μrel = 0.75 (Tg = 1.37 h) for 10 generations. We then changed the growth rate to μrel = 0.15 (Tg = 6.87 h) and allowed the culture to reach the steady state. These two growth rates were chosen because within the host and most anatomical niches there is likely to be a change in the availability of nutrients during colonization and the transition from health to disease. The initial analysis revealed that when grown under continuous culture, S. aureus WCH-SK2 produced a mixture of large colonies and SCV colonies, and the proportion of SCVs increased dramatically from 1 to 3% (μrel = 0.75) of the population to 20% at μrel = 0.15 (Fig. 2A).
FIG 1.

Growth requirements of S. aureus strain WCH-SK2. Growth-limiting nutrients were determined by changing the concentrations of arginine, glutamic acid, and proline (A) and then glucose (B) in CDM. The final concentration of ingredients in the CDM used is shown in Table S1 in the supplemental material. The growth increased proportionally to the increased concentrations of the three amino acids (AA) and glucose.
FIG 2.

The growth rate in CDM alters colony type. Colonies observed from S. aureus WCH-SK2 cells grown in batch culture were large pigmented colonies, and with a growth rate of μrel = 0.75, the pigmentation was reduced. A growth rate of μrel = 0.15 produced small, nonpigmented colonies ranging in size from 3 mm to <1 mm. The growth in a chemostat over time naturally selects for SCV cells in the population.
Determination of the MIC of methylglyoxal.
The MIC of methylglyoxal was assayed using the broth microdilution technique for MIC determination for antibiotics. In brief, 5 ×105 CFU were inoculated into each well (of a 96-well plate) containing 250 μl of CDM, and the added methylglyoxal was diluted with a dilution factor of 1/4. The MIC value was recorded as the lowest concentration at which there was no visible bacterial growth. One hundred microliters from the wells was plated on tryptone soya agar (TSA), and the number of CFU/ml was assessed.
Continuous culture growth in chemostat.
Starter cultures of S. aureus WCH-SK2 were grown in 20 ml of TSB as described above. This was directly inoculated into the chemostat (BioFlo C30; New Brunswick Scientific, Edison, NJ), with a working volume of 365 ml. The culture was grown under batch conditions to assess the maximum growth rate during log phase. The medium pump was then switched on to initiate continuous culture. Samples were taken from the chemostat and plated onto TSB agar after serial dilution to determine cellular viability (CFU/ml). When the culture had reached the steady state (10 generations), the growth rate (μ) was equal to the dilution rate (D) of the medium flowing into the culture vessel. Based on the equation Tg = ln2/D, the medium flow rate was initially set to 182.5 ml/h, where D = 0.50 h−1 and Tg = 1.37 h (μrel = 0.75), and the culture was allowed to reach the steady state once again. The dilution rate was then reduced to give μrel = 0.15 (D = 0.10 h−1 and Tg = 6.87 h). This lower growth rate was used as the best representation of the expected in vivo growth (37).
Samples were taken each day and were plated to determine CFU/ml and assess colony phenotype and for scanning electron microscopy (SEM) analysis and biofilm assays. The initial study monitored the growth for 10 days, moving from batch culture to continuous culture at the higher growth rate followed by the low growth rate. The subsequent experiment monitored the bacteria over 60 days, adding the methylglyoxal chemical stress at day 30 (0.0078%). This concentration was a sublethal dose in the batch culture. Methylglyoxal was added to the growth medium reservoir, and therefore the concentration slowly increased in the culture vessel over time. At day 10 after methylglyoxal was added at 0.0078%, the concentration in the growth medium was increased to 0.031% methylglyoxal (which is higher than the MIC). The growth and phenotype of the bacteria were monitored until day 60.
SCV characterization.
Quantitative assessment of SCVs was performed by measuring the colony size after plating on TSA plates after 48 to 72 h of incubation. SCVs were defined variants producing colonies with a diameter of <1 mm (1/5 to 1/10 the normal size of colonies) with reduced pigmentation and hemolytic activity (38). The percentage of SCVs within the population was determined by the number of SCVs per total number of colonies (CFU/ml). The colonies with a size of >1 mm with pigmentation were recorded as normal colonies.
Species identification of SCVs.
To confirm the identity of small colonies as S. aureus and not contaminants, 16S rRNA gene sequencing was applied to SCVs and normal colonies. The genomic DNA of these colonies was extracted and purified using the Wizard Genome DNA purification kit (Promega). The Universal 16S rRNA gene primers used for PCR were 27F (AGAGTTTGATCMTGGCTCAG) and 1492R (TACGGYTACCTTGTTACGACTT). The PCR products were purified and sequenced by the IMVS Royal Adelaide Hospital. The sequences obtained were checked on BLAST/NCBI to determine the identification. All cells were identified as S. aureus.
Determination of reverted SCVs.
SCVs were tested for reversion by subculturing individual colonies onto TSA plates overnight under non-stress conditions (37°C, 48 to 72 h). The capacity of the SCV colonies to revert to normal large colonies was recorded if there was the change in size (diameter of >1 mm, pigmented, and hemolytic). Colonies that matched the SCV criteria underwent several cycles of subculturing on TSA plates. Stable SCVs were assessed as remaining as SCVs after 10 cycles of subculturing.
SEM.
Scanning electron microscopy (SEM) was performed on bacterial samples taken at the described at time points from the chemostat and compared to batch-grown cells and cells stressed with methylglyoxal. The bacterial cells were filtered through 0.2-μm-pore Whatman filter, and the filter paper was then fixed with fixative solution (4% paraformaldehyde, 1.25% glutaraldehyde in phosphate-buffered saline [PBS] plus 4% sucrose [pH 7.2]). Filter papers were then washed with PBS–4% sucrose and postfixed for 1 h with 0.1% osmium tetraoxide before undergoing 10-min dehydration steps in 70%, 90%, and 100% ethanol baths. The samples were then dried (Bal-tec critical point dryer CPD030) and mounted on a stub for coating with platinum. The images were then examined using a Phillips XL30 field emission scanning electron microscope.
Biofilm assay.
The ability of the different cells to form a biofilm was assayed using a standard 96-well polystyrene plate assay (2). Mid-exponential-phase (OD600, 0.5 to 0.8) S. aureus cells were inoculated into 250 μl TSB and incubated for 24 h at 37°C with shaking. Planktonic cells were removed by washing, and biofilm cells were visualized by staining with 0.1% crystal violet. Ethanol-acetone (20:80 [vol/vol]) was added, and OD630 values were read (Biotek EL808 spectrophotometer). Each sample was performed with eight replicates. The values presented are an average, and errors are presented as the standard deviation (SD).
Determination of components of the extracellular matrix.
To identify the extracellular DNA (eDNA), SYTO 9 (green) and propidium iodide (PI [red]) (BacLight bacterial viability kit; Molecular Probes, Life Technologies) were mixed and diluted following the manufacturer's instructions. Alexa 488-conjugated lectins (Molecular Probes, Life Technologies) were used to characterize the polysaccharide component of the EPS matrix. This consisted of 5 fluorescently conjugated lectins, including concanavalin A (50 μg/ml), lectin GS-II from Griffonia simplicifolia (100 μg/ml), wheat germ agglutinin (100 μg/ml), lectin PNA (50 μg/ml) from Arachis hypogaea (peanut), and lectin SBA (50 μg/ml) from Glycine max (soybean). Three microliters of PI was added per ml of the lectin cocktail to differentiate between eDNA and polysaccharide.
After staining, samples were incubated in the dark for 15 to 30 min and then were applied as spots on glass slides and viewed with an Olympus IX-70 microscope using a 100× objective. Fluorescence and phase-contrast images were captured, and false colors were merged with the Metamorph software program (version 7.7.3.0; Molecular Devices).
Transcriptomics.
Whole-cell gene expression profiles were compared using transcriptome sequencing (RNA-seq) techniques. Samples included batch culture cells, the SCV population from the chemostat with 0.0078% methylglyoxal, and the stable SCV cells produced from the chemostat with 0.031% methylglyoxal. To prevent RNA from degradation, cells were directly added to RNAProtect (Qiagen). The ratio used was 1:1 1/5 of the total cell culture volume to RNAProtect. This was left on ice for 2 h before being centrifuged (4,000 × g) for 5 min at 4°C, and then the supernatant was discarded. The cell pellet was kept at −80°C for RNA extraction. RNA was extracted using RNAeasy minikit according to the RNAeasy minikit standard protocol (Qiagen). The RNA quality of the samples was checked with the Agilent Bioanalyzer according to the Agilent RNA 6000 Nano kit standard protocol: samples were loaded into the RNA Nano chip and run using an Agilent 2100 Bioanalyzer. For each sample, three biological replicates of cell growth, harvesting, and RNA extraction were performed. The RNA was pooled and provided to the Adelaide Cancer Genomic Research Facility (Adelaide Australia) for library preparation and sequencing (RNA-seq) using the Ion Proton platform (Ion Torrent; Life Technologies).
The analysis pipeline used Bowtie2 (39) to align reads from both samples to the S. aureus NCTC8325 reference genome (GenBank accession no. NC_007795), followed by processing with SAMtools and BEDTools to generate a mapped read count for the reference genes from each sample. Differential expression analysis was performed using the R program within the package edgeR and DESeq. Based on the analysis with R, the genes that were statistically significant using the Benjamini-Hochberg procedure with a false discovery rate controlled at <0.1 were recorded in the Results section.
Membrane proteomics.
The growing cells sampled from the chemostat were pelleted by centrifugation (8,000 × g) for 10 min at 4°C and then washed twice with ice-cold (TBS) buffer (50 mM Tris, 150 mM NaCl [pH 8.0]). The cell pellets were resuspended in lysis buffer containing 50 mM Tris, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 U RNase, and 40 U DNase (Sigma, St. Louis, MO). The cells were then lysed using two cycles through a French pressure cell (SLM Aminco, Inc., Australia) at 12,000 lb/in2, and the lysate was kept at room temperature for 30 min and then centrifuged twice (8,000 × g at 4°C for 10 min). The supernatant was collected and stored at −20°C. The protein concentration was measured using a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA).
The protein extract was pelleted by centrifugation at 45,000 × g for 1 h to pellet the membranes. The supernatant was discarded, and the crude membrane protein was resuspended in 1 ml of carbonate buffer (200 mM Na2CO3 [pH 11.0]) and then incubated for 1 h on ice with intermittent mixing every 15 min using an insulin syringe. Following incubation, solid urea was added to a concentration of 8 M, and the protein reduction was performed at 50°C in 5 mM tris(2-carboxyethyl) phosphine hydrochloride for 30 min followed by alkylation in 10 mM iodoacetamide in the dark for 15 min at room temperature. Proteinase K was then added in an enzyme/protein ratio of 1:50 followed by incubation for 15 to 18 h in a ThermoMixer (37°C at 200 rpm). One volume of 10% acetonitrile in water was added before the solution was cooled on ice for 15 min. Following centrifugation for 1 h (45,000 × g at 4°C), the supernatant was discarded. To remove residual urea, the pellet was rinsed with 50 mM triethylammonium bicarbonate (TEAB [pH 7.8]) buffer and again centrifuged (45,000 × g at 4°C for 1 h). For the chymotrypsin digest, the pellet was resuspended in 200 μl of digestion buffer (50 mM TEAB [pH 7.8], 10 mM CaCl2, 0.5% RapiGest [Waters, Milford, MA]) before 4 μg (>40 U/mg) chymotrypsin (Sigma) was added. The digestion was carried out while shaking (200 rpm) at 30°C for 6 to 10 h. The sample was centrifuged (20,000 × g) three times for 15 min at 4°C, each time keeping the supernatant that contained the peptides produced by the chymotryptic digest. Two microliters of isotope-coded protein label 0 (ICPL0) was added to the digested peptides from the naturally stable SCVs (nsSCVs) at 0.031% methylglyoxal, and 2 μl of ICPL6 (ICPL duplex kit; Serva, Germany) was added to the digested peptides from cells from batch culture. Argon was used to overlay both samples. The samples were vortexed for 30 s and then sonicated for 1 min. The samples were then incubated for an hour at room temperature. The samples were incubated for another hour at room temperature. Two microliters of the STOP solution (Serva ICPL duplex kit) was added, and the mixture was incubated at 20 min to destroy excess reagent.
The two ICPL-labeled samples were then pooled and cleaned using a C18 spin column. The resulting peptide mixture was then prepared for liquid chromatography-tandem mass spectrometry (LC-MS/MS) by centrifugal lyophilization and resuspension in 0.1% trifluoroacetic acid (TFA)–5% CH3CN.
Peptides were separated on a high-performance liquid chromatography (HPLC) system (Thermo Scientific) using a Thermo Scientific separation column (Acclaim PepMap RSLC) (C18; pore size, 100 Å; particle size, 2 μm; 75-μm inside diameter [i.d.] by 15-cm length) and a Thermo Scientific Acclaim PepMap100 trapping column (C18; pore size, 100 Å; particle size, 3 μm; 75-μm i.d. by 2-cm length). The HPLC system was coupled to a LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Berman, Germany), using the following buffer system: buffer A, 2% acetonitrile (ACN) plus 0.1% formic acid (FA) in water; buffer B, 80% ACN plus 0.1% FA in water. For in-line desalting and concentration, 2 μl of the sample was loaded onto the trap column and then washed for 5 min with 100% buffer A at a flow rate of 5 μl/min. Peptides were eluted at a flow rate of 300 nl/min with the following gradient: 4% buffer B for 10 min, gradient to 40% B over 115 min, gradient to 60% B over 10 min, gradient to 90% B over 5 min, 90% B for 5 min, gradient from 90% to 4% B in 30 s, and 4% B for 30 min. The effluent from the HPLC column was directly electrosprayed into the mass spectrometer. The LTQ Orbitrap XL instrument was operated in data-dependent mode to automatically switch between full-scan MS and MS/MS acquisition. Instrument control was through Thermo Tune Plus and Xcalibur software (Thermo Scientific). Full-scan MS spectra were acquired over the m/z range 300 to 2,000. In each scan, the six most abundant peptide ions with charge states of ≥2 were sequentially isolated and subjected to collision-induced dissociation (CID).
The raw data file was first converted into the open mzXML format and subsequently to mgf using MSConvert. The mgf file containing CID spectra was then submitted to MASCOT (version 2.3.02; Matrix Science, Inc., Boston, MA) for peptide identification. The mzXML file was processed for determination of ICPL6/ICPL0 ratios using ICPL-ESI Quant (Max Planck Institute for Biochemistry).
RNA-seq accession number.
The RNA-seq data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (55) and are accessible through GEO Series accession number GSE63750.
RESULTS AND DISCUSSION
The development of S. aureus SCV is linked to cell-cell interactions.
We previously screened a set of 79 S. aureus isolates for their ability to form SCVs (40). This includes 72 clinical isolates (Women's and Children's Hospital; WCH-SK1 to WCH-SK72) and 7 reference strains (strains previously studied: the USA300 linage strain JE2, Mu50, Mu3, NCTC8325, COL, Newman, and Sanger 252). We used a range of chemicals that would be present in some contexts in the host-pathogen environment: methylglyoxal, glycolaldehyde, glyceraldehyde, acrolein, paraquat, hydrogen peroxide, and S-nitrosoglutathione (GSNO). Under these conditions, 26.6% of the strains induced biofilm under stress, and 60.3% of the strains switched to SCV forms under stress (40). We now have observed the cells (by SEM) of the strains that did induce an SCV cell type when treated with the different chemical stresses (see Fig. S1 to S3 in the supplemental material). We unusually observed that the activities associated with the development of SCV were cell-cell interactions (or an extracellular matrix). This occurred in more than 55% of the SCV strains observed. We chose one of these strains (WCH-SK2) for a deeper analysis of the development of SCV and a characterization of the SCV state.
The growth requirements of S. aureus WCH-SK2.
We chose one strain (WCH-SK2) to study its development of SCV and then to maintain the SCV cells for analysis. To achieve this, we aimed to grow the cells under conditions that permitted control of growth parameters and growth rate, continuous culture in a chemostat. We needed to study the cellular and physiological characteristics of the S. aureus clinical isolate WCH-SK2 under specific growth conditions. We then aimed to use growth-phase-independent and nutrient-limiting conditions in a chemically defined medium (CDM) in a chemostat. This initially required the determination of its growth requirements. The CDM used was modified from HHW (35) (see Table S1 in the supplemental material) by iteratively removing potential carbon, nitrogen, and energy sources followed by the controlled addition of particular nutrients while measuring changes in cellular yield (Fig. 1). This defined a specific CDM to enable the growth rate of S. aureus WCH-SK2 to be accurately adjusted during continuous culture in a chemostat. The amino acids arginine, proline, and glutamic acid were essential for growth, and when supplied in higher concentrations could also be used to produce energy (ATP). Alternatively, when the three amino acids were present in low concentrations, the glucose concentration could also be adjusted to increase cellular yield and maintain growth rate (Fig. 1).
Steady-state growth of S. aureus WCH-SK2.
We initially grew WCH-SK2 as a batch culture in the chemostat until the log phase had been achieved, measuring growth rate and other features of the growth (Materials and Methods). The medium pump was then turned on, and growth was allowed to reach the steady state (10 generations). Within a host niche, bacterial generation times are broadly thought to range from 2 to 24 h (37), and consequently bacterial systems that are functional are very likely to be vastly different from those seen when the cells are growing near their maximum growth rate in batch culture (41). We initially simply controlled the growth rate (using a high growth rate of μrel = 0.75 and a low growth rate of μrel = 0.15) of S. aureus WCH-SK2 and observed the effect on the colonies formed. Controlling the growth rate altered the predominant colony type of the population (Fig. 2). Cells grown in the steady state at a high growth rate did not produce SCVs.
Long-term growth of S. aureus WCH-SK2 under limiting conditions.
It is acknowledged that successful colonization by S. aureus requires growth in the host environment for prolonged periods. We therefore undertook an analysis of growth of S. aureus WCH-SK2 in a long-term, steady-state study using low nutrients and a low growth rate and then introducing methylglyoxal, a resident chemical stress. Methylglyoxal is inevitably present in the host-pathogen milieu of toxic chemicals, and this is with or without an immune response (42).
We maintained the cells in steady-state (at μrel = 0.15) over 209 generations (60 days), adding the chemical stress (methylglyoxal) at day 30 (Fig. 3). We had previously determined in batch culture studies the MIC of WCH-SK2 to methylglyoxal (Table 1). Over the first 30 days, the population changed in colony type from large (4 to 5 mm in diameter) and either slightly pigmented or nonpigmented to a mixture of nonpigmented colonies with diameters of ca. 3 mm to smaller colonies with diameters ranging from 1 to 2 mm (Fig. 3). The proportion of SCVs within the bacterial population stabilized at ca. 20% by day 30 (Fig. 3). We added methylglyoxal at two concentrations. A concentration below the MIC (0.0078%) was added, and while there was little effect on cellular viability over the next 20 days (69.5 generations in Fig. 3A), there was an increase in SCVs within the population. At day 50, the concentration of methylglyoxal was increased to a level exceeding the MIC (0.031%) (Fig. 3A). At this concentration, there was a large increase in SCVs. The addition of the chemical stress appeared to cause an initial fall in cell viability before recovering. This suggests that there had been a selection for a cell type that favored cells with an ability to continue to grow under these conditions with a concentration of methylglyoxal above the MIC. Despite this stress, there was continued growth, now with a percentage of SCVs of almost 80% and cells covered in an extracellular matrix (Fig. 3A). Even before the addition of methylglyoxal on day 30, there was some observed extracellular matrix (Fig. 3C). It should be noted that we have performed a separate study using a continuous culture growth analysis with a high growth rate: there was little or no production of SCVs, and cells were killed by the addition of methylglyoxal at the higher concentration (0.03% [data not shown]). The SCV cells taken from bacterial population in the chemostat within this time frame reverted during subculturing to the parental cell type. However, after addition of methylglyoxal (post-day 50), the SCVs did not revert and remained as SCVs over several subcultures: these cells are a stable SCV cell type. The combination of stresses favored S. aureus to be in this stable SCV state. The process we have applied (consistent with colonization and infection), involving low nutrients and low growth rate, has enabled the inherently slow-growing SCVs to compete in the population, and further stresses (methylglyoxal and time) have selected for a specific cell type to become dominant in the population. The stable SCV cells did not appear as smaller cells (Fig. 3E), but after specifically taking the smaller colonies from the population (at day 55), we observed that these stable SCV cells had a lower growth rate (Fig. 4A).
FIG 3.

Prolonged steady-state growth induces SCV in S. aureus WCH-SK2. (A) Growth observed over 60 days in continuous culture. Cell numbers (CFU/ml) were enumerated each day, as was the percentage of SCVs in the population (blue bars). Data are given as means ± standard deviation (SD) (n = 3 replicates). Methylglyoxal (0.0078%) was added at day 30 (red bars). At day 50, methylglyoxal was increased to 0.031% (green bars). Photographs show representative colonies on TSA plates (right panel) and cells by SEM at 12,000× (middle panel) and 35,000× (left panel), as observed from day 1 (B), day 20 (C), day 35 (D), and day 50 (E).
TABLE 1.
MIC of methylglyoxal for S. aureus WCH-SK2 in batch culture
| Methylglyoxal MIC (% [wt/vol]) | Mean OD630 | Viabilitya |
|---|---|---|
| 0.5 | 0.052 | − |
| 0.25 | 0.051 | − |
| 0.125 | 0.053 | − |
| 0.06 | 0.052 | − |
| 0.03 | 0.055 | − |
| 0.015 | 0.06 | + |
| 0.008 | 0.11 | + |
| 0.004 | 0.26 | + |
| 0.002 | 0.48 | + |
| 0.001 | 0.45 | + |
| 0.0005 | 0.45 | + |
| 0 | 0.46 | + |
−, not viable; +, viable.
FIG 4.

The S. aureus WCH-SK2 SCV cells from the chemostat in the presence of methylglyoxal produce stable SCVs. (A) After addition of 0.031% methylglyoxal to the chemostat (day 50), the population was represented by approximately 80% SCVs (SCV-type colonies) and larger colonies (Non-SCV colonies [still nonpigmented colonies]). These were separately assessed. Growth was assayed in complex, rich medium (TSB) and the CDM used in the chemostat. The SCV-type colonies displayed a low growth rate. (B) After several cycles of subculture, the SCV type cells remained as small colonies on agar plates (left panels); these stable SCVs are the stable SCV cell type. The cells appeared the same size as the non-SCV cells but maintained the unique extracellular matrix (as viewed by SEM at 35,000× [right panel]).
The S. aureus WCH-SK2 stable SCV cells are covered in an extracellular matrix.
Having induced and then generated a stable SCV cell type in WCH-SK2, we were then uniquely able to study its characteristics. It should be noted that the other strains (such as NCTC8325) that showed a chemically induced SCV form and cell-cell interactions (or extracellular matrix [see Fig. S1 to S3 in the supplemental material]) could not accurately be assayed as they reverted to their non-SCV form during the experiments. Cell samples were taken from the chemostat each day of the 60-day experiment, and examination of the cells (SEM) revealed the formation of an extracellular matrix (Fig. 3B to E). The stable SCV cell community (at days 50 to 60) was embedded within this matrix (Fig. 3E). This extracellular matrix was maintained during subsequent subculturing (on TSB agar plates) and was observed in the smaller colonies sampled and noted to be stable beyond day 50 (Fig. 4B). This is a different feature from previous observations on SCVs. There are some reports from studies using a menadione-auxotrophic SCV suggesting increased cell aggregation by S. aureus SCVs. Singh and coworkers showed an increase in polysaccharide intercellular adhesin (PIA) in the SCV (43), which was tightly coupled to biofilm production under the growth conditions they used. Thymidine-dependent SCVs have also been observed under SEM and showed a linking between cells that was interpreted as impaired cell separation (9). The results from both of these reports are different from what we have seen from steady-state growth with a prolonged time frame. In contrast to these results, the stable SCV cells that were allowed to grow under steady-state conditions possess a distinct extracellular matrix covering a population of aggregated cells (Fig. 3E). All of these studies are in vitro analyses, and it should be kept in mind they represent an aspect of the complex nature of the conditions that exist in the host.
The SCV extracellular matrix is eDNA and protein, not polysaccharide.
We assessed the composition of the SCV extracellular matrix for DNA, polysaccharide, and then intra/extracellular DNA (Fig. 5). eDNA is clearly associated with this matrix, but polysaccharide was only linked to cells and certainly not the matrix. This is a distinction from other similar cell aggregates and most S. aureus biofilms (43). Treatment of the stable SCV cells with either DNase or proteinase dispersed the cell aggregate and removed the matrix (as seen in SEM and by DNA-specific staining) (Fig. 6). It is noteworthy that these enzymes separately removed the matrix, suggesting an essential interplay by these components. This matrix and these cells are clearly distinct from what is seen in biofilms. The EPS of S. aureus biofilms is known to be composed of eDNA and polysaccharides (specifically, PIA [44]), although polysaccharide is not always present. Other studies have shown that in different biofilm phenotypes, there is expression of adhesive proteins, such as biofilm-associated protein Bap (45), accumulation-associated protein Aap, surface proteins SasG and SasC, and fibronectin binding proteins, FnBPA and FnBPB.
FIG 5.
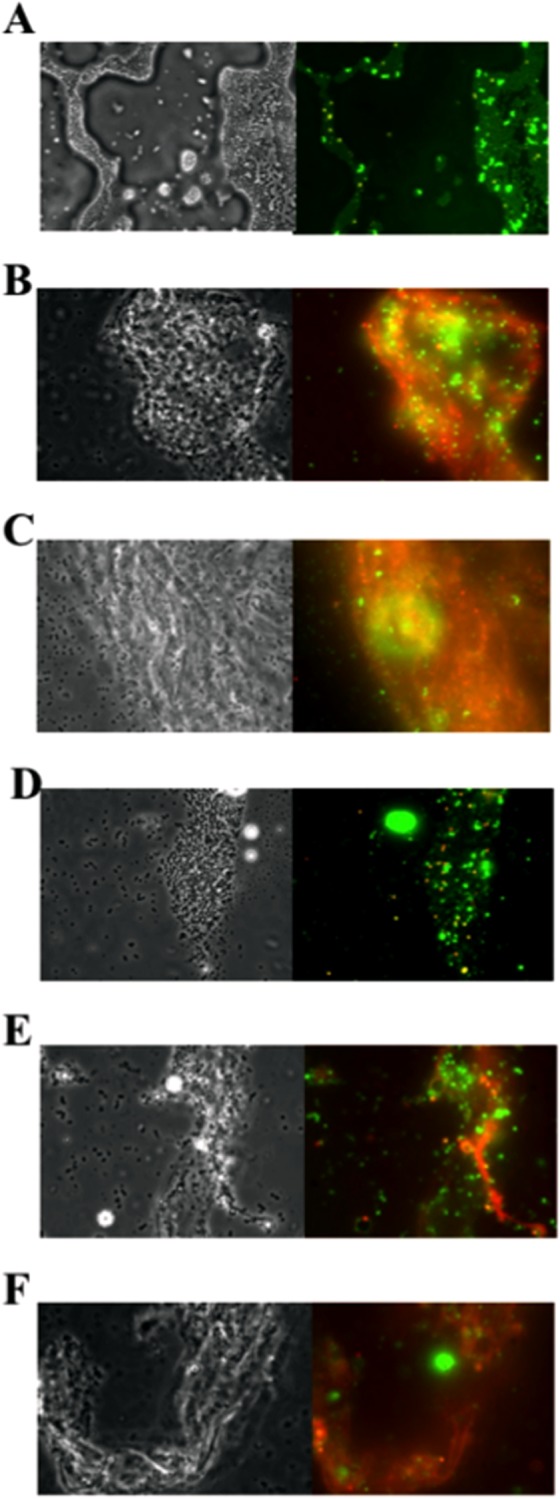
The extracellular matrix of stable SCV cells contains eDNA. S. aureus WCH-SK2 cells were observed under phase-contrast microscopy (100× [right panel]) and stained for matrix (visualized by fluorescence microscopy [left panel]). Cells were taken from batch culture (A), on day 30 with 0.0078% methylglyoxal (B), and on day 50 with 0.031% methylglyoxal (C). Cells were stained with propidium iodide (PI [red]) to identify dead cells or eDNA and SYTO 9 (green) to identify live cells. Yellow or orange indicates a mixture of these components. Batch culture cells (D), cells with 0.0078% methylglyoxal (E), and cells with 0.031% methylglyoxal (F) were then stained with PI and Alexa 488-conjugated lectins for polysaccharides. Green indicates the presence of polysaccharides. The predominate red color reveals eDNA with limited polysaccharide.
FIG 6.

The extracellular matrix of the stable SCV cells contains eDNA and protein. The stable SCV S. aureus WCH-SK2 cells were grown in batch culture, and the presence of its extracellular matrix was confirmed before treatment with increasing amounts of either DNase or proteinase K. Cells were observed under phase-contrast microscopy (100× [left panel]) and stained with PI (red) and SYTO 9 (green) for fluorescence microcopy (middle panel). The cells were also examined under SEM (12,000× [right panel]). The extracellular matrix is seen before treatment and is increasingly removed by addition of either DNase or proteinase K. The number of viable cells (CFU/ml) remains constant during treatment.
Stable SCV cells are a distinct cell type but are linked to the biofilm phenotype.
The stable SCV cells were assayed for biofilm formation and did show an increased biofilm formation that was reversed by removing the matrix (Fig. 7). While the stable SCV cells (which were growing planktonically in the chemostat beyond day 50 in continuous culture) represent a distinct lifestyle, they perhaps act as a precursor stage for a biofilm phenotype. Supporting this, the chemostat was left beyond day 60 for observation and not sampling, and when observed at day 68 (15.1 generations beyond the final sample point at day 60), there was a dramatic shift in cells in the chemostat to those that were pigmented and with cell aggregates or flocs and the distinct formation of a biofilm throughout the apparatus (data not shown).
FIG 7.

The S. aureus WCH-SK2 stable SCV cells are linked to biofilm formation. The abilities of WCH-SK2 cells to produce a biofilm were compared when grown in batch culture or taken from the chemostat (i.e., the stable SCV population, labeled “nsSCVs,” and then specifically the small colonies from this population subcultured through several cycles, labeled “Small colonies”). The stable SCV cells were also assayed after treatment with increasing amounts of DNase and proteinase K.
The changes that are defining the switch to stable SCV.
Using a transcriptomics approach (on batch culture-grown cells compared to the chemostat SCVs (day 30) and the stable SCVs (day 55) (Fig. 8; see Tables S2 to S5 in the supplemental material), we determined there was an induction in stable SCV cells of pathways associated with metabolism (especially amino acid transport, biosynthesis, and catabolism) and surface proteins. Importantly, there was no induction of any of the previously reported adhesive proteins for biofilm cells, as mentioned above, or indeed those proteins previously reported in S. aureus SCVs (46–48). Recently a large, 1.1-MDa, (10,000 amino acids [aa]) surface protein known as the host-extracellular matrix binding protein, EmbP or Ebh, was identified as expressed in biofilm of Staphylococcus epidermidis (49), and although not shown in S. aureus biofilm, it is involved in S. aureus virulence (50). In the stable SCV cells, there was a large increase (log2 5-fold) in Ebh expression. Furthermore, ebh mutants were reported as larger cells, and Ebh has therefore been linked to the cell size of S. aureus by its interference with peptidoglycan biosynthesis and stability (50). While the stable SCV cells do not appear to be smaller cells, in terms of aggregation and as a potential feature of the stable SCV extracellular matrix, the induction of this large surface protein is significant. In addition, we prepared membrane protein fractions from batch culture-grown cells and chemostat-derived stable SCV cells, and these were analyzed and compared (see Table S6 in the supplemental material). In this membrane proteomics, Ebh showed an almost 5-fold increase in the stable SCV compared to batch-grown cells—a validation of the transcriptomics data. There were changes in the expression of other membrane proteins that may also form part of the stable SCV matrix.
FIG 8.

Changes in the S. aureus WCH-SK2 transcriptional profile through transition from batch culture cells to SCVs and then a stable SCV population. Shown is a transcriptomics comparison of the whole-cell gene expression in both the SCV population (cells taken from the chemostat with 0.0078% methylglyoxal [top panels]) and the stable SCV cell type (0.031% methylglyoxal [bottom panels]) compared to batch culture cells. Genes with a significant differential expression were placed in a functional category. The values represent the numbers within a category as a percentage of the total number of genes with change in expression within a set: either upregulated (left panels) or downregulated (right panels).
The profile of metabolic genes we have identified and indeed the induction of surface proteins, such as Ebh, have not previously been seen in SCVs and are different from those of previous reports (46–48). It is reasonable to postulate that Ebh is indeed a core factor in the SCV lifestyle (perhaps, in particular the stable SCV cell type) but was missed in previous studies. Furthermore, it is potentially a factor in the unique extracellular matrix linked to the stable SCV.
Also, previously not reported for SCVs, the stable SCV cells included upregulation of the epiABC gene cluster, suggestive of its production of the lantibiotic (small, nonribosomal peptide) epidermin. Type A (I) lantibiotics act similarly to bacteriocins and have a similar effect on sensitive bacteria, including other Gram-positive bacteria and S. aureus itself. They have multiple modes of bactericidal activity, mainly through the targeting of lipid II (51). Slow-growing and stable SCV cells could therefore have an inherent resistance (in addition to the resistance that is mediated by an export pump, which was also induced in the stable SCV cells). This indicates that during prolonged colonization, the stable SCVs have the ability to persist in an anatomical niche and they use lantibiotics to target their rapidly growing counterparts, ensuring a reduced host-cell immune response. In addition, stable SCV cells have downregulation in hemolysins, leukocins, capsule, and pigment (see Tables S2 to S5 in the supplemental material). These are all factors that interact or induce immune cell responses. Previous studies have shown that there is a quorum-sensing system, the accessory gene regulator (agr) system, in S. aureus, and this is central in the control of specific virulence factors (52–54). There have been studies that report mutations in the agr system and indeed mutations through the course of infection (54). In this context, we have sequenced the DNA across this region in WCH-SK2 cells taken from batch culture before the 60-day study and then from a sample taken at day 55. The agr DNA sequence aligned 100% to those from reference strains, and there was no mutation in this system as a consequence of the prolonged growth under limiting conditions for 60 days (see Fig. S4 in the supplemental material). The phenotypic variations we have reported are therefore not a consequence of mutations in agr. Taken together, these data portray this stable SCV lifestyle as a quasi-dormant state with altered energy metabolism and cell-surface components, potentially acting as a stage before biofilm formation (Fig. 9).
FIG 9.

Model for the development of the stable SCV cell type in S. aureus. Over time, and with the stress of limiting growth conditions, low growth rate, and then the presence of a resident chemical stress, there is an increase in SCVs. This cell type is further developed to a stable SCV cell type, growing with a concentration of chemical stress above the calculated MIC. These are enclosed in a matrix, and they induce the expression of lantibiotics (removing the parental S. aureus cells) and repress the factors that would stimulate an immune response. Further time under this stress switches the cells to a biofilm.
Previous models of persistence by S. aureus have suggested the generation of SCVs has been based on low growth rate, low membrane potential, and lack of transport of antibiotics into the cell (38). We have applied growth parameters replicating those encountered during the time frame for colonization in the host, and using a clinical isolate, we have generated a stable SCV cell type. Characterization of the properties of these cells indicated that the cells remained at the same size but displayed a lower growth rate. The cells were covered in an extracellular matrix, and the molecular characteristics highlighted a quasi-dormant lifestyle, hidden from the host's normal responses, indicative of persister cells. These observations may be central to the organism's continued survival and persistence in the host.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Adelaide Microscopy for assistance with various microscopy experiments. We also acknowledge R. Morona and members of the Morona Research Group for use and help with fluorescence microscopy. Transcriptomics was performed at the Adelaide Cancer Genomic Research Facility. A. Tikhomirova provided support with RNA-seq data analysis. We acknowledge the assistance of J. Eddes with proteomics.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02702-14.
REFERENCES
- 1.Lewis K. 2010. Persister cells. Annu Rev Microbiol 64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 2.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 3.Archer NK, Mazaitis MJ, Costerton JW, Leid JG, Powers ME, Shirtliff ME. 2011. Staphylococcus aureus biofilms: properties, regulation, and roles in human disease. Virulence 2:445–459. doi: 10.4161/viru.2.5.17724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arciola CR, Campoccia D, Speziale P, Montanaro L, Costerton JW. 2012. Biofilm formation in Staphylococcus implant infections. A review of molecular mechanisms and implications for biofilm-resistant materials. Biomaterials 33:5967–5982. doi: 10.1016/j.biomaterials.2012.05.031. [DOI] [PubMed] [Google Scholar]
- 5.Kolle W, Hetsch W. 1906. Die experimentelle Bakteriologie und die Infektionskrankheiten mit besonderer Berücksich-tigung der Immunitätslehre. Ein Lehrbuch für Studierende, Ärzte und Medizinalbeamte. Urban & Schwarzenberg, Berlin, Germany. [Google Scholar]
- 6.Goudie J, Goudie R. 1955. Recurrent infections by a stable dwarf-colony variant of Staphylococcus aureus. J Clin Pathol 8:284–287. doi: 10.1136/jcp.8.4.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wise RI, Spink WW. 1954. The influence of antibiotics on the origin of small colonies (G variants), of Micrococcus pyogenes var. Aureus1. J Clin Invest 33:1611–1622. doi: 10.1172/JCI103041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kahl BC, Belling G, Becker P, Chatterjee I, Wardecki K, Hilgert K, Cheung AL, Peters G, Herrmann M. 2005. Thymidine-dependent Staphylococcus aureus small-colony variants are associated with extensive alterations in regulator and virulence gene expression profiles. Infect Immun 73:4119–4126. doi: 10.1128/IAI.73.7.4119-4126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kahl BC, Belling G, Reichelt R, Herrmann M, Proctor RA, Peters G. 2003. Thymidine-dependent small-colony variants of Staphylococcus aureus exhibit gross morphological and ultrastructural changes consistent with impaired cell separation. J Clin Microbiol 41:410–413. doi: 10.1128/JCM.41.1.410-413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaudaux P, Francois P, Bisognano C, Kelley WL, Lew DP, Schrenzel J, Proctor RA, McNamara PJ, Peters G, Von Eiff C. 2002. Increased expression of clumping factor and fibronectin-binding proteins by hemB mutants of Staphylococcus aureus expressing small colony variant phenotypes. Infect Immun 70:5428–5437. doi: 10.1128/IAI.70.10.5428-5437.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Eiff C, McNamara P, Becker K, Bates D, Lei X-H, Ziman M, Bochner BR, Peters G, Proctor RA. 2006. Phenotype microarray profiling of Staphylococcus aureus menD and hemB mutants with the small-colony-variant phenotype. J Bacteriol 188:687–693. doi: 10.1128/JB.188.2.687-693.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Proctor RA, Kriegeskorte A, Kahl B, Becker K, Löffler B, Peters G. 2014. Staphylococcus aureus small colony variants (SCVs): a road map for the metabolic pathways involved in persistent infections. Front Cell Infect Microbiol 4:99. doi: 10.3389/fcimb.2014.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, Peters G. 2006. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat Rev Microbiol 4:295–305. doi: 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- 14.Al Laham N, Rohde H, Sander G, Fischer A, Hussain M, Heilmann C, Mack D, Proctor R, Peters G, Becker K, von Eiff C. 2007. Augmented expression of polysaccharide intercellular adhesin in a defined Staphylococcus epidermidis mutant with the small-colony-variant phenotype. J Bacteriol 189:4494–4501. doi: 10.1128/JB.00160-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao W, Chua K, Davies JK, Newton HJ, Seemann T, Harrison PF, Holmes NE, Rhee H-W, Hong J-I, Hartland EL, Stinear TP, Howden BP. 2010. Two novel point mutations in clinical Staphylococcus aureus reduce linezolid susceptibility and switch on the stringent response to promote persistent infection. PLoS Pathog 6:e1000944. doi: 10.1371/journal.ppat.1000944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitchell G, Fugère A, Pépin Gaudreau K, Brouillette E, Frost EH, Cantin AM, Malouin F. 2013. SigB is a dominant regulator of virulence in Staphylococcus aureus small-colony variants. PLoS One 8:e65018. doi: 10.1371/journal.pone.0065018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atalla H, Gyles C, Mallard B. 2011. Staphylococcus aureus small colony variants (SCVs) and their role in disease. Anim Health Res Rev 12:33–45. doi: 10.1017/S1466252311000065. [DOI] [PubMed] [Google Scholar]
- 18.Kahl BC. 2010. Impact of Staphylococcus aureus on the pathogenesis of chronic cystic fibrosis lung disease. Int J Med Microbiol 300:514–519. doi: 10.1016/j.ijmm.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Tan NCW, Cooksley CM, Roscioli E, Drilling AJ, Douglas R, Wormald PJ, Vreugde S. 2014. Small-colony variants and phenotype switching of intracellular Staphylococcus aureus in chronic rhinosinusitis. Allergy 69:1364–1371. doi: 10.1111/all.12457. [DOI] [PubMed] [Google Scholar]
- 20.Kahl BC, Duebbers A, Lubritz G, Haeberle J, Koch HG, Ritzerfeld B, Reilly M, Harms E, Proctor RA, Herrmann M, Peters G. 2003. Population dynamics of persistent Staphylococcus aureus isolated from the airways of cystic fibrosis patients during a 6-year prospective study. J Clin Microbiol 41:4424–4427. doi: 10.1128/JCM.41.9.4424-4427.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cole AM, Wu M, Kim Y-H, Ganz T. 2000. Microanalysis of antimicrobial properties of human fluids. J Microbiol Methods 41:135–143. doi: 10.1016/S0167-7012(00)00140-8. [DOI] [PubMed] [Google Scholar]
- 22.van Belkum A, Melles DC, Nouwen J, van Leeuwen WB, van Wamel W, Vos MC, Wertheim HFL, Verbrugh HA. 2009. Co-evolutionary aspects of human colonisation and infection by Staphylococcus aureus. Infect Genet. Evol 9:32–47. doi: 10.1016/j.meegid.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 23.Libberton B, Coates RE, Brockhurst MA, Horsburgh MJ. 2014. Evidence that intraspecific trait variation among nasal bacteria shapes the distribution of Staphylococcus aureus. Infect Immun 82:3811–3815. doi: 10.1128/IAI.02025-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nouwen J, Boelens H, van Belkum A, Verbrugh H. 2004. Human factor in Staphylococcus aureus nasal carriage. Infect Immun 72:6685–6688. doi: 10.1128/IAI.72.11.6685-6688.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalhoff A. 1985. Differences between bacteria grown in vitro and in vivo. J Antimicrob Chemother 15:175–195. doi: 10.1093/jac/15.suppl_A.175. [DOI] [PubMed] [Google Scholar]
- 26.Jay SJ, Johanson WG Jr, Pierce AK, Reisch JS. 1976. Determinants of lung bacterial clearance in normal mice. J Clin Invest 57:811–817. doi: 10.1172/JCI108356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johanson WG Jr, Jay SJ, Pierce AK. 1974. Bacterial growth in vivo an important determinant of the pulmonary clearance of Diplococcus pneumoniae in rats. J Clin Invest 53:1320–1325. doi: 10.1172/JCI107679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nugent KM, Pesanti EL. 1982. Nonphagocytic clearance of Staphylococcus aureus from murine lungs. Infect Immun 36:1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown PS, Pope CE, Marsh RL, Qin X, McNamara S, Gibson R, Burns JL, Deutsch G, Hoffman LR. 2014. Directly sampling the lung of a young child with cystic fibrosis reveals diverse microbiota. Ann Am Thorac Soc 11:1049–1055. doi: 10.1513/AnnalsATS.201311-383OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Opijnen T, Camilli A. 2012. A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res 22:2541–2551. doi: 10.1101/gr.137430.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goerke C, Wolz C. 2010. Adaptation of Staphylococcus aureus to the cystic fibrosis lung. Int J Med Microbiol 300:520–525. doi: 10.1016/j.ijmm.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Hirschhausen N, Block D, Bianconi I, Bragonzi A, Birtel J, Lee JC, Dübbers A, Küster P, Kahl J, Peters G, Kahl BC. 2013. Extended Staphylococcus aureus persistence in cystic fibrosis is associated with bacterial adaptation. Int J Med Microbiol 303:685–692. doi: 10.1016/j.ijmm.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 33.Ferenci T. 2006. A cultural divide on the use of chemostats. Microbiology 152:1247–1248. doi: 10.1099/mic.0.28651-0. [DOI] [PubMed] [Google Scholar]
- 34.Ferenci T, Robert KP. 2007. Bacterial physiology, regulation and mutational adaptation in a chemostat environment. Adv Microb Physiol 53:169–229, 315. doi: 10.1016/S0065-2911(07)53003-1. [DOI] [PubMed] [Google Scholar]
- 35.Hussain M, Hastings JGM, White PJ. 1991. A chemically defined medium for slime production by coagulase-negative staphylococci. J Med Microbiol 34:143–147. doi: 10.1099/00222615-34-3-143. [DOI] [PubMed] [Google Scholar]
- 36.Monod J. 1949. The growth of bacterial cultures. Annu Rev Microbiol 3:371–394. doi: 10.1146/annurev.mi.03.100149.002103. [DOI] [Google Scholar]
- 37.Socransky SS, Manganielli AD, Propas D, Orum U, van Houte J. 1977. Bacteriological studies of developing supragingival dental plaque. J Periodontal Res 12:90–106. doi: 10.1111/j.1600-0765.1977.tb00112.x. [DOI] [PubMed] [Google Scholar]
- 38.Proctor RA, van Langevelde P, Kristjansson M, Maslow JN, Arbeit RD. 1995. Persistent and relapsing infections associated with small-colony variants of Staphylococcus aureus. Clin Infect Dis 20:95–102. doi: 10.1093/clinids/20.1.95. [DOI] [PubMed] [Google Scholar]
- 39.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bui LMG, Turnidge JD, Kidd SP. 2 October 2014. The induction of Staphylococcus aureus biofilm formation or small colony variants is a strain-specific response to host-generated chemical stresses. Microbes Infect doi: 10.1016/j.micinf.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 41.Ellwood DC, Phipps PJ, Hamilton IR. 1979. Effect of growth rate and glucose concentration on the activity of the phosphoenolpyruvate phosphotransferase system in Streptococcus mutans Ingbritt grown in continuous culture. Infect Immun 23:224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalapos MP. 2008. The tandem of free radicals and methylglyoxal. Chem Biol Interact 171:251–271. doi: 10.1016/j.cbi.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 43.Singh R, Ray P, Das A, Sharma M. 2010. Enhanced production of exopolysaccharide matrix and biofilm by a menadione-auxotrophic Staphylococcus aureus small-colony variant. J Med Microbiol 59:521–527. doi: 10.1099/jmm.0.017046-0. [DOI] [PubMed] [Google Scholar]
- 44.Cramton SE, Gerke C, Schnell NF, Nichols WW, Götz F. 1999. The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect Immun 67:5427–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cucarella C, Solano C, Valle J, Amorena B, Lasa Í Penadés JR. 2001. Bap, a Staphylococcus aureus surface protein involved in biofilm formation. J Bacteriol 183:2888–2896. doi: 10.1128/JB.183.9.2888-2896.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dean MA, Olsen RJ, Long SW, Rosato AE, Musser JM. 2014. Identification of point mutations in clinical Staphylococcus aureus strains that produce small-colony variants auxotrophic for menadione. Infect Immun 82:1600–1605. doi: 10.1128/IAI.01487-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kohler C, von Eiff C, Peters G, Proctor RA, Hecker M, Engelmann S. 2003. Physiological characterization of a heme-deficient mutant of Staphylococcus aureus by a proteomic approach. J Bacteriol 185:6928–6937. doi: 10.1128/JB.185.23.6928-6937.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kriegeskorte A, König S, Sander G, Pirkl A, Mahabir E, Proctor RA, von Eiff C, Peters G, Becker K. 2011. Small colony variants of Staphylococcus aureus reveal distinct protein profiles. Proteomics 11:2476–2490. doi: 10.1002/pmic.201000796. [DOI] [PubMed] [Google Scholar]
- 49.Christner M, Franke GC, Schommer NN, Wendt U, Wegert K, Pehle P, Kroll G, Schulze C, Buck F, Mack D, Aepfelbacher M, Rohde H. 2010. The giant extracellular matrix-binding protein of Staphylococcus epidermidis mediates biofilm accumulation and attachment to fibronectin. Mol Microbiol 75:187–207. doi: 10.1111/j.1365-2958.2009.06981.x. [DOI] [PubMed] [Google Scholar]
- 50.Cheng AG, Missiakas D, Schneewind O. 2014. The giant protein Ebh is a determinant of Staphylococcus aureus cell size and complement resistance. J Bacteriol 196:971–981. doi: 10.1128/JB.01366-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bastos MCF, Ceotto H, Coelho MLV, Nascimento JS. 2009. Staphylococcal antimicrobial peptides: relevant properties and potential biotechnological applications. Curr Pharm Biotechnol 10:38–61. doi: 10.2174/138920109787048580. [DOI] [PubMed] [Google Scholar]
- 52.Laabei M, Jamieson WD, Massey R, Jenkins ATA. 2014. Staphylococcus aureus interaction with phospholipid vesicles—a new method to accurately determine accessory gene regulator (agr) activity. PLoS One 9:e87270. doi: 10.1371/journal.pone.0087270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pader V, James EH, Painter KL, Wigneshweraraj S, Edwards AM. 2014. The Agr quorum-sensing system regulated fibronectin binding but not hemolysis in the absence of a functional electron transport chain. Infect Immun 82:4337–4347. doi: 10.1128/IAI.02254-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tuchscherr L, Buzzola FR, Alvarez LP, Lee JC, Sordelli DO. 2008. Antibodies to capsular polysaccharide and clumping factor A prevent mastitis and the emergence of unencapsulated and small-colony variants of Staphylococcus aureus in mice. Infect Immun 76:5738–5744. doi: 10.1128/IAI.00874-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.