

Abstract
TcdB is one of the key virulence factors of Clostridium difficile that is responsible for causing serious and potentially fatal colitis. The toxin contains at least two enzymatic domains: an effector glucosyltransferase domain for inactivating host Rho GTPases and a cysteine protease domain for the delivery of the effector domain into host cytosol. Here, we describe a novel intrabody approach to examine the role of these enzymes of TcdB in cellular intoxication. By screening a single-domain heavy chain (VHH) library raised against TcdB, we identified two VHH antibodies, 7F and E3, that specifically inhibit TcdB cysteine protease and glucosyltransferase activities, respectively. Cytoplasmic expression of 7F intrabody in Vero cells inhibited TcdB autoprocessing and delayed cellular intoxication, whereas E3 intrabody completely blocked the cytopathic effects of TcdB holotoxin. These data also demonstrate for the first time that toxin autoprocessing occurs after cysteine protease and glucosyltransferase domains translocate into the cytosol of target cells. We further determined the role of the enzymatic activities of TcdB in in vivo toxicity using a sensitive systemic challenge model in mice. Consistent with these in vitro results, a cysteine protease noncleavable mutant, TcdB-L543A, delayed toxicity in mice, whereas glycosyltransferase-deficient TcdB demonstrated no toxicity up to 500-fold of the 50% lethal dose (LD50) when it was injected systemically. Thus, glucosyltransferase but not cysteine protease activity is critical for TcdB-mediated cytopathic effects and TcdB systemic toxicity, highlighting the importance of targeting toxin glucosyltransferase activity for future therapy.
INTRODUCTION
Clostridium difficile is an anaerobic Gram-positive bacterial species that can induce serious and potentially fatal inflammatory disease of the colon and is the most prevalent cause of antibiotic-associated diarrhea and pseudomembranous colitis in nosocomial settings (1, 2). Disease in patients with C. difficile infection is strongly associated with the two exotoxins, TcdA and TcdB (3). Both toxins are large, homologous single-chain proteins that contain at least four distinct domains (4–6): the N terminus glucosyltransferase domain (GTD), a cysteine protease domain (CPD), a translocation domain (TD), and a C terminus receptor binding domain (RBD; also known as combined repetitive oligopeptides, or CROPs). A recent study suggests that there might also be an additional receptor binding region besides the N-terminal CROP region (7) although the specific region has yet to be identified. Both toxins exert cytopathic effects that include cell rounding after disruption of the actin cytoskeleton and tight junctions in human colonocytes (8, 9). Toxin exposure may also trigger potent cytotoxic and inflammatory effects leading to mucosal cell death, diarrhea, and colitis associated with C. difficile infections (10, 11). TcdB appears to be more clinically relevant for C. difficile virulence as it is invariably associated with clinically isolated pathogenic strains (12–14). The high potency of TcdB is attributed in part to the efficient enzymatic activities of its GTD and CPD domains (15, 16).
The exact method of toxin entry into target cells remains unknown, but a molecular model of the toxin mode of action is emerging (17). Initially, the CROPs are thought to bind to some unknown molecules on the cell surface, facilitating toxin entry into cells via receptor-mediated endocytosis (18–20). Once the endosome is acidified, the toxins undergo a conformational change (21), inserting the transmembrane region into the endosomal membrane and translocating the CPD and GTD into the cytosol (22, 23). Finally, the cysteine protease self-cleaves the GTD, releasing it from the rest of the toxin (24, 25). Once in the cytosol, free GTD inactivates Rho GTPases, leading to the intoxication of host cells and resulting in cell rounding and apoptosis (8, 11, 26, 27). Evidence that GTD release into the cytoplasm is mediated by CPD activity is largely based on in vitro studies. This autoproteolytic activity in TcdA and TcdB is mediated by allosteric cofactors, inositol hexakis- and heptakisphosphate (InsP6 and InsP7) (24, 28, 29). We along with others have demonstrated using cysteine protease activity-deficient TcdB mutants, as well as a noncleavable TcdA or TcdB, that blocking the release of GTD into the host cell cytosol delays, but does not prevent, the cytopathic and cytotoxic activities of TcdA or TcdB (30, 31). Kim et al. reported glucosyltransferase-independent disruption of focal adhesion formation (32) and production of reactive oxygen species (33) in colonocytes induced by TcdA. Most recently, several studies have indicated that neither the CPD nor GTD enzymatic activities of TcdB are required for cellular intoxication at high toxin doses (34–36), whereas the hydrophobic region in the translocation domain and GTD are important for the rapid induction of cell death (37, 38). These studies utilize toxin mutagenesis, which is well known to alter protein active-site specificity or conformational integrity (39). More importantly, clinical relevance of toxin mutants needs to be validated in animal disease models.
VHHs are characterized as a class of functional variable heavy-chain immunoglobulins that lack light chains and are produced by camelid animals, such as alpacas (40, 41). The VH regions of these VHHs are similar to conventional VH domains but have unique sequence and structural characteristics. VHHs are small (∼15 kDa), easy to produce and manipulate genetically, and generally more stable than conventional antibody (Ab) fragments (42–44). VHHs bind to antigen targets with an affinity equivalent to conventional IgG heavy chain-only antibody fragments (45). Because of their small size, VHHs are often found to have unusual epitope specificities, particularly an improved capability to bind to active-site pockets in enzymes in order to inhibit their functions (46, 47). VHHs have been reported in the treatment of toxin-mediated disease (48, 49). VHHs against the two C. difficile toxins have also been generated which possessed toxin-neutralizing activity (42, 50) and potent therapeutic efficacy against fulminant C. difficile infection (51).
In this study, we explored the potential of using VHHs to investigate the virulence role of the TcdB enzymatic machinery. We performed library screening and identified novel VHHs that specifically inhibit glucosyltransferase activity and CPD-mediated autoprocessing of wild-type TcdB. By utilizing these VHHs as intrabodies, we demonstrated that while inhibition of either enzymatic activity in TcdB attenuated its toxicity, glucosyltransferase was crucial in mediating such cytopathic responses, whereas CPD-mediated autoprocessing regulates the potency of the toxin activity. Moreover, in a systemic toxemia model we found that the toxin glucosyltransferase activity, but not that of cysteine protease, was an absolute necessity for the systemic toxicity of TcdB in mice.
MATERIALS AND METHODS
Ethics statement.
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal experiments performed in this study were reviewed and approved by the IACUC committee at Tufts University Cummings School of Veterinary Medicine (protocol 2008-GR20) or at the University of Maryland School of Medicine (protocol D120301).
C. difficile toxins, immunogens, and immunization.
Bioactive full-length recombinant TcdB (52) and glucosyltransferase-deficient mutant aTcdB (TcdB-W102A D288N) (53) were purified using His tag affinity chromatography as described previously (52). Adult male and female alpacas were immunized with purified recombinant aTcdB (50 to 100 μg) subcutaneously up to five times at intervals of no less than 3 weeks with alum adjuvant (with CpG in the primary immunization). Blood samples were collected prior to each immunization for IgG titer determination. Five days after the final boost, peripheral blood lymphocytes (PBLs) were harvested.
Construction and screening of the VHH library.
The VHH library generated from PBLs obtained from aTcdB-immunized alpacas has been described previously (51). Panning for VHH-displayed phage was achieved by pulldown methods using biotinylated TcdB. TcdB was biotinylated using a Pierce EZ-Link NHS-PEG4 Biotin kit (Pierce Biotechnology, Rockford, IL) per the manufacturer's instructions. For panning, biotinylated toxin was incubated with 50 μl of phage library in 4% dry milk in phosphate-buffered saline (PBS) for 1 h, followed by incubation with streptavidin beads preblocked with 4% dry milk in PBS for 30 min. Beads were washed 10 times with PBS, phages were eluted with 0.2 M glycine (pH 2.4), and the buffer was neutralized with 1 M Tris-HCl (pH 7.4). Three decreasing concentrations of toxin (from 500 ng to 6.2 ng) were used in successive panning cycles to increase the stringency of selection for toxin binding. Eluted phages were then used to infect bacteria (Escherichia coli ER2738), which were plated on LB-ampicillin-tetracycline plates. Phage clones were screened for binding by enzyme-linked immunosorbent assay (ELISA) against wild-type TcdB. Unique VHH clones displaying the strongest ELISA results were sequenced and expressed as described below.
VHH expression and purification.
Selected VHH coding sequences were cloned into the pET32b expression vector (Novagen) for cytosolic expression of VHHs fused to thioredoxin in E. coli Rosetta-gami 2(DE3)/pLacI (Novagen). VHH monomers contain a carboxyl-terminal epitope tag (E tag) for detection and a hexahistidine tag (His tag) for purification. The E. coli culture pellet (from 100 ml of culture) was resuspended in 5 ml of lysis buffer, and cells were disrupted by a One-Shot cell disruptor (Constant Systems, Kennesaw, GA). The supernatant was passed through a 0.2-μm-pore-size sterile syringe filter (VWR) before being passed through a nickel-charged Hi Trap chelating high-performance column (GE Healthcare). Purification of recombinant His-tagged VHHs from bacterial lysate was performed by Ni-affinity chromatography.
Identifying VHHs binding to native GTD from TcdB.
To screen for GTD-binding VHHs, the chimeric toxin TxA-Bgt consisting of the GTD of TcdB and the CPD, TD, and RBD from TcdA was generated. To generate the chimera TxA-Bgt, a unique BamHI site was introduced in between the GTD and CPD without changing the sequence of amino acids in both pHis-TcdA and pHis-TcdB by overlap PCR. The gene encoding GTD of TcdB was introduced into pHis-TcdA through BsrGI/BamHI digestion to generate the plasmid pHis-TxA-Bgt. The plasmid was used to transform Bacillus megaterium, and TxA-Bgt was expressed and purified using methods described previously (52). TxA-Bgt is fully cytotoxic to CT26 cells, indicating that the protein contains a functional GTD from TcdB. Purified VHHs were screened against TxA-Bgt using standard ELISA. Specific binding of the VHHs against GTD was further verified by immunoblotting against autoprocessed TcdB in the presence of InsP6 as described below.
InsP6-induced autocleavage of toxins.
InsP6-induced autocleavage of toxins was carried out as described previously (29, 54). TcdB was diluted in 10 mM Tris (pH 7.5) buffer to a concentration of 10 μg/ml in a final volume of 20 μl in the presence or absence of VHHs (10 μg/ml). The reaction was initiated by addition of InsP6 (10 μM), and the mixture was incubated for 2 h. Reactions were stopped by the addition of 5 μl of 5× SDS sample buffer, and products were analyzed by standard Western blotting using VHHs or alpaca anti-TcdB polyclonal antibodies (generated in this laboratory) or anti-E tag antibodies to visualize VHHs.
Glucosyltransferase activity of toxins.
GTD activity of TcdB was measured by the ability of GTD to glucosylate Rho GTPase Rac1 in a cell-free assay (30, 54). For this assay, the cytosolic fractions of Vero cells were incubated with TcdB (10 μg/ml) or TcdB plus VHH (100 μg/ml) at 37°C for 30 min. The reaction was terminated by the addition of SDS sample buffer and heating the mixture at 100°C for 5 min before the product was loaded onto a 12% SDS-PAGE gel. An antibody that specifically recognizes the nonglucosylated form of Rac1 (clone 102; BD Bioscience) was used to assess Rac1 glucosylation in a standard Western blot analysis. Anti-β-actin (clone AC-40; Sigma) antibody was also used to detect β-actin as a loading control.
ELISAs.
For ELISAs, plates were coated with 0.5 μg/ml of TcdB overnight at 4°C. Plates were blocked with 5% milk (100 μl/well) in PBS and incubated with serial dilutions of antibodies (VHH Abs or VHH plus peptide at 100 μl/well; concentrations as indicated in the figure legends) in PBS–0.1% (vol/vol) Tween 20 (PBST) at room temperature (RT) for 1 h. The plates were washed with PBST and incubated with a goat anti-E tag-IgG-horseradish peroxidase (HRP) conjugate (Bethyl Laboratories, Montgomery, TX) at RT for 1 h for VHH titration. After three washes with PBST, TMB (3,3′,5,5′-tetramethylbenzidine) substrate (100 μl/well; KPL, Gaithersburg, MD) was added to each well for 5 min. The reaction was stopped by the addition of 50 μl/well 1 M H2SO4 and read on a Bio-Rad plate reader (Hercules, CA) at 450 nm. Reported values are representative of three independent experiments.
Cytopathic effect and cytotoxicity.
Human ileocecal adenocarcinoma cell line HCT-8 and African green monkey kidney Vero cells (ATCC, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) with 10% fetal bovine serum, 1 mM sodium pyruvate, 2 mM l-glutamine, 100 U/ml penicillin, and 40 μg/ml streptomycin sulfate. Cytopathic assays were carried out as previously described (53). Serially diluted VHHs and toxins were premixed using toxin at a concentration of 0.2 ng/ml before being added to each well, and cell rounding was measured by phase-contrast microscopy. Cytotoxicity of Vero cells was measured by a lactate dehydrogenase (LDH) cytotoxicity kit (Pierce) according to the manufacturer's instructions. Vero cells were exposed to wild-type TcdB, autoprocessing mutant TcdB-L543A, or glucosyltransferase-deficient TcdB-W102A D288N at either 10 or 1 ng/ml for different days before the supernatant was harvested for LDH assays.
Intracellular expression of VHH intrabodies and inhibition of cellular intoxication.
To express functional VHHs in the cytosol of host cells, red fluorescent protein (RFP)-VHH fusions were constructed within pCS2-mRFP-N3 vectors (Addgene). Vero cells were transfected with 4 μg of pCS2-mRFP-VHH fusion plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Empty pCS2-mRFP-N3 plasmid vectors were used as a control. Transfected Vero cells were incubated for 24 to 48 h, RFP expression was determined by fluorescence microscopy, and VHH expression from cytosolic fractions was analyzed by Western blotting and ELISA. To test whether intrabody activity can block cytopathic effects in cells, Vero cells transfected with VHH vectors were incubated with TcdB (1 ng/ml), and cell rounding was analyzed as described above. In some experiments, cells were exposed to an autoprocessing-deficient mutant, TcdB-L543A (kindly provided by Aimee Shen, University of Vermont), at 10 ng/ml before cell rounding was analyzed. Cell rounding in both transfected and nontransfected cells was quantitated as described previously (30).
Systemic challenge of mice with purified toxins.
Six-week-old CD1 mice were purchased from Jackson Laboratory (MI, USA) and housed in a dedicated pathogen-free facility. Mice were handled and cared for in accordance with Institutional Animal Care and Use Committee guidelines. For systemic challenge, purified wild-type TcdB, TcdB-L543A, and aTcdB (TcdB-W102A D288N) in PBS were injected intraperitoneally into mice at either 20 ng or 100 ng per mouse for TcdB and TcdB-L543A and at 10 μg per mouse for aTcdB. Control mice were injected with PBS only. Five mice were used per group and were closely monitored for signs of systemic disease as described previously (55), and moribund animals were sacrificed.
Statistical analysis.
Data were analyzed by Kaplan-Meier survival analysis with a log rank test of significance, analysis of variance (ANOVA), and one-way (ANOVA) followed by Bonferroni posttests using the Prism statistical software program. Results are expressed as means ± standard errors of means. A P value of <0.05 was regarded as indicating a significant difference between groups.
RESULTS
VHHs 7F and E3 bind to GTD of TcdB.
In order to identify and characterize VHHs that inhibit C. difficile toxin enzymatic activities, we generated a TcdB-specific VHH phage library and screened for VHHs that bound strongly to functional domains of the toxin (51). The VHHs were overexpressed in E. coli, purified, and further analyzed for domain-specific binding efficiency (51). A chimeric toxin, TxA-Bgt, was generated that consisted of the GTD of TcdB (amino acids [aa]1 to 543) with the rest of the domains derived from TcdA. TxA-Bgt was fully cytotoxic, with activity similar to that of the wild-type TcdA in cultured Vero cells, suggesting that it has a native form of GTD. We identified four VHHs (5D, E3, 7F, and C6) (51) that bound with high affinity to TxA-Bgt; two clones, 7F and E3, showed selective binding to TcdB, but not TcdA, by ELISA (Fig. 1A and B). Western blot analysis confirmed that 7F and E3 bound specifically to the TcdB GTD domain because both of the antibodies recognized the full-length toxin (270 kDa; aa 1 to 2366) and the inositol hexakisphosphate (InsP6)-GTD cleavage fragment (63 kDa; aa 1 to 543) but not the C-terminal fragment lacking the GTD (207 kDa; aa 544 to 2366) (Fig. 1C). Thus, the binding epitopes for VHHs 7F and E3 are contained within the GTD of TcdB.
FIG 1.

VHHs 7F and E3 bind to the GTD of TcdB. (A and B) VHHs 7F and E3 were tested for specific binding to both TcdA and TcdB by ELISA. Toxin-coated 96-well plates were incubated with serial dilutions of 7F or E3. Goat anti-E tag-IgG-HRP was used as a secondary antibody. Overall binding was determined by absorbance at 450 nm (optical density at 450 nm [OD450]). (C) 7F and E3 binding to TcdB by Western blotting. Specific 7F, E3, monoclonal antibody recognizing the C-terminal portion of TcdB (MoAb), or alpaca polyserum binding to holotoxin TcdB (polyclonal Abs) were assessed by immunoblotting. The bands represent full-length TcdB (∼270 kDa), the C-terminal fragment of cleaved TcdB (aa 544 to 2366, ∼ 207 kDa), and the cleaved GTD (aa 1 to 543, ∼63 kDa).
7F and E3 inhibit TcdB-induced Rac glucosylation and autocleavage, respectively.
TcdB exerts its cytopathic effects via glucosylation of Rho family GTPases in the cytoplasm in a GTD-dependent manner (8, 26). To analyze whether VHH binding to GTD interferes with this critical step in cellular intoxication, both E3 and 7F were assessed for inhibiting activity in a cell-free Rac1 glucosylating assay. Wild-type TcdB induced Rac1 glucosylation in Vero cell lysates (Fig. 2A). This activity was inhibited by addition of E3 but not by 7F or the other VHHs tested (Fig. 2A). We next studied whether InsP6-mediated autoprocessing of TcdB is inhibited by VHH addition. Western blot analysis demonstrated that InsP6 induced TcdB autocleavage, with release of the 63-kDa GTD from the full-length toxin, whereas addition of 7F abolished this autocleavage (Fig. 2B). E3 did not block this autocatalytic cleavage (Fig. 2C). Thus, 7F and E3 specifically inhibit CPD-mediated autocleavage and glucosyltransferase activity, respectively. We utilized these VHH tools to study the importance of GTD and CPD enzymatic activities in cellular intoxication.
FIG 2.

E3 and 7F inhibit glucosyltransferase and autoprocessing activities, respectively. (A) E3, but not 7F or other tested VHHs, affects the glucosyltransferase activity of TcdB. Vero cell lysates were analyzed for Rac1 glucosylation by immunoblotting after exposure to wild-type TcdB and/or E3 or by 7F VHHs. A monoclonal antibody (clone 102) that binds only to nonglucosylated Rac1 was used as a probe. β-Actin was used as an equal loading control. (B) 7F blocks the autocatalytic cleavage of GTD from full-length TcdB. TcdB was incubated in the presence or absence of 7F and InsP6. The autocleavage and release of GTD from full-length TcdB were assessed by immunoblotting using a monoclonal antibody against GTD. (C) E3 does not block the autocleavage of GTD from full-length TcdB. (D) The peptide consisting of aa 520 to 543 binds specifically to 7F. Ninety-six-well plates were coated with the peptide consisting of aa 520 to 543 or aa 422 to 439 and incubated with serial dilutions (0.32 to 1,000 ng/ml) of 7F or control VHH 5D. Goat anti-E tag-IgG-HRP was used as a secondary antibody. (E) The peptide consisting of aa 520 to 543 significantly reduces binding of 7F to GTD. Ninety-six-well plates were coated with 0.5 μg/ml TcdB and incubated with 7F or 5D VHH alone (100 or 10 ng/ml) or with the VHHs plus the peptide consisting of aa 520 to 543 or aa 422 to 439. Goat anti-E tag-IgG HRP was used as a secondary antibody. *, P < 0.05; ns, not significant.
7F binds to an amino acid sequence of GTD directly adjacent to the CPD cleavage site.
Because 7F shows binding specificity to the GTD of TcdB yet inhibits autoprocessing activity associated with the toxin's cysteine protease domain, we further investigated the mechanism of autocleavage inhibition. In silico studies of 7F binding epitopes on TcdB predicted antibody binding in a GTD region immediately adjacent to the CPD cleavage site between aa 543 and 544 (56). To test this, we synthesized the peptide SFDDARAKAQFEEYKRNYFEGSL consisting of TcdB aa 520 to 543 and measured this for specific binding to 7F by ELISA. As a control we used peptide NDFNTTTNTFIDSIMAEA consisting of TcdB aa 422 to 439. As shown in Fig. 2D, 7F bound to aa 520 to 543 in a dose-dependent fashion, whereas the control VHH 5D that also binds GTD (51) did not. Neither 7F nor 5D bound to the control peptide, aa 422 to 439 (Fig. 2D), suggesting that the binding of 7F to aa 520 to 543 is specific. Epitope specificity was further confirmed by competitive peptide inhibition of 7F binding to TcdB (Fig. 2E). The specific (aa 520 to 543) or control (aa 422 to 439) peptide was added to 7F and control VHH 5D before the peptides were analyzed for specific binding to TcdB by ELISA. The peptide consisting of aa 520 to 543 significantly reduced 7F binding, whereas it had no effect on 5D-specific binding to TcdB. As expected, the control peptide consisting of aa 422 to 439 had no inhibitory effects on the binding of either 7F or 5D to TcdB. These data demonstrate that 7F does not inhibit cysteine protease activity directly, but binding to the toxin region directly adjacent to the CPD autocleavage site prevents autoprocessing and release of free GTD.
FIG 7.
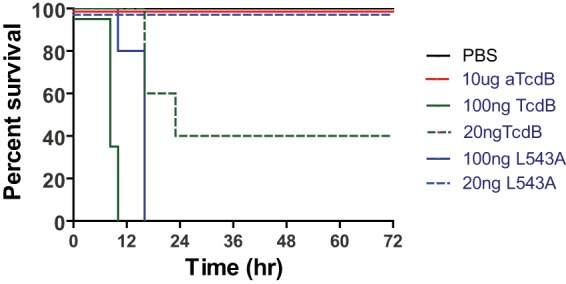
Mouse mortality after systemic inoculation of wild-type and mutant TcdB. Intraperitoneal inoculation of mice with 100 ng of either wild-type TcdB or TcdB-L543A resulted in 100% mortality although the time to full mortality was delayed in mice treated with TcdB-L543A compared to those treated with wild-type TcdB (log rank test; P < 0.01). Inoculation of mice with 20 ng of TcdB led to 60% mortality, whereas the same dose of TcdB-L543A did not cause any mortality; although all mice displayed signs of systemic disease, they eventually recovered (P < 0.05). Mice inoculated with a glucosyltransferase-deficient TcdB variant (aTcdB) at a dose of 10 μg/mouse showed no sign of disease, and all mice survived (P < 0.01).
Expressing functional intracellular VHHs (intrabodies).
Because both 7F and E3 have the capacity to inhibit the functional consequences of the two respective enzymatic domains of TcdB, we tested whether the VHHs could be expressed as intrabodies within the cytosol of host cells to study the intracellular mode of toxin action. To demonstrate that both 7F and E3 are functionally expressed within the cytosol of target cells, RFP-VHH fusion vectors were transiently transfected into Vero cells. Cells were lysed, and cytosolic fractions were tested for VHH binding to TcdB. Cytosolic fractions from 7F-, C6-, and E3-transfected cells showed dose-dependent binding to TcdB but not to TcdA by ELISA (Fig. 3A). Intact VHHs accumulated at equivalent levels in these respective cytosolic fractions when expressed in Vero cells (Fig. 3B). More importantly, both E3 and 7F intrabodies significantly reduced TcdB-induced Rac 1 glucosylation, whereas control intrabodies 5D and C6 had no inhibitory effect (Fig. 3B and C). Thus, VHHs can be successfully transfected to produce neutralizing intrabodies in cultured cells that are sensitive to TcdB intoxication.
FIG 3.

E3 and 7F can be expressed as intrabodies and maintain their inhibitory functions. (A) Cytosolic fractions from cells transfected with GTD-specific VHHs show strong binding to TcdB. Vero cells transfected with VHH-encoding plasmids were lysed, and cytosolic fractions were tested for specific binding to TcdB by ELISA. TcdB-coated 96-well plates (colored lines) were incubated with serial dilutions of cytosolic fractions. TcdA-coated wells and cytosolic fractions from untransfected cells (cells only) (black lines) were used as negative controls. (B) Effects of 7F and E3 intrabodies on TcdB-induced Rac1 glucosylation. Vero cells were transfected with a panel of fusion plasmids containing genes for RFP-fused VHH monomers before treatment with TcdB. Cytosolic fractions of transfected Vero cells were assessed by Western blotting for the presence of VHH intrabodies and for Rac1 glucosylation status. Antibodies specific for VHHs or the nonglucosylated form of Rac1 (clone 102) were used for visualization. Total Rac1 (clone 23A8) levels were determined as a loading control. (C) Densitometry analysis of percentage of nonglucosylated Rac1 relative to total Rac1 from the three experiments shown in panel B. *, P < 0.05; ns, not significant.
7F delays TcdB cytopathic activity by inhibiting toxin autoprocessing.
To examine whether inhibition of TcdB enzymatic functions prevents intoxication, both HCT-8 and Vero cells were transiently transfected with either 7F, E3, or GTD-specific VHH control (C6 and 5D) fusion plasmids before exposure to 1 ng/ml TcdB. Because the VHH fusion plasmids also contain functional red fluorescent protein (RFP), fluorescent and phase-contrast microscopy images were simultaneously recorded, and cytopathic effects of TcdB were quantified in transfected (white/red cells) and nontransfected (dark) cells in the same toxin-exposed wells (Fig. 4). In control experiments without TcdB exposure, no aberrant cell morphology resulted from transfection with intrabodies (Fig. 4A, B, and C). Following intoxication, TcdB rapidly induced cell rounding in C6- or 5D-transfected and nontransfected Vero control cells (Fig. 4A). In contrast, cytopathic effects were not recorded in cells successfully transfected with 7F or E3, whereas nontransfected cells in the same wells rounded (Fig. 4A).
FIG 4.

E3 eliminates cytopathic effects induced by TcdB, but 7F only delays their onset. (A) Cells transfected with either E3 or 7F intrabodies do not show cytopathic effects after 1 h of TcdB exposure. Vero cells were transfected with empty control plasmid vectors (cell alone) or with fusion plasmids containing 7F, E3, C6, or 5D. Cells were assessed for cytopathic effects using a combination of phase-contrast microscopy and fluorescence microscopy after exposure with TcdB (1 ng/ml). For these pictures, dark cells represent untransfected cells while white cells represent cells successfully transfected with fusion plasmids. (B) HCT-8 cells were transfected with E3, 7F, or control 5D fusion plasmids before exposure to TcdB (1 ng/ml) for 150 min. (C) 7F intrabodies only delay the onset of TcdB-mediated cytopathic effects, whereas E3 blocks these effects. Vero cells were transfected with either 7F, E3, or control (5D) fusion plasmids before exposure to TcdB (1 ng/ml) and assessed for cytopathic effects over time using a combination of phase-contrast microscopy and fluorescence microscopy. In the images in panels B and C, untransfected cells appear dark while successfully transfected cells appear red. (D) Quantitative analysis of TcdB-induced cytopathic effects. After TcdB exposure, the percentages of affected cells were determined by counting rounded cells in the total cell population in either untransfected or transfected cells from five random microscopy fields. *, P < 0.05 between transfected and untransfected cells in the same wells.
We have recently reported that blockage of GTD autoprocessing as a result of toxin mutagenesis delays but does not completely inhibit TcdB cytopathic effects (30). To examine whether cell cytotoxicity is similarly delayed after inhibition of toxin autocleavage by 7F intrabody, we monitored cell rounding in VHH-transfected cells with time after TcdB intoxication (Fig. 4B, C, and D). Nontransfected and VHH 5D-transfected HCT-8 and Vero cells began rounding within 40 min, with virtually all cells showing cytopathic effects by 1.5 h (Fig. 4B and C). In contrast, cells transfected with 7F intrabody did not show any cell rounding until 1.5 to 2 h after TcdB exposure, with most cells showing cytopathic effects by 2.5 h (Fig. 4C). Quantitative analysis of transfected and nontransfected cells in the same toxin-exposed wells demonstrated that 7F intrabody significantly delayed cell rounding up to 1.5 h after TcdB exposure (Fig. 4D). No significant differences were recorded after this time.
To confirm that the delay in TcdB-mediated cytopathic effects was due to binding of 7F to GTD and prevention of toxin autocleavage inside cells, we utilized an autoprocessing-deficient TcdB mutant in the cellular cytotoxicity assays. In this mutant, the leucine at the substrate recognition site for CPD has been replaced with an alanine (L543A), thus preventing enzymatic release of GTD from TcdB (30, 57). This single amino acid mutation did not change the binding affinity of 7F to the toxin as the wild type and mutant TcdB bound similarly to 7F by ELISA (Fig. 5A). To investigate the effects of 7F intrabody on TcdB-L543A-mediated intoxication, Vero cells were transfected with VHH plasmids prior to intoxication with 10 ng/ml of TcdB-L543A. 7F did not exhibit any significant inhibitory effects on TcdB-L543A-induced cell rounding over control 5D transfections (Fig. 5B). Thus, 7F intrabody does not inhibit or further delay cellular cytotoxicity by TcdB-L543A. Since 7F is expressed in the cytosol and does not access endosomes, our data provide the first experimental demonstration that toxin autoprocessing occurs following exposure to the host cytosol.
FIG 5.

The inhibition of cytopathic effects is due to the inhibition of enzyme activities of TcdB. (A) 7F binds similarly to wild-type TcdB and mutant TcdB-L543A. Ninety-six-well plates were coated with 7F and incubated with serial dilutions of either TcdB or TcdB-L543A (LA). Goat anti-E tag-IgG-HRP was used as a secondary antibody. Overall binding was determined by absorbance at 450 nm (OD450). (B) Vero cells were transfected with either 7F, E3, or control (5D) fusion plasmids before exposure to TcdB-L543A (10 ng/ml) and assessed for cytopathic effects over time using a combination of phase-contrast microscopy and fluorescence microscopy. In these pictures, dark cells represent untransfected cells, while red cells represent successfully transfected cells. (C) Quantitative analysis of TcdB-induced cytopathic effects. After TcdB exposure, the percentages of affected cells were determined by counting rounded cells in the total cell population in either untransfected or transfected cells from five random microscopy fields. *, P < 0.05 between transfected and untransfected cells in the same wells.
E3 intrabody inhibits TcdB cytopathic activity.
Interestingly, cells transfected with E3 intrabody (inhibiting GTD activity) did not demonstrate cytopathic effects upon either wild-type or autoprocessing-deficient TcdB at any of the times investigated (Fig. 4 and 5B and C). The E3 transiently transfected cells never rounded with over 24 h of toxin exposure during the entire experiment period. This protective effect of E3 intrabody correlated well with a glucosyltransferase-deficient TcdB mutant (TcdB-W102A D288N, designated aTcdB), which fails to exhibit cytotoxicity at either significantly higher toxin concentrations over 72 h of incubation time (53) or at 1 ng/ml (Fig. 6A) or 10 ng/ml (Fig. 6B). On the other hand, low doses of TcdB and TcdB-L543A induced cytopathic effects on host cells and eventually led to cell death at comparable levels after longer times of toxin exposure (Fig. 6). To confirm that E3 is targeting the GTD catalytic site and is not interfering with toxin autocleavage, cytopathic effects in E3-transfected cells remained significantly blocked after addition of TcdB-L543A (Fig. 5B and C). These data strongly support the idea that TcdB-mediated cytopathic effects are dependent on the toxin glucosyltransferase activity but that prevention of autoprocessing only delays the onset of cytopathic effects that eventually lead to cell death.
FIG 6.

Cytotoxicity of wild-type and mutant TcdB. Vero cells in 96-well plates were exposed to 1 or 10 ng/ml of TcdB, TcdB-L543A (LA), or TcdB-W102A D288N (aTcdB) for 1 to 4 days. Supernatants from each well were harvested, and LDH activity in supernatants was measured. The percentage of cytotoxicity was determined by the ratio of LDH release values to the maximum LDH release under the same treatment. The LDH release values represent the average of three experiments in which three replicates were averaged. Error bars indicate the standard errors among the values obtained from the three experiments.
Systemic toxicity by TcdB requires functional glucosyltransferase activity.
To investigate the pathophysiological validity of our in vitro toxin findings, we used a well-characterized systemic TcdB toxemia model (55, 58) to test enzyme-deficient toxin mutants. Mice challenged intraperitoneally with mutant TcdB demonstrated significantly attenuated systemic virulence responses. At a TcdB 50% lethal dose (LD50) (20 ng/mouse), no mortality was evident with TcdB-L543A although all mice showed signs of systemic disease (Fig. 7). At a higher lethal toxin dose (100 ng/mouse), both wild-type TcdB- and TcdB-L543A-challenged animals developed fulminant disease although a significant delay in clinical symptoms was evident with the toxin autocleavage mutant (Fig. 7). In contrast, no clinical symptoms were evident with up to 10 μg/mouse of aTcdB (500-fold TcdB LD50) (Fig. 7). These data demonstrate that the autoprocessing-deficient TcdB-L543A is attenuated, whereas the TcdB glucosyltransferase mutant is completely atoxic in vivo.
DISCUSSION
The major virulence factors of the C. difficile toxins are the glucosyltransferases (8, 59). Both toxins also contain a cysteine protease domain that facilitates delivery of the glucosyltransferase domain into the target cell (24, 25). Recent in vitro studies have challenged this consensus by reporting enzyme-independent cytopathic and cytotoxicity activities of the toxins by assessing the effects of TcdB mutants on different cell lines (30, 31, 34–36). A major limitation of these studies is the general failure to validate experimental findings using an independent approach that does not alter toxin active-site specificity or conformational integrity. In this study, we describe a novel intrabody approach that critically tests the role of the glucosyltransferase and cysteine protease in TcdB virulence. Our studies demonstrated a regulatory role of the cysteine protease of TcdB in cellular intoxication and an essential role of the toxin glucosyltransferase in its cytopathic effects and systemic toxicity.
To examine whether the toxin glucosyltransferase and autoprocessing are required for in vitro cytotoxicity, we utilized specific VHH intrabodies as biological tools that inhibit the native form of TcdB enzymatic machinery inside host cells. After a library screening, we identified two unique VHHs, 7F and E3, that bind with high affinity to GTD yet inhibit distinct TcdB enzymatic activities located in different functional domains. Specifically, E3 interferes with the ability of TcdB to glucosylate Rac1 in cell-free assays, whereas 7F blocks toxin autocleavage by allosteric cofactor InsP6. The inhibitory action of 7F binding likely impedes toxin self-cleavage by interfering with catalytic processing of substrate in the CPD. Computer simulations indicated that steric hindrance may play a role by binding an epitope immediately juxtaposed to the cleavage site on the interdomain linker arm (aa 520 to 543), which we confirmed experimentally. When expressed at high levels in both Vero and HCT-8 cells, 7F attenuated the cytopathic response to TcdB, whereas E3 expression completely abolished cell rounding. These studies provide both the first evidence that intracellular autocleavage and release of GTD regulate the toxin's activity and independent experimental validation of the key virulence role of glucosyltransferase in wild-type TcdB. Our study also demonstrates more generally a role for VHH technology in assaying the virulence functions of intact holotoxins. Antitoxin VHH intrabodies have been explored as potential therapeutics to neutralize intracellular toxins (60); this intrabody approach has not previously been utilized to study toxin intracellular trafficking and mechanisms of action. Given the relatively small size, high solubility, and specificity of VHHs (42–44), the use of intrabodies may represent a powerful new approach to study the intracellular mode of action of the C. difficile toxins. Indeed, the current virulence model shows that toxins are actively internalized and that following acidification in endosomal compartments, the N terminus is translocated into the cytosol. However, it is unknown whether CPD-mediated autoprocessing occurs before or after the translocation of the toxins. It is reasonable to assume that InsP6 or other activators may accumulate in both cytosol and endosome to concentrations that are sufficient to cause activation of the cysteine protease leading to autoprocessing. Because the intrabodies are expressed in the cytosol and are unlikely reach into endosomes, our data thus provide experimental evidence indicating that autoprocessing occurs after toxin translocation from endosomes into the cytosol.
In this study, we chose low to medium doses of TcdB (1 to 10 ng/ml) to treat the cultured cells since TcdB is an extremely potent toxin. TcdB induces cytopathic effects on most cultured cells in a dose range of pg/ml and has an LD50 of 1 μg/kg in CD1 mice. Several recent reports demonstrated that TcdB induces rapid cell death that is independent of enzymatic activities of the toxin (34–38). Such a glucosyltransferase-independent activity is, however, dependent upon exposing cultured cells or tissue explants at high doses (at μg/ml ranges) of TcdB. The exact mechanism of the glucosyltransferase-independent cell death induced by TcdB is not clear, but it appears that the cell death relies on the pore-forming activity of TcdB (38). Interestingly, Wohlan et al. found that the cytotoxicity induced by high doses of TcdB is dependent upon the translocation of GTD into host cells (37). Therefore, at this stage we cannot dismiss the possibility that the GTD or CPD may possess other unknown functions that are important for cellular intoxication and C. difficile infection. Because the toxin enzymatic domains are conserved over divergent microbial species, it seems likely that they serve an important functional role. Pathogenic bacteria generally have evolved to rapidly eliminate biological functions that are unnecessary for pathogenesis, survival, or gaining a competitive advantage. Studies in animal models are therefore urgently warranted to delineate the clinical relevance of these in vitro findings.
Although an animal intestinal disease model testing enzymatically deficient holotoxins has not yet been established, a systemic TcdB toxemia model has been reported (53, 58). Because TcdB disseminates systemically during C. difficile infection and causes toxemia (55, 61, 62), we investigated systemic toxicity of wild-type and mutant toxins by challenging mice intraperitoneally with defined toxin concentrations. The TcdB LD50 in mice is 20 ng (1 μg kg−1), with 100 ng causing fulminant disease in all animals. We confirmed that the noncleavable mutant TcdB-L543A is attenuated in vivo as no mice developed fulminant disease at the TcdB LD50. However, this mutant is still inherently toxic since all mice developed severe toxemia when 100 ng of TcdB-L543A was administered although onset of clinical disease was significantly delayed. This result is consistent with recently reported in vitro findings that toxin autoprocessing is not an essential requirement for cytotoxicity but regulates its potency (30). A recent report that TcdB from hypervirulent strains is more cytotoxic than TcdB from historical strains because of its increased efficiency in autoprocessing supports this hypothesis (16). In contrast, glucosyltransferase-deficient TcdB was not cytotoxic in mice up to a dose 500-fold higher than the LD50 of wild-type TcdB. This is consistent with our previous finding that both glucosyltransferase-deficient TcdA and TcdB lost their systemic toxicity (53). The in vivo data thus demonstrate that TcdB autoprocessing is not essential for TcdB systemic toxicity, whereas glucosyltransferase activity is an absolute requirement.
Our previous results (30, 53) and results from this study further clarify the role of CPD-mediated autoprocessing and glucosyltransferase in TcdB's cytopathic effects and in vivo toxicity, and an updated model for domain activation can be proposed (Fig. 8). After TcdB endocytosis, both GTD and CPD are translocated through the endosomal membrane into the cytosol, presumably through pores created by the toxin transmembrane domain. Membrane-associated InsP7 and cytosolic InsP6 bind to the CPD, thus activating the cleavage of GTD in a CPD-dependent process. Liberated GTD is able to bind to Rho GTPases throughout the cell cytosol, inducing rapid cytopathic effects and cytotoxicity. However, when the CPD activity is low or there is a deficiency in autoprocessing (whether by genetic mutation, physical blockage by intrabodies, or some other mechanism), the full-length toxin stays tethered to the membrane of endosomes after their translocation. While tethered, the toxin GTD may only interact with and modify smaller numbers of Rho GTPases, such as those bound to membranes or those within the cytosol that contact the endosome, as proposed earlier by Kreimeyer et al. (31). The reduced amount of GTPase glucosylation that occurs in this scenario delays the onset of toxin-mediated cytopathic effects as more time is required for glucosylation to reach critical levels. These data support the conclusion that CPD-mediated autoprocessing of C. difficile toxins is not necessary for cytopathic effects and systemic toxicity of the toxin but acts only to accelerate the rate in which hosts become intoxicated, an action dependent upon glucosyltransferase activity of the toxins.
FIG 8.
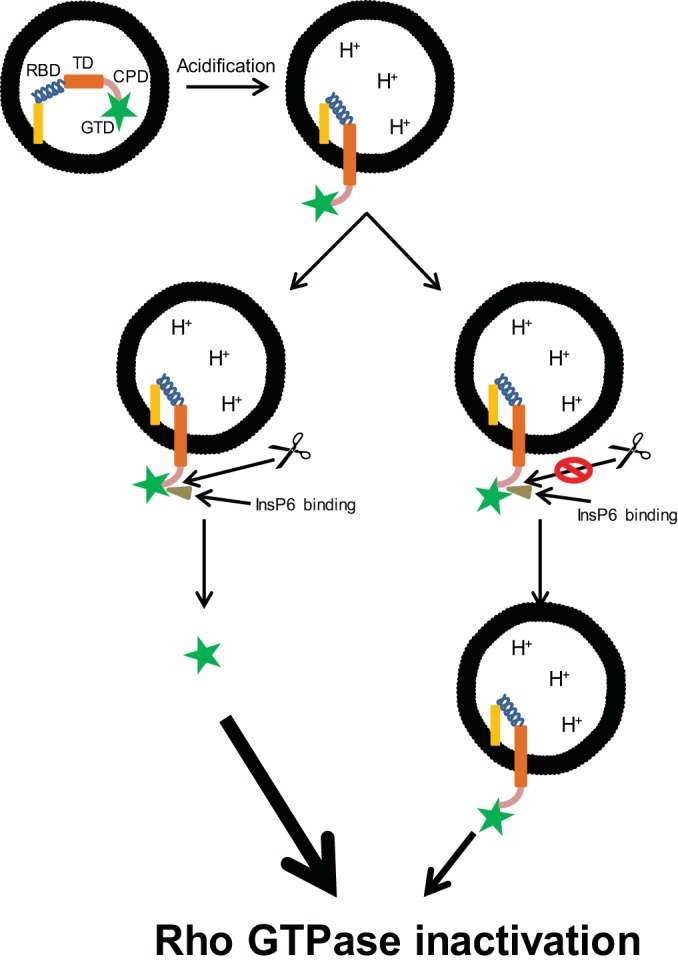
Revised proposed model for the mechanism of action of C. difficile toxins. C. difficile toxins endocytose upon receptor binding, and both GTD and CPD are translocated through the endosomal membrane into the cytoplasm. Cytoplasmic InsP6 binds to the toxins and activates GTD cleavage via CPD, liberating GTD into the cytoplasm. However, depending on the potency of CPD activities or in the presence of CPD or autoprocessing inhibitors, the cleavage and release of GTD are reduced or blocked, and GTDs of the toxins remain tethered to the endosomal membrane. Both liberated and tethered GTDs target host small Rho GTPases for glucosylation but at different cellular compartments and rates of action.
In summary, we utilized two specific intrabodies (E3 and 7F) as novel tools to study TcdB enzymatic functions and demonstrated a key role of glucosyltransferase and a regulatory role of cysteine protease in TcdB virulence. Using a systemic toxin challenge model, we consistently demonstrated that although TcdB cysteine protease activity potentiates disease pathogenesis, in vivo potency is critically dependent on the glucosyltransferase activity, highlighting the importance of targeting this enzyme activity for future therapy.
ACKNOWLEDGMENTS
This work was supported by awards R01AI088748, R01DK084509, R56AI99458, and U19 AI109776 to H.F. and awards R01DK56338, R21DK096323, and U54 AI057159 to T.C.S., funded by the National Institute of Allergy and Infectious Diseases and National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health (NIH).
REFERENCES
- 1.Kelly CP, LaMont JT. 2008. Clostridium difficile—more difficult than ever. N Engl J Med 359:1932–1940. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 2.Zilberberg MD. 2009. Clostridium difficile-related hospitalizations among US adults, 2006. Emerg Infect Dis 15:122–124. doi: 10.3201/eid1501.080793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jank T, Aktories K. 2008. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol 16:222–229. doi: 10.1016/j.tim.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Albesa-Jove D, Bertrand T, Carpenter EP, Swain GV, Lim J, Zhang J, Haire LF, Vasisht N, Braun V, Lange A, von Eichel-Streiber C, Svergun DI, Fairweather NF, Brown KA. 2010. Four distinct structural domains in Clostridium difficile toxin B visualized using SAXS. J Mol Biol 396:1260–1270. doi: 10.1016/j.jmb.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 6.Pruitt RN, Chambers MG, Ng KK, Ohi MD, Lacy DB. 2010. Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc Natl Acad Sci U S A 107:13467–13472. doi: 10.1073/pnas.1002199107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schorch B, Song S, van Diemen FR, Bock HH, May P, Herz J, Brummelkamp TR, Papatheodorou P, Aktories K. 2014. LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc Natl Acad Sci U S A 111:6431–6436. doi: 10.1073/pnas.1323790111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. 1995. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375:500–503. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- 9.Nam HJ, Kang JK, Kim SK, Ahn KJ, Seok H, Park SJ, Chang JS, Pothoulakis C, Lamont JT, Kim H. 2010. Clostridium difficile toxin A decreases acetylation of tubulin, leading to microtubule depolymerization through activation of histone deacetylase 6, and this mediates acute inflammation. J Biol Chem 285:32888–32896. doi: 10.1074/jbc.M110.162743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pothoulakis C, Lamont JT. 2001. Microbes and microbial toxins: paradigms for microbial-mucosal interactions II. The integrated response of the intestine to Clostridium difficile toxins. Am J Physiol Gastrointest Liver Physiol 280:G178–G183. [DOI] [PubMed] [Google Scholar]
- 11.Qa'Dan M, Ramsey M, Daniel J, Spyres LM, Safiejko-Mroczka B, Ortiz-Leduc W, Ballard JD. 2002. Clostridium difficile toxin B activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells. Cell Microbiol 4:425–434. doi: 10.1046/j.1462-5822.2002.00201.x. [DOI] [PubMed] [Google Scholar]
- 12.Lyras D, O'Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 14.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 15.Chaves-Olarte E, Weidmann M, Eichel-Streiber C, Thelestam M. 1997. Toxins A and B from Clostridium difficile differ with respect to enzymatic potencies, cellular substrate specificities, and surface binding to cultured cells. J Clin Invest 100:1734–1741. doi: 10.1172/JCI119698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lanis JM, Hightower LD, Shen A, Ballard JD. 2012. TcdB from hypervirulent Clostridium difficile exhibits increased efficiency of autoprocessing. Mol Microbiol 84:66–76. doi: 10.1111/j.1365-2958.2012.08009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giesemann T, Egerer M, Jank T, Aktories K. 2008. Processing of Clostridium difficile toxins. J Med Microbiol 57:690–696. doi: 10.1099/jmm.0.47742-0. [DOI] [PubMed] [Google Scholar]
- 18.von Eichel-Streiber C, Sauerborn M, Kuramitsu HK. 1992. Evidence for a modular structure of the homologous repetitive C-terminal carbohydrate-binding sites of Clostridium difficile toxins and Streptococcus mutans glucosyltransferases. J Bacteriol 174:6707–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ho JG, Greco A, Rupnik M, Ng KK. 2005. Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc Natl Acad Sci U S A 102:18373–18378. doi: 10.1073/pnas.0506391102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papatheodorou P, Zamboglou C, Genisyuerek S, Guttenberg G, Aktories K. 2010. Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS One 5:e10673. doi: 10.1371/journal.pone.0010673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qa'Dan M, Spyres LM, Ballard JD. 2000. pH-induced conformational changes in Clostridium difficile toxin B. Infect Immun 68:2470–2474. doi: 10.1128/IAI.68.5.2470-2474.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barth H, Pfeifer G, Hofmann F, Maier E, Benz R, Aktories K. 2001. Low pH-induced formation of ion channels by Clostridium difficile toxin B in target cells. J Biol Chem 276:10670–10676. doi: 10.1074/jbc.M009445200. [DOI] [PubMed] [Google Scholar]
- 23.Pfeifer G, Schirmer J, Leemhuis J, Busch C, Meyer DK, Aktories K, Barth H. 2003. Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J Biol Chem 278:44535–44541. doi: 10.1074/jbc.M307540200. [DOI] [PubMed] [Google Scholar]
- 24.Reineke J, Tenzer S, Rupnik M, Koschinski A, Hasselmayer O, Schrattenholz A, Schild H, von Eichel-Streiber C. 2007. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446:415–419. doi: 10.1038/nature05622. [DOI] [PubMed] [Google Scholar]
- 25.Egerer M, Giesemann T, Jank T, Satchell KJ, Aktories K. 2007. Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J Biol Chem 282:25314–25321. doi: 10.1074/jbc.M703062200. [DOI] [PubMed] [Google Scholar]
- 26.Hofmann F, Busch C, Prepens U, Just I, Aktories K. 1997. Localization of the glucosyltransferase activity of Clostridium difficile toxin B to the N-terminal part of the holotoxin. J Biol Chem 272:11074–11078. doi: 10.1074/jbc.272.17.11074. [DOI] [PubMed] [Google Scholar]
- 27.Nottrott S, Schoentaube J, Genth H, Just I, Gerhard R. 2007. Clostridium difficile toxin A-induced apoptosis is p53-independent but depends on glucosylation of Rho GTPases. Apoptosis 12:1443–1453. doi: 10.1007/s10495-007-0074-8. [DOI] [PubMed] [Google Scholar]
- 28.Savidge TC, Urvil P, Oezguen N, Ali K, Choudhury A, Acharya V, Pinchuk I, Torres AG, English RD, Wiktorowicz JE, Loeffelholz M, Kumar R, Shi L, Nie W, Braun W, Herman B, Hausladen A, Feng H, Stamler JS, Pothoulakis C. 2011. Host S-nitrosylation inhibits clostridial small molecule-activated glucosylating toxins. Nat Med 17:1136–1141. doi: 10.1038/nm.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Egerer M, Giesemann T, Herrmann C, Aktories K. 2009. Autocatalytic processing of Clostridium difficile toxin B. Binding of inositol hexakisphosphate. J Biol Chem 284:3389–3395. doi: 10.1074/jbc.M806002200. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Shi L, Yang Z, Feng H. 2013. Cytotoxicity of Clostridium difficile toxin B does not require cysteine protease-mediated autocleavage and release of the glucosyltransferase domain into the host cell cytosol. Pathog Dis 67:11–18. doi: 10.1111/2049-632X.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kreimeyer I, Euler F, Marckscheffel A, Tatge H, Pich A, Olling A, Schwarz J, Just I, Gerhard R. 2011. Autoproteolytic cleavage mediates cytotoxicity of Clostridium difficile toxin A. Naunyn Schmiedebergs Arch Pharmacol 383:253–262. doi: 10.1007/s00210-010-0574-x. [DOI] [PubMed] [Google Scholar]
- 32.Kim H, Rhee SH, Pothoulakis C, LaMont JT. 2009. Clostridium difficile toxin A binds colonocyte Src causing dephosphorylation of focal adhesion kinase and paxillin. Exp Cell Res 315:3336–3344. doi: 10.1016/j.yexcr.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim H, Rhee SH, Kokkotou E, Na X, Savidge T, Moyer MP, Pothoulakis C, LaMont JT. 2005. Clostridium difficile toxin A regulates inducible cyclooxygenase-2 and prostaglandin E2 synthesis in colonocytes via reactive oxygen species and activation of p38 MAPK. J Biol Chem 280:21237–21245. doi: 10.1074/jbc.M413842200. [DOI] [PubMed] [Google Scholar]
- 34.Chumbler NM, Farrow MA, Lapierre LA, Franklin JL, Haslam DB, Goldenring JR, Lacy DB. 2012. Clostridium difficile toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog 8:e1003072. doi: 10.1371/journal.ppat.1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farrow MA, Chumbler NM, Lapierre LA, Franklin JL, Rutherford SA, Goldenring JR, Lacy DB. 2013. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc Natl Acad Sci U S A 110:18674–18679. doi: 10.1073/pnas.1313658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donald RG, Flint M, Kalyan N, Johnson E, Witko SE, Kotash C, Zhao P, Megati S, Yurgelonis I, Lee PK, Matsuka YV, Severina E, Deatly A, Sidhu M, Jansen KU, Minton NP, Anderson AS. 2013. A novel approach to generate a recombinant toxoid vaccine against Clostridium difficile. Microbiology 159:1254–1266. doi: 10.1099/mic.0.066712-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wohlan K, Goy S, Olling A, Srivaratharajan S, Tatge H, Genth H, Gerhard R. 2014. Pyknotic cell death induced by Clostridium difficile TcdB: Chromatin condensation and nuclear blister are induced independently of the glucosyltransferase activity. Cell Microbiol 16:1678–1692. doi: 10.1111/cmi.12317. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Z, Park M, Tam J, Auger A, Beilhartz GL, Lacy DB, Melnyk RA. 2014. Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. Proc Natl Acad Sci U S A 111:3721–3726. doi: 10.1073/pnas.1400680111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Carrion-Vazquez M, Oberhauser AF, Marszalek PE, Fernandez JM. 2000. Point mutations alter the mechanical stability of immunoglobulin modules. Nat Struct Biol 7:1117–1120. doi: 10.1038/81964. [DOI] [PubMed] [Google Scholar]
- 40.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R. 1993. Naturally occurring antibodies devoid of light chains. Nature 363:446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 41.Arbabi Ghahroudi M, Desmyter A, Wyns L, Hamers R, Muyldermans S. 1997. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett 414:521–526. doi: 10.1016/S0014-5793(97)01062-4. [DOI] [PubMed] [Google Scholar]
- 42.Hussack G, Arbabi-Ghahroudi M, van Faassen H, Songer JG, Ng KK, MacKenzie R, Tanha J. 2011. Neutralization of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding domain. J Biol Chem 286:8961–8976. doi: 10.1074/jbc.M110.198754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dumoulin M, Conrath K, Van Meirhaeghe A, Meersman F, Heremans K, Frenken LG, Muyldermans S, Wyns L, Matagne A. 2002. Single-domain antibody fragments with high conformational stability. Protein Sci 11:500–515. doi: 10.1110/ps.34602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arbabi-Ghahroudi M, Tanha J, MacKenzie R. 2009. Isolation of monoclonal antibody fragments from phage display libraries. Methods Mol Biol 502:341–364. doi: 10.1007/978-1-60327-565-1_20. [DOI] [PubMed] [Google Scholar]
- 45.Koide A, Tereshko V, Uysal S, Margalef K, Kossiakoff AA, Koide S. 2007. Exploring the capacity of minimalist protein interfaces: interface energetics and affinity maturation to picomolar KD of a single-domain antibody with a flat paratope. J Mol Biol 373:941–953. doi: 10.1016/j.jmb.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Genst E, Silence K, Decanniere K, Conrath K, Loris R, Kinne J, Muyldermans S, Wyns L. 2006. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc Natl Acad Sci U S A 103:4586–4591. doi: 10.1073/pnas.0505379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koch-Nolte F, Reyelt J, Schossow B, Schwarz N, Scheuplein F, Rotherburg S, Haag F, Alzogaray V, Cauerhff A, Goldbaum FA. 2007. Single domain antibodies from llama effectively and specifically block T cell ecto-ADP-ribosyltransferase ART2.2 in vivo. FASEB J 21:3490–3498. doi: 10.1096/fj.07-8661com. [DOI] [PubMed] [Google Scholar]
- 48.Adams H, Brummelhuis W, Maassen B, van Egmond N, El Khattabi M, Detmers F, Hermans P, Braam B, Stam J, Verrips T. 2009. Specific immuno capturing of the staphylococcal superantigen toxic-shock syndrome toxin-1 in plasma. Biotechnol Bioeng 104:143–151. doi: 10.1002/bit.22365. [DOI] [PubMed] [Google Scholar]
- 49.Tremblay JM, Mukherjee J, Leysath CE, Debatis M, Ofori K, Baldwin K, Boucher C, Peters R, Beamer G, Sheoran A, Bedenice D, Tzipori S, Shoemaker CB. 2013. A single VHH-based toxin neutralizing agent and an effector antibody protects mice against challenge with Shiga toxins 1 and 2. Infect Immun 81:4592–4603. doi: 10.1128/IAI.01033-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hussack G, Arbabi-Ghahroudi M, Mackenzie CR, Tanha J. 2012. Isolation and characterization of Clostridium difficile toxin-specific single-domain antibodies. Methods Mol Biol 911:211–239. doi: 10.1007/978-1-61779-968-6_14. [DOI] [PubMed] [Google Scholar]
- 51.Yang Z, Schmidt D, Liu W, Li S, Shi L, Sheng J, Chen K, Yu H, Tremblay JM, Chen X, Piepenbrink KH, Sundberg EJ, Kelly CP, Bai G, Shoemaker CB, Feng H. 2014. A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice. J Infect Dis 210:964–972. doi: 10.1093/infdis/jiu196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang G, Zhou B, Wang J, He X, Sun X, Nie W, Tzipori S, Feng H. 2008. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol 8:192. doi: 10.1186/1471-2180-8-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang H, Sun X, Zhang Y, Li S, Chen K, Shi L, Nie W, Kumar R, Tzipori S, Wang J, Savidge T, Feng H. 2012. A chimeric toxin vaccine protects against primary and recurrent Clostridium difficile infection. Infect Immun 80:2678–2688. doi: 10.1128/IAI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Shi L, Li S, Yang Z, Standley C, ZhuGe R, Savidge T, Wang X, Feng H. 2013. A segment of 97 amino acids within the translocation domain of Clostridium difficile toxin B is essential for toxicity. PLoS One 8:e58634. doi: 10.1371/journal.pone.0058634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steele J, Chen K, Sun X, Zhang Y, Wang H, Tzipori S, Feng H. 2012. Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J Infect Dis 205:384–391. doi: 10.1093/infdis/jir748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rupnik M, Pabst S, Rupnik M, von Eichel-Streiber C, Urlaub H, Soling HD. 2005. Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiology 151:199–208. doi: 10.1099/mic.0.27474-0. [DOI] [PubMed] [Google Scholar]
- 57.Shen A, Lupardus PJ, Gersch MM, Puri AW, Albrow VE, Garcia KC, Bogyo M. 2011. Defining an allosteric circuit in the cysteine protease domain of Clostridium difficile toxins. Nat Struct Mol Biol 18:364–371. doi: 10.1038/nsmb.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lanis JM, Heinlen LD, James JA, Ballard JD. 2013. Clostridium difficile 027/BI/NAP1 encodes a hypertoxic and antigenically variable form of TcdB. PLoS Pathog 9:e1003523. doi: 10.1371/journal.ppat.1003523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Just I, Selzer J, von Eichel-Streiber C, Aktories K. 1995. The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J Clin Invest 95:1026–1031. doi: 10.1172/JCI117747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alzogaray V, Danquah W, Aquirre A, Urrutia M, Berquer P, Garcia Vescovi E, Haag F, Koch-Nolte F, Goldbaum FA. 2011. Single-domain llama antibodies as specific intracellular inhibitors of SpvB, the actin ADP-ribosylating toxin of Salmonella typhimurium. FASEB J 25:526–534. doi: 10.1096/fj.10-162958. [DOI] [PubMed] [Google Scholar]
- 61.He X, Wang J, Steele J, Sun X, Nie W, Tzipori S, Feng H. 2009. An ultrasensitive rapid immunocytotoxicity assay for detecting Clostridium difficile toxins. J Microbiol Methods 78:97–100. doi: 10.1016/j.mimet.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qualman SJ, Petric M, Karmali MA, Smith CR, Hamilton SR. 1990. Clostridium difficile invasion and toxin circulation in fatal pediatric pseudomembranous colitis. Am J Clin Pathol 94:410–416. [DOI] [PubMed] [Google Scholar]