

Abstract
Klebsiella pneumoniae is an important pathogen that causes hospital-acquired septicemia and is associated with the recent emergence of community-acquired pyogenic liver abscess (PLA). Clinical typing suggests that K. pneumoniae infections originate from the gastrointestinal reservoir. However, the underlying mechanism remains unknown. Here, we have sought to determine how K. pneumoniae penetrates the intestinal barrier. We identified that bacteremia and PLA clinical isolates adhered to and invaded intestinal epithelial cells. Internalization of K. pneumoniae in three different human colonic cell lines was visualized by confocal microscopy and three-dimensional (3D) imaging. Using a Transwell system, we demonstrated that these K. pneumoniae isolates translocated across a polarized Caco-2 monolayer. No disruptions of transepithelial electrical resistance and altered distribution of tight junction protein ZO-1 or occludin were observed. Therefore, K. pneumoniae appeared to penetrate the intestinal epithelium via a transcellular pathway. Using specific inhibitors, we characterized the host signaling pathways involved. Inhibition by cytochalasin D and nocodazole suggested that actin and microtubule cytoskeleton were both important for K. pneumoniae invasion. A Rho inhibitor, ML141, LY294002, and an Akt1/2 inhibitor diminished K. pneumoniae invasion in a dose-dependent manner, indicating that Rho family GTPases and phosphatidylinositol 3-kinase (PI3K)/Akt signaling were required. By a mouse model of gastrointestinal colonization, in vivo invasion of K. pneumoniae into colonic epithelial cells was demonstrated. Our results present evidence to describe a possible mechanism of gastrointestinal translocation for K. pneumoniae. Cell invasion by manipulating host machinery provides a pathway for gut-colonized K. pneumoniae cells to penetrate the intestinal barrier and access extraintestinal locations to cause disease.
INTRODUCTION
Klebsiella pneumoniae, a Gram-negative enteric bacterium, is a common pathogen that causes hospital-acquired urinary tract infections (UTIs), septicemia, and pneumonia (1). A new type of community-acquired K. pneumoniae infection that is associated with pyogenic liver abscess (PLA) has emerged worldwide, especially in East Asian countries (2–5). This disease is often complicated by metastatic infections, such as meningitis and endophthalmitis. An important virulence factor of K. pneumoniae is the capsule, an extracellular polysaccharide structure that protects bacteria from lethal serum factors and phagocytosis. There are at least 79 capsular types that have been defined, and an association of capsular type with disease severity has been observed (6, 7). Strains with the K1 and K2 capsular types have been identified as the predominant virulent types and are prevalent in K. pneumoniae PLA (6, 8, 9).
The intestine is one of the major reservoirs of K. pneumoniae. A seroepidemiological survey of K. pneumoniae cells that have colonized the gastrointestinal tract showed that capsular types K1 and K2 were the most prevalent and were responsible for 9.8% of a total of 592 fecal isolates from healthy Chinese adults in Asian countries (10). Epidemiological studies have suggested that most K. pneumoniae infections are preceded by colonization of the gastrointestinal tract (11–15). Clinical capsular typing and pulsed-field gel electrophoresis analysis revealed a similarity in strain serotypes and genotypes of K. pneumoniae fecal isolates from healthy carriers and patients with liver abscess (13). A direct association between extended-spectrum β-lactamase (ESBL)-producing K. pneumoniae strains detected in the gut microbiota and isolates responsible for bloodstream infections was also implied. Studies of ESBL-producing K. pneumoniae strains demonstrated the genetic relatedness among outbreak isolates obtained from prior colonization events and subsequent diseases (14, 15). A hypothesis that gut-derived K. pneumoniae causes infections has been proposed; however, the potential mechanistic details involved have not been elucidated.
The intestinal mucosa is lined by an epithelial cell layer that provides a tight barrier that protects against microbial pathogens (16). There are two general routes that microbes use to penetrate the intestinal epithelium and enter into lymph nodes or the systemic circulation: the transcellular (intracellular) and the paracellular (intercellular) pathways (17–19). In the transcellular pathway, well-studied enteropathogens such as Salmonella, Shigella, Listeria, and Yersinia species invade nonphagocytic epithelial cells by subverting host cytoskeleton dynamics (20). In the paracellular pathway, bacteria such as Vibrio cholerae (21), Campylobacter jejuni (22), and Pseudomonas aeruginosa (23) perturb epithelial integrity to facilitate bacterial translocation by disrupting the cell tight junctions (TJs), the structures between epithelial cells that control paracellular permeability.
How K. pneumoniae interacts with the host intestinal epithelium during pathogenesis and the mechanism of potential intestinal translocation remain unclear. K. pneumoniae is classically regarded to be an extracellular pathogen. Nevertheless, internalization of a UTI isolate and a pneumonia isolate into epithelial cells have been described (24–26). The capsule of K. pneumoniae has been proposed as a bacterial factor that impedes cell adherence and invasion (27). Little is known about the host factors involved. In this study, we investigated interactions between intestinal epithelial cells and clinical K. pneumoniae strains that cause systemic infections. We used human intestinal cells to identify K. pneumoniae bacteremia and PLA isolates that adhered to and invaded intestinal epithelial cells. A Transwell system was employed to assess K. pneumoniae translocation across intestinal monolayers. Using specific cell inhibitors, the host signaling pathways involved in K. pneumoniae invasion were further determined.
MATERIALS AND METHODS
Bacterial strains and cell culture.
K. pneumoniae clinical strains that caused systemic infections were isolated from the blood of patients (28, 29); strains NTUH-K2044 and A21 were PLA isolates, and strains Ca0401, Ca0438, and 5721 were bacteremia isolates. Klebsiella capsular types were determined using cps-PCR (30). The cell-invasive bacterium Salmonella enterica serovar Typhimurium ATCC 14028 and the noninvasive bacterium Escherichia coli HB101 were used in invasion and translocation assays (31, 32). For comparison, commensal E. coli strain TVGHEC01, a human stool isolate provided by Yi-Tsung Lin from the Taipei Veterans General Hospital (TVGH), was tested. Bacteria were grown in Luria-Bertani (LB) broth at 37°C with shaking or on solidified LB–1.5% agar plates. When necessary, the medium was supplemented with 50 μg/ml kanamycin. Three human enterocyte-like cell lines were cultured in this study. Caco-2 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% nonessential amino acids. HCT-8 cells were maintained in RPMI 1640 medium supplemented with 2 mM glutamine, 1 mM pyruvate, and 10% FBS. HT-29 cells were maintained in McCoy's 5a medium supplemented with 2 mM glutamine and 10% FBS. All cells were grown in a humidified 5% CO2 atmosphere at 37°C. All culture media and supplements were obtained from Gibco/BRL (Gaithersburg, MD).
Adhesion and invasion assays.
Adhesion and invasion assays were performed mainly according to the methods described previously (27, 33). For adhesion assays, Caco-2 cells in 24-well plates (∼5 × 105 cells per well) were prewashed with Hanks' balanced salt solution (HBSS). Mid-log-phase K. pneumoniae cells (A600, 0.4 to 0.6) in FBS-free DMEM were added to each well at a multiplicity of infection (MOI) of 50 bacteria/cell. After centrifugation at 200 × g for 5 min, plates were incubated for 20 min in a humidified 5% CO2 atmosphere at 37°C. After incubation, wells were washed with HBSS three times, and bacteria were released by the addition of 0.2% Triton X-100 (Sigma-Aldrich, St. Louis, MO). For invasion assays, K. pneumoniae cells in plates containing DMEM and 10% FBS were added to Caco-2 cells, incubated for 1.5 h, and washed twice with HBSS, followed by a second incubation for 1.5 h in fresh medium containing 100 μg/ml gentamicin to kill extracellular bacteria. Finally, cells were incubated again and lysed with 0.2% Triton X-100. Recovered bacteria were quantified by plating appropriate dilutions on LB agar plates and counting CFU. The number of adherent or intracellular bacteria is expressed as a proportion of the inoculum. All assays were conducted in duplicate and were repeated independently at least two times. When necessary, data are presented with the value of intracellular bacteria normalized for the number that adhered.
For inhibitor studies, Caco-2 cells were incubated with individual inhibitors 1 h prior to infection and the presence of inhibitors was maintained throughout the duration of the invasion period. The inhibitors that were analyzed were related to eukaryotic cytoskeleton dynamics or signal transduction: cytochalasin D (Sigma-Aldrich), nocodazole (Sigma-Aldrich), ML141 (Merck Millipore, Darmstadt, Germany), LY294002 (Merck Millipore), PP1 (Merck Millipore), genistein (Merck Millipore); Rho inhibitor I (Cytoskeleton, Denver, CO), Rac1 inhibitor (Merck Millipore), and Akt1/2 kinase inhibitor (Merck Millipore). The concentrations were chosen based on usage in previous studies (24, 34–39). Bacterial and cell viability were found not to be affected by the inhibitors, as checked by plating experiments and trypan blue exclusion. Caco-2 cells were treated with equivalent amounts of dimethyl sulfoxide (DMSO) (Sigma-Aldrich) as a control.
For intracellular survival experiments, invasion assays using Caco-2 cells were performed as described above. Following an invasion period of 1.5 h, infected cells were washed with HBSS, and then fresh medium containing 100 μg/ml gentamicin was added. Incubated cells were lysed at various times as indicated, and intracellular bacteria were plated for CFU counting.
Translocation assays and TEER measurements.
To assess the potential ability of K. pneumoniae cells to penetrate the intestinal epithelium, translocation assays were performed based on the method described previously (32, 33, 40, 41). Caco-2 cells can form tight polarized monolayers and be used as a model of the intestinal epithelium (32, 33, 40, 42). Cells were grown onto Transwell inserts (3-μm pore size, 0.33-cm2 filtration area) (Merck Millipore) for 15 days to form a tight monolayer and differentiate to generate TJs, assessed by transepithelial electrical resistance (TEER) measurements with a Millicell ERS-2 V-Ohm meter (Merck Millipore). Caco-2 monolayers were considered to be fully differentiated when a stable TEER of >300 Ω/cm2 was obtained (40, 42). Bacteria at 1 × 107 CFU were added to the apical side of monolayers in the upper chambers of a Transwell insert. After 1 h of incubation in a humidified 5% CO2 atmosphere at 37°C, viable bacteria in the upper and lower chambers were determined by plating appropriate dilutions on LB agar plates. The number of translocated bacteria recovered in the lower chambers was expressed as the proportion of the bacterial count in the upper chamber (41). When necessary, data are presented with the value of translocated bacteria normalized for the number that adhered. The experiments were conducted in duplicate and repeated independently two times.
For comparison, EGTA (Sigma-Aldrich) was used to disrupt TJs (42). Caco-2 cell monolayers were treated with 10 mM EGTA in complete medium for 1 h at 37°C in a humidified 5% CO2 atmosphere.
Immunofluorescence and microscopy.
The plasmid pCRII-TOPO with a gene encoding green fluorescent protein (pCRII-TOPO::GFP) was electroporated into K. pneumoniae (43). To visualize potential cell internalization of K. pneumoniae by fluorescence microscopy, Caco-2 cells (∼5 × 105 cells per well) grown on 12-mm round cover glasses in 24-well plates were infected for 2 h with K. pneumoniae Ca0438 carrying pCRII-TOPO::GFP. Cells were washed with phosphate-buffered saline (PBS) (Gibco/BRL), fixed with 2% paraformaldehyde for 15 min, and permeabilized with 0.2% Triton X-100 for 1 min. The actin cytoskeleton was stained with rhodamine-labeled phalloidin (Invitrogen, Carlsbad, CA) for 15 min, and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (AppliChem GmbH, Darmstadt, Germany) for 5 min at room temperature.
For immunofluorescence staining of TJ proteins, Caco-2 cells (∼1 × 105 cells) were grown on 12-mm round cover glasses in 24-well plates for 15 days to form monolayers and generate TJs (44). K. pneumoniae Ca0438 cells carrying the plasmid pCRII-TOPO::GFP were added to Caco-2 monolayers as described above. After fixation and permeabilization, cells were incubated in 3% bovine serum albumin (BSA) (Sigma-Aldrich) in PBS as a blocking solution for 1 h, washed with PBS, and then incubated with the following primary antibodies for 1 h at 37°C: rabbit polyclonal anti-ZO-1 (Invitrogen) or anti-occludin (Invitrogen). Following additional washes with PBS, cells were incubated with Alexa Fluor 594 anti-rabbit IgG (Invitrogen) for 30 min at room temperature.
Images were initially observed using a Zeiss Axioplan 2 fluorescence microscope. To determine the potential intracellular location of K. pneumoniae, confocal microscopy and three-dimensional (3D) image analysis were performed. Images were captured by a Leica SP5 confocal microscope with 405-, 488-, and 543-nm lasers. Serial confocal z-stacks were taken with steps of 0.4 μm, and images were combined to observe cell-associated bacteria. When necessary, 3D images were analyzed by XYZ sections using the software Volocity 6.1 (PerkinElmer, Waltham, MA).
Animal experiments.
To establish intestinal colonization, 5-week-old female BALB/cByl mice were intragastrically infected with 109 CFU K. pneumoniae Ca0438 in 100 μl 0.95% saline solution. After 4 weeks, fecal samples from mice were plated on deoxycholate hydrogen sulfide lactose (DHL) agar (Eiken Chemical, Tokyo, Japan), the selective medium for enterobacteria, and K. pneumoniae colonies were confirmed using K2 cps-PCR (30). K. pneumoniae-colonized mice were sacrificed 4 weeks after inoculation. Colons were fixed in 4% paraformaldehyde, OCT embedded, and cryosectioned by the Laboratory Animal Center of the National Taiwan University College of Medicine. All animal experiments were conducted in a manner consistent with the ethical requirements of the Animal Care Committee at the National Taiwan University. For immunostaining and microscopy, specimens were permeabilized with 0.2% Triton X-100 and blocked with 3% BSA for 1 h. K. pneumoniae Ca0438 was immunostained with rabbit polyclonal anti-K2 capsule antibody (LTK BioLaboratories, Taipei, Taiwan) for 1 h and then Alexa Fluor 488-labeled anti-rabbit IgG (Invitrogen) for 30 min. The actin cytoskeleton was stained with rhodamine-phalloidin and DNA stained with DAPI as described above.
Statistical analyses.
Results are expressed as means ± standard errors of the means (SEM). Data were analyzed using a Student t test with a 95% confidence interval. A P value of <0.05 was considered significant. Graphpad Prism software was used for statistical analysis (San Diego, CA).
RESULTS
Adhesion of K. pneumoniae to human intestinal epithelial cells.
We initially analyzed five K. pneumoniae clinical isolates for adhesion to intestinal epithelial cells. A bacteremia-inducing strain, Ca0438, showed a relatively higher adhesion rate than other K. pneumoniae strains (Fig. 1A). While the other strains adhered to Caco-2 cells with rates of <0.16%, Ca0438 adhered with a rate of 1.46% ± 0.18%. The intimate attachment of K. pneumoniae Ca0438 onto the host cell surface was observed using immunofluorescence microscopy (Fig. 1B). The capsular type of Ca0438 was determined as K2 by cps-PCR (30). This indicated that K. pneumoniae strains causing systemic infections were able to closely adhere to intestinal epithelial cells. Since Ca0438 adhered to intestinal cells better, it appeared to be a useful strain to study interactions between K. pneumoniae and intestinal epithelial cells.
FIG 1.

Adhesion of K. pneumoniae to human intestinal epithelial cells. (A) Adhesion assays of K. pneumoniae clinical isolates (as indicated) to Caco-2 cells. The adhesion rate was expressed as the proportion of the inoculum that adhered. Each bar indicates the mean ± SEM. For Ca0438 and all other strains tested, P is <0.05. (B) Immunofluorescence micrograph illustrating the attachment of K. pneumoniae Ca0438 cells to the cell surface (indicated by arrows). Cultured Caco-2 cells were infected by K. pneumoniae Ca0438 cells carrying pCRII-TOPO::GFP (green) for 30 min. The actin cytoskeleton was stained with rhodamine-phalloidin (red). Scale bar, 10 μm.
Invasion and intracellular replication of K. pneumoniae in human intestinal epithelial cells.
We next investigated potential cell invasion of K. pneumoniae clinical isolates. Gentamicin-based invasion assays showed that some K. pneumoniae isolates could invade Caco-2 cells. K. pneumoniae Ca0438 invaded cells with a higher recovery rate of intracellular bacteria than other tested K. pneumoniae strains (see Fig. S1 in the supplemental material for results without normalization for bacterial adhesion). After normalization for adhesion, the invasion rates (invasion/adhesion) of strains Ca0438, 5721, and A21 were significantly higher than that of noninvasive E. coli strain HB101 (Fig. 2A). The invasion/adhesion rates of Ca0438 and 5721 were also significantly higher than that of commensal E. coli strain TVGHEC01. Therefore, these K. pneumoniae clinical isolates appeared to be able to invade nonphagocytic epithelial cells.
FIG 2.

Invasion and intracellular replication of K. pneumoniae in human intestinal epithelial cells. (A) Invasion assays using different clinical isolates in Caco-2 cells. The invasion rate was expressed after normalization for adhesion. Each bar indicates the mean ± SEM. *, P < 0.05. (B) Three-dimensional image analysis showing the invasion of K. pneumoniae Ca0438 in different human intestinal epithelial cell lines. Cultured cells were infected by K. pneumoniae Ca0438 carrying pCRII-TOPO::GFP (green) for 2 h. The actin cytoskeleton and DNA were stained with rhodamine-phalloidin (red) and DAPI (blue). Images from confocal microscopy with z-stacking were analyzed by XY, XZ, and YZ sections. Intracellular K. pneumoniae cells are indicated by white arrows. Scale bar, 10 μm. (C) Intracellular survival of K. pneumoniae Ca0438 within Caco-2 cells. Cells were infected by K. pneumoniae Ca0438 for 1.5 h. After being washed with HBSS, cells were incubated in the presence of gentamicin for various periods of time as indicated and then lysed for the recovery of intracellular bacteria on LB agar. Data are presented as means ± SEM.
We further performed confocal fluorescence microscopy and 3D analysis to confirm cell internalization. The presence of intracellular K. pneumoniae within Caco-2 cells was clearly observed for Ca0438 (Fig. 2B, left panel) and 5721 (see Fig. S2 in the supplemental material). To determine whether this observation was peculiar to one cell line or not, cells from two other human colonic lines, HCT-8 and HT-29, were tested. Invasion of K. pneumoniae Ca0438 into both HCT-8 and HT-29 cells was found (Fig. 2B, the middle and right panels). Therefore, K. pneumoniae entry was not peculiar to Caco-2 cells. These results verified invasion by K. pneumoniae of human intestinal epithelial cells.
We investigated the recovery over time of intracellular K. pneumoniae cells in intestinal epithelial cells. Ca0438 survived and replicated within Caco-2 cells for a period of time (Fig. 2C). The number of intracellular K. pneumoniae increased during the first 6 h. Thereafter, the number of intracellular bacteria declined over time and, after 48 h, decreased to ∼31% of the intracellular bacteria recovered after 1 h of incubation in the presence of gentamicin. After 72 h, only ∼2% of the bacteria recovered after 1 h of gentamicin incubation remained viable.
K. pneumoniae translocates across polarized intestinal cell monolayers.
Potential translocation of K. pneumoniae across the gastrointestinal epithelial barrier was investigated using transepithelial translocation assays. Caco-2 cells can form tight polarized monolayers and differentiate to generate TJs and a brush border, mimicking the human intestinal epithelium (33, 40). K. pneumoniae Ca0438, 5721, and A21 cells were found to translocate across Caco-2 monolayers and were recovered from the lower chamber of the Transwell device. The translocation rate of Ca0438 without normalization for bacterial adhesion was higher than those of other tested strains (see Fig. S3 in the supplemental material for the proportion of the inoculum that translocated). Nevertheless, the translocation/adhesion rates (normalized for adhesion) of Ca0438, 5721, and A21 were all significantly higher than those of E. coli HB101 and the commensal E. coli strain TVGHEC01 (Fig. 3A). K. pneumoniae Ca0438 and 5721 were blood isolates from the patients with bacteremia, and A21 was from the patient with PLA. Our results indicated that a K. pneumoniae strain causing systemic infections was able to penetrate a polarized intestinal monolayer.
FIG 3.

Translocation of K. pneumoniae cells across polarized intestinal cell monolayers. (A) Translocation assays using human Caco-2 cell monolayers. The translocation rate was expressed after normalization for adhesion. Each bar indicates the mean ± SEM. For Ca0438 and HB101, 5721 and HB101, A21 and HB101, Ca0438 and TVGHEC01, 5721 and TVGHEC01, and A21 and TVGHEC01, all P values are <0.05. (B) TEER measurements of Caco-2 cell monolayers during K. pneumoniae infection at the time points indicated. For comparison, cells were incubated with HB101 or EGTA (10 mM), a chemical that disrupted cell TJs. Three independent experiments were conducted. Data are presented as means ± SEM. (C) Representative immunofluorescence micrographs illustrating cell TJs during K. pneumoniae Ca0438 infection. Differentiated Caco-2 cells were infected by K. pneumoniae Ca0438 cells carrying pCRII-TOPO::GFP (green) or were incubated with EGTA for 1 h. Two major TJ proteins, ZO-1 and occludin, were immunostained with rabbit anti-ZO-1 (upper graphs) or anti-occludin (lower graphs) and the secondary antibody Alexa Fluor 594 anti-rabbit IgG (red). Scale bar, 10 μm.
The integrity of epithelial cell monolayers during K. pneumoniae translocation was assessed by TEER measurements. When infected by K. pneumoniae Ca0438, 5721, or A21, TEER values did not change significantly, similar to that observed for incubation with E. coli HB101 (Fig. 3B). Addition of EGTA, which destroyed TJs, induced a reduction of TEER. Therefore, the integrity of epithelial cell monolayers was not significantly perturbed by K. pneumoniae when bacterial translocation occurred. We also examined cell TJs after K. pneumoniae Ca0438 infection using immunofluorescence microscopy. Two major TJ proteins, ZO-1 and occludin, were immunostained and observed. During Ca0438 infection, the expression and distribution of ZO-1 and occludin between adjacent cells were similar to those of control uninfected Caco-2 cells (Fig. 3C). In contrast, EGTA induced disruption of TJs and alterations in ZO-1 and occludin localization. Therefore, K. pneumoniae did not induce obvious changes of ZO-1 or occludin proteins in Caco-2 cell monolayers during bacterial translocation.
Based on these results, we suggest that K. pneumoniae may translocate across the intestinal epithelium without dramatically disrupting monolayer integrity and TJs.
Host Rho GTPases and the PI3K/Akt pathway are required for K. pneumoniae invasion of intestinal epithelial cells.
We sought to identify the host factors involved in K. pneumoniae invasion. Several bacterial pathogens, such as Salmonella, Shigella, Listeria, and Yersinia, have evolved strategies to subvert eukaryotic cytoskeletal function to invade host cells (20). Actin microfilaments and microtubules are the major components of cytoskeleton. We found that cytochalasin D and nocodazole, which disrupted actin and microtubule polymerization, respectively, inhibited Ca0438 invasion in a dose-dependent manner (Fig. 4). In the presence of 0.1 μM cytochalasin D, the number of intracellular bacteria decreased to 8.5% of that under mock invasion conditions. In the presence of 30 μM nocodazole, the number of intracellular bacteria was reduced to 8.4% of that under mock invasion conditions. No significant effect on K. pneumoniae invasion by equivalent DMSO was observed (data not shown). These results indicated that host cytoskeleton actin and microtubule were both required for K. pneumoniae invasion of intestinal epithelial cells.
FIG 4.

Eukaryotic cytoskeleton is important for K. pneumoniae invasion. Invasion assays of K. pneumoniae Ca0438 using Caco-2 cells were performed in the presence of compounds that interfered with host actin (cytochalasin D) or microtubules (nocodazole). Mock invasion in the absence of inhibitors was defined as 100% relative invasiveness. Data are presented as means ± SEM. *, P < 0.05 by Student's t test.
Eukaryotic cytoskeletal dynamics is regulated by a number of signaling proteins. To characterize the mechanism of K. pneumoniae Ca0438 invasion, we explored specific host signaling pathways involved. The potential effects of specific inhibitors previously reported to interfere with cell signaling and bacterial invasion were examined (34–39): Rho inhibitor I (the C3 exoenzyme of Clostridium botulinum—an inhibitor for RhoA, RhoB, and RhoC), ML141 (an inhibitor for Cdc42 and Rac1), Rac1 inhibitor, genistein (an inhibitor for protein tyrosine kinase), PP1 (an inhibitor for Src kinase), and LY294002 (an inhibitor for phosphatidylinositol 3-kinase [PI3K]). In the invasion assays, Rho inhibitor I, ML141, and LY294002 reduced Ca0438 invasion in a dose-dependent manner (Fig. 5A). No significant inhibition on Ca0438 invasion by genistein and PP1 was found (data not shown). The cell GTPases Rho, Cdc42, and Rac all belong to the Rho protein family. Inhibition by Rho inhibitor I and ML141 indicated the involvement of Rho family GTPases. As Rac1 inhibitor alone did not have a significant effect (Fig. 5A), Rho and Cdc42 but possibly not Rac1 play an important role in K. pneumoniae invasion of intestinal epithelial cells.
FIG 5.

Effects of eukaryotic signaling inhibitors on K. pneumoniae invasion. Invasion assays of K. pneumoniae Ca0438 using Caco-2 cells were performed in the presence of cell signaling inhibitors as indicated. Rho inhibitor I (RhoA, RhoB, and RhoC inhibitor), ML141 (Cdc42/Rac1 inhibitor), LY294002 (PI3K inhibitor), and Akt1/2 inhibitor reduced Ca0438 invasion. Mock invasion in the absence of inhibitors was defined as 100% relative invasiveness. Data are presented as means ± SEM. *, P < 0.05 by Student's t test.
Inhibition by LY294002 suggested the involvements of PI3K in K. pneumoniae invasion. A major downstream effector of PI3K is Akt, a serine-threonine kinase that regulates the activity of many other target molecules (45). We further found that Akt1/2 inhibitor (an inhibitor for Akt1 and Akt2) caused the decrease of Ca0438 invasion in a dose-dependent manner (Fig. 5B). Therefore, Akt kinase also contributed to K. pneumoniae Ca0438 invasion. The PI3K/Akt pathway is possibly required for K. pneumoniae invasion.
K. pneumoniae invades intestinal epithelial cells in vivo.
K. pneumoniae is classically considered to be an extracellular pathogen. K. pneumoniae in vivo invasion of intestinal epithelial cells has never been clearly shown. To investigate the interactions between colonized K. pneumoniae and gastrointestinal epithelium, we established a gastrointestinal colonization model in mice. The 50% lethal dose (LD50) of K. pneumoniae Ca0438 following intragastric inoculation of BALB/cByl mice was determined to be >1 × 108 CFU during the 4-week observation period. By intragastric inoculation with 1 × 109 CFU in mice, the presence of K. pneumoniae Ca0438 in the fecal samples collected at 4 weeks postinoculation was found (∼104 CFU/g), with confirmation of colony morphology and K2 cps-PCR. The colons of mice were cryosectioned and stained for immunofluorescence microscopy. Most of the K. pneumoniae Ca0438 cells were present in the lumen of the colons (Fig. 6A). Notably, we also observed intracellular K. pneumoniae Ca0438 in the colonic epithelial cells (Fig. 6B). This indicated that K. pneumoniae was able to invade epithelial cells in vivo during colonization in the host colon.
FIG 6.

K. pneumoniae invades intestinal epithelial cells in vivo. Mice were intragastrically inoculated with K. pneumoniae Ca0438 for gastrointestinal colonization. The colon of colonized mice was cryosectioned and immunostained using rhodamine-phalloidin for actin (red), DAPI for nuclei (blue), and rabbit anti-K2 capsule and Alexa Fluor 488 anti-rabbit IgG for K. pneumoniae (green). (A) Representative immunofluorescence micrographs showing K. pneumoniae Ca0438 in the lumen of the colon (indicated by white arrows). Scale bar, 10 μm (B) Three-dimensional image analysis showing intracellular K. pneumoniae Ca0438 (indicated by white arrows). Images from confocal microscopy with z-stacking were analyzed by XY, XZ, and YZ sections. Scale bars, 10 μm.
DISCUSSION
K. pneumoniae can cause nosocomial septicemia and a new type of community-acquired PLA. Drug-resistant K. pneumoniae remains a problem, mainly due to the increasing frequency of ESBL production (46) and carbapenem-resistant K. pneumoniae (47). Antibiotic selection pressure is not sufficient to explain the persistence and spread of this pathogen. Epidemiological studies suggest that K. pneumoniae infections could originate from gastrointestinal reservoirs. Here, we provide direct evidence showing translocation of K. pneumoniae across the polarized intestinal barrier. Our results support the hypothesis that K. pneumoniae from human reservoirs has the ability to invade and penetrate the intestinal epithelium. New insight into a possible mechanism for how gastrointestinal K. pneumoniae causes extraintestinal infections is offered, as illustrated in Fig. 7.
FIG 7.
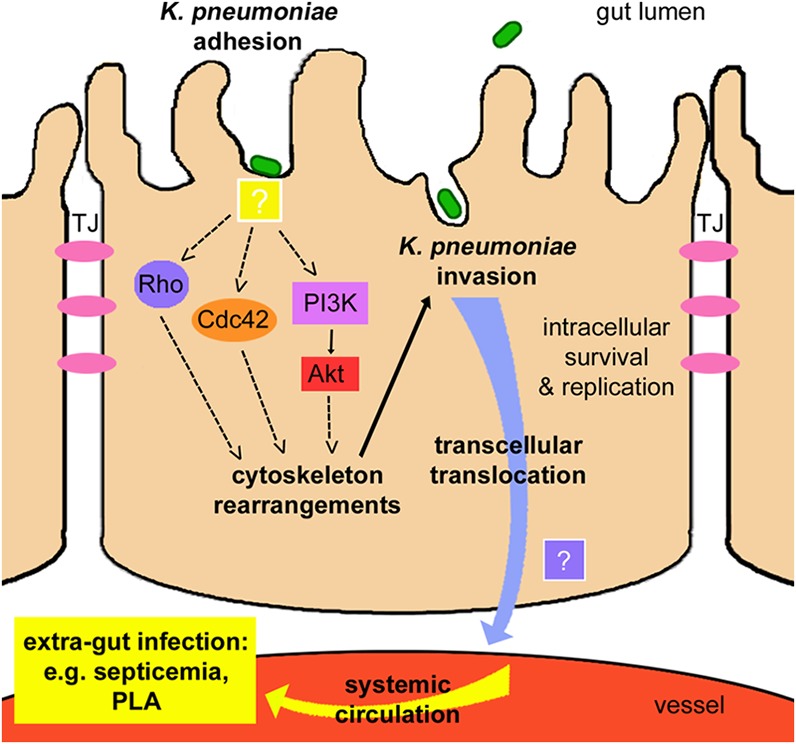
Model for intestinal translocation of K. pneumoniae. Gastrointestinal K. pneumoniae translocates across the intestinal epithelium via a transcellular mechanism by active bacterial invasion. Adhesion and interaction between K. pneumoniae and intestinal epithelial cells (with unidentified factors) trigger the cellular signal transduction cascades. Host Rho family GTPases Rho and Cdc42 and the PI3K/Akt pathway are activated to induce cell cytoskeleton rearrangements, which lead to engulfment and invasion of bacteria. Intracellular K. pneumoniae cells can survive, replicate, and exit the cell at the basal membrane (by an unidentified mechanism). Transcellular passage allows K. pneumoniae to penetrate the intestinal barrier, enter systemic circulation, and access extraintestinal locations to cause disease.
The intestinal mucosa with polarized monolayers of enterocytes provides a barrier that isolates the host from luminal microbes and antigens. We suggest that K. pneumoniae can employ a transcellular pathway by active bacterial invasion for intestinal epithelial translocation. Our data demonstrated cell invasion and epithelial translocation of the bacteremia and PLA isolates Ca0438, 5721, and A21. No significant decreases in TEER or changes in TJs were observed during epithelial translocation of K. pneumoniae. Therefore, these clinical K. pneumoniae strains possibly penetrated monolayers without causing obvious disruptions of monolayer integrity or paracellular permeability. K. pneumoniae belongs to human microbiota, and the gut resident strains have been shown to share genotypes similar to those the liver abscess and bacteremia strains (13-15). We suggest that this transcellular translocation mechanism is exploited generally by K. pneumoniae strains from the gut flora to cause systemic infections. Transcellular translocation across gastrointestinal epithelium has been suggested for the commensal bacteria E. coli and Proteus mirabilis (48). Different E. coli strains may employ different pathways to interact with or transverse the epithelium. For example, commensal translocating E. coli strain C25, a human gut isolate widely used to study gastrointestinal translocation, was shown to induce reduction in intestinal epithelial resistance, alterations in tight junction protein localization, and interruption of paracellular permeability (32). A new potential E. coli pathovar, adherent and invasive E. coli (AIEC), was found to adhere and invade intestinal epithelial cells via type 1 pili, and enhanced expression of fimbriae promoted AIEC translocation through M cell monolayers (49). Whether K. pneumoniae strains causing different types of infections can use different pathways requires investigation. Which bacterial genes in K. pneumoniae bacteremia and PLA strains are responsible for epithelial invasion and translocation also needs to be further characterized.
Three-dimensional (3D) image analysis in our study clearly showed the internalization of K. pneumoniae cells within enterocytes, implying a role of cell invasion in K. pneumoniae translocation across the intestinal epithelium. For the first time, we have demonstrated K. pneumoniae cells' in vivo ability to invade intestinal epithelial cells by using a colonization model in mice. We used a strain with better adhesion ability (Ca0438) as a demonstration model strain. The bacteremia isolate Ca0438 belongs to K2 capsular type, which is one of the representative capsular types in the gastrointestinal colonization strains. The LD50 of Ca0438 (>1 × 108 CFU) appeared to be higher than that in PLA isolates (e.g., NTUH-K2044 [1 × 105 CFU]) (50). We inoculated Ca0438 with a maximum of 1 × 109 CFU to increase the probability of observing intracellular K. pneumoniae cells. Virulence factors like serum and phagocytosis resistance and iron acquisition should be different between Ca0438 (a K2 Canadian strain) and NTUH-K2044 (a hypermucoid K1 Asian strain). The overall virulence in NTUH-K2044 was higher than that of Ca0438, although Ca0438 had a higher invasion rate (Fig. 2A). Our in vivo observation suggested K. pneumoniae invaded intestinal cells in a low frequency. As K. pneumoniae belongs to the already colonized (adhered) intrinsic microbiota in human intestine, the invasion rate would not be as high as those of extrinsic invasive pathogens. In agreement with this, gentamicin protection assays showed that K. pneumoniae Ca0438 invaded Caco-2 cells at a rate (∼1.8 × 10−4 of the inoculum) lower than that of the invasive pathogen S. Typhimurium (∼1.09 × 10−2 of the inoculum). A survey of invasion of clinical extraintestinal pathogenic E. coli (ExPEC) strains of Intestine-407 cells showed the invasion rates of most collected strains ranged from 10−5 to 10−3 of the inoculum (51). We showed the invasion rate of K. pneumoniae was significantly higher than that of commensal E. coli strain TVGHEC01. Therefore, K. pneumoniae appeared to invade the intestinal epithelium at a rate lower than those of extrinsic invasive pathogens but not the commensal bacteria. Similarly, our data showed that Ca0438 translocated across Caco-2 monolayers at a rate of ∼4.8 × 10−5 of the inoculum, which was lower than the reported rate of ∼1 × 10−2 of the inoculum for S. Typhimurium across Caco-2 monolayers (52). We found that the translocation rate of K. pneumoniae was higher than that of commensal E. coli strain TVGHEC01. For comparison, studies of commensal E. coli strain C25 have reported translocation rates of ∼2 × 10−5 of the inoculum across Caco-2 monolayers (53) and ∼1 × 10−7 of the inoculum across T84 colonic cell monolayers (32). The intestinal translocation ability of K. pneumoniae could be more comparable to those of intrinsic bacteria rather than the extrinsic pathogens.
Our data showed that cells of the bacteremia-inducing K. pneumoniae strains survived and replicated within intestinal epithelial cells. K. pneumoniae Ca0438 replicated within intestinal epithelial cells during the first 6 h after the addition of gentamicin and survived for ∼72 h. One study of S. Typhimurium showed an increased number of intracellular bacteria within 24 h inside MDCK epithelial cells (54). Another study also reported that a Salmonella enterica serovar Typhi clinical isolate increased within 24 h inside HeLa cells (55). A K. pneumoniae UTI isolate in a previous study was reported to replicate inside bladder epithelial cells within 4 h and survived for at least 96 h (24). However, a K. pneumoniae pneumonia isolate was shown to be unable to survive and replicate within human lung epithelial cells (25). The time course of intracellular multiplication of K. pneumoniae appeared not to be as long as those of extrinsic invasive pathogens within epithelial cells. Intracellular localization of K. pneumoniae could help the bacteria to escape host immune attack and establish a population in the gut. Although invasion may usually occur in a low frequency, we infer that under some circumstances, K. pneumoniae invasion could be enhanced. One possibility is that K. pneumoniae regulates the expression of capsule to affect cell adhesion and invasion (27). Promoted cell invasion provides a route for epithelial translocation, allowing gastrointestinal K. pneumoniae to access the bloodstream for systemic circulation, disseminate in the host, approach other organs, and cause disease.
The host-signaling cascades can be exploited by some pathogens to facilitate their internalization into host cells. We found the dual requirements of eukaryotic actin and microtubules for K. pneumoniae Ca0438 invasion, in agreement with a previous study of a UTI strain by Oelschlaeger and Tall (24). Our results further identified the requirements of host Rho family GTPases and PI3K/Akt signaling pathway in intestinal invasion of K. pneumoniae. These host proteins are involved in cell cytoskeleton regulation and are the popular targets hijacked by several enteroinvasive bacteria. Rho family GTPases are small GTP-binding proteins as molecular switches that control cell proliferation and differentiation, regulation of the actin cytoskeleton, membrane trafficking, and nuclear transport (56). Two major mechanisms to modulate the activity of Rho family GTPases have been described in several invasive pathogens (20, 57). The “trigger mechanism” is employed by Shigella and Salmonella spp. by injecting bacterial effector proteins into host cells via type III secretion systems. Bacterial proteins interact to activate Cdc42 and Rac1, inducing downstream actin rearrangements and membrane ruffling that result in the engulfment and internalization of bacteria. By the “zipper mechanism,” Yersinia pseudotuberculosis and Listeria monocytogenes invade cells via engagement of bacterial ligands and host receptors on the plasma membrane that elicits downstream cellular signaling events, such as the activation of Rac1, and ultimately leads to actin reassembly. How K. pneumoniae initiates Rho GTPase signaling remains unknown. What K. pneumoniae factors are involved and how the factors interact with Rho GTPases are worth further investigation.
In addition to Rho GTPases, our data showed a requirement for eukaryotic PI3K and Akt for K. pneumoniae invasion. In response to stimulation, regulatory protein PI3K recruits a variety of downstream proteins and has been implicated in many cell processes, including actin polymerization (45). PI3K/Akt signaling has also been reported to promote microtubule stabilization (58). Therefore, K. pneumoniae possibly modulates PI3K and Akt to control both actin and microtubule dynamics during the process of invasion. Pathogens such as L. monocytogenes (59), E. coli K1 (60), P. aeruginosa (61), and Streptococcus pneumoniae (62) have been shown to activate PI3K/Akt signaling to promote internalization to cells. Here, K. pneumoniae represents a new example of a bacterial species that modulates PI3K/Akt signaling. Cell protein-tyrosine kinases and Src kinase have been documented to contribute to invasion of Listeria (63), Salmonella (64), Shigella (65) and E. coli (38). Differently, our data showed that the inhibition of protein-tyrosine kinases by genistein or Src kinase by PP1 had no significant effect on K. pneumoniae invasion.
In summary, our study provides evidence to describe a possible mechanism for gastrointestinal K. pneumoniae penetration of the epithelial barrier to cause systemic infection. K. pneumoniae can invade nonphagocytic enterocytes, likely by hijacking the eukaryotic signaling machinery to control downstream cytoskeleton dynamics. The related signaling networks and bacterial factors that interact with cellular proteins are worth further characterization. Understanding how K. pneumoniae interacts with host cells and the underlying mechanisms involved will provide further insight into K. pneumoniae pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Hsien-Ho Lin at the Institute of Epidemiology and Preventive Medicine, National Taiwan University, for kind help with statistical analysis. We thank Po-An Su for help with animal experiments and the staff of the imaging core at the First Core Labs, NTUCM, for assistance with microscopy.
This study was supported by grants from the National Science Council, National Taiwan University, the Liver Disease Prevention and Treatment Research Foundation, and the National Taiwan University Hospital in Taiwan.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02345-14.
REFERENCES
- 1.Podschun RUU. 1998. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev 11:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ko WC, Paterson DL, Sagnimeni AJ, Hansen DS, Von Gottberg A, Mohapatra S, Casellas JM, Goossens H, Mulazimoglu L, Trenholme G, Klugman KP, McCormack JG, Yu VL. 2002. Community-acquired Klebsiella pneumoniae bacteremia: global differences in clinical patterns. Emerg Infect Dis 8:160–166. doi: 10.3201/eid0802.010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang CC, Yen CH, Ho MW, Wang JH. 2004. Comparison of pyogenic liver abscess caused by non-Klebsiella pneumoniae and Klebsiella pneumoniae. J Microbiol Immunol Infect 37:176–184. [PubMed] [Google Scholar]
- 4.Lederman ER, Crum NF. 2005. Pyogenic liver abscess with a focus on Klebsiella pneumoniae as a primary pathogen: an emerging disease with unique clinical characteristics. Am J Gastroenterol 100:322–331. doi: 10.1111/j.1572-0241.2005.40310.x. [DOI] [PubMed] [Google Scholar]
- 5.Chung DR, Lee SS, Lee HR, Kim HB, Choi HJ, Eom JS, Kim JS, Choi YH, Lee JS, Chung MH, Kim YS, Lee H, Lee MS, Park CK. 2007. Emerging invasive liver abscess caused by K1 serotype Klebsiella pneumoniae in Korea. J Infect 54:578–583. doi: 10.1016/j.jinf.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Mizuta K, Ohta M, Mori M, Hasegawa T, Nakashima I, Kato N. 1983. Virulence for mice of Klebsiella strains belonging to the O1 group: relationship to their capsular (K) types. Infect Immun 40:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu CR, Lin TL, Pan YJ, Hsieh PF, Wang JT. 2013. Isolation of a bacteriophage specific for a new capsular type of Klebsiella pneumoniae and characterization of its polysaccharide depolymerase. PLoS One 8:e70092. doi: 10.1371/journal.pone.0070092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fung CP, Chang FY, Lee SC, Hu BS, Kuo BI, Liu CY, Ho M, Siu LK. 2002. A global emerging disease of Klebsiella pneumoniae liver abscess: is serotype K1 an important factor for complicated endophthalmitis? Gut 50:420–424. doi: 10.1136/gut.50.3.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Struve C, Bojer M, Nielsen EM, Hansen DS, Krogfelt KA. 2005. Investigation of the putative virulence gene magA in a worldwide collection of 495 Klebsiella isolates: magA is restricted to the gene cluster of Klebsiella pneumoniae capsule serotype K1. J Med Microbiol 54:1111–1113. doi: 10.1099/jmm.0.46165-0. [DOI] [PubMed] [Google Scholar]
- 10.Lin YT, Siu LK, Lin JC, Chen TL, Tseng CP, Yeh KM, Chang FY, Fung CP. 2012. Seroepidemiology of Klebsiella pneumoniae colonizing the intestinal tract of healthy Chinese and overseas Chinese adults in Asian countries. BMC Microbiol 12:13. doi: 10.1186/1471-2180-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montgomerie JZ. 1979. Epidemiology of Klebsiella and hospital-associated infections. Rev Infect Dis 1:18. [DOI] [PubMed] [Google Scholar]
- 12.De Champs C, Sauvant MP, Chanal C, Sirot D, Gazuy N, Malhuret R, Baguet JC, Sirot J. 1989. Prospective survey of colonization and infection caused by expanded-spectrum-beta-lactamase-producing members of the family Enterobacteriaceae in an intensive care unit. J Clin Microbiol 27:2887–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fung CP, Lin YT, Lin JC, Chen TL, Yeh KM, Chang FY, Chuang HC, Wu HS, Tseng CP, Siu LK. 2012. Klebsiella pneumoniae in gastrointestinal tract and pyogenic liver abscess. Emerg Infect Dis 18:1322–1325. doi: 10.3201/eid1808.111053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pena C, Pujol M, Ardanuy C, Ricart A, Pallares R, Linares J, Ariza J, Gudiol F. 1998. Epidemiology and successful control of a large outbreak due to Klebsiella pneumoniae producing extended-spectrum beta-lactamases. Antimicrob Agents Chemother 42:53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Almeida VC, Pessoa-Silva CL, Sampaio JL, Gontijo Filho PP, Teixeira LM, Moreira BM. 2005. Genetic relatedness among extended-spectrum beta-lactamase-producing Klebsiella pneumoniae outbreak isolates associated with colonization and invasive disease in a neonatal intensive care unit. Microb Drug Resist 11:21–25. doi: 10.1089/mdr.2005.11.21. [DOI] [PubMed] [Google Scholar]
- 16.Walker RI, Owen RL. 1990. Intestinal barriers to bacteria and their toxins. Annu Rev Med 41:393–400. doi: 10.1146/annurev.me.41.020190.002141. [DOI] [PubMed] [Google Scholar]
- 17.Sears CL. 2000. Molecular physiology and pathophysiology of tight junctions. V. Assault of the tight junction by enteric pathogens. Am J Physiol Gastrointest Liver Physiol 279:G1129–G1134. [DOI] [PubMed] [Google Scholar]
- 18.Kazmierczak BI, Mostov K, Engel JN. 2001. Interaction of bacterial pathogens with polarized epithelium. Annu Rev Microbiol 55:407–435. doi: 10.1146/annurev.micro.55.1.407. [DOI] [PubMed] [Google Scholar]
- 19.Balzan S, de Almeidaq Uadros C, de Cleva R, Zilberstein B, Cecconello I. 2007. Bacterial translocation: overview of mechanisms and clinical impact. J Gastroenterol Hepatol 22:464–471. doi: 10.1111/j.1440-1746.2007.04933.x. [DOI] [PubMed] [Google Scholar]
- 20.Cossart P, Sansonetti PJ. 2004. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304:242–248. doi: 10.1126/science.1090124. [DOI] [PubMed] [Google Scholar]
- 21.Wu Z, Nybom P, Magnusson KE. 2000. Distinct effects of Vibrio cholerae haemagglutinin/protease on the structure and localization of the tight junction-associated proteins occludin and ZO-1. Cell Microbiol 2:11–17. doi: 10.1046/j.1462-5822.2000.00025.x. [DOI] [PubMed] [Google Scholar]
- 22.Backert S, Boehm M, Wessler S, Tegtmeyer N. 2013. Transmigration route of Campylobacter jejuni across polarized intestinal epithelial cells: paracellular, transcellular or both? Cell Commun Signal 11:72. doi: 10.1186/1478-811X-11-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okuda J, Hayashi N, Okamoto M, Sawada S, Minagawa S, Yano Y, Gotoh N. 2010. Translocation of Pseudomonas aeruginosa from the intestinal tract is mediated by the binding of ExoS to an Na,K-ATPase regulator, FXYD3. Infect Immun 78:4511–4522. doi: 10.1128/IAI.00428-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oelschlaeger TA, Tall BD. 1997. Invasion of cultured human epithelial cells by Klebsiella pneumoniae isolated from the urinary tract. Infect Immun 65:2950–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cortes G, Alvarez D, Saus C, Alberti S. 2002. Role of lung epithelial cells in defense against Klebsiella pneumoniae pneumonia. Infect Immun 70:1075–1080. doi: 10.1128/IAI.70.3.1075-1080.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosen DA, Pinkner JS, Jones JM, Walker JN, Clegg S, Hultgren SJ. 2008. Utilization of an intracellular bacterial community pathway in Klebsiella pneumoniae urinary tract infection and the effects of FimK on type 1 pilus expression. Infect Immun 76:3337–3345. doi: 10.1128/IAI.00090-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sahly H, Podschun R, Oelschlaeger TA, Greiwe M, Parolis H, Hasty D, Kekow J, Ullmann U, Ofek I, Sela S. 2000. Capsule impedes adhesion to and invasion of epithelial cells by Klebsiella pneumoniae. Infect Immun 68:6744–6749. doi: 10.1128/IAI.68.12.6744-6749.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chuang YP, Fang CT, Lai SY, Chang SC, Wang JT. 2006. Genetic determinants of capsular serotype K1 of Klebsiella pneumoniae causing primary pyogenic liver abscess. J Infect Dis 193:645–654. doi: 10.1086/499968. [DOI] [PubMed] [Google Scholar]
- 29.Hsu CR, Lin TL, Chen YC, Chou HC, Wang JT. 2011. The role of Klebsiella pneumoniae rmpA in capsular polysaccharide synthesis and virulence revisited. Microbiology 157:3446–3457. doi: 10.1099/mic.0.050336-0. [DOI] [PubMed] [Google Scholar]
- 30.Pan YJ, Fang HC, Yang HC, Lin TL, Hsieh PF, Tsai FC, Keynan Y, Wang JT. 2008. Capsular polysaccharide synthesis regions in Klebsiella pneumoniae serotype K57 and a new capsular serotype. J Clin Microbiol 46:2231–2240. doi: 10.1128/JCM.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenshine I, Ruschkowski S, Foubister V, Finlay BB. 1994. Salmonella typhimurium invasion of epithelial cells: role of induced host cell tyrosine protein phosphorylation. Infect Immun 62:4969–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zareie M, Riff J, Donato K, McKay DM, Perdue MH, Soderholm JD, Karmali M, Cohen MB, Hawkins J, Sherman PM. 2005. Novel effects of the prototype translocating Escherichia coli, strain C25 on intestinal epithelial structure and barrier function. Cell Microbiol 7:1782–1797. doi: 10.1111/j.1462-5822.2005.00595.x. [DOI] [PubMed] [Google Scholar]
- 33.Louwen R, Nieuwenhuis EE, van Marrewijk L, Horst-Kreft D, de Ruiter L, Heikema AP, van Wamel WJ, Wagenaar JA, Endtz HP, Samsom J, van Baarlen P, Akhmanova A, van Belkum A. 2012. Campylobacter jejuni translocation across intestinal epithelial cells is facilitated by ganglioside-like lipooligosaccharide structures. Infect Immun 80:3307–3318. doi: 10.1128/IAI.06270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benedek O, Nagy G, Emody L. 2004. Intracellular signalling and cytoskeletal rearrangement involved in Yersinia pestis plasminogen activator (Pla) mediated HeLa cell invasion. Microb Pathog 37:47–54. doi: 10.1016/j.micpath.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 35.Luck SN, Bennett-Wood V, Poon R, Robins-Browne RM, Hartland EL. 2005. Invasion of epithelial cells by locus of enterocyte effacement-negative enterohemorrhagic Escherichia coli. Infect Immun 73:3063–3071. doi: 10.1128/IAI.73.5.3063-3071.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Q, Zhao WD, Zhang K, Fang WG, Hu Y, Wu SH, Chen YH. 2010. PI3K-dependent host cell actin rearrangements are required for Cronobacter sakazakii invasion of human brain microvascular endothelial cells. Med Microbiol Immunol 199:333–340. doi: 10.1007/s00430-010-0168-8. [DOI] [PubMed] [Google Scholar]
- 37.Surviladze Z, Waller A, Strouse JJ, Bologa C, Ursu O, Salas V, Parkinson JF, Phillips GK, Romero E, Wandinger-Ness A, Sklar LA, Schroeder C, Simpson D, Noth J, Wang J, Golden J, Aube J. 16 December 2010. A potent and selective inhibitor of Cdc42 GTPase. Probe Reports from the NIH Molecular Libraries Program. National Center for Biotechnology Information, Bethesda, MD. [PubMed] [Google Scholar]
- 38.Liu W, Zhao WD, Yan JC, Ren ZY, Fang WG, Zhu L, Shang DS, Chen YH. 2010. Involvement of Src tyrosine kinase in Escherichia coli invasion of human brain microvascular endothelial cells. FEBS Lett 584:27–32. doi: 10.1016/j.febslet.2009.10.090. [DOI] [PubMed] [Google Scholar]
- 39.Maruvada R, Kim KS. 2012. IbeA and OmpA of Escherichia coli K1 exploit Rac1 activation for invasion of human brain microvascular endothelial cells. Infect Immun 80:2035–2041. doi: 10.1128/IAI.06320-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finlay BB, Falkow S. 1990. Salmonella interactions with polarized human intestinal Caco-2 epithelial cells. J Infect Dis 162:1096–1106. doi: 10.1093/infdis/162.5.1096. [DOI] [PubMed] [Google Scholar]
- 41.Harvey P, Battle T, Leach S. 1999. Different invasion phenotypes of Campylobacter isolates in Caco-2 cell monolayers. J Med Microbiol 48:461–469. doi: 10.1099/00222615-48-5-461. [DOI] [PubMed] [Google Scholar]
- 42.Baker NT, Graham LL. 2010. Campylobacter fetus translocation across Caco-2 cell monolayers. Microb Pathog 49:260–272. doi: 10.1016/j.micpath.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 43.Pan YJ, Lin TL, Hsu CR, Wang JT. 2011. Use of a Dictyostelium model for isolation of genetic loci associated with phagocytosis and virulence in Klebsiella pneumoniae. Infect Immun 79:997–1006. doi: 10.1128/IAI.00906-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim KP, Loessner MJ. 2008. Enterobacter sakazakii invasion in human intestinal Caco-2 cells requires the host cell cytoskeleton and is enhanced by disruption of tight junction. Infect Immun 76:562–570. doi: 10.1128/IAI.00937-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buttrick GJ, Wakefield JG. 2008. PI3-K and GSK-3: Akt-ing together with microtubules. Cell Cycle 7:2621–2625. doi: 10.4161/cc.7.17.6514. [DOI] [PubMed] [Google Scholar]
- 46.Nijssen S, Florijn A, Bonten MJ, Schmitz FJ, Verhoef J, Fluit AC. 2004. Beta-lactam susceptibilities and prevalence of ESBL-producing isolates among more than 5000 European Enterobacteriaceae isolates. Int J Antimicrob Agents 24:585–591. doi: 10.1016/j.ijantimicag.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 47.Nordmann P, Cuzon G, Naas T. 2009. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect Dis 9:228–236. doi: 10.1016/S1473-3099(09)70054-4. [DOI] [PubMed] [Google Scholar]
- 48.Wells CL, Erlandsen SL. 1991. Localization of translocating Escherichia coli, Proteus mirabilis, and Enterococcus faecalis within cecal and colonic tissues of monoassociated mice. Infect Immun 59:4693–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chassaing B, Etienne-Mesmin L, Bonnet R, Darfeuille-Michaud A. 2013. Bile salts induce long polar fimbriae expression favouring Crohn's disease-associated adherent-invasive Escherichia coli interaction with Peyer's patches. Environ Microbiol 15:355–371. doi: 10.1111/j.1462-2920.2012.02824.x. [DOI] [PubMed] [Google Scholar]
- 50.Hsieh PF, Lin TL, Lee CZ, Tsai SF, Wang JT. 2008. Serum-induced iron-acquisition systems and TonB contribute to virulence in Klebsiella pneumoniae causing primary pyogenic liver abscess. J Infect Dis 197:1717–1727. doi: 10.1086/588383. [DOI] [PubMed] [Google Scholar]
- 51.Martinez-Medina M, Mora A, Blanco M, Lopez C, Alonso MP, Bonacorsi S, Nicolas-Chanoine MH, Darfeuille-Michaud A, Garcia-Gil J, Blanco J. 2009. Similarity and divergence among adherent-invasive Escherichia coli and extraintestinal pathogenic E. coli strains. J Clin Microbiol 47:3968–3979. doi: 10.1128/JCM.01484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kortman GA, Boleij A, Swinkels DW, Tjalsma H. 2012. Iron availability increases the pathogenic potential of Salmonella typhimurium and other enteric pathogens at the intestinal epithelial interface. PLoS One 7:e29968. doi: 10.1371/journal.pone.0029968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Macutkiewicz C, Carlson G, Clark E, Dobrindt U, Roberts I, Warhurst G. 2008. Characterisation of Escherichia coli strains involved in transcytosis across gut epithelial cells exposed to metabolic and inflammatory stress. Microbes Infect 10:424–431. doi: 10.1016/j.micinf.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 54.Leung KY, Finlay BB. 1991. Intracellular replication is essential for the virulence of Salmonella typhimurium. Proc Natl Acad Sci U S A 88:11470–11474. doi: 10.1073/pnas.88.24.11470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yabuuchi E, Ikedo M, Ezaki T. 1986. Invasiveness of Salmonella typhi strains in HeLa S3 monolayer cells. Microbiol Immunol 30:1213–1224. doi: 10.1111/j.1348-0421.1986.tb03050.x. [DOI] [PubMed] [Google Scholar]
- 56.Etienne-Manneville S, Hall A. 2002. Rho GTPases in cell biology. Nature 420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 57.Finlay BB. 2005. Bacterial virulence strategies that utilize Rho GTPases. Curr Top Microbiol Immunol 291:1–10. [DOI] [PubMed] [Google Scholar]
- 58.Onishi K, Higuchi M, Asakura T, Masuyama N, Gotoh Y. 2007. The PI3K-Akt pathway promotes microtubule stabilization in migrating fibroblasts. Genes Cells 12:535–546. doi: 10.1111/j.1365-2443.2007.01071.x. [DOI] [PubMed] [Google Scholar]
- 59.Mansell A, Khelef N, Cossart P, O'Neill LA. 2001. Internalin B activates nuclear factor-kappa B via Ras, phosphoinositide 3-kinase, and Akt. J Biol Chem 276:43597–43603. doi: 10.1074/jbc.M105202200. [DOI] [PubMed] [Google Scholar]
- 60.Zhao WD, Liu W, Fang WG, Kim KS, Chen YH. 2010. Vascular endothelial growth factor receptor 1 contributes to Escherichia coli K1 invasion of human brain microvascular endothelial cells through the phosphatidylinositol 3-kinase/Akt signaling pathway. Infect Immun 78:4809–4816. doi: 10.1128/IAI.00377-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kierbel A, Gassama-Diagne A, Mostov K, Engel JN. 2005. The phosphoinositol-3-kinase-protein kinase B/Akt pathway is critical for Pseudomonas aeruginosa strain PAK internalization. Mol Biol Cell 16:2577–2585. doi: 10.1091/mbc.E04-08-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Agarwal V, Hammerschmidt S. 2009. Cdc42 and the phosphatidylinositol 3-kinase-Akt pathway are essential for PspC-mediated internalization of pneumococci by respiratory epithelial cells. J Biol Chem 284:19427–19436. doi: 10.1074/jbc.M109.003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Velge P, Bottreau E, Kaeffer B, Yurdusev N, Pardon P, Van Langendonck N. 1994. Protein tyrosine kinase inhibitors block the entries of Listeria monocytogenes and Listeria ivanovii into epithelial cells. Microb Pathog 17:37–50. doi: 10.1006/mpat.1994.1050. [DOI] [PubMed] [Google Scholar]
- 64.Wells CL, Jechorek RP, Kinneberg KM, Debol SM, Erlandsen SL. 1999. The isoflavone genistein inhibits internalization of enteric bacteria by cultured Caco-2 and HT-29 enterocytes. J Nutr 129:634–640. [DOI] [PubMed] [Google Scholar]
- 65.Mounier J, Popoff MR, Enninga J, Frame MC, Sansonetti PJ, Van Nhieu GT. 2009. The IpaC carboxyterminal effector domain mediates Src-dependent actin polymerization during Shigella invasion of epithelial cells. PLoS Pathog 5:e1000271. doi: 10.1371/journal.ppat.1000271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.