

Abstract
As an obligate intracellular pathogen, the apicomplexan parasite Toxoplasma gondii evades immune system-mediated clearance by undergoing stage differentiation to persist indefinitely in susceptible hosts. Previously, we found that mice deficient in the ectoenzyme CD73, which generates adenosine in the extracellular matrix, were resistant to chronic toxoplasmosis after oral infection with T. gondii. Resistance in CD73 knockout mice was due to a delay in parasite differentiation in the central nervous system (CNS). To further clarify the role of CD73 and extracellular adenosine in T. gondii pathogenesis, we infected wild-type (WT) and CD73−/− mice with T. gondii cysts systemically by the intraperitoneal (i.p.) route. In contrast to oral infection, i.p. infected CD73−/− mice were highly susceptible to immune-mediated pathology, with significantly increased infiltration of neutrophils and T cells into the peritoneal cavity. Administration of the broad-spectrum adenosine receptor agonist 5′-N-ethylcarboxamidoadenosine (NECA) protected CD73−/− mice against T. gondii-induced immunopathology, suggesting that the absence of CD73-generated adenosine led to the increased susceptibility in these mice. Peritoneal exudate cells from infected CD73−/− mice produced higher levels of the inflammatory mediators nitric oxide, tumor necrosis factor alpha (TNF-α), and interleukin-1β (IL-1β), without enhanced parasite killing or clearance. Bone marrow chimeras established that CD73 expression in both hematopoietic and nonhematopoietic compartments contributes to limiting T. gondii-induced immunopathology. In addition, mice deficient in the adenosine receptor A2A were more susceptible to immunopathology during intraperitoneal infection with T. gondii than WT mice. Thus, extracellular adenosine is a key immune regulator that limits collateral tissue damage due to an intracellular pathogen and promotes host survival.
INTRODUCTION
Toxoplasma gondii is an obligate intracellular protozoan pathogen with broad host range and tissue tropism. During infection, T. gondii tachyzoites invade host cells and replicate rapidly before inducing a potent immune response that controls parasite numbers through direct killing. The immune response is dominated by interleukin-12 (IL-12) priming and subsequent interferon gamma (IFN-γ) release by both innate and adaptive immune cells. Although a robust immune response is critical for controlling parasite-induced pathology, the immune response itself can cause severe pathology, and this balance may underscore differences in genetic susceptibility to toxoplasmosis among different hosts.
Infection by pathogens such as T. gondii cause local as well as systemic inflammation. While inflammation is necessary to clear infection, it is often costly to the host. If not properly regulated, inflammation can result in extensive collateral tissue damage. Purinergic signaling is a well-conserved pathway by which the local immune response is orchestrated through the autocrine and paracrine generation of ATP and its metabolites (1). During an inflammatory response, ATP from damaged cells, platelets, and cells undergoing oxidative stress is released into the extracellular space. This extracellular ATP amplifies the inflammatory immune response through the recruitment and activation of immune cells such as T cells, neutrophils, and macrophages. ATP is dephosphorylated to ADP and AMP by the ectoapyrase CD39 (2). AMP is further converted to extracellular adenosine by the ecto-5′-nucleotidase CD73. Extracellular adenosine regulates the immune response to prevent excessive cellular damage (3, 4). Cells in the local environment that express any of the four G-protein-coupled adenosine receptors (A1, A2A, A2B, and A3) can respond to local tissue damage (5). The type of response following adenosine receptor (AR) activation is likely to depend on the responding cell and the adenosine receptor(s) that it expresses. For instance, adenosine signaling through the A1 receptor promotes neutrophil chemotaxis to sites of infection and their adhesion to inflamed endothelium, while signaling through the A2A receptor inhibits adhesion (6, 7). Adenosine inhibits the production of proinflammatory factors such as tumor necrosis factor alpha (TNF-α), IL-1β, and reactive oxygen species (ROS) by monocytes and dendritic cells and is important for T regulatory cell suppressor function and the generation of anti-inflammatory cytokines (8–10). Therefore, adenosine regulates inflammation and at the same time acts as a cell damage signal to promote cell migration to sites of tissue injury.
Extracellular adenosine in association with the regulation of local inflammation is well documented. Indeed, mice deficient in CD73 exhibit several defects in immunoregulation following diverse inflammatory challenges. CD73−/− mice are more prone to sepsis (3), dextran sulfate sodium (DSS)-induced inflammatory bowel disease (4), lung injury (11), and tissue ischemia, hypoxia, and inflammation (9, 12, 13). On the other hand, deletion or inhibition of CD73 may have therapeutic potential in cancer (14–16), multiple sclerosis (17), and chronic Toxoplasma gondii infection (18). Thus, defining the role of this pleiotropic enzyme in inflammation and immunity may offer promising therapeutic targets.
Interestingly, purines, including adenosine, play an important role in T. gondii biology. Like other apicomplexa, T. gondii needs host purines, such as adenosine, to effectively complete its life cycle, as it cannot synthesize its own. We have previously documented the role of adenosine in the chronic stage of T. gondii infection by the peroral route (18). We demonstrated that mice deficient in CD73 were resistant to toxoplasmosis. This resistance was due to a defect in parasite differentiation in central nervous system (CNS) tissue. In this study, we identify for the first time the critical role of CD73-generated adenosine in regulating the immune response to T. gondii infection in an acute systemic model of toxoplasmosis.
MATERIALS AND METHODS
Mice.
C57BL/6 mice (wild type [WT]) were purchased from Jackson Laboratories. CD73−/− mice were obtained from Linda Thompson at the Oklahoma Medical Research Foundation (OMRF). A1AR−/− and A2AAR−/− mice were obtained from Jurgen Schnermann (NIH NIDDK, Bethesda, MD) and Jiang-Fan Chen (Boston University School of Medicine, Boston, MA), respectively. Mice were bred and housed under specific-pathogen-free conditions in the animal facility at Cornell University. All procedures performed on mice were approved by the Cornell University animal review committee.
Toxoplasma gondii infection.
Wild-type, CD73−/−, A1AR−/−, and A2AAR−/− mice were infected with 10, 20, or 40 T. gondii ME49 cysts or 1,000 strain RH tachyzoites by intraperitoneal (i.p.) injection in 0.2 ml phosphate-buffered saline (PBS). Survival rates and weight loss were monitored daily. In some experiments, groups of mice were euthanized at 3, 6, 8, and 9 days postinfection, and tissues (spleen, mesenteric lymph nodes [MLNs], liver, pancreas, lungs, and brains), peritoneal exudate cells (PECs), and/or blood was collected for further processing.
Histology.
Terminally anesthetized mice were perfused with ice-cold PBS, and tissues were collected for histology. Abdominal fat pads were fixed in 10% formalin and processed for histology.
Treatment with adenosine receptor agonists and antagonists.
Mice infected with 20 ME49 cysts as described above were treated with the adenosine analogue 5′-N-ethylcarboxamidoadenosine (NECA; Tocris Biosciences, Minneapolis, MN, USA). NECA was solubilized in dimethyl sulfoxide (DMSO), resuspended in PBS, and then injected into CD73−/− mice i.p. daily (starting 3 days before infection) at a 20 μM concentration in 0.1 ml PBS. The same final concentration of DMSO was used as a vehicle control.
Isolation of peritoneal exudate cells.
Naive and infected mice were euthanized by inhalation of isoflurane prior to cell collection. Eight milliliters of ice-cold PBS was injected into the peritoneal cavity of each mouse, followed by 2 min of palpitation to dislodge adherent peritoneal cells. The cells were then collected by aspiration with a needle and syringe. Peritoneal cells were centrifuged at 400 × g for 5 min and resuspended in RPMI medium (Cellgro) containing 10% (vol/vol) fetal calf serum, 1,000 U/ml penicillin, 1,000 μg/ml streptomycin, 25 mM HEPES, 1 mM l-glutamine, and 50 μM beta-mercaptoethanol.
Isolation of spleen and mesenteric lymph node cells.
Mesenteric lymph nodes and spleens were excised from euthanized mice and processed aseptically to generate single-cell suspensions. Spleens were further treated to remove red blood cells by using ACK lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 100 μM EDTA). Cells were resuspended in complete RPMI medium.
Ex vivo flow cytometry.
Single-cell suspensions were incubated with antibody cocktails in fluorescence-activated cell sorter (FACS) buffer (0.5% bovine serum albumin [BSA], 0.009% NaN3 in PBS) for 15 min at 4°C to 8°C. Prior to cell staining, antibody binding to the Fc receptor was blocked with 5 μg/ml rat anti-mouse CD16/CD32 antibody (FcBlock; BD Biosciences). Cells were washed and resuspended in FACS buffer, and samples were then acquired on a FACSCanto II instrument by using FACSDiva software. Analysis of flow cytometry data was performed with FlowJo software.
Cytospins.
Peritoneal exudate cells were labeled with CD45-allophycocyanin (APC) and 1A8-fluorescein isothiocyanate (FITC) and then adhered to slides by using a Shandon Cytospin 4 cytocentrifuge. Slides were fixed with 2% paraformaldehyde and then mounted with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI). Slides were imaged with a Zeiss Axio Imager M1 fluorescence microscope system.
Cell culture.
Spleen, mesenteric lymph node, and peritoneal exudate cells were resuspended to 5 ×106 cells per ml medium and cultured at 37°C in a 5% CO2 humidified incubator for 48 h with 20 μg/ml of soluble tachyzoite antigen (STAg). Cell-free supernatants were collected and stored at −20°C until analysis.
Cytokine ELISAs.
Cell culture supernatants and sera from naive and infected mice were analyzed for IL-1β, TNF-α, IL-12, and IFN-γ by using Ready-SET-GO enzyme-linked immunosorbent assay (ELISA) kits from eBioscience, according to the manufacturer's instructions.
Measurement of nitrite production.
Nitric oxide levels in serum and cell culture supernatants were quantified by measuring the presence of nitrite using Griess reagent (19). Briefly, an equal volume of cell culture supernatant was incubated with Griess reagent (0.1% N-1-napthylethylenediamine dihydrochloride premixed with 1% sulfanilamide in 5% phosphoric acid) and read at 550 nm in a BioTek plate reader. A sodium nitrite standard curve was used to determine nitrite levels.
Bone marrow chimeras.
WT and CD73-deficient mice were lethally irradiated to ablate bone marrow (BM) cells (450 rads twice, 4 h apart). Mice were then reconstituted with bone marrow cells from congenic WT or CD73−/− donor mice. Each mouse received 106 BM cells intravenously (i.v.) and were then placed on treatment with acidified water containing sulfamethoxazole-trimethoprim (Bactrim) for 10 days to reduce the risk of opportunistic infection. After 8 to 10 weeks, mice were infected with 20 T. gondii ME49 cysts i.p. and monitored for survival and weight loss.
Adoptive transfer.
RAG1−/− mice were reconstituted with 107 total spleen leukocytes per mouse from WT or CD73−/− hosts by retro-orbital injection. One week later, mice were infected with 20 ME49 cysts of T. gondii i.p. and monitored for morbidity and mortality.
Statistical analysis.
Data were analyzed for statistical significance by using GraphPad Prism 5 software. Kaplan-Meier survival curves were generated to compare mortality between groups of mice by using log-rank tests. Weight loss, parasite burden, and cell composition data were analyzed for statistical significance by using one-way repeated-measures analysis of variance (ANOVA) with Bonferroni posttests or Student's t tests where applicable. Statistical significance was defined as a P value of ≤0.05 (asterisks in the figures indicate increased significance).
RESULTS
Intraperitoneal infection with Toxoplasma gondii is lethal in CD73−/− hosts.
We set out to investigate the role of CD73-generated adenosine, a potent immunomodulator, in T. gondii infection and immunity. Mice infected with T. gondii via the intraperitoneal route develop systemic inflammation, characterized by the recruitment and activation of leukocytes in the peritoneal cavity, increased numbers of Th1 cytokines in serum, and the release of acute-phase mediators such as nitric oxide by responding cells (20, 21). In the absence of CD73, we found a significant and lethal dysregulation of the immune response to T. gondii, with increased morbidity and mortality in CD73−/− hosts infected with both the avirulent type II ME49 strain (Fig. 1A and B) and the highly virulent type I RH strain (Fig. 1C and D). To determine whether the increased morbidity and mortality in CD73−/− mice were not restricted to the ME49 strain, we infected mice with the more virulent RH strain, in parallel with the avirulent ME49 strain. Although the kinetics of infection are very different between the two strains, the overall patterns of increased susceptibility, morbidity, and mortality (weight loss) in CD73−/− mice were quite similar (Fig. 1A to D). Indeed, gross pathology of the peritoneal cavity of ME49 strain-infected mice revealed extensive fatty tissue necrosis in CD73−/− mice (Fig. 1E). Histological sections of abdominal fat pads showed acute inflammation and infiltration of cells in infected CD73−/− mice only (Fig. 1F). This finding suggests that T. gondii infection in the absence of CD73-generated adenosine induced the infiltration of immune cells to inflammatory sites (22).
FIG 1.

CD73−/− mice are more susceptible to acute toxoplasmosis than wild-type mice. CD73 knockout (CD73−/−) mice and wild-type littermates were infected with T. gondii cysts by i.p. injection. (A and B) Survival curve (A) and weight loss (B) following infection with the ME49 strain. (C and D) Survival curve (C) and weight loss (D) following infection with the RH strain. (E) Gross pathology. (F) Hematoxylin-and-eosin-stained sections of abdominal fat pads of ME49 strain-infected mice 8 days after infection (n = 4 to 5 mice per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data from one of three independent experiments are shown.
Susceptibility to T. gondii is associated with increased infiltration of neutrophils and T lymphocytes.
To investigate the cause of the increased morbidity and mortality observed in the absence of CD73, we characterized the immune response to T. gondii i.p. infection in WT and CD73−/− hosts. About one-third (33%) of naive CD45+ resident peritoneal cells expressed CD73, as determined by flow cytometry (Fig. 2A). The major cell population expressing CD73 was B cells, at 21%, which is approximately one-quarter of the total CD19+ B cell population, at 81.9%, as determined by flow cytometry (Fig. 2A). Both CD4+ and CD8+ T cells were mainly CD73+, although overall there were fewer T cells than other cell types in the peritoneal resident cell compartment. T. gondii infection led to the infiltration of CD45+ cells in the peritoneal cavity, with a steady increase in the absolute number of cells over the course of acute infection (days 0 to 8) in both mouse strains (Fig. 2B). However, the composition of the peritoneal exudate cells changed dramatically during this 8-day course of infection. In uninfected mice, resident peritoneal exudate cells were ∼60% B cells, 20 to 30% macrophages, and 10 to 20% T cells (Fig. 2C). During the course of infection in both mouse strains, the proportion of B cells decreased, and neutrophil, T cell, and NK cell numbers increased dramatically both proportionally and in absolute numbers (Fig. 2B and C). In contrast to WT mice, the cell composition in the peritoneal cavity of infected CD73−/− mice remained dominated by T cells and kidney-shaped neutrophil cells even 8 days after infection (Fig. 2C and D). The slow resolution of inflammation in CD73−/− mice is consistent with the tissue pathology associated with i.p. infection in these mice.
FIG 2.

Cell composition comparison of peritoneal exudate cells from infected wild-type and CD73−/− mice. Cells were collected by peritoneal lavage from wild-type and CD73−/− mice infected i.p. with 20 T. gondii ME49 cysts and stained for immune cell markers for flow cytometric and cytospin analyses. (A) Flow cytometric analysis of peritoneal exudate cells from a naive WT mouse. (B) Absolute PEC counts. (C) Cell composition on days 2, 4, and 8 quantified by flow cytometry, gated on CD45-positive leukocytes, and identified with anti-murine antibodies recognizing CD19 (B cells), CD3 (T cells), NK1.1 (NK cells), or Ly6-G (1A8) (neutrophils). (D) Cytospin analysis of cells stained for CD45 and 1A8 (neutrophils). Arrows indicate kidney-shaped neutrophils. ***, P < 0.001 (n = 3 to 4 per group per time point; data shown are from one of three independent experiments).
Increased levels of inflammatory mediators in the absence of extracellular adenosine.
To determine if the increased cellular infiltration and immunopathology observed for T. gondii-infected CD73−/− mice was due to increases in the levels of inflammatory mediators, we analyzed sera and cell culture supernatants of WT and CD73−/− mice infected with T. gondii i.p. Although not statistically significant, we found that CD73−/− mice showed a consistent pattern of elevated levels of inflammatory mediators such as IL-1β (spleen, PECs, and MLNs) and TNF-α (spleen) compared to WT mice (Fig. 3A and B). In addition, peritoneal exudate cells isolated from CD73−/− mice produced significantly increased levels of nitric oxide (Fig. 3C). CD73−/− mice also produced elevated IFN-γ levels in serum (Fig. 3D), consistent with the increased T cell numbers seen in the peritoneal cavity (Fig. 2B).
FIG 3.

Dysregulated cytokine responses in CD73−/− mice. (A and B) Spleen, peritoneal exudate, or mesenteric lymph node cells isolated from WT and CD73−/− mice that were infected with 20 ME49 cysts for 8 days were cultured in vitro with soluble T. gondii antigens (20 μg/ml) for 48 h and then assayed for IL-1β (A) and TNF-α (B) production by ELISA. Serum was collected and analyzed. (C) Peritoneal exudate cell nitric oxide production measured by using Griess reagent. (D) Serum interferon gamma. **, P < 0.01; ***, P < 0.001 (n = 3 to 4 mice per group; data shown are from one of three experiments).
Parasite control in CD73-deficient mice is limited.
We next investigated whether the heightened immune response in CD73-deficient mice promoted better parasite control. We collected peritoneal exudate cells, splenic leukocytes, and mesenteric lymph node cells from mice at 2, 4, and 8 days postinfection and assessed parasite burden by flow cytometry. In the peritoneal exudate cells, both CD11b+ and CD11b− cells were found to harbor T. gondii tachyzoites during the acute stage of infection in both WT and CD73−/− mice (Fig. 4). Interestingly, more cells from the mesenteric lymph nodes of CD73-deficient mice were infected with T. gondii than in their WT counterparts, suggesting that the parasite was able to disseminate more systemically in these animals. Nevertheless, during chronic infection, we found that surviving CD73−/− mice recovered, and although the values were not statistically significant, these mice did show a reduced parasite burden in their brains (see Fig. S1 in the supplemental material). This finding is consistent with our previously reported finding that CD73 promotes T. gondii persistence in the CNS during chronic infection (18).
FIG 4.

Parasite dissemination during acute infection. Mice were infected with 20 cysts of ME49 and euthanized 2, 4, or 8 days later. Peritoneal exudate cells, mesenteric lymph node cells, and spleen leukocytes were collected and stained for CD11b and T. gondii p30 to quantify parasitized cells by flow cytometry (n = 3 per group per time point).
Treatment of CD73−/− mice with the adenosine receptor agonist NECA inhibits susceptibility to T. gondii infection.
Since CD73 has both enzymatic and signaling properties, we next determined which functional absence led to the susceptibility of CD73−/− mice to T. gondii infection. We infected WT and CD73−/− mice with T. gondii ME49 cysts i.p., simultaneously treated a group of CD73−/− mice with the nonhydrolyzable adenosine receptor analogue 5′-N-ethylcarboxamidoadenosine (NECA), and then monitored mice for weight loss and morbidity. As we observed previously, T. gondii-infected CD73−/− mice lost more weight than and exhibited increased morbidity compared to WT mice (Fig. 5A). We found that NECA, which has a high affinity for adenosine receptors and mediates adenosine receptor signaling but does not participate in the purine salvage pathway, protected CD73−/− mice from T. gondii-induced pathology compared to CD73−/− mice treated with the vehicle only (Fig. 5A). This finding suggested that the susceptibility of CD73−/− hosts to T. gondii infection via the intraperitoneal route was due mainly to an absence of extracellular adenosine. We next determined whether the decreased morbidity observed for NECA-treated CD73−/− mice was due to a decreased parasite burden. Interestingly, the elevated parasite burden observed for CD73−/− mice was not ablated by NECA treatment, even though NECA treatment protected these mice from T. gondii-induced morbidity (Fig. 5B). This finding reinforces our hypothesis that the absence of CD73-generated adenosine resulted in immunopathology in mice as a response to T. gondii infection, leading to increased collateral tissue damage and high morbidity and mortality rates.
FIG 5.

CD73-generated adenosine protects mice against inflammation via adenosine receptor signaling. (A) Weight loss following infection in WT and CD73−/− mice treated with the vehicle control (DMSO only) compared to that in CD73−/− mice treated with the adenosine analogue NECA. Data from one of two independent experiments are shown. (B) Parasite burden determined by quantitative real-time PCR of the T. gondii B1 gene at 8 days postinfection. Results are the averages of data from two experiments (n = 3 to 5 mice per group). gDNA, genomic DNA. **, P < 0.01; ***, P < 0.001; n.s., not significant.
A2A adenosine receptor knockout mice recapitulate the susceptibility to i.p. infection observed for CD73-deficient hosts.
Although all four adenosine receptors participate in modulating the immune response, the A2A receptor in particular has been shown to regulate various aspects of inflammation, from recruitment of leukocytes to effector functions of the innate and adaptive immune responses (23–25). In a recent study, ablation of the A2A receptor on immune cells rendered mice susceptible to a more severe form of experimental autoimmune encephalomyelitis (26). To determine which adenosine receptor played a dominant role in protection against T. gondii-induced immunopathology, we infected A1AR and A2AAR knockout mice and compared their susceptibilities to those of WT and CD73−/− mice. We found that while A1AR−/− mice had an outcome of disease similar to that of WT mice, A2AAR-deficient mice mirrored the susceptibility observed for CD73−/− mice (Fig. 6). From this, we concluded that the ability of CD73−/− mice to more readily succumb to T. gondii immunopathology was due to the absence of A2A adenosine receptor signaling.
FIG 6.
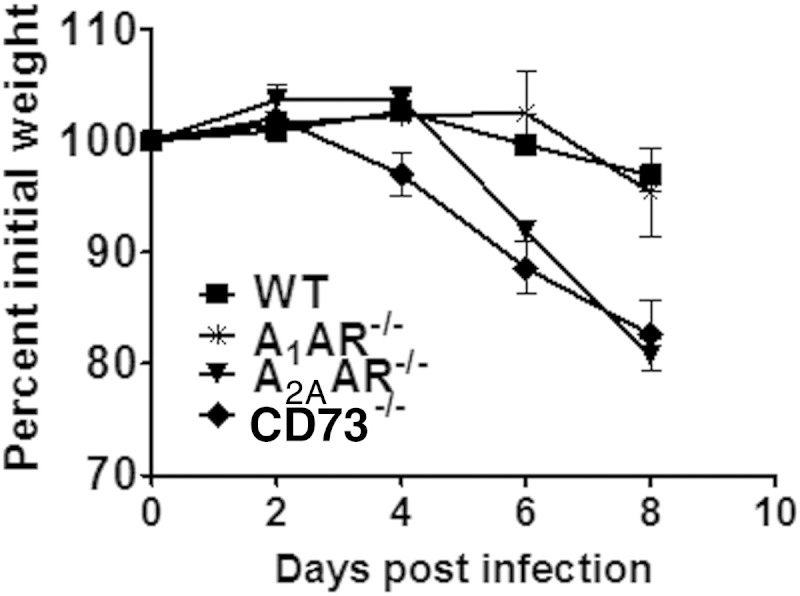
The A2A adenosine receptor mediates the protective effects of extracellular adenosine. Wild-type, CD73−/−, A1 adenosine receptor knockout (A1AR−/−), or A2A adenosine receptor knockout (A2AAR−/−) mice were infected with 20 ME49 cysts of T. gondii and monitored for weight loss. Data from one of two independent experiments are shown.
Hematopoietic and nonhematopoietic cells contribute to CD73-generated adenosine to limit inflammation.
Lymphocyte subsets and cells of the myeloid lineage are major sources of CD73-generated adenosine (25). Endothelial cells, epithelial cells, and fibroblasts are all known to express CD73, and this expression has been shown to promote lymphocyte extravasation and accumulation at sites of inflammation (17, 27). To determine the contributions of these two compartments to the critical release of CD73-derived extracellular adenosine, we generated chimeric mice, in which WT and CD73−/− mice were depleted of bone marrow cells through sublethal irradiation, and then rescued them with the adoptive transfer of bone marrow cells from WT or CD73−/− donors. After infecting these mice with T. gondii ME49, we monitored survival and weight loss. We found that all CD73−/− mice died by day 10 after infection, whether reconstituted with CD73+ or CD73− bone marrow cells (Fig. 7A), which correlated with a loss of body weight (Fig. 7B). This finding suggests that CD73 expression in the nonhematopoietic lineage is critical for preventing immunopathology induced by T. gondii systemic infection. Interestingly, WT mice reconstituted with CD73-deficient cells were partially susceptible, losing more weight than and exhibiting increased morbidity and mortality compared to WT mice reconstituted with CD73+ bone marrow cells (Fig. 7A). These mice also succumbed later than CD73−/− mice, and 1 out of 5 mice survived the acute stage of disease. Although the loss in body weight was delayed, it was substantial compared to that of WT mice (Fig. 7B). This finding suggests that early during infection, adenosine generated by nonhematopoietic cells is indispensable for controlling disease, while adenosine generated by immune cells may be critical later during the acute stage of disease. However, infection of RAG1−/− mice, which lack T and B lymphocytes, reconstituted with either WT or CD73−/− splenocytes before infection with T. gondii established that CD73 expression was not critical for T or B cells to mediate protection (see Fig. S2 in the supplemental material). Further research is ongoing to determine the immune cell compartment responsible for CD73-generated adenosine in T. gondii infection.
FIG 7.
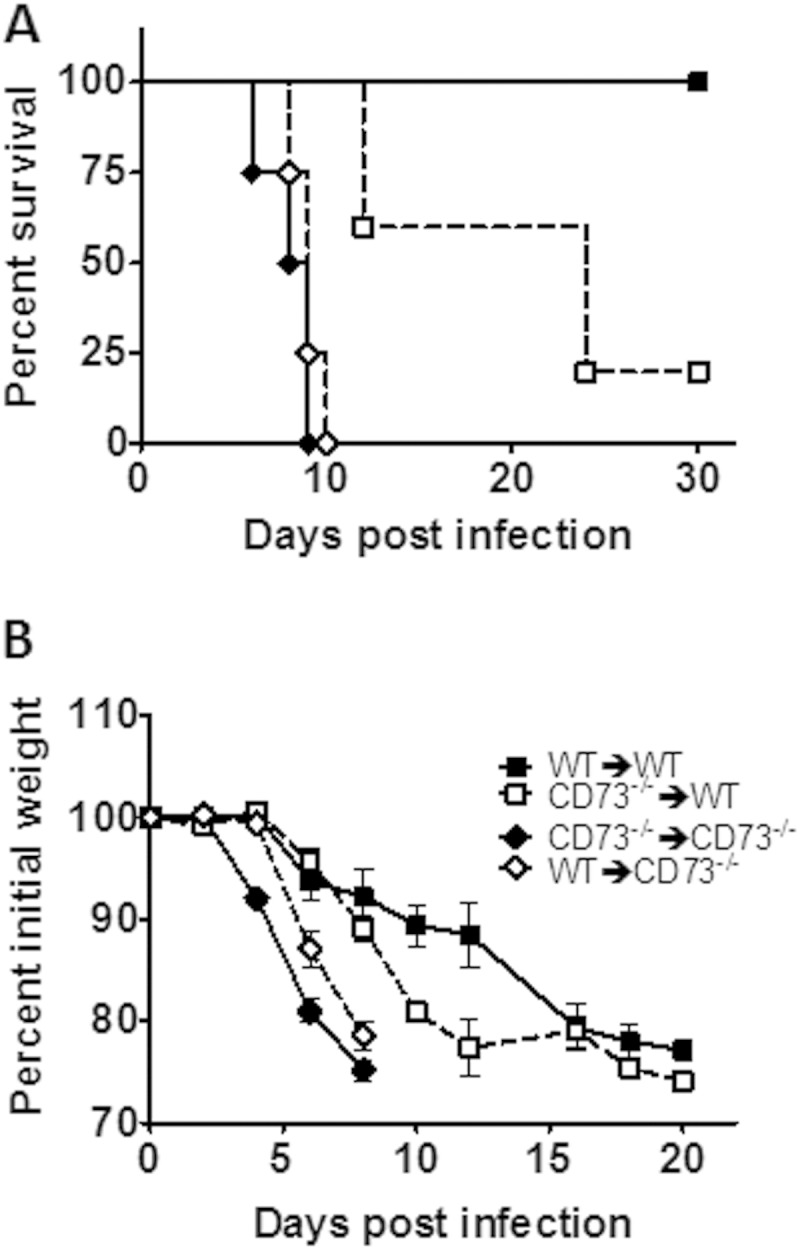
Hematopoietic and nonhematopoietic cells contribute CD73-generated adenosine to limit inflammation. Bone marrow chimeric mice were generated as described in Materials and Methods, infected with T. gondii ME49 cysts i.p., and then monitored for survival (A) and weight loss (B). Data from one of two independent experiments are shown.
DISCUSSION
Infection with T. gondii via the intraperitoneal route induces rapid immune cell infiltration and a robust immune response, resulting in extensive tissue damage in mice. The genetics of both the host and the parasite influence the outcome of infection. The immune response to T. gondii is dominated by a proinflammatory, Th1-skewed response that is critical for both resistance to and clearance of the parasite but is also detrimental to the host if not tightly regulated. In this study, we found that the ectoenzyme CD73 was critical for limiting immunopathology and for survival of mice infected with T. gondii via the intraperitoneal route. The protective effects of CD73 were mediated by the generation of extracellular adenosine, which prevented a proinflammatory cascade. Cells from CD73−/− mice also secreted higher levels of proinflammatory mediators such as IL-1β, TNF-α, and nitric oxide, all of which are known to mediate bystander immunopathology. Thus, the cytokine dysregulation that we observed in the absence of CD73 may promote collateral tissue damage during the acute response to infection.
We found that the A2A adenosine receptor was the major receptor implicated in protection against hyperinflammatory cellular infiltration in CD73-deficient mice. In the absence of CD73, mice may exhibit a proinflammatory phenotype as a consequence of a lack of extracellular adenosine either directly or indirectly through the buildup of ATP, which is known to act as a danger signal and promotes inflammation. A deficit in CD73 enzymatic activity could lead to not only adenosine depletion but also upstream ATP accumulation. ATP is known to activate the NLRP3 inflammasome via P2X receptor signaling, leading to the release of active IL-1β via caspase-1 processing of pro-IL-1β (28). It is possible that in CD73-deficient mice, a buildup of ATP could lead to inflammasome activation either via P2X7 signaling or through a lack of antagonism by adenosine, which could explain the observed increase in the level of IL-1β. However, the results with A2A receptor knockout mice, which appear to largely recapitulate the susceptibility observed for CD73−/− mice, suggest a dominant role for adenosine receptor signaling, at least during the early stage of infection. Since A2A receptor knockout mice have a functional A2B receptor, it is possible that the delay in weight loss observed for these mice reveals a role for the A2B receptor early during infection.
Our data indicate that the A2A adenosine receptor is largely responsible for the limiting effect of adenosine in systemic inflammation induced by T. gondii i.p. infection. Mice deficient in the A2A receptor exhibited immunopathology similar to that of mice lacking CD73. We also determined that adenosine receptor signaling is critical for curtailing the acute stage of the immune response to T. gondii. For example, treatment with NECA, a nonhydrolyzable adenosine analogue that does not participate in the purine salvage pathway, protected CD73−/− mice from the almost lethal immunopathology. These findings are in contrast to our previous work showing that adenosine as a purine source, and not its signaling function, is required for T. gondii survival during chronic infection. This suggests that while there is a complex relationship between adenosine and T. gondii, adenosine can serve to protect the host during both the acute and chronic stages of infection. During the acute stage of infection, adenosine signaling is required to curtail inflammation and limit collateral tissue damage, while its depletion after acute infection can inhibit the parasite's ability to form long-lived cysts (18).
It is interesting that the route of infection with T. gondii has a striking effect on the outcome of infection in CD73−/− mice. These mice are resistant to the acute phase of oral infection yet are highly susceptible to intraperitoneal infection. Similar effects may be seen in comparisons of different mouse strains, suggesting that host genetic factors could impact the outcome of infection (29, 30). For example, BALB/c mice are resistant to both oral and intraperitoneal infection, while C57BL/6 mice are highly susceptible to oral infection but relatively resistant to intraperitoneal infection. Moreover, even within the same genetic background, the route of infection can give rise to different responses, as the local immune system may differ in cell composition, with a tendency to polarize the response toward a Th1, a Th2, or even a regulatory phenotype (29). It is likely that the regulation of the immune response in the peritoneal cavity requires the presence of adenosine to limit pathology, while other compartments (the gastrointestinal [GI] tract or the CNS during chronic infection) may have multiple or redundant mechanisms to control immunopathology. Notably, the blood-brain barrier prevents neutrophils from accumulating in the CNS, whereas the peritoneal cavity does not have this additional barrier to leukocyte hyperinfiltration.
Aside from reducing proinflammatory cascades, extracellular adenosine is also implicated in promoting anti-inflammatory factors such as IL-10 and the induction of suppressor T cells. In particular, A2B and A2A signaling induces IL-10 release from macrophages and microglia (8, 10, 31). It is also possible that other regulatory factors, such as IL-4 and transforming growth factor β (TGF-β), contribute to the CD73-mediated protective effects of extracellular adenosine in T. gondii i.p. infection. It would be interesting to determine if CD73-generated adenosine promotes the generation of anti-inflammatory mediators necessary for limiting pathology during i.p. infection with T. gondii.
In addition to inducible nitric oxide synthase (iNOS)-generated nitric oxide, interferon gamma induces other protective factors to limit T. gondii replication. In infected mouse cells, treatment with IFN-γ induces the accumulation of IRG (IFN-inducible GTPase) proteins around the parasitophorous vacuole, leading to membrane disruption and parasite killing (32, 33). In human cells, the enzyme indoleamine 2,3-dioxygenase (IDO) degrades l-tryptophan, which has been shown to suppress T. gondii proliferation in various cells, including macrophages, astrocytes, epithelial cells, and fibroblasts (34–36). IDO activity in the CNS is thought to be of greater importance than nitric oxide production by IFN-γ-induced iNOS in human host cells. Thus, it would be interesting to determine the role of adenosine receptor signaling in IDO-mediated control of T. gondii infection in human macrophages or fibroblasts. Yet another pathway that may intersect with the extracellular adenosine-T. gondii interaction is the production of reactive oxygen species (ROS). ROS production can be induced by extracellular ATP in T. gondii-infected macrophages (37), and since it is plausible that levels of ATP or its metabolites ADP and AMP are elevated in CD73−/− mice, it would be informative to test the contribution of this pathway to the control of T. gondii cyst formation in CD73-deficient hosts.
In conclusion, in this study, we identified a host genetic factor that may be implicated in controlling immune cell-mediated pathology associated with a protozoan infection. Based on the ubiquitous expression of CD73 in the brain and the important role of extracellular adenosine in modulating the immune response and maintaining functional barrier integrity in the GI tract, the blood-brain barrier, and other important intersections of the immune response (4, 38) with the external environment and the first lines of defense against dangerous pathogens, CD73 and extracellular adenosine present potential targets for therapeutic interventions in many ways.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants R01 NS053011 (to M.S.B.) and R01 NS05301-04S4 (to D.A.M.) from the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02536-14.
REFERENCES
- 1.Junger WG. 2011. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 11:201–212. doi: 10.1038/nri2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deaglio S, Robson SC. 2011. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv Pharmacol 61:301–332. doi: 10.1016/B978-0-12-385526-8.00010-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hasko G, Csoka B, Koscso B, Chandra R, Pacher P, Thompson LF, Deitch EA, Spolarics Z, Virag L, Gergely P, Rolandelli RH, Nemeth ZH. 2011. Ecto-5′-nucleotidase (CD73) decreases mortality and organ injury in sepsis. J Immunol 187:4256–4267. doi: 10.4049/jimmunol.1003379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bynoe MS, Waickman A, Mahamed DA, Mueller C, Mills JH, Czopik A. 2012. CD73 is critical for the resolution of murine colonic inflammation. J Biomed Biotechnol 2012:260983. doi: 10.1155/2012/260983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linden J. 2001. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol 41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 6.Barletta KE, Ley K, Mehrad B. 2012. Regulation of neutrophil function by adenosine. Arterioscler Thromb Vasc Biol 32:856–864. doi: 10.1161/ATVBAHA.111.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eltzschig HK, Weissmuller T, Mager A, Eckle T. 2006. Nucleotide metabolism and cell-cell interactions. Methods Mol Biol 341:73–87. doi: 10.1385/1-59745-113-4:73. [DOI] [PubMed] [Google Scholar]
- 8.Le Moine O, Stordeur P, Schandene L, Marchant A, de Groote D, Goldman M, Deviere J. 1996. Adenosine enhances IL-10 secretion by human monocytes. J Immunol 156:4408–4414. [PubMed] [Google Scholar]
- 9.Jian R, Sun Y, Wang Y, Yu J, Zhong L, Zhou P. 2012. CD73 protects kidney from ischemia-reperfusion injury through reduction of free radicals. APMIS 120:130–138. doi: 10.1111/j.1600-0463.2011.02827.x. [DOI] [PubMed] [Google Scholar]
- 10.Hasko G, Szabo C, Nemeth ZH, Kvetan V, Pastores SM, Vizi ES. 1996. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 2647 macrophages and in endotoxemic mice. J Immunol 157:4634–4640. [PubMed] [Google Scholar]
- 11.Volmer JB, Thompson LF, Blackburn MR. 2006. Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol 176:4449–4458. doi: 10.4049/jimmunol.176.7.4449. [DOI] [PubMed] [Google Scholar]
- 12.Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP. 2004. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med 200:1395–1405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blume C, Felix A, Shushakova N, Gueler F, Falk CS, Haller H, Schrader J. 2012. Autoimmunity in CD73/ecto-5′-nucleotidase deficient mice induces renal injury. PLoS One 7:e37100. doi: 10.1371/journal.pone.0037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salmi M, Jalkanen S. 2012. Host CD73 impairs anti-tumor immunity. Oncoimmunology 1:247–248. doi: 10.4161/onci.1.2.18310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang B. 2012. CD73 promotes tumor growth and metastasis. Oncoimmunology 1:67–70. doi: 10.4161/onci.1.1.18068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yegutkin GG, Marttila-Ichihara F, Karikoski M, Niemela J, Laurila JP, Elima K, Jalkanen S, Salmi M. 2011. Altered purinergic signaling in CD73-deficient mice inhibits tumor progression. Eur J Immunol 41:1231–1241. doi: 10.1002/eji.201041292. [DOI] [PubMed] [Google Scholar]
- 17.Mills JH, Thompson LF, Mueller C, Waickman AT, Jalkanen S, Niemela J, Airas L, Bynoe MS. 2008. CD73 is required for efficient entry of lymphocytes into the central nervous system during experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 105:9325–9330. doi: 10.1073/pnas.0711175105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahamed DA, Mills JH, Egan CE, Denkers EY, Bynoe MS. 2012. CD73-generated adenosine facilitates Toxoplasma gondii differentiation to long-lived tissue cysts in the central nervous system. Proc Natl Acad Sci U S A 109:16312–16317. doi: 10.1073/pnas.1205589109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. 1982. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 126:131–138. doi: 10.1016/0003-2697(82)90118-X. [DOI] [PubMed] [Google Scholar]
- 20.Goldszmid RS, Caspar P, Rivollier A, White S, Dzutsev A, Hieny S, Kelsall B, Trinchieri G, Sher A. 2012. NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity 36:1047–1059. doi: 10.1016/j.immuni.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez Y, Rosado JD, Vega L, Elizondo G, Estrada-Muniz E, Saavedra R, Juarez I, Rodriguez-Sosa M. 2010. The unexpected role for the aryl hydrocarbon receptor on susceptibility to experimental toxoplasmosis. J Biomed Biotechnol 2010:505694. doi: 10.1155/2010/505694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yarovinsky F, Hieny S, Sher A. 2008. Recognition of Toxoplasma gondii by TLR11 prevents parasite-induced immunopathology. J Immunol 181:8478–8484. doi: 10.4049/jimmunol.181.12.8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okusa MD, Linden J, Huang L, Rieger JM, Macdonald TL, Huynh LP. 2000. A(2A) adenosine receptor-mediated inhibition of renal injury and neutrophil adhesion. Am J Physiol Renal Physiol 279:F809–F818. [DOI] [PubMed] [Google Scholar]
- 24.Sharma AK, Laubach VE, Ramos SI, Zhao Y, Stukenborg G, Linden J, Kron IL, Yang Z. 2010. Adenosine A2A receptor activation on CD4+ T lymphocytes and neutrophils attenuates lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg 139:474–482. doi: 10.1016/j.jtcvs.2009.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, Kuchroo VK, Strom TB, Robson SC. 2007. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mills JH, Kim DG, Krenz A, Chen JF, Bynoe MS. 2012. A2A adenosine receptor signaling in lymphocytes and the central nervous system regulates inflammation during experimental autoimmune encephalomyelitis. J Immunol 188:5713–5722. doi: 10.4049/jimmunol.1200545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, Zhang B. 2011. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest 121:2371–2382. doi: 10.1172/JCI45559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Latz E. 2010. The inflammasomes: mechanisms of activation and function. Curr Opin Immunol 22:28–33. doi: 10.1016/j.coi.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson AM. 1984. Strain-dependent, route of challenge-dependent, murine susceptibility to toxoplasmosis. Z Parasitenkd 70:303–309. doi: 10.1007/BF00927816. [DOI] [PubMed] [Google Scholar]
- 30.McLeod R, Eisenhauer P, Mack D, Brown C, Filice G, Spitalny G. 1989. Immune responses associated with early survival after peroral infection with Toxoplasma gondii. J Immunol 142:3247–3255. [PubMed] [Google Scholar]
- 31.Koscso B, Csoka B, Selmeczy Z, Himer L, Pacher P, Virag L, Hasko G. 2012. Adenosine augments IL-10 production by microglial cells through an A2B adenosine receptor-mediated process. J Immunol 188:445–453. doi: 10.4049/jimmunol.1101224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor GA, Feng CG, Sher A. 2004. p47 GTPases: regulators of immunity to intracellular pathogens. Nat Rev Immunol 4:100–109. doi: 10.1038/nri1270. [DOI] [PubMed] [Google Scholar]
- 33.Butcher BA, Greene RI, Henry SC, Annecharico KL, Weinberg JB, Denkers EY, Sher A, Taylor GA. 2005. p47 GTPases regulate Toxoplasma gondii survival in activated macrophages. Infect Immun 73:3278–3286. doi: 10.1128/IAI.73.6.3278-3286.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murray HW, Szuro-Sudol A, Wellner D, Oca MJ, Granger AM, Libby DM, Rothermel CD, Rubin BY. 1989. Role of tryptophan degradation in respiratory burst-independent antimicrobial activity of gamma interferon-stimulated human macrophages. Infect Immun 57:845–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daubener W, Remscheid C, Nockemann S, Pilz K, Seghrouchni S, Mackenzie C, Hadding U. 1996. Anti-parasitic effector mechanisms in human brain tumor cells: role of interferon-gamma and tumor necrosis factor-alpha. Eur J Immunol 26:487–492. doi: 10.1002/eji.1830260231. [DOI] [PubMed] [Google Scholar]
- 36.Oberdorfer C, Adams O, MacKenzie CR, De Groot CJ, Daubener W. 2003. Role of IDO activation in anti-microbial defense in human native astrocytes. Adv Exp Med Biol 527:15–26. doi: 10.1007/978-1-4615-0135-0_2. [DOI] [PubMed] [Google Scholar]
- 37.Correa G, Marques da Silva C, de Abreu Moreira-Souza AC, Vommaro RC, Coutinho-Silva R. 2010. Activation of the P2X(7) receptor triggers the elimination of Toxoplasma gondii tachyzoites from infected macrophages. Microbes Infect 12:497–504. doi: 10.1016/j.micinf.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 38.Mills JH, Alabanza LM, Mahamed DA, Bynoe MS. 2012. Extracellular adenosine signaling induces CX3CL1 expression in the brain to promote experimental autoimmune encephalomyelitis. J Neuroinflammation 9:193. doi: 10.1186/1742-2094-9-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.