

Abstract
Bacterial tyrosine kinases and their cognate protein tyrosine phosphatases are best known for regulating the biosynthesis of polysaccharides. Moreover, their roles in the stress response, DNA metabolism, cell division, and virulence have also been documented. The aim of this study was to investigate the pathogenicity and potential mechanisms of virulence dependent on the tyrosine kinase BceF and phosphotyrosine phosphatase BceD of the cystic fibrosis opportunistic pathogen Burkholderia contaminans IST408. The insertion mutants bceD::Tp and bceF::Tp showed similar attenuation of adhesion and invasion of the cystic fibrosis lung epithelial cell line CFBE41o- compared to the parental strain B. contaminans IST408. In the absence of bceD or bceF genes, B. contaminans also showed a reduction in the ability to translocate across polarized epithelial cell monolayers, demonstrated by a higher transepithelial electrical resistance, reduced flux of fluorescein isothiocyanate-labeled bovine serum albumin, and higher levels of tight junction proteins ZO-1, occludin, and claudin-1 present in monolayers exposed to these bacterial mutants. Furthermore, bceD::Tp and bceF::Tp mutants induced lower levels of interleukin-6 (IL-6) and IL-8 release than the parental strain. In conclusion, although the mechanisms of pathogenicity dependent on BceD and BceF are not understood, these proteins contribute to the virulence of Burkholderia by enhancement of cell attachment and invasion, disruption of epithelial integrity, and modulation of the proinflammatory response.
INTRODUCTION
Proteins belonging to the bacterial tyrosine kinase (BY-kinase) family have no eukaryotic homologues and were first described as being involved in the biosynthesis of polysaccharides in several bacterial systems. BY-kinases contain Walker A and Walker B ATP-binding motifs that bind ATP and use it to promote tyrosine phosphorylation. This posttranslational modification can be reversed by the action of protein tyrosine phosphatases (PTPs), whose genes are usually located near the genes encoding BY-kinases. Cycles of tyrosine phosphorylation and dephosphorylation by the actions of BY-kinases and PTP proteins, respectively, have been shown to control the amount of and/or molecular weight of the polysaccharide (1, 2). In addition, several studies have shown that BY-kinases are able to interact with other proteins that are not connected to polysaccharide biosynthesis, promoting their phosphorylation and thereby controlling their activity and, in some cases, their cellular location (1, 3). Therefore, BY-kinases are implicated in several cell functions, such as DNA metabolism, resistance to stress, and cellular division, among others (4–7). Regarding bacterial tyrosine phosphatases, they can be grouped in the families of the eukaryotic-like phosphatases (PTPs) and dual-specific phosphatases, the low-molecular-weight protein tyrosine phosphatases (LMW-PTPs), and the polymerase-histidinol phosphatases (PHPs) (8). Despite many of them being involved in polysaccharide production (2), several have been implicated in host-bacteria interactions (8). Examples include the following: the YopH protein tyrosine phosphatase of Yersinia pseudotuberculosis, which promotes cytoskeletal rearrangements and inhibition of phagocytosis (9); SptP from Salmonella enterica serovar Typhimurium and StpA from Salmonella enterica serovar Typhi, which disrupt the host cell cytoskeleton (10, 11); MptpA and MptpB from Mycobacterium tuberculosis, which inhibit phagosome maturation (12, 13) and subvert the host immune response (14); LipA of Listeria monocytogenes, which interferes with the actin cytoskeleton (15); and the nonfunctional tyrosine phosphatase Dpm of Burkholderia cenocepacia, which has been implicated in the delayed maturation of bacteria-containing vacuoles in macrophages (16). All these characterized tyrosine phosphatases, acting as effector proteins, are secreted into eukaryotic cells and, except for MptpA and Dpm, which are LMW-PTPs, all of the other mentioned enzymes are members of the PTP family (8, 16).
Bacteria belonging to the Burkholderia cepacia complex are ubiquitously found in natural environments but also occur as contaminants in man-made products, such as pharmaceuticals, cosmetics, and disinfectants (17). They are opportunistic pathogens, particularly for cystic fibrosis (CF) patients. Although a transient infection of the respiratory tract may occur for some patients, the acquisition of B. cepacia complex most typically results in chronic infection (17, 18). Depending on the B. cepacia complex strain, this colonization ranges from asymptomatic to a rapid decline of lung function characterized by a necrotizing pneumonia and the development of septicemia known as cepacia syndrome (19). Burkholderia cenocepacia and Burkholderia multivorans are the predominant species in infected CF patients (20, 21), but an increasing number of outbreaks caused by B. cepacia complex species such as Burkholderia contaminans has been reported (22–24). B. cepacia complex species are also intrinsically resistant to several antibiotics and able to form biofilms, making their eradication from both lungs and clinical devices very difficult (25–27). In vitro studies have shown that during infection of lung epithelial cells, B. cepacia complex isolates adhere to the apical surface of the epithelium, forming microcolonies followed by cell invasion and disruption of tight junction integrity, promoting bacterial translocation to the basolateral surface via paracytosis (28–30). The ability of B. cepacia complex strains to cross the epithelium paracellularly or transcellularly, penetrating the airway barriers, is associated with the ability of these bacteria to cause cepacia syndrome (31, 32).
Many B. cepacia complex virulence factors have been characterized and implicated in virulence or persistence of the infection (reviewed in reference 33). Among these virulence factors is the exopolysaccharide (EPS) cepacian, which has been found to be produced by clinical and environmental Burkholderia species, including B. cepacia complex and non-B. cepacia complex species (reviewed in reference 34). Among the proteins required for cepacian biosynthesis, the BY-kinase BceF and the LMW-PTP BceD, which are conserved among the Burkholderia genus, seem to have a central role in the regulation of cepacian production (35, 36). In addition, the CF clinical isolate Burkholderia contaminans (formerly B. cepacia) strain IST408 bceF mutant showed lower swarming and swimming motilities, failed to produce biofilms on abiotic surfaces, was less resistant to stress conditions such us UV irradiation and heat shock stress, showed attenuation of virulence in Galleria mellonella, and was completely avirulent in a chronic granulomatous disease gp91phox−/− mouse infection model (7, 35, 37). As posttranslational modifications mediated by BceF and possibly BceD proteins are implicated in several cellular processes, including virulence properties, we investigated the importance of both proteins in the interaction of B. cepacia complex bacteria with host cells. Namely, we used a cystic fibrosis CFBE41o- lung epithelial cell line model to test adhesion, invasion, tight junction disruption, and immune stimulation by the CF clinical isolate B. contaminans IST408 and its isogenic derivative bceD and bceF mutants.
MATERIALS AND METHODS
Bacterial strains and media.
The bacterial strains used in this study were the highly mucoid genetically modifiable CF clinical isolate Burkholderia contaminans IST408 and its derivative insertion mutants bceD::Tp, bceE::Tp, and bceF::Tp (7, 35) and Escherichia coli strains NCIB9415 (NCIMB, United Kingdom) and DH5α (38). By using recA-based genomovar-specific primer pairs for gene amplification followed by restriction fragment length polymorphism analysis with HaeIII, strain IST408 was considered for several years to belong to the species B. cepacia (genomovar I, taxon K). Recently, with the introduction of the multilocus sequence typing (MLST) method (and sequencing of the genes atpD, gltB, gyrB, recA, lepA, phaC, and trpP), this strain was relocated to the species B. contaminans (according to the Burkholderia MLST database, it is ST96). This strain has been used as a model for cepacian biosynthesis (34). All strains were routinely cultured in Lennox broth (LB) at 37°C. Growth medium was supplemented with antibiotics when required to maintain the selective pressure at the following concentrations: for B. contaminans strains, trimethoprim at 100 μg/ml and chloramphenicol at 100 μg/ml; for E. coli strains, chloramphenicol at 25 μg/ml and kanamycin at 50 μg/ml.
DNA manipulation techniques.
Genomic DNA was extracted from B. contaminans IST408 by using the DNeasy blood and tissue kit (Qiagen), following the manufacturer's instructions. Plasmid DNA isolation and purification, DNA restriction, agarose gel electrophoresis, and E. coli transformation were carried out using standard procedures (39). B. contaminans electrocompetent cells were transformed by electroporation using a Bio-Rad Gene Pulser II system (200 Ω, 25 μF, 2.5 kV) and grown overnight before being plated on selective medium. Triparental conjugation to B. contaminans strains was performed using the helper plasmid pRK2013 (40).
Plasmid construction and complementation experiments.
Complementation of B. contaminans mutant strains was performed by expressing bceD, bceE, or bceF from the promoter of the bce operon directing the biosynthesis of cepacian (41) present in the replicative vector pBBR1MCS (42). To prepare the plasmids expressing each of the genes, the bce promoter and each gene region were amplified by PCR using B. contaminans IST408 genomic DNA as the template. The primers used to amplify the bce promoter region were Pbce-Fw (GATAAGCTTCTCCTCGATTGAAGT [restriction sites are shown in italics]) and Pbce-Rev (GTGCATATGCTTCGATTCAAACGT). The primers used to amplify the bceD, bceE, and bceF genes were bceD-Fw (GCTCATATGCGGAACATCCTGATCGTCT), bceD-Rev (CACTCTAGAACAGGCTGACAGGAAAGTCG), bceE-Fw (GAACATATGCTGAAACGCCCGATG), bceE-Rev (TGATCTAGAGGAGCAGCTGGCCGAGGA), bceF-Fw (GAACATATGGTGAACACGCAAGCGAAA), and bceF-Rev (TTATCTAGAATGCGGATCAGGCGCTCA). The amplified promoter region was restricted with HindIII/NdeI, and the gene regions were restricted with NdeI/XbaI and ligated to the HindIII/XbaI sites of the intermediate pUK21 vector. Each fragment containing the bce promoter and one of the gene regions was then cloned into the pBBR1MCS vector. The plasmids were named pLM136-2, pLM127-13, and pLM135-6 and carried the bceD, bceF, and bceE genes, respectively; they were confirmed by DNA sequencing. The empty vector or the vector with the inserted gene was introduced into the corresponding mutant or parental strain by triparental conjugation.
Cell line culture.
The epithelial cell line used was CFBE41o-, derived from a patient homozygous for the cystic fibrosis transmembrane conductance regulator (CFTR) ΔF508 mutation, which leads to defects in chloride ion and water transport across the cell membrane (43). CFBE41o- cells were routinely maintained in fibronectin/Vitrogen-coated flasks in Eagle's modified minimum essential medium (MEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS; Lonza), 1% (vol/vol) nonessential amino acids (Gibco), l-glutamine (2 mM; Sigma-Aldrich), and 1% (vol/vol) penicillin-streptomycin (Sigma-Aldrich) (44) in a humidified atmosphere at 37°C with 5% CO2.
Host cell attachment.
Prior to bacterial infection, epithelial cells were seeded onto fibronectin-coated 24-well plates (Orange Scientific) at 4 × 105 cells/well in supplemented MEM without antibiotics and incubated overnight at 37°C in a 5% CO2 atmosphere. Bacterial strains were grown in LB until the optical density at 640 nm (OD640) reached 0.6 (log phase) and were used to infect epithelial cells at a multiplicity of infection (MOI) of 10 (10 bacteria to 1 epithelial cell). Bacteria were applied to the seeded CFBE41o- cells in MEM supplemented medium, and the plates were centrifuged at 700 × g for 5 min, to facilitate bacterial attachment. The plates were then incubated for 30 min at 37°C in an atmosphere of 5% CO2. Each well was washed vigorously three times with phosphate-buffered saline (PBS) to remove unbound bacteria. Cells were lysed with buffer (0.01 M PBS, 20 mM EDTA, 0.5% Triton X-100; pH 7.4) for 30 min at 4°C. Adhesion of bacteria was quantified by CFU counts after plating serial dilutions onto LB agar and incubation for 24 h at 37°C. E. coli NCIB9415 was used as a negative control. Duplicates of each strain were performed per assay, and the results presented were obtained from three independent experiments. Results are expressed as the percentage of adhesion, which was calculated as the CFU recovered divided by the CFU applied to the epithelial cells, multiplied by 100.
Confocal laser scanning microscopy assays.
To visualize attachment to epithelial cells, bacteria were stained by using plasmid pIN29, which encodes the red fluorescent protein DsRed (45). This plasmid was introduced into parental strain B. contaminans IST408 or the bceD::Tp, bceE::Tp, and bceF::Tp mutants by electroporation. Bacterial adhesion (MOI, 50) was performed as described above, but glass coverslips were placed into 24-well plates before cell seeding. At the end of the adhesion experiment, coverslips with cells were washed with PBS. Immunostaining experiments were performed as previously described (46). For these experiments, samples were fixed with 3.7% (vol/vol) formaldehyde for 20 min, quenched with 50 mM NH4Cl for 10 min, immersed in 0.2% (vol/vol) Triton X-100 for 5 min, and saturated with 5% (wt/vol) bovine serum albumin (BSA) during 30 min. Samples were then incubated with primary mouse E-cadherin antibody (1:100; Santa Cruz Biotechnology) for 1 h 30 min, followed by washing with PBS. The secondary antibody was a polyclonal goat anti-mouse antibody coupled to Alexa Fluor 488 (1:500; Santa Cruz Biotechnology).
Samples were examined on a Leica TCS SP5 inverted microscope (model DMI6000; Leica Microsystems CMS GmbH, Mannheim, Germany) with a 63×, 1.2-numerical aperture water apochromatic objective (47). Orthogonal projections were generated with ImageJ software (http://rsb.info.nih.gov/ij/).
Invasion of epithelial cells.
Studies of invasion of epithelial cells were performed as described previously (28). B. contaminans strains were applied to 4 × 105 CFBE41o- cells in MEM supplemented medium (MOI, 10). The plates were centrifuged at 700 × g for 5 min to facilitate bacterial attachment and incubated under standard conditions (37°C, 5% CO2). After 2 h, the MEM was removed and 500 μl of ciprofloxacin (2 mg/ml) was added to kill extracellular bacteria. After 2 h of incubation with antibiotic, cell monolayers were washed three times with PBS and incubated in lysis buffer for 30 min at 4°C. The number of intracellular bacteria was determined by CFU counts on LB agar. The product of the last PBS wash before cell lysis was also plated onto LB agar to ensure that antibiotic killing activity was greater than 99.99%. Invasion was expressed as a percentage and was calculated as the CFU recovered divided by the CFU applied to the cells, multiplied by 100. Duplicates experiments for each strain were performed per assay, and the results are the mean values from three independent experiments.
Growth rates and ciprofloxacin MIC determinations.
Growth rates of the strains under study were determined in LB and MEM statically at 37°C in a 5% CO2 humidified atmosphere, and growth was measured by monitoring the OD640 for 24 h. Growth rates were calculated from the exponential phase of growth from at least three independent experiments. To determine ciprofloxacin MICs, serial dilutions of Mueller-Hinton medium with ciprofloxacin ranging from 0 to 256 μg/ml were inoculated with the parental and mutant strains for 24 h at 37°C under a 5% CO2 atmosphere, and growth was determined by measuring the OD640.
Bacterial infection of polarized lung epithelial cells.
The disruption of tight junction integrity by B. contaminans strains was evaluated based on changes in transepithelial electrical resistance (TER) in polarized CFBE41o- cells. To obtain polarized epithelia, the cells were seeded at a density of 6 × 105 cells/well onto Transwell permeable supports (12-mm well; pore size, 0.4 μm; Costar, Corning, NY), which were precoated with Vitrogen (Nutacon, Netherlands). The FBS was replaced by UltraserG 24 h after seeding (48). Cells were incubated for another 24 h before removal of the medium in the apical chamber, grown for 6 days with an air-liquid interface, and fed basolaterally on alternate days. The functional integrity of the epithelial monolayers was confirmed by measuring the transepithelial electrical resistance with an epithelial volt-ohmmeter (EVOM; World Precision Instruments). The monolayers were infected (MOI, 10) by resuspending bacteria (log phase) in antibiotic-free supplemented MEM, which was applied onto the apical surface. The TER was measured at different time points for up to 6 h. E. coli infection and a well containing MEM only were used as controls of junction integrity during the assays and to correct the resistance values obtained.
Epithelial permeability.
The permeability of cell polarized monolayers was quantified by measuring the flux of fluorescein isothiocyanate (FITC)-labeled BSA across the epithelial layer, as described previously (30). Log-phase bacteria (MOI, 10) suspended in antibiotic-free supplemented MEM containing 0.1% (wt/vol) FITC-labeled BSA (Sigma-Aldrich) were inoculated onto the apical surface of the cell monolayer. The medium in the basolateral chamber was initially free of FITC-labeled BSA. At 4 and 6 h after infection, 50 μl of medium was collected from the basolateral chamber and fluorescence was measured with a multimode microplate reader (FilterMax F5; Molecular Devices). The coefficient of permeability (Papp) was calculated as described previously (49).
Western blot analyses.
To confirm that the strains under study had different abilities to disrupt tight junctions, polarized epithelial cell extracts from cell layers incubated with B. contaminans, E. coli (MOI, 10), or medium alone were prepared and ZO-1, occludin, and claudin-1 levels were analyzed by Western blotting, with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) used as loading control protein. Briefly, polarized CFBE41o- cells were lysed 4 h postinfection by adding RIPA buffer (Sigma-Aldrich) and a protease inhibitor cocktail (Thermo Scientific) to the filters, followed by scraping to recover cell lysate. The protein content of the lysates was quantified by using a Nanodrop spectrophotometer, and proteins were applied to 6% SDS-PAGE gels (for ZO-1 and occludin) or 12% gels for claudin-1. Proteins were subsequently transferred to a nitrocellulose membrane by using a Trans-Blot Turbo transfer system (Bio-Rad) with the standard program Mix MW (25 V, 1.3 A, 7 min). Membranes were blocked with Tris-buffered saline solution containing 5% (wt/vol) skim milk and 0.5% (vol/vol) Tween 20 (TBS-T) for 1 h. Blots were incubated overnight at 4°C with primary antibodies against ZO-1 or claudin-1 (Invitrogen), occludin (BD Biosciences), or GAPDH (Santa Cruz Biotechnology), using a dilution of 1:1,000 for anti-occludin and 1:300 for the remaining antibodies. Membranes were washed three times with TBS-T and incubated with 1:2,000-diluted goat anti-mouse serum (Thermo Scientific) for ZO-1, occludin, and GAPDH detection and with goat anti-rabbit serum (Santa Cruz Biotechnology) for claudin-1 detection. The secondary antibodies were conjugated to horseradish peroxidase. Following 1 h of incubation at room temperature, membranes were washed 5 times with TBS-T. Proteins were detected by the use of ECL reagent (Amersham Biosciences), and chemiluminescence was captured by using a charge-coupled-device camera (Fusion Solo; Vilber Lourmat). The density of each individual band was normalized against GAPDH by densitometry using ImageJ software. The results are expressed as the percentage of ZO-1, occludin, or claudin-1 under each condition, compared with the control (uninfected cells).
Proinflammatory responses of CFBE41o- cells after exposure to B. contaminans.
CFBE41o- cells were seeded into 24-well plates and incubated under standard conditions for 24 h in supplemented MEM with heat-inactivated FBS. Cells were washed with PBS and incubated with bacteria (MOI, 10). After 24 h, supernatants were collected and centrifuged at 6,000 × g for 15 min. The samples obtained were analyzed using enzyme-linked immunosorbent assay (ELISA) MAX Deluxe kits (BioLegend), following the manufacturer's instructions.
Lipopolysaccharide analysis.
Lipopolysaccharide (LPS) was obtained from bacteria by microextraction using proteinase K digestion of proteins as described previously (50). Briefly, 1.5-ml aliquots of bacterial cultures with an OD640 of 0.7 were harvested and washed with PBS. The pellet was solubilized in 50 μl of lysis buffer (2% [wt/vol] SDS, 4% [vol/vol] 2-mercaptoethanol, 10% [vol/vol] glycerol, 1 M Tris-Cl [pH 6.8], and 0.1% [wt/vol] bromophenol blue). Bacterial proteins were then digested by 1-h incubation at 60°C with 10 μl of proteinase K (2.5 mg/ml). Ten-microliter LPS fractions were analyzed by 15% SDS-PAGE, and the LPS pattern was revealed by silver nitrate staining according to standard protocols (51).
Statistical analysis.
All quantitative data were obtained from at least three independent assays which included duplicates for each strain. Error propagation was used to calculate standard errors, and one-way analysis of variance (ANOVA) statistics were used to determine P values, with parental B. contaminans IST408 as a reference group. Differences were considered statistically significant if the P value was lower than 0.05.
RESULTS
Mutant strains defective in bceD or bceF genes are affected in adhesion and invasion of lung epithelial CFBE41o- cells.
B. contaminans IST408 and the phosphotyrosine phosphatase bceD::Tp mutant produce the exopolysaccharide cepacian, although the mutant produces 25% less EPS and it possibly has a lower molecular mass (35). In contrast, the tyrosine kinase bceF::Tp mutant is unable to produce exopolysaccharide. To exclude that the possible differences between the tyrosine kinase bceF::Tp mutant and the parental strain are due to EPS production and not to the presence/absence of the BceF kinase, the EPS-defective bceE::Tp mutant (7) was also included in this study. The gene bceE encodes an outer membrane protein homologous to Wza of E. coli, which is known to oligomerize and create a channel through which the polysaccharide is exported (34).
To test differences in adhesion, the CFBE41o- cells were challenged with the bacterial strains under study at an MOI of 10. Adherent bacteria were quantified by CFU counts after eukaryotic cell lysis and plating onto LB agar. The results showed that the percentage of adherent bacteria was higher in the parental B. contaminans IST408 and EPS-defective bceE::Tp mutant strains than in the phosphotyrosine phosphatase-defective bceD::Tp or tyrosine kinase bceF::Tp mutant strains (P < 0.001 in both cases), which had a percentage of adhesion slightly above that of the E. coli strain used as a negative control (Fig. 1A). Complementation experiments of the bceD::Tp and bceF::Tp mutants that entailed expression of the bceD and bceF genes, respectively, from the bce promoter present in a modified pBBR1MCS vector rescued the mutant adhesion phenotypes to levels similar to the parental strain, B. contaminans IST408 (Fig. 1A).
FIG 1.

Cellular adhesion and invasion. (A) Adhesion of B. contaminans IST408 parental strain harboring pBBR1MCS and of mutants (bceD::Tp/pBBR1MCS, bceE::Tp/pBBR1MCS, and bceF::Tp/pBBR1MCS), complemented mutants (bceD::Tp/pbceD, bceE::Tp/pbceE, bceF::Tp/pbceF), and E. coli NCIB9415 to CFBE41o- lung epithelial cells, using an MOI of 10. Data are the mean of three independent experiments. A difference for which the P value was <0.001 (indicated with an asterisk) was considered statistically significant compared to B. contaminans IST408/pBBR1MCS. pBBR, pBBR1MCS. (B) Fluorescence confocal microscopy images of CFBE41o- lung epithelial cells infected with B. contaminans strains (MOI, 50). Orthogonal projections of the confocal stack are also shown for the parental B. contaminans IST408 and bceD::Tp mutant. Eukaryotic cell plasma membrane was stained with an antibody to E-cadherin by using Alexa Fluor 488 (green). Bacteria were labeled with DsRed. (C) Invasion of CFBE41o- epithelial cells by B. contaminans IST408/pBBR1MCS, mutants bceD::Tp/pBBR1MCS, bceE::Tp/pBBR1MCS, and bceF::Tp/pBBR1MCS, or complemented mutants (bceD::Tp/pbceD, bceE::Tp/pbceE, or bceF::Tp/pbceF) (MOI, 10). The results are from three independent experiments. The significance levels of the differences between the mutants and the parental strain were analyzed by one-way ANOVA. *, P < 0.003. (D) Growth rates of B. contaminans IST408, bceD::Tp, bceE::Tp, and bceF::Tp mutants grown in LB or MEM at 37°C under an atmosphere of 5% CO2. Growth was determined by OD640 measurements, and data are the averages of at least three independent experiments.
Fluorescence confocal microscopy experiments were also performed to assess bacterial adhesion after 30 min of incubation. From the data shown in Fig. 1A, the average number of bceD::Tp or bceF::Tp mutant cells per CFBE41o- cell was 0.25. To ensure that these mutant cells were easily detected, the MOI was increased to 50. Ten independent fields were examined per sample, and an image of each strain (Fig. 1B) confirmed the general low adhesion levels shown in Fig. 1A. The confocal images of the monolayers infected with B. contaminans IST408 and the bceE::Tp mutant allowed visualization of bacteria attached to the cell surface. In contrast, the bceD::Tp and bceF::Tp mutants were scarcely visible (Fig. 1B). To show that bacteria were mostly outside epithelial cells, orthogonal projections of B. contaminans IST408 and the bceD::Tp mutant were produced (Fig. 1B). To evaluate whether the differences in bacterial adhesion had an effect on intracellular invasion, epithelial cells and each bacterial strain were allowed to interact for 2 h, followed by treatment with ciprofloxacin to eliminate extracellular bacteria. The results obtained indicated that in comparison to the parental strain IST408, the bceE::Tp mutant showed similar invasion levels, while the tyrosine kinase bceF::Tp and the phosphatase bceD::Tp mutants were about 8 times less invasive (P < 0.003) (Fig. 1C). Complementation of mutants restored intracellular invasion to levels similar to the parental strain. The average number of invading bacteria per CFBE41o- cell was 0.020 (±0.005 [standard error]) and 0.022 (±0.005) for the parental strain and the bceE::Tp mutant, respectively, and 0.003 (±0.001) for the bceD::Tp and bceF::Tp mutants. These values are within the range obtained for other B. cepacia complex strains (28). To exclude that the difference in invasion was due to unequal growth rates or susceptibility to ciprofloxacin, additional experiments were performed. As shown in Fig. 1D, no significant differences in the growth rate, either in LB or MEM (37°C, 5% CO2), were observed between the parental isolate and the bceD::Tp, bceE::Tp, and bceF::Tp mutants. The MIC for ciprofloxacin, measured in Mueller-Hinton medium at 37°C and 5% CO2, was 1 μg/ml for all the strains, showing that all had the same susceptibility to this antibiotic.
BceD and BceF proteins are required for tight junction disruption.
To assess the integrity of infected polarized cell monolayers, we measured transepithelial electrical resistance and the transepithelial flux of FITC-labeled BSA. For the first approach, the polarized epithelial cells with a TER higher than 200 Ω were challenged with each B. contaminans strain or E. coli at an MOI of 10, and changes in the TER were measured over time. After 4 h of infection, a significant reduction of TER was observed in the epithelial cells infected with B. contaminans IST408 or the bceE::Tp mutant (Fig. 2A). In contrast, monolayers challenged with either the phosphotyrosine phosphatase bceD::Tp or the tyrosine kinase bceF::Tp mutants showed a lower impact on tight junction integrity than did the parental strain (Fig. 2A). In trans complementation of bceD::Tp and bceF::Tp mutants restored TER to levels close to that of the parental strain. Even though all B. contaminans strains tested seemed to interfere with tight junction integrity, the phosphotyrosine phosphatase bceD::Tp and the tyrosine kinase bceF::Tp mutants clearly showed a phenotype attenuation over 6 h relative to B. contaminans IST408 results (P < 0.001) (Fig. 2A).
FIG 2.
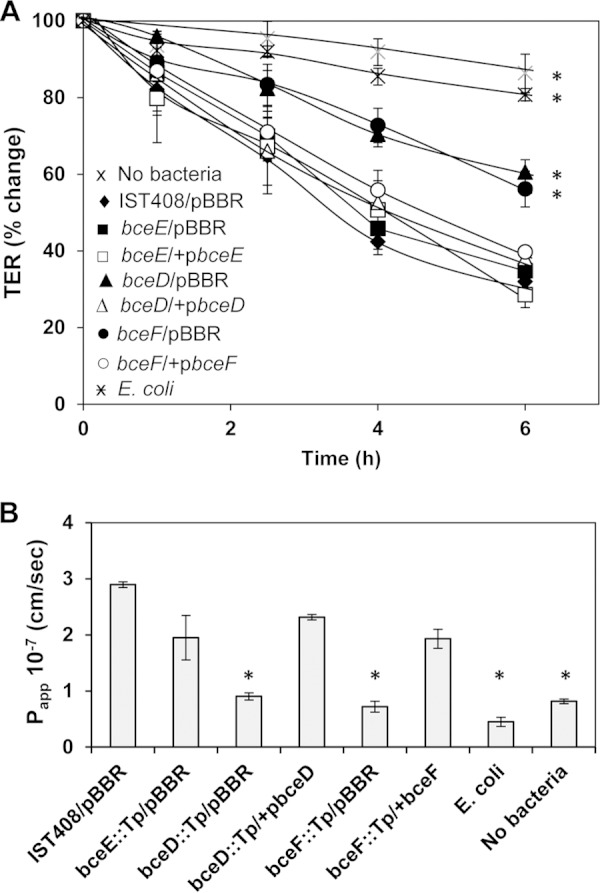
Integrity of polarized CFBE41o- monolayers upon exposure to B. contaminans strains. (A) TER was measured over time in polarized cells infected with the parental IST408/pBBR1MCS, its derivative mutants bceD::Tp, bceE::Tp, and bceF::Tp, or complemented mutants bceD::Tp/pbceD, bceE::Tp/pbceE, bceF::Tp/pbceF, and E. coli NCIB9415, or in uninfected cells. Data are the percent change (± the standard error of the mean) from the initial TER at each time point. (B) Permeability of cell monolayers (Papp) was determined by measuring the flux of FITC-labeled BSA from apical to basolateral media. All infections were done at an MOI of 10, and data represent the means of three independent experiments. Asterisks indicate values that were significantly different from the ones obtained for polarized cells infected with IST408/pBBR1MCS (P < 0.05).
To further demonstrate attenuation by the bceD::Tp and bceF::Tp mutants of the compromise of the epithelial barrier, we measured the diffusion of FITC-labeled BSA from the apical to basolateral compartments. By 6 h postinfection, significantly more FITC-labeled BSA was detected in the basolateral medium of B. contaminans IST408, bceE::Tp, and complemented mutants than was observed with the bceD::Tp or bceF::Tp mutant, E. coli, or uninfected control polarized monolayers. These differences were clearly visible based on the Papp calculated 4 h postinfection (Fig. 2B). The transepithelial flux of FITC-labeled BSA in the presence of bceD::Tp and bceF::Tp mutants was comparable to that for E. coli-infected cell monolayers or uninfected cell monolayers (below 1 × 10−7 cm/s). In contrast, the FITC-labeled BSA flux across cell monolayers infected with parental IST408, bceE::Tp, or complemented mutants was in the range of 2 × 10−7 to 3 × 10−7 cm/s (Fig. 2B).
Effect of the B. contaminans bceD or bceF mutants on tight junction protein levels.
It has been shown that a dramatic drop in the TER indicates opening of the tight junction complexes and, consequently, the disruption of epithelial integrity (28, 30, 52). To confirm the disruption of these tight junction complexes by B. contaminans, immunodetection of zonula occludens (ZO-1), occludin, and claudin-1 proteins, which play a key role in organization and regulation of tight junctions, was performed (Fig. 3A and C). The relative levels of ZO-1, occludin, or claudin-1 were determined by densitometry using ImageJ analysis and normalized against the intensities obtained with the anti-GAPDH antibody. The percentage of proteins detected was calculated against uninfected epithelia, which was considered 100%. The results presented in Fig. 3B show that the ZO-1 protein level was very low in cells infected with the parental B. contaminans IST408 (32.3% ± 13.9%) and EPS-defective bceE::Tp mutant (18.3% ± 4.8%), but the epithelial monolayers infected with the BY-kinase or phosphotyrosine phosphatase mutants still had significant amounts of ZO-1, with the bceF::Tp mutant having 59.7% ± 14.0% and the bceD::Tp 66.9% ± 2.0% of the protein remaining. Mutant complementation decreased significantly the amount of ZO-1 to levels similar to those with the parental strain and the bceE::Tp mutant. No significant changes were observed when polarized cells were infected with E. coli (98.2% ± 12.1%). Western blot analyses for claudin-1 and occludin showed reductions to about 40% of their levels in the polarized monolayers infected with the parental B. contaminans IST408 or bceE::Tp mutant (Fig. 3C and D), while the levels of both proteins in monolayers infected with the bceD::Tp or bceF::Tp mutants remained slightly lower than in the uninfected control monolayers. The immunoblots for occludin detection showed two bands. According to Kim and collaborators (30), the highest band should be the hyperphosphorylated form, with the lowest representing a reduced phosphorylation level. Despite the total reduced levels of occludin in monolayers infected with parental IST408 or the bceE::Tp mutant, we did not see any differences in the relative levels of each of the two bands compared to the uninfected polarized cells.
FIG 3.

Western blot analysis of infected polarized CFBE41o- monolayers. Total cell extracts were analyzed for estimation of ZO-1 and GAPDH levels (A) and occludin, claudin-1, and GAPDH levels (C) in CFBE41o- monolayers after 4 h of exposure to each of the strains under study. Densitometry analysis of ZO-1 expression (B) and occludin and claudin-1 expression (D) were determined by using ImageJ software. Numbers in the Western blot lanes (A and C) correspond to the strain names indicated in charts B and D. Band intensities for ZO-1, claudin-1, and occludin, determined from three independent experiments, were normalized against GAPDH and are expressed as the mean percent change ± the standard deviation, relative to results in control uninfected cells. Asterisks indicate values that were significantly different than the ones obtained for cells infected with the parental strain, B. contaminans IST408 (P < 0.05).
Mutant strains defective in bceD and bceF genes inhibit IL-8 and IL-6 release.
Proinflammatory cytokines have been shown to be increased in lungs of children with CF (53) and are associated with a poorer prognosis, as the accumulation of IL-6 and IL-8 in lung tissue leads to an exacerbation of the immune system and tissue damage (54). To evaluate whether the tyrosine kinase bceF::Tp and the phosphatase bceD::Tp mutants were able to trigger the same immune response as the parental IST408 strain, we quantified IL-6 and IL-8 production by CFBE41o- cells after 24 h of bacterial infection. The results obtained showed that both IL-6 and IL-8 production levels were higher in epithelial cells infected with the parental IST408 or with the bceE::Tp mutant than in the cells infected with the mutants bceD::Tp and bceF::Tp or the control without bacteria (Fig. 4A and B). Complementation of the bceD::Tp and bceF::Tp mutants under study rescued the production of both cytokines to levels similar to those in epithelial cells infected with the parental strain.
FIG 4.

Bacterial strain LPS profiles and secretion of cytokines by CFBE41o- epithelial cells following exposure to B. contaminans. (A and B) Cytokine levels of IL-6 (A) and IL-8 (B) were measured by ELISA, and results represent the averages of three independent assays. Asterisks indicate values that were significantly different from the ones obtained for uninfected cells (P < 0.006). (C) SDS-PAGE of lipopolysaccharide extracted from the indicated B. contaminans strains. Protein-free cell extracts were separated in 15% SDS-PAGE gels, and LPS was visualized by silver staining.
As previous expression profiling of the bceF::Tp mutant showed that several genes involved in LPS biosynthesis are differentially expressed (7), we analyzed the LPS pattern of the different strains via SDS-PAGE. Under the tested conditions, there was no difference between the LPS patterns of the parental versus the mutant strains (Fig. 4C), excluding the possibility that the differential cytokine levels released by the epithelial cells was caused by major structural changes or in the quantity of the LPS produced by the bacteria.
DISCUSSION
Most of the research on CF B. cepacia complex pathogenicity has been dedicated to B. cenocepacia, since this species, together with B. multivorans, accounts for approximately 80% of B. cepacia complex infections worldwide (20). However, several reports have shown that other B. cepacia complex species, among them B. contaminans, are also implicated in transient or chronic infections, poor prognosis, and transmission between patients (20, 21, 55). To gain insights into the B. contaminans interaction with host cells, we investigated the role of a BY-kinase and an LMW-PTP in this process, as some of their homologues are known to interfere with host signaling (8). Previous expression profiling and phenotype characterization of the parental strain B. contaminans IST408 and the bceD::Tp and the bceF::Tp mutants (7, 35) indicated that BceD and BceF are involved in multiple phenotypic characteristics, including motility, biofilm formation, EPS biosynthesis, resistance to stress conditions, and virulence in the Galleria mellonella model. These observations suggest the existence of a complex regulatory network where the tyrosine phosphorylation state of a target protein(s), mediated by the BY-kinase BceF, possibly in conjunction with the PTP BceD, plays a fundamental role. Although several BceF homologues have been characterized and their roles in the regulation of protein function and cellular location demonstrated (3), little is known about their importance in host-pathogen interactions. As an example, analysis of the E. coli O157:H7 phosphotyrosine proteome showed that phosphorylation of a tyrosine residue of the regulator SspA positively affected expression and secretion of T3SS proteins and formation of A/E lesions on HeLa cell monolayers (56). Similarly, a few LMW-PTP proteins homologous to BceD are known to interfere in several host signaling pathways (8, 16). Accordingly, in our work we tested the importance of BceF and BceD in the pathogenesis of B. contaminans infection by using the CFBE41o- cell line as a model. An important observation is that the absence of the BY-kinase or the LMW-PTP phosphatase is associated with similar phenotypes while interacting with CFBE41o- cells. This suggests that BceF and BceD, by having global regulatory functions, may act on the same unknown bacterial pathway that ultimately leads to the adhesion/invasion and tight junction disruption phenotypes observed here. The three-dimensional structures of BY-kinases suggest that the phosphatase may be critical for the cycling of the kinase between monomers and higher-order oligomers (57). In this situation, not having BceF or having it in an inactive monomeric/oligomeric state due to the absence of BceD would perhaps result in the same cellular outcome regarding interactions with host cells.
The B. cepacia complex infection process starts with adhesion of bacteria to the apical surface of epithelial cells (29), but little is known about the structures involved in this host-bacterium interaction. Although some adhesin proteins of B. cenocepacia have been implicated in attachment to epithelial cells (46, 58, 59), no studies on the putative adhesins of B. contaminans IST408 have been done. Previous expression profiling (under growth conditions dissimilar from the ones tested here) of the bceF mutant compared to the IST408 parental strain showed decreased expression of a putative autotransporter adhesin homologue to BCAS0321 of B. cenocepacia J2315 and at least eight genes encoding putative outer membrane porins (7). Therefore, further experiments are needed to attribute a possible role of B. contaminans IST408 adhesin/Omp proteins to the CFBE41o- cell adhesion phenotype. Studies also have shown that in some B. cepacia complex species, the invasion process starts with the formation of microcolonies and of biofilms reassembling structures in the mucous layer that eventually reach the epithelial surface (29). Previous data on bceD::Tp and bceF::Tp mutant biofilm formation showed that they form less biofilm on abiotic surfaces than the parental strain or the bceE::Tp mutant (7, 35). This phenotype correlates with the adhesion/invasion ability of the B. contaminans tested strains toward the CFBE41o- cells in this study, suggesting that the formation of biofilm-like structures on the cell surface might be important for B. contaminans cellular invasion.
Some species of the B. cepacia complex invade eukaryotic cells via a membrane-bound vacuole, where bacteria have been shown to survive by escaping from late endosomes and replicating within epithelial cells (26, 32, 60). Besides cellular invasion, B. cepacia complex species have been shown to transverse the respiratory epithelium paracellularly, which may be associated with the B. cepacia complex ability to disseminate beyond the lungs at a higher frequency than other CF pathogens, causing cepacia syndrome (31). Accordingly, B. cepacia complex migration to the basolateral surface of epithelium is associated with a strong decrease of TER due to the loss of cell junction integrity (29). Duff and collaborators have shown that 4 different species within the B. cepacia complex are able to disrupt epithelial monolayer integrity and translocate through to the basolateral side of lung epithelial monolayers (28). In our study, B. contaminans parental strain IST408 also dramatically reduced transepithelial resistance and increased the permeability of polarized CFBE41o- monolayers. In contrast, the abilities of bceD::Tp and bceF::Tp mutants to impair tight junction integrity were closer to those of the negative controls. Bacterial pathogens can interfere with epithelial tight junction integrity by different mechanisms, including changes at the expression level or phosphorylation level or dislocation of tight junction proteins (61–63). In B. cepacia complex bacteria, representative isolates of three epidemic lineages of B. cenocepacia have been shown to transverse polarized 16HBE41o- respiratory epithelial cells (non-CF) in vitro by dephosphorylation and dissociation of occludin from the tight junction complex, but no difference was observed for ZO-1 protein (30). In another study with environmental B. cenocepacia strains, no apparent effect on the tight junction protein ZO-1 was observed in non-CF polarized 16HBE41o- cells, while in CF polarized cells CFBE41o- cells, the amount of ZO-1 and its localization were altered (52). In our study, we observed a reduction of ZO-1, occludin, and claudin-1 levels and these were more pronounced in parental B. contaminans IST408 and the bceE::Tp mutant than in the bceD::Tp and bceF::Tp mutants. The mechanism by which BceD and BceF affect tight junction integrity is unknown. BceF is an inner membrane protein, and its role in this disruption is most likely indirect. Regarding BceD, this protein could be secreted and interfere with host cell functions. Nevertheless, a recent study by Andrade and Valvano showed that under the in vitro experimental conditions tested, BceD was not detected in the growth medium (16). Despite this result, it cannot be excluded that, in the presence of CFBE41o- cells, BceD may be secreted.
The development of cepacia syndrome is also associated with increased levels of proinflammatory cytokines in blood and sputum (64), particularly with a large amount of IL-8 (53), which attracts neutrophils and macrophages into the CF lung. These cells of the immune system release free radicals and proteolytic enzymes to eliminate bacteria, but these mechanisms have been shown to be inefficient against B. cepacia complex bacteria and contribute to a further increase in lung damage and to the establishment of chronic inflammation (64). In this work, we have shown that in the absence of BceD and BceF proteins, B. contaminans induces a lower proinflammatory response. Although the LPS profiles of the wild-type and mutant strains were comparable, it cannot be excluded that modifications of lipid A acylation or phosphorylation levels might occur. Expression profiling analysis of the B. contaminans bceF::Tp mutant indicated an upregulation of the lpxO gene, which encodes a putative dioxygenase required for acyl chain hydroxylation (7), suggesting that different degrees of 2-hydroxylation might be present with bceF::Tp mutant and wild-type strains. Modified lipid A structures with different phosphorylation or acylation patterns elicit different host immune responses (65, 66). Nevertheless, Palfreyman and coauthors showed that B. cepacia complex cell-free supernatants can also increase IL-8 production, suggesting that other factors besides LPS can enhance the immune response (67). The reduced inflammatory response could also be due to the lower level of attachment to host cells and a concomitant lack of direct stimulation of epithelial cells. Further investigation into the adhesins and host pattern recognition receptors involved will help elucidate the mechanism. Taken together, our results indicate that the BceF and BceD proteins have an important role in B. contaminans virulence, since a range of critical events involved in the infection of CF lung epithelial cells was compromised in both mutants. Moreover, since the BceF and BceD proteins have homologues in both Gram-negative and Gram-positive bacteria, their involvement in virulence might be a common feature among pathogens.
ACKNOWLEDGMENTS
We acknowledge Dieter Gruenert from the University of California, San Francisco, for the kind donation of the CFBE41o- epithelial cell line.
This work was supported by FEDER and Fundação para a Ciência e a Tecnologia, Portugal (project PTDC/QUI-BIQ/118260/2010 to L.M.M. and PEsT-OE/EQB/LA0023/2011) and postdoctoral grants to A.S.F. and I.N.S. A.S.F. also acknowledges a grant from the European Society of Clinical Microbiology and Infectious Diseases and a scholarship from the Federation of European Biochemical Societies. S. McClean and M. Callaghan are members of the EU COST Action BM1003: Microbial cell surface determinants of virulence as targets for new therapeutics in cystic fibrosis.
REFERENCES
- 1.Bechet E, Guiral S, Torres S, Mijakovic I, Cozzone AJ, Grangeasse C. 2009. Tyrosine-kinases in bacteria: from a matter of controversy to the status of key regulatory enzymes. Amino Acids 37:499–507. doi: 10.1007/s00726-009-0237-8. [DOI] [PubMed] [Google Scholar]
- 2.Cozzone AJ. 2005. Role of protein phosphorylation on serine/threonine and tyrosine in the virulence of bacterial pathogens. J Mol Microbiol Biotechnol 9:198–213. doi: 10.1159/000089648. [DOI] [PubMed] [Google Scholar]
- 3.Jers C, Pedersen MM, Paspaliari DK, Schütz W, Johnsson C, Soufi B, Macek B, Jensen PR, Mijakovic I. 2010. Bacillus subtilis BY-kinase PtkA controls enzyme activity and localization of its protein substrates. Mol Microbiol 77:287–299. doi: 10.1111/j.1365-2958.2010.07227.x. [DOI] [PubMed] [Google Scholar]
- 4.Lacour S, Bechet E, Cozzone AJ, Mijakovic I, Grangeasse C. 2008. Tyrosine phosphorylation of the UDP-glucose dehydrogenase of Escherichia coli is at the crossroads of colanic acid synthesis and polymyxin resistance. PLoS One 3:e3053. doi: 10.1371/journal.pone.0003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petranovic D, Michelsen O, Zahradka K, Silva C, Petranovic M, Jensen PR, Mijakovic I. 2007. Bacillus subtilis strain deficient for the protein-tyrosine kinase PtkA exhibits impaired DNA replication. Mol Microbiol 63:1797–1805. doi: 10.1111/j.1365-2958.2007.05625.x. [DOI] [PubMed] [Google Scholar]
- 6.Mijakovic I, Petranovic D, Macek B, Cepo T, Mann M, Davies J, Jensen PR, Vujaklija D. 2006. Bacterial single-stranded DNA-binding proteins are phosphorylated on tyrosine. Nucleic Acids Res 34:1588–1596. doi: 10.1093/nar/gkj514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreira AS, Silva IN, Oliveira VH, Becker JD, Givskov M, Ryan RP, Fernandes F, Moreira LM. 2013. Comparative transcriptomic analysis of the Burkholderia cepacia tyrosine kinase bceF mutant reveals a role in tolerance to stress, biofilm formation, and virulence. Appl Environ Microbiol 79:3009–3020. doi: 10.1128/AEM.00222-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitmore SE, Lamont RJ. 2012. Tyrosine phosphorylation and bacterial virulence. Int J Oral Sci 4:1–6. doi: 10.1038/ijos.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bliska JB. 2000. Yop effectors of Yersinia spp. and actin rearrangements. Trends Microbiol 8:205–208. doi: 10.1016/S0966-842X(00)01738-8. [DOI] [PubMed] [Google Scholar]
- 10.Murli S, Watson RO, Galán JE. 2001. Role of tyrosine kinases and the tyrosine phosphatase SptP in the interaction of Salmonella with host cells. Cell Microbiol 3:795–810. doi: 10.1046/j.1462-5822.2001.00158.x. [DOI] [PubMed] [Google Scholar]
- 11.Arricau N, Hermant D, Waxin H, Popoff MY. 1997. Molecular characterization of the Salmonella typhi StpA protein that is related to both Yersinia YopE cytotoxin and YopH tyrosine phosphatase. Res Microbiol 148:21–26. doi: 10.1016/S0923-2508(97)81896-7. [DOI] [PubMed] [Google Scholar]
- 12.Koul A, Choidas A, Treder M, Tyagi AK, Drlica K, Ullrich Y SA. 2000. Cloning and characterization of secretory tyrosine phosphatases of Mycobacterium tuberculosis. J Bacteriol 182:5425–5432. doi: 10.1128/JB.182.19.5425-5432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castandet J, Prost JF, Peyron P, Astarie-Dequeker C, Anes E, Cozzone AJ, Griffiths G, Maridonneau-Parini I. 2005. Tyrosine phosphatase MptpA of Mycobacterium tuberculosis inhibits phagocytosis and increases actin polymerization in macrophages. Res Microbiol 156:1005–1013. doi: 10.1016/j.resmic.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 14.Zhou B, He Y, Zhang X, Xu J, Luo Y, Wang Y, Franzblau SG, Yang Z, Chan RJ, Liu Y, Zheng J, Zhang Z-Y. 2010. Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc Natl Acad Sci U S A 107:4573–4578. doi: 10.1073/pnas.0909133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kastner R, Dussurget O, Archambaud C, Kernbauer E, Soulat D, Cossart P, Decker T. 2011. LipA, a tyrosine and lipid phosphatase involved in the virulence of Listeria monocytogenes. Infect Immun 79:2489–2498. doi: 10.1128/IAI.05073-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrade A, Valvano MA. 2014. A Burkholderia cenocepacia gene encoding a non-functional tyrosine phosphatase is required for the delayed maturation of the bacteria-containing vacuoles in macrophages. Microbiology 160:1332–1345. doi: 10.1099/mic.0.077206-0. [DOI] [PubMed] [Google Scholar]
- 17.Coutinho CP, Dos Santos SC, Madeira A, Mira NP, Moreira AS, Sá-Correia I. 2011. Long-term colonization of the cystic fibrosis lung by Burkholderia cepacia complex bacteria: epidemiology, clonal variation, and genome-wide expression alterations. Front Cell Infect Microbiol 1:12. doi: 10.3389/fcimb.2011.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramsay KA, Butler CA, Paynter S, Ware RS, Kidd TJ, Wainwright CE, Bell SC. 2013. Factors influencing acquisition of Burkholderia cepacia complex organisms in patients with cystic fibrosis. J Clin Microbiol 51:3975–3980. doi: 10.1128/JCM.01360-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isles A, Maclusky I, Corey M, Gold R, Prober C, Fleming P, Levison H. 1984. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J Pediatr 104:206–210. doi: 10.1016/S0022-3476(84)80993-2. [DOI] [PubMed] [Google Scholar]
- 20.Lipuma JJ. 2010. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev 23:299–323. doi: 10.1128/CMR.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drevinek P, Mahenthiralingam E. 2010. Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin Microbiol Infect 16:821–830. doi: 10.1111/j.1469-0691.2010.03237.x. [DOI] [PubMed] [Google Scholar]
- 22.Martin M, Christiansen B, Caspari G, Hogardt M, von Thomsen AJ, Ott E, Mattner F. 2011. Hospital-wide outbreak of Burkholderia contaminans caused by prefabricated moist washcloths. J Hosp Infect 77:267–270. doi: 10.1016/j.jhin.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 23.Moehring RW, Lewis SS, Isaacs PJ, Schell WA, Thomann WR, Althaus MM, Hazen KC, Dicks KV, Lipuma JJ, Chen LF, Sexton DJ. 2014. Outbreak of bacteremia due to Burkholderia contaminans linked to intravenous fentanyl from an institutional compounding pharmacy. JAMA Intern Med 174:606–612. doi: 10.1001/jamainternmed.2013.13768. [DOI] [PubMed] [Google Scholar]
- 24.Martina P, Bettiol M, Vescina C, Montanaro P, Mannino MC, Prieto CI, Vay C, Naumann D, Schmitt J, Yantorno O, Lagares A, Bosch A. 2013. Genetic diversity of Burkholderia contaminans isolates from cystic fibrosis patients in Argentina. J Clin Microbiol 51:339–344. doi: 10.1128/JCM.02500-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aaron SD, Ferris W, Henry DA, Speert DP, Macdonald NE. 2000. Multiple combination bactericidal antibiotic testing for patients with cystic fibrosis infected with Burkholderia cepacia. Am J Respir Crit Care Med 161:1206–1212. doi: 10.1164/ajrccm.161.4.9907147. [DOI] [PubMed] [Google Scholar]
- 26.Caraher E, Duff C, Mullen T, Mc Keon S, Murphy P, Callaghan M, McClean S. 2007. Invasion and biofilm formation of Burkholderia dolosa is comparable with Burkholderia cenocepacia and Burkholderia multivorans. J Cyst Fibros 6:49–56. doi: 10.1016/j.jcf.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Sass A, Marchbank A, Tullis E, Lipuma JJ, Mahenthiralingam E. 2011. Spontaneous and evolutionary changes in the antibiotic resistance of Burkholderia cenocepacia observed by global gene expression analysis. BMC Genomics 12:373. doi: 10.1186/1471-2164-12-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duff C, Murphy PG, Callaghan M, McClean S. 2006. Differences in invasion and translocation of Burkholderia cepacia complex species in polarised lung epithelial cells in vitro. Microb Pathog 41:183–192. doi: 10.1016/j.micpath.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Schwab U, Leigh M, Ribeiro C, Yankaskas J, Burns K, Gilligan P, Sokol P, Boucher R. 2002. Patterns of epithelial cell invasion by different species of the Burkholderia cepacia complex in well-differentiated human airway epithelia. Infect Immun 70:4547–4555. doi: 10.1128/IAI.70.8.4547-4555.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JY, Sajjan US, Krasan GP, LiPuma JJ. 2005. Disruption of tight junctions during traversal of the respiratory epithelium by Burkholderia cenocepacia. Infect Immun 73:7107–7112. doi: 10.1128/IAI.73.11.7107-7112.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McClean S, Callaghan M. 2009. Burkholderia cepacia complex: epithelial cell-pathogen confrontations and potential for therapeutic intervention. J Med Microbiol 58:1–12. doi: 10.1099/jmm.0.47788-0. [DOI] [PubMed] [Google Scholar]
- 32.Saldías MS, Valvano MA. 2009. Interactions of Burkholderia cenocepacia and other Burkholderia cepacia complex bacteria with epithelial and phagocytic cells. Microbiology 155:2809–2817. doi: 10.1099/mic.0.031344-0. [DOI] [PubMed] [Google Scholar]
- 33.Leitão JH, Sousa SA, Ferreira AS, Ramos CG, Silva IN, Moreira LM. 2010. Pathogenicity, virulence factors, and strategies to fight against Burkholderia cepacia complex pathogens and related species. Appl Microbiol Biotechnol 87:31–40. doi: 10.1007/s00253-010-2528-0. [DOI] [PubMed] [Google Scholar]
- 34.Ferreira AS, Silva IN, Oliveira VH, Cunha R, Moreira LM. 2011. Insights into the role of extracellular polysaccharides in Burkholderia adaptation to different environments. Front Cell Infect Microbiol 1:00016. doi: 10.3389/fcimb.2011.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferreira AS, Leitão JH, Sousa SA, Cosme AM, Sá-Correia I, Moreira LM. 2007. Functional analysis of Burkholderia cepacia genes bceD and bceF, encoding a phosphotyrosine phosphatase and a tyrosine autokinase, respectively: role in exopolysaccharide biosynthesis and biofilm formation. Appl Environ Microbiol 73:524–534. doi: 10.1128/AEM.01450-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferreira AS, Leitão JH, Silva IN, Pinheiro PF, Sousa SA, Ramos CG, Moreira LM. 2010. Distribution of cepacian biosynthesis genes among environmental and clinical Burkholderia strains and role of cepacian exopolysaccharide in resistance to stress conditions. Appl Environ Microbiol 76:441–450. doi: 10.1128/AEM.01828-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sousa SA, Ulrich M, Bragonzi A, Burke M, Worlitzsch D, Leitão JH, Meisner C, Eberl L, Sá-Correia I, Döring G. 2007. Virulence of Burkholderia cepacia complex strains in gp91phox−/− mice. Cell Microbiol 9:2817–2825. doi: 10.1111/j.1462-5822.2007.00998.x. [DOI] [PubMed] [Google Scholar]
- 38.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York, NY. [Google Scholar]
- 40.Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A 76:1648–1652. doi: 10.1073/pnas.76.4.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moreira LM, Videira PA, Sousa SA, Leitão JH, Cunha MV, Sá-Correia I. 2003. Identification and physical organization of the gene cluster involved in the biosynthesis of Burkholderia cepacia complex exopolysaccharide. Biochem Biophys Res Commun 312:323–333. doi: 10.1016/j.bbrc.2003.10.118. [DOI] [PubMed] [Google Scholar]
- 42.Kovach ME, Phillips RW, Elzer PH, Roop RM II, Peterson KM. 1994. pBBR1MCS: a broad-host-range cloning vector. Biotechniques 16:800–802. [PubMed] [Google Scholar]
- 43.Goncz KK, Feeney L, Gruenert DC. 1999. Differential sensitivity of normal and cystic fibrosis airway epithelial cells to epinephrine. Br J Pharmacol 128:227–233. doi: 10.1038/sj.bjp.0702772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gruenert DC, Willems M, Cassiman JJ, Frizzell RA. 2004. Established cell lines used in cystic fibrosis research. J Cyst Fibros 3:191–196. doi: 10.1016/j.jcf.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 45.Vergunst AC, Meijer AH, Renshaw SA, O'Callaghan D. 2010. Burkholderia cenocepacia creates an intramacrophage replication niche in zebrafish embryos, followed by bacterial dissemination and establishment of systemic infection. Infect Immun 78:1495–1508. doi: 10.1128/IAI.00743-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mil-Homens D, Leça MI, Fernandes F, Pinto SN, Fialho AM. 2014. Characterization of BCAM0224, a multifunctional trimeric autotransporter from the human pathogen Burkholderia cenocepacia. J Bacteriol 196:1968–1979. doi: 10.1128/JB.00061-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pinto SN, Silva LC, de Almeida RFM, Prieto M. 2008. Membrane domain formation, interdigitation, and morphological alterations induced by the very long chain asymmetric C24:1 ceramide. Biophys J 95:2867–2879. doi: 10.1529/biophysj.108.129858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kunzelmann K, Schwiebert EM, Zeitlin PL, Kuo WL, Stanton BA, Gruenert DC. 1993. An immortalized cystic fibrosis tracheal epithelial cell line homozygous for the delta F508 CFTR mutation. Am J Respir Cell Mol Biol 8:522–529. doi: 10.1165/ajrcmb/8.5.522. [DOI] [PubMed] [Google Scholar]
- 49.Stutts MJ, Boucher RC, Bromberg PA, Gatzy JT. 1981. Effects of ammonium and nitrate salts on ion transport across the excised canine trachea. Toxicol Appl Pharmacol 60:91–105. doi: 10.1016/0041-008X(81)90139-3. [DOI] [PubMed] [Google Scholar]
- 50.Hitchcock PJ, Brown TM. 1983. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol 154:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai CM, Frasch CE. 1982. A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal Biochem 119:115–119. doi: 10.1016/0003-2697(82)90673-X. [DOI] [PubMed] [Google Scholar]
- 52.Bevivino A, Pirone L, Pilkington R, Cifani N, Dalmastri C, Callaghan M, Ascenzioni F, McClean S. 2012. Interaction of environmental Burkholderia cenocepacia strains with cystic fibrosis and non-cystic fibrosis bronchial epithelial cells in vitro. Microbiology 158:1325–1333. doi: 10.1099/mic.0.056986-0. [DOI] [PubMed] [Google Scholar]
- 53.Dean TP, Dai Y, Shute JK, Church MK, Warner JO. 1993. Interleukin-8 concentrations are elevated in bronchoalveolar lavage, sputum, and sera of children with cystic fibrosis. Pediatr Res 34:159–161. doi: 10.1203/00006450-199308000-00010. [DOI] [PubMed] [Google Scholar]
- 54.Gibson RL, Burns JL, Ramsey BW. 2003. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168:918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 55.Cunha MV, Leitão JH, Mahenthiralingam E, Vandamme P, Lito L, Barreto C, Salgado MJ, Sá-Correia I. 2003. Molecular analysis of Burkholderia cepacia complex isolates from a Portuguese cystic fibrosis center: a 7-year study. J Clin Microbiol 41:4113–4120. doi: 10.1128/JCM.41.9.4113-4120.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hansen A-M, Chaerkady R, Sharma J, Díaz-Mejía JJ, Tyagi N, Renuse S, Jacob HKC, Pinto SM, Sahasrabuddhe NA, Kim M-S, Delanghe B, Srinivasan N, Emili A, Kaper JB, Pandey A. 2013. The Escherichia coli phosphotyrosine proteome relates to core pathways and virulence. PLoS Pathog 9:e1003403. doi: 10.1371/journal.ppat.1003403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Collins RF, Beis K, Clarke BR, Ford RC, Hulley M, Naismith JH, Whitfield C. 2006. Periplasmic protein-protein contacts in the inner membrane protein Wzc form a tetrameric complex required for the assembly of Escherichia coli group 1 capsules. J Biol Chem 281:2144–2150. doi: 10.1074/jbc.M508078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mil-Homens D, Fialho AM. 2012. A BCAM0223 mutant of Burkholderia cenocepacia is deficient in hemagglutination, serum resistance, adhesion to epithelial cells and virulence. PLoS One 7:e41747. doi: 10.1371/journal.pone.0041747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sajjan U, Wu Y, Kent G, Forstner J. 2000. Preferential adherence of cable-piliated Burkholderia cepacia to respiratory epithelia of CF knockout mice and human cystic fibrosis lung explants. J Med Microbiol 49:875–885. [DOI] [PubMed] [Google Scholar]
- 60.Sajjan US, Yang JH, Hershenson MB, LiPuma JJ. 2006. Intracellular trafficking and replication of Burkholderia cenocepacia in human cystic fibrosis airway epithelial cells. Cell Microbiol 8:1456–1466. doi: 10.1111/j.1462-5822.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 61.Petit L, Gibert M, Gourch A, Bens M, Vandewalle A, Popoff MR. 2003. Clostridium perfringens epsilon toxin rapidly decreases membrane barrier permeability of polarized MDCK cells. Cell Microbiol 5:155–164. doi: 10.1046/j.1462-5822.2003.00262.x. [DOI] [PubMed] [Google Scholar]
- 62.Simonovic I, Rosenberg J, Koutsouris A, Hecht G. 2000. Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell Microbiol 2:305–315. doi: 10.1046/j.1462-5822.2000.00055.x. [DOI] [PubMed] [Google Scholar]
- 63.Wu Z, Nybom P, Magnusson KE. 2000. Distinct effects of Vibrio cholerae haemagglutinin/protease on the structure and localization of the tight junction-associated proteins occludin and ZO-1. Cell Microbiol 2:11–17. doi: 10.1046/j.1462-5822.2000.00025.x. [DOI] [PubMed] [Google Scholar]
- 64.Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, Berger M. 1995. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med 152:2111–2118. doi: 10.1164/ajrccm.152.6.8520783. [DOI] [PubMed] [Google Scholar]
- 65.Kong Q, Six D, Liu Q, Gu L, Wang S, Alamuri P, Raetz C, Curtiss R III. 2012. Phosphate groups of lipid A are essential for Salmonella enterica serovar Typhimurium virulence and affect innate and adaptive immunity. Infect Immun 80:3215–3224. doi: 10.1128/IAI.00123-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nielsen H, Birkholz S, Andersen L, Moran A. 1994. Neutrophil activation by Helicobacter pylori lipopolysaccharides. J Infect Dis 170:135–139. doi: 10.1093/infdis/170.1.135. [DOI] [PubMed] [Google Scholar]
- 67.Palfreyman RW, Watson ML, Eden C, Smith AW. 1997. Induction of biologically active interleukin-8 from lung epithelial cells by Burkholderia (Pseudomonas) cepacia products. Infect Immun 65:617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]