

Abstract
Myeloid-derived suppressor cells (MDSCs) were recently found to accumulate in the lungs during Pneumocystis pneumonia (PcP). Adoptive transfer of these cells caused lung damage in recipient mice, suggesting that MDSC accumulation is a mechanism of pathogenesis in PcP. In this study, the phagocytic activity of alveolar macrophages (AMs) was found to decrease by 40% when they were incubated with MDSCs from Pneumocystis-infected mice compared to those incubated with Gr-1+ cells from the bone marrow of uninfected mice. The expression of the PU.1 gene in AMs incubated with MDSCs also was decreased. This PU.1 downregulation was due mainly to decreased histone 3 acetylation and increased DNA methylation caused by MDSCs. MDSCs were found to express high levels of PD-L1, and alveolar macrophages (AMs) were found to express high levels of PD-1 during PcP. Furthermore, PD-1 expression in AMs from uninfected mice was increased by 18-fold when they were incubated with MDSCs compared to those incubated with Gr-1+ cells from the bone marrow of uninfected mice. The adverse effects of MDSCs on AMs were diminished when the MDSCs were pretreated with anti-PD-L1 antibody, suggesting that MDSCs disable AMs through PD-1/PD-L1 ligation during PcP.
INTRODUCTION
Pneumocystis pneumonia (PcP) is an opportunistic disease in immunocompromised patients. Although alveolar macrophages (AMs) play an important role in the clearance of microorganisms in the lungs, they are defective in phagocytosis (1, 2), and their number is decreased during PcP (3–8). A cause of this AM number decrease is increased apoptosis due to elevated levels of intracellular polyamines (9, 10). The causes for AM dysfunction during PcP are not clear; one possible cause is downregulation of the transcription factor PU.1 (11), as it regulates the expression of many macrophage receptors (11–15). The mechanism of PU.1 downregulation is unknown.
We have recently found that myeloid-derived suppressor cells (MDSCs) accumulate in the lungs during PcP (16), and that adoptive transfer of MDSCs from mice with PcP causes lung damage in the recipient mice (16). MDSCs are a heterogeneous population of bone marrow-derived myeloid progenitor cells and immature myeloid cells. In health, these cells quickly differentiate into mature granulocytes, macrophages, or dendritic cells. This differentiation is blocked in certain conditions, such as cancer, various infectious diseases, sepsis, trauma, and some autoimmune diseases (17). MDSCs have the morphology of monocytes or granulocytes; thus, they are classified as monocytic and granulocytic MDSCs. In mice, MDSCs coexpress Gr-1 and CD11b (αM-integrin) (18). In humans, MDSCs are HLA-DR− or HLA-DRlow and CD11b+, CD33+, or CD15+ (19). MDSCs are immunosuppressive and have been shown to suppress the functions of NK cells, T cells, and B cells (20, 21). The suppressive activity of MDSCs appears to be inversely related to the expression of the programmed death 1 protein (PD-1), as MDSCs from PD-1−/− mice are more immunosuppressive than those from wild-type mice (20).
PD-1 (CD279) is a coinhibitory molecule. As with CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) and BTLA (B- and T-lymphocyte attenuator), a major function of PD-1 is to prevent the activated T cells from becoming overzealous, leading to adverse inflammatory responses and organ damage (22–24). PD-1 is a membrane protein of the CD28 family. It is expressed on the surfaces of many immune cells, including CD4+ T cells, CD8+ T cells, NK T cells, B cells, and monocytes (22, 24, 25). Its ligand PD-L1 (CD274), also called B7 homolog 1 (B7-H1), is a type I transmembrane protein and is constitutively expressed on T cells, B cells, macrophages, and dendritic cells (22, 24, 25).
During persistent antigen exposure, antigen-specific CD8+ T cells may lose their effector functions, such as proliferation and cytokine production; this phenomenon is referred to as CD8+ T-cell exhaustion (26). The PD-1/PD-L1 signaling pathway plays a major role in the generation of exhausted CD8+ T cells in numerous settings, including cancer and chronic viral infections of human immunodeficiency virus (HIV), hepatitis C virus (HCV), hepatitis B virus (HBV), lymphocytic choriomeningitis virus (LCMV), and simian immunodeficiency virus (SIV) (27–33). The PD-1/PD-L1 signaling pathway also is involved in immune tolerance, as PD-1−/− knockout leads to autoimmune encephalomyelitis, lupus-like syndrome (34), or dilated cardiomyopathy in mice (35–37). One intronic single-nucleotide polymorphism of the PD-1 gene is correlated with the development of systemic lupus erythematosus in Europeans and Mexicans (38). The PD-1/PD-L1 signaling pathway also modulates the function of regulatory T cells (Treg), as blockade of the PD-1/PD-L1 pathway abrogates Treg-mediated immune tolerance in mice (39, 40).
Although the suppressive effects of MDSCs on T cells have been studied extensively, it is unknown whether MDSCs have any adverse effects on macrophages. Since MDSCs express PD-L1 (17, 41, 42) and macrophages have been shown to express PD-1 (43–47), we tested the hypothesis that MDSCs interact with AMs through PD-1/PD-L1 ligation, causing PU.1 downregulation and defects in phagocytosis during PcP.
MATERIALS AND METHODS
Animal model of PcP.
C57BL/6 mice were obtained from Harlan (Indianapolis, IN). All animals used in this study were female, 18 to 20 g in weight. The study was approved by the Indiana University Animal Care and Use Committee and carried out under the supervision of veterinarians. Immunosuppression of mice was achieved by intraperitoneal injection of 0.3 mg anti-CD4 (L3T4) monoclonal antibody (MAb; clone GK1.5; Harlan, Indianapolis, IN) to each mouse once a week to deplete CD4+ cells until the mice were sacrificed. One week after the initial injection, each mouse was transtracheally instilled with 2 × 106 Pneumocystis murina organisms in 20 μl sterile phosphate-buffered saline (PBS). The Pneumocystis organisms used as the inoculum were obtained from heavily infected mouse lungs and isolated as previously described (8). Tetracycline (0.73 g/liter) was added to the drinking water to prevent bacterial infections in mice.
Isolation of AMs.
After being anesthetized by intramuscular injection of 30 μl ketamine cocktail (ketamine hydrochloride, 80 mg/ml; acepromazine, 1.76 mg/ml; atropine, 0.38 μg/ml), each Pneumocystis-infected mouse was lavaged with sterile saline (1 ml at a time) through an intratracheal catheter until a total of 10 ml lavage fluid was recovered, as described previously (8). The bronchoalveolar lavage (BAL) fluid was centrifuged at 300 × g for 10 min to pellet cells. The cells in each pellet were resuspended in 1 ml PBS containing 0.5% bovine serum albumin (BSA). CD11c+ AMs were isolated from mice that had been infected with Pneumocystis for 5 weeks, as the number of these cells is dramatically decreased afterward (unpublished observation). These cells were separated from total BAL fluid cells using biotin-anti-mouse CD11c antibody (117303; BioLegend) and anti-biotin magnetic microbeads (130-090-485; Miltenyi Biotech). To ensure the purity of the AM preparation, this isolation procedure was repeated twice.
Isolation and confirmation of MDSCs.
MDSCs from the BAL fluids of Pneumocystis-infected mice and the bone marrow of immunosuppressed, uninfected mice were isolated using the myeloid-derived suppressor cell isolation kit (Miltenyi Biotec, Auburn, CA), which contains anti-Gr-1 antibody-conjugated, magnetically activated cell sorting (MACS) microbeads, as described previously (16). To ensure that the isolated MDSCs were not contaminated with Pneumocystis organisms, this isolation procedure was repeated twice. The cells isolated from bone marrow were mainly Gr-1+ cells (referred to as Gr1BM cells here), as there were few MDSCs in bone marrow. The identity of the isolated MDSCs (CD11b+, Gr-1+) was confirmed by microscopy, flow cytometry, and T-cell proliferation inhibition assay as described previously (16).
Coculture of AMs with MDSCs.
A total of 1 × 105 AMs isolated from healthy, uninfected mice were incubated with 5 × 105 MDSCs isolated from mice that had been infected with Pneumocystis for 5 weeks overnight at 37°C and 5% CO2 in 0.6 ml of RPMI 1640 medium containing 10% fetal bovine serum (FBS) and 10 ng/ml of mouse granulocyte-macrophage colony-stimulating factor (GM-CSF; Invitrogen), which is required to maintain the viability of MDSCs in culture (18). Since GM-CSF regulates the expression of dectin-1 (48), it also is essential for the phagocytic activity of AMs. The 5:1 MDSC-to-AM ratio was determined empirically. As a negative control, an aliquot of the same AMs was incubated with a 5-fold excess of Gr1BM cells from uninfected, immunosuppressed mice. The isolated Gr1BM cells were maintained in the same culture medium as that for MDSCs. Gr1BM cells were used as the negative control, because they are precursors of MDSCs. Although MDSCs from the lungs of uninfected mice would be the most appropriate cells for negative controls, few or none of such cells exist in these mice. To block PD-1/PD-L1 signaling, MDSCs were incubated with anti-mouse PD-L1 (2 μg/ml; 124301; BioLegend) antibody at 37°C for 2 h before the overnight coculture with AMs. As the negative control, MDSCs were treated with rat IgG2b (400601; BioLegend).
Phagocytosis assay.
After an overnight coculture with AMs, MDSCs were removed using anti-Gr-1 antibody-conjugated magnetic microbeads and a MACS separation column (Miltenyi Biotec, Auburn, CA). AMs in the flowthrough fraction were pelleted by centrifugation and then resuspended in 50 μl of RPMI 1640 medium containing 10% FBS and 20 μg of fluorescein-conjugated zymosan A beads. The mixture was placed on a coverslip and incubated at 37°C in a moisture chamber for 1 h. After washing off nonadherent cells and free zymosan beads, one drop of ProLong Gold antifade reagent containing 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) was added to the coverslip that was then placed on a slide. Fluorescent zymosan beads, instead of Pneumocystis organisms, were used as the substrate for phagocytosis assays, because the defect in AM phagocytosis during PcP is a general one and is not specific to Pneumocystis organisms (49). Since the major component of zymosan beads is β-glucan, they resemble the cyst form of Pneumocystis. In addition, zymosan beads are commonly used as the substrate for phagocytosis assays (50–52). Each sample first was examined under a fluorescence microscope, and the number of zymosan beads phagocytosed by a total of 100 AMs was determined by confocal microscopy. The cells were examined at multiple focal planes to distinguish the zymosan beads that were phagocytosed from those that attached on the surface of AMs.
RNA isolation and real-time PCR.
Total RNA was isolated from AMs using TRIzol reagent (Invitrogen) and reverse transcribed into cDNA using the iScript kit (Bio-Rad). For AMs that were coincubated with MDSCs or Gr1BM cells, these cells were removed using anti-Gr-1 antibody-conjugated magnetic microbeads and a MACS separation column (Miltenyi Biotec, Auburn, CA) before RNA isolation. SYBR green-based real-time quantitative PCR (qPCR) was performed using the following primers: 5′-CCTGCTTCACCACCTTCTTGA-3′ and 5′-TGTGTCCGTCGTGGATCTGA-3′ for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-TCTTGCCGTAGTTGCGCAG-3′ and 5′-GCATCTGGTGGGTGGACAA-3′ for PU.1, 5′-GCCCTAGCTGTCTTCTGCTC-3′ and 5′-TCCTTCAGAGTGTCGTCCTTG-3′ for PD-1, and 5′-TATGGTGGTGCCGACTACAA-3′ and 5′-TGGCTCCCAGAATTACCAAG-3′ for PD-L1. The GAPDH cDNA was coamplified in each reaction to serve as the internal control, and fold change in PU.1, PD-1, or PD-L1 expression was calculated using the equation 2−ΔΔCT, where CT is threshold cycle.
ChIP.
A total of 5 × 105 of AMs were incubated with 2.5 × 106 MDSCs or Gr1BM cells as described above. After removal of MDSCs or Gr1BM cells using anti-Gr-1 antibody-conjugated magnetic microbeads and a MACS separation column (Miltenyi Biotec, Auburn, CA), AMs were treated with 0.1% formaldehyde in RPMI 1640 medium containing 10% FBS for 10 min at room temperature to cross-link histone proteins to DNA. The fixation was quenched by adding a 1/10 volume of 1.25 M glycine and incubating for 5 min. Cells were pelleted by centrifugation at 300 × g for 10 min at 4°C and then lysed in 100 μl lysis buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, and 1% SDS). Each cell lysate was diluted by adding 400 μl 1× PBS and sonicated at 30-s intervals for 8 cycles (sonic dismembrator model 300; Fisher). After sonication, insoluble cell debris was removed by centrifugation at 11,000 × g for 10 min at 4°C. The supernatant of each sample was diluted with equal volumes of 2× radioimmunoprecipitation assay (RIPA) buffer (1.8% NaCl, 20 mM Tris, pH 7.6, 0.2% SDS, 0.2% Triton X-100, 0.2% deoxycholate, and 10 mM EDTA) and divided into four aliquots to perform chromatin immunoprecipitation (ChIP).
One of these 4 aliquots was used as the input control in which no antibody was added. The other 3 aliquots were incubated with 0.5 μg of anti-H3K4me3 (ab8580; Abcam), 2 μg of anti-H3Ac (06-599; Millipore), or 2 μg of anti-H3K27me3 (07-449; Millipore) at 4°C with continuous rotation for 2 h. Protein A-agarose beads (10 μl) then were added to precipitate the antibody-bound chromatin. Each precipitate was dissolved in 50 μl of distilled H2O and digested with proteinase K (20 μg), followed by phenol-chloroform extraction to purify DNA. The purified DNA was precipitated with isopropanol and then dissolved in 50 μl of water. Real-time PCR was performed to amplify the promoter region of PU.1 and both the 3′ and 5′ sides of its upstream regulatory element (3′URE and 5′URE, respectively), which is located approximately 14 kb upstream from the PU.1 transcription start site (53). The SYBR green PCR master mix (TaKaRa) and the following primers were used for the PCR assays: 5′-GGGAGGCAGAGCACACATG-3′ and 5′-GTTTCCACATCGGCAGCAG-3′ for 3′URE, 5′-GCCCAGGCTAGGGAAGTTTG-3′ and 5′-GAGAGCAGAGCACTTCATGGCTA-3′ for 5′URE, and 5′-GTAGCGCAAGAGATTTATGCAAAC-3′ and 5′-GCACAAGTTCCTGATTTTATCGAA-3′ for the promoter region. Each PCR was performed in triplicate, and 1% of input DNA was analyzed in a manner identical to that for the control.
The average CT value of each ChIP reaction was used to calculate percent input using the equation 100 × 2(adjusted input CT − ChIP CT) (http://www.invitrogen.com/site/us/en/home/Products-and-Services/Applications/epigenetics-noncoding-rna-research/Chromatin-Remodeling/Chromatin-Immunoprecipitation-ChIP/chip-analysis.html), where adjusted input CT (adjusted to 100%) was calculated as the raw input CT minus 6.664 (i.e., log2 of 100). Data were presented as the ratio of percent input of ChIP with anti-H3K4me3 (activating modification) to that of ChIP with anti-H3K27me3 (suppressing modification) and ChIP with anti-H3Ac (activating modification) to that of ChIP with anti-H3K27me3 (suppressing modification).
DNA methylation assay.
The EpiTect II DNA methylation enzyme kit (335452; Qiagen) was used to assess CpG methylation of the PU.1 gene. Genomic DNA was isolated from AMs incubated with Gr1BM cells or MDSCs using the DNeasy blood and tissue kit (69504; Qiagen). Each DNA sample was divided into 4 aliquots (62.5 ng each) that then were subjected to mock (no enzyme), methylation-sensitive (MSRE), methylation-dependent (MDRE), and double (MSRE and MDRE) restriction endonuclease digestion according to the instructions of the kit. The enzyme reaction mixtures were mixed directly with qPCR master mix and the primer pair 5′-GTAGCGCAAGAGATTTATGCAAAC-3′ and 5′-GCACAAGTTCCTGATTTTATCGAA-3′ to amplify a region (100 bp) of the PU.1 promoter. The resulting CT values were entered into the data analysis spreadsheet of the kit, which automatically calculated the relative amount of methylated and unmethylated DNA.
Flow cytometry.
Fluorescent anti-mouse antibodies, including fluorescein isothiocyanate (FITC)-Gr-1 (108405), phycoerythrin (PE)-CD11b (101207), PE-PD-1 (109103), PE-PD-L1 (124307), and Alexa Fluor 647-CD11c (117314), were purchased from BioLegend (San Diego, CA). After an incubation in 100 μl of 1× PBS containing 0.5% bovine serum albumin for 1 h, AMs, MDSCs, and control Gr1BM cells were stained with appropriate antibodies on ice for 1 h and then examined with a BD FACSCalibur flow cytometer (BD Biosciences) as described previously (16). Separate sets of cells were stained with PE-labeled IgG isotype antibody (400607; BioLegend) to control for background fluorescence. The flow cytometry data generated were analyzed with FlowJo software (Tree Star, Ashland, OR).
Western blot analysis.
Proteins were separated by 10% SDS-PAGE and then transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-P; EMD Millipore). The membrane was blocked in TBST (20 mM Tris-HCl, 500 mM NaCl, 0.05% Tween 20, pH 7.5) containing 5% BSA. Subsequently, the membrane was probed with anti-mouse PD-1 (Sigma-Aldrich) and anti-mouse GAPDH (BioLegend) antibody in TBST containing 5% BSA. The binding of the primary antibodies was detected with horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz Biotechnology). After washing, the membrane was incubated with the Amersham ECL Advance Western blotting detection reagent (GE Healthcare).
Statistics.
Data are expressed as means ± standard deviations. An unpaired two-tailed Student's t test was used to evaluate the difference in data from two different samples, including the following: phagocytosis of AMs incubated with MDSCs versus those incubated with Gr1BM cells; levels of PU.1 expression, histone modification, or DNA methylation in AMs incubated with MDSCs versus levels in those incubated with Gr1BM cells; PD-L1 expression in MDSCs versus that in Gr1BM cells; levels of PD-1 expression in AMs incubated with MDSCs versus levels in those incubated with Gr1BM cells; and levels of suppression of AM phagocytosis or PU.1 expression in AMs by the MDSCs pretreated with anti-PD-L1 versus levels for those pretreated with control IgG. The difference was considered significant if the P value was <0.05.
RESULTS
AMs incubated with MDSCs became defective in phagocytosis.
To investigate the effect of MDSCs on the phagocytic activity of AMs, AMs from uninfected mice were cocultured with MDSCs and then assayed for their phagocytic activity. The purity of the AM preparation was examined by microscopy, and all cells on the slides were found to have the typical macrophage morphology (Fig. 1). AMs were incubated with MDSCs from mice that had been infected with Pneumocystis for 5 weeks or with control Gr1BM cells at a ratio of 5:1 (5 × 105 MDSCs or Gr1BM to 1 × 105 AMs) overnight and analyzed for their ability to phagocytose zymosan beads. Results of confocal microscopy showed that AMs incubated with MDSCs phagocytized an average of 4.1 ± 1.2 zymosan beads per cell, and that those incubated with Gr1BM cells phagocytized an average of 6.6 ± 0.8 zymosan beads per cell. These results indicated a 40% decrease in the phagocytic activity of AMs when they were incubated with MDSCs from Pneumocystis-infected mice compared to those incubated with Gr1BM cells (P = 0.038) (Fig. 2A to C).
FIG 1.

Purity of isolated AMs. AMs were isolated from BAL fluids of immunosuppressed, uninfected mice (AMs/L3T4) and Pneumocystis-infected mice (AMs/PcP) using biotin-anti-mouse CD11c antibody and anti-biotin magnetic microbeads. An aliquot of the purified cells was cytospun on a slide; the cells were stained with Giemsa stain and examined by microscopy.
FIG 2.
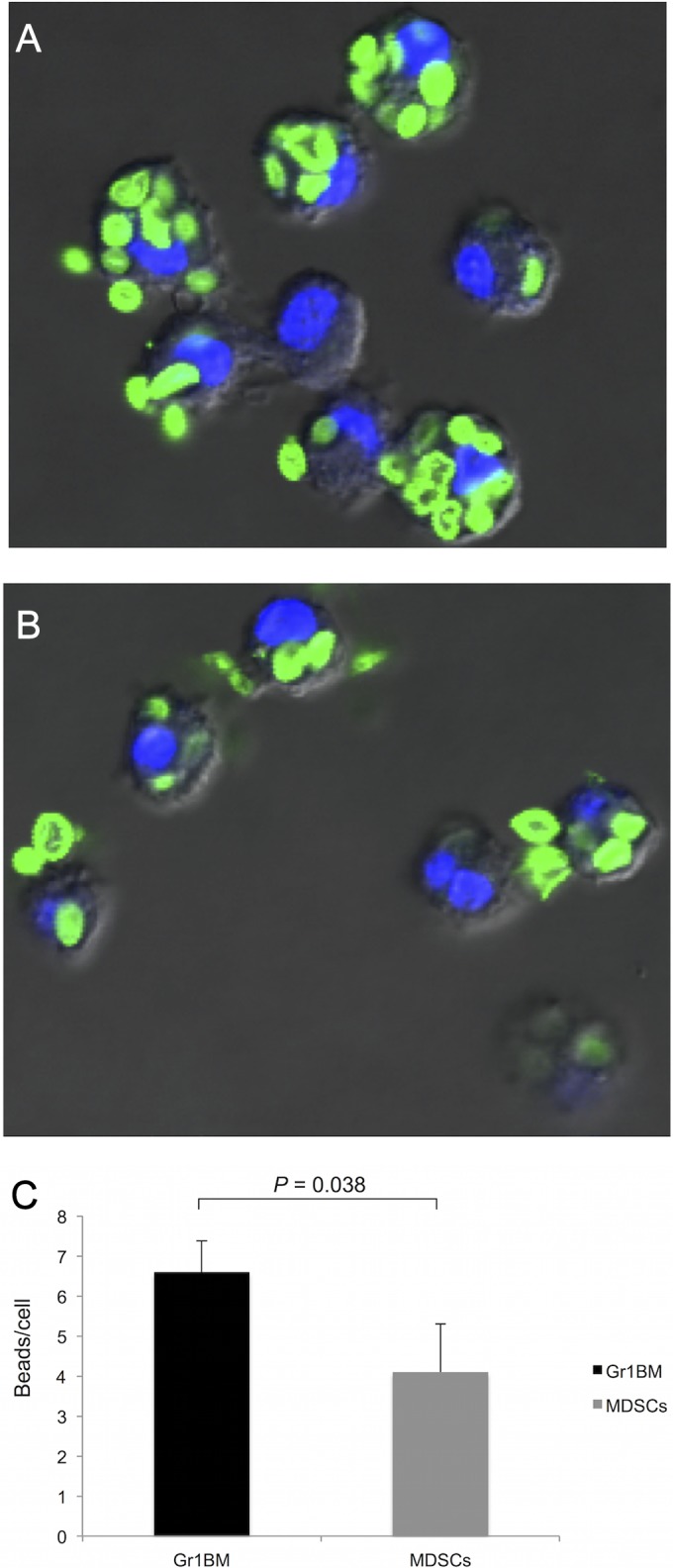
Reduced phagocytic activity in AMs incubated with MDSCs. AMs cocultured with control Gr1BM cells (A) or MDSCs (B) overnight were incubated with fluorescein-conjugated zymosan beads for 1 h. The nuclei of AMs were counterstained with DAPI. (C) The number of zymosan beads phagocytosed by AMs incubated with MDSCs or Gr1BM cells were counted under a confocal microscope. Data are presented as means ± SD from three independent experiments.
PU.1 expression was decreased in AMs incubated with MDSCs.
Since PU.1 positively regulates the expression of many macrophage receptors, we hypothesized that the defect in phagocytosis of AMs incubated with MDSCs was due to PU.1 downregulation. To test this hypothesis, AMs incubated overnight with MDSCs from Pneumocystis-infected mice or Gr1BM cells were assessed for PU.1 expression. PU.1 expression in AMs that were not incubated with either MDSCs or Gr1BM cells was determined to serve as a control. Total RNA was isolated from the AMs and analyzed by PU.1 real-time RT-PCR (qRT-PCR) in triplicate. The PU.1 expression in AMs incubated with Gr1BM and in AMs incubated with MDSCs were 1.25-fold ± 0.19-fold and 0.36-fold ± 0.04-fold, respectively, relative to that in AMs without coincubation with either Gr1BM cells or MDSCs (P = 0.0014) (Fig. 3). This result indicated that coincubation with MDSCs caused a downregulation in PU.1 expression in AMs by approximately 70%.
FIG 3.

Decreased PU.1 expression in AMs incubated with MDSCs. AMs from uninfected mice were cocultured with MDSCs or Gr1BM cells at a ratio of 1:5 for 16 h in a 37°C incubator with 5% CO2. After removing MDSCs and Gr1BM cells with anti-Gr-1 antibody-conjugated magnetic microbeads, total RNA of AMs was isolated, and PU.1 mRNA levels were determined by real-time PCR. The level of PU.1 expression in AMs that were not incubated with MDSCs or Gr1BM cells was set as 1, and that in AMs incubated with either type of cell was compared to it. Data are presented as means ± SD from three independent experiments.
MDSCs affected PU.1 expression in AMs at the epigenetic level.
Experiments were performed to test the hypothesis that PU.1 downregulation in AMs incubated with MDSCs was due to epigenetic changes. ChIP assays were performed to examine histone modifications on the PU.1 gene. AMs incubated overnight with MDSCs from Pneumocystis-infected mice or control Gr1BM cells from uninfected mice were examined. As seen in Fig. 4, the ratios of percent input of H3K4me3 to that of H3K27me3 of the 3′URE, 5′URE, and promoter of PU.1 in AMs incubated with Gr-1+ cells were 1.9, 1.4, and 1.4, respectively, and those of AMs incubated with MDSCs were 1.2, 1.35, and 1.2, respectively. The difference in the methylation of H3K4 and H3K27 in the 3′URE between AMs incubated with MDSCs and those incubated with Gr1BM cells was statistically significant (P = 0.045) (Fig. 4). A profound decrease in H3Ac in AMs incubated with MDSCs was observed compared to that in AMs incubated with Gr1BM cells. The ratios of the percent input of H3Ac to that of H3K27me3 of the 3′URE, 5′URE, and promoter of PU.1 in AMs incubated with Gr1BM cells were 1.7, 2.1, and 3.0, respectively, and those of AMs incubated with MDSCs were 0.4, 0.8, and 1.5, respectively (Fig. 4). Therefore, incubation of AMs with MDSCs caused a 76, 62, and 50% decrease in H3Ac in the 3′URE (P = 0.032), 5′URE (P = 0.019), and promoter (P = 0.004) of PU.1, respectively, compared to levels of those incubated with Gr-1+ cells. Results of DNA methylation assays showed that CpG methylation of the PU.1 gene was 77.3% ± 1.73% in AMs incubated with Gr1BM cells but was 91.9% ± 0.9% in those incubated with MDSCs (Fig. 5).
FIG 4.

Increased histone deacetylation of PU.1 gene in AMs incubated with MDSCs. AMs from uninfected mice were incubated with MDSCs or Gr1BM cells overnight. After removing MDSCs and Gr1BM cells with anti-Gr-1 antibody-conjugated magnetic microbeads, the AMs were treated with 0.1% formaldehyde to cross-link histone proteins to DNA, lysed, and sonicated to generated chromatin fragments. Chromatin immunoprecipitation was performed using anti-H3K4me3, anti-H3ac, and anti-H3K27me3 antibodies in separate reactions. DNA in the precipitated chromatin was isolated and used as the template for real-time PCR to amplify the 3′URE, 5′URE, and promoter regions of the PU.1 gene. The CT values obtained were used to determine the ratios of percent input of H3K4me3 to H3K27me3 and H3Ac to H3K27me3. Data are presented as means ± SD from three independent experiments.
FIG 5.

Increased DNA methylation of PU.1 promoter in AMs incubated with MDSCs. AMs from uninfected mice were incubated with MDSCs or Gr1BM cells overnight. After removing MDSCs and Gr1BM, AM genomic DNA was isolated and assessed for CpG methylation by digestion with methylation-dependent and methylation-sensitive restriction enzymes using the EpiTect methyl II enzyme kit (Qiagen). Real-time PCR then was performed to amplify a 100-bp region of the PU.1 promoter. The resulting CT values were entered into the data analysis spreadsheet of the kit to calculate the relative amount of methylated DNA in each sample. Data are presented as means ± SD from three independent experiments.
PD-1 expression was increased in AMs from PcP mice.
To determine whether the PD-1/PD-L1 pathway is activated in AMs during PcP, the expression of PD-1 in AMs was examined. Real-time RT-PCR was performed on RNA isolated from AMs of mice infected for 5 weeks. RNA from the AMs of immunosuppressed, uninfected mice was analyzed in an identical manner to serve as the control. The PD-1 expression in AMs (AMs/PcP) from Pneumocystis-infected mice was found to be 105-fold higher than in those (AMs/L3T4) from immunosuppressed, uninfected mice (P = 0.007) (Fig. 6A). To confirm this result, PD-1 expression on the surface of AMs from uninfected and infected mice was examined by flow cytometry using antibodies against PD-1 and CD11c (Fig. 6B); the latter is a marker of AMs (54). Western blotting with anti-PD-1 antibody also was performed to further confirm PD-1 overexpression in AMs (Fig. 6C). Results showed that AMs from Pneumocystis-infected mice had a higher level of PD-1 expression than immunosuppressed, uninfected mice.
FIG 6.
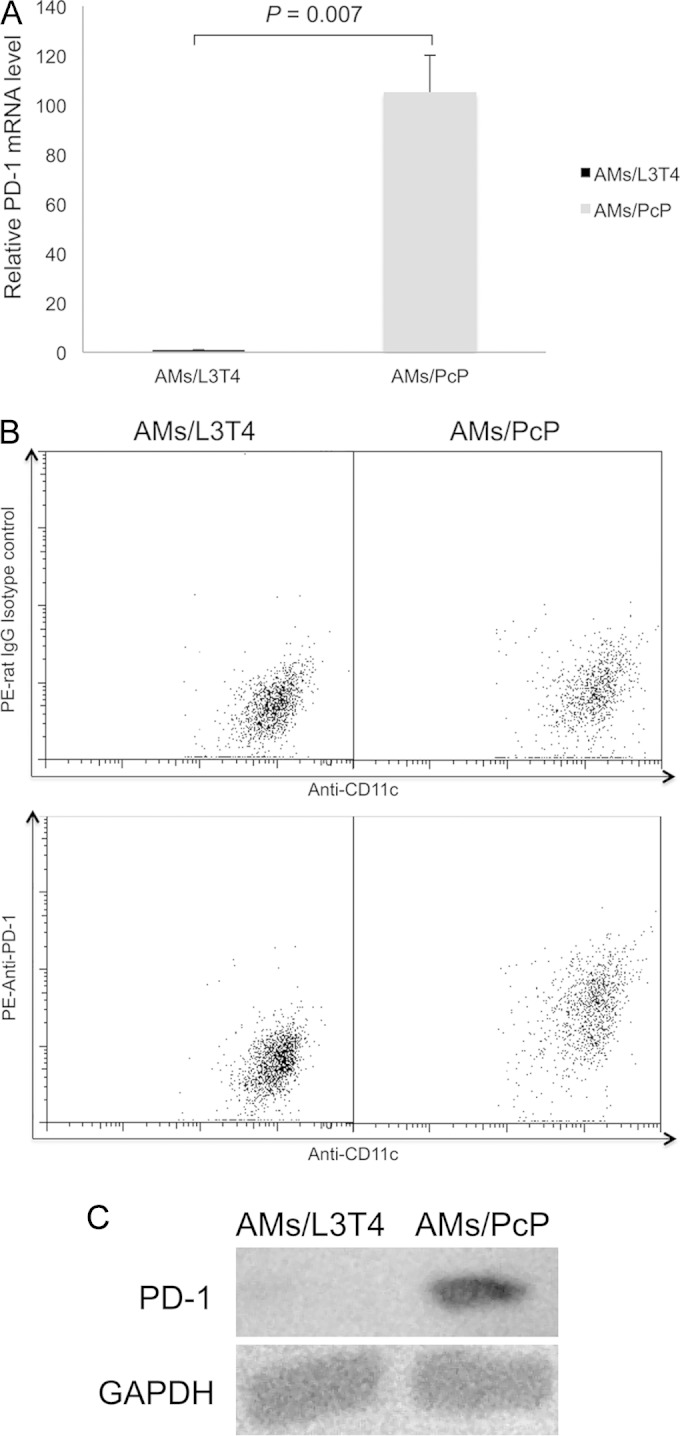
Increased PD-1 expression in AMs from PcP mice. AMs (AMs/PcP) were isolated from PcP mice at 5 weeks post-Pneumocystis infection. Control AMs (AMs/L3T4) were from uninfected mice immunosuppressed by weekly injection of anti-CD4 (L3T4) antibody. (A) Total RNA was isolated from AMs, and PD-1 gene expression was determined by real-time RT-PCR. The average PD-1 expression level in AMs/L3T4 was set as 1, and that in AMs/PcP was compared to it. Data are presented as means ± SD from three independent experiments. (B) The AMs were examined by flow cytometry using anti-CD11c (Alexa Flour 647 conjugated), rat IgG isotype control (phycoerythrin [PE] conjugated), and anti-PD-1 (phycoerythrin conjugated) antibodies. The result shown is representative of three independent experiments. (C) PD-1 expression in AMs from PcP mice was further confirmed by Western blotting using anti-PD-1 antibody. The expression of GAPDH was examined similarly as a loading control.
MDSCs from PcP mice expressed high levels of PD-L1.
Real-time RT-PCR was performed to determine PD-L1 mRNA levels in MDSCs from mice infected with Pneumocystis for 5 weeks. PD-L1 mRNA in Gr1BM cells from the bone marrow of immunosuppressed, uninfected mice was analyzed in an identical manner to serve as the control. PD-L1 expression in MDSCs was found to be 62-fold higher than that in Gr1BM cells from uninfected mice (P = 0.0055) (Fig. 7A). This result was consistent with those of flow cytometry, which also showed a higher level of PD-L1 expression on the surface of MDSCs from Pneumocystis-infected mice than on that of Gr-1+ cells from uninfected mice (Fig. 7B).
FIG 7.
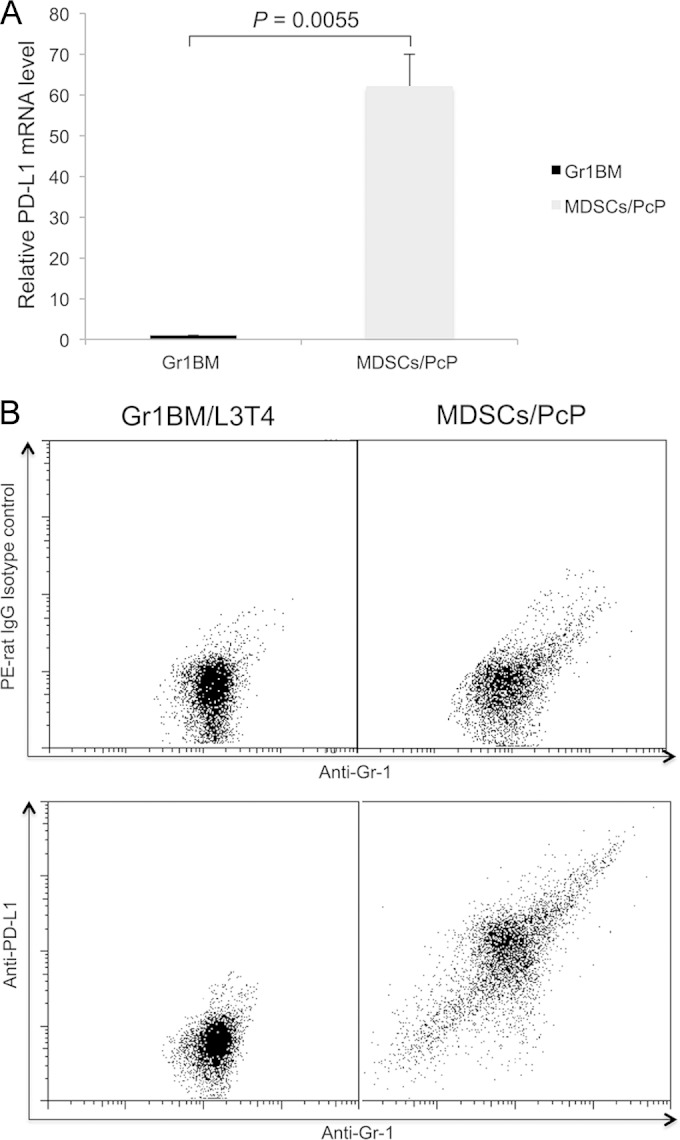
Increased PD-L1 expression in MDSCs from PcP mice. MDSCs (MDSCs/PcP) were isolated from PcP mice at 5 weeks post-Pneumocystis infection. Control Gr1BM cells were isolated from uninfected mice immunosuppressed by weekly injection of anti-CD4 (L3T4) antibody. (A) Total RNA was isolated from the cells, and PD-L1 gene expression was determined by real-time RT-PCR. The average PD-L1 expression level in Gr1BM cells was set as 1, and that in MDSCs was compared to it. Data are presented as means ± SD from three independent experiments. (B) The cells were examined by flow cytometry using anti-Gr-1 and anti-PD-L1 antibodies. The result shown is representative of three independent experiments.
PD-1 expression was increased in AMs incubated with MDSCs.
To explore the possibility that PD-1 expression in AMs is caused by MDSCs, PD-1 mRNA levels in AMs cocultured with MDSCs or control Gr1BM cells overnight were measured by real-time RT-PCR. PD-1 expression levels in AMs incubated with Gr1BM and in those incubated with MDSCs were 1.15-fold ± 0.1-fold and 18.33-fold ± 2.55-fold, respectively, of that in AMs without coincubation with either type of cell (P = 0.007). This result indicated an 18-fold increase in PD-1 mRNA levels when AMs were incubated with MDSCs compared to those incubated with Gr1BM cells (Fig. 8A). This result was confirmed by flow cytometry, which showed that AMs incubated with MDSCs had increased PD-1 expression on the cell surface compared with that of AMs incubated with control Gr1BM cells (Fig. 8B).
FIG 8.
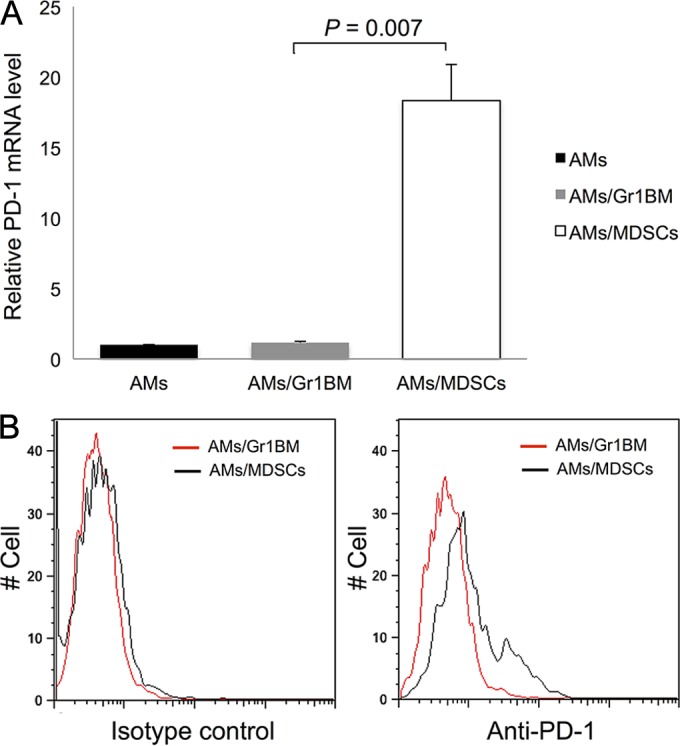
Increased PD-1 expression in AMs incubated with MDSDs. AMs isolated from uninfected mice were cocultured with MDSCs or Gr1BM cells at a ratio of 1:5 for 16 h in a 37°C incubator with 5% CO2. (A) After removing MDSCs and Gr1BM cells with anti-Gr-1 antibody-conjugated magnetic microbeads, total RNA was isolated from AMs of each group. PD-1 gene expression was determined by real-time RT-PCR. The level of AMs/Gr1BM was set as 1, and that in AMs/MDSCs was compared to it. Data are presented as means ± SD from three independent experiments. (B) AMs incubated with MDSCs or Gr1BM cells were analyzed for surface PD-1 expression by flow cytometry.
Blockade of PD-1 signaling with anti-PD-L1 antibody abolished the adverse effects of MDSCs on AMs.
To investigate whether the PD-1/PD-L1 signaling pathway played a role in the inhibitory effect of MDSCs on AMs, MDSCs were treated with anti-PD-L1 antibody or control IgG for 2 h before they were incubated with AMs overnight. The AMs then were analyzed for their PU.1 mRNA levels and ability to phagocytose zymosan beads. As shown in Fig. 9A, PU.1 expression levels in AMs cocultured with MDSCs that were pretreated with anti-PD-L1 antibody or control IgG were 1-fold ± 0.25-fold and 0.41-fold ± 0.16-fold, respectively, relative to that of AMs without coincubation with MDSCs (P = 0.016). This result indicated that blockade of PD-1/PD-L1 signaling reduced the suppressive effect of MDSCs on PU.1 expression in AMs. In addition, pretreatment of MDSCs with anti-PD-L1 antibody reduced their ability to inhibit the phagocytic function of AMs. Results of confocal microscopy showed that AMs incubated with these MDSCs phagocytosed an average of 7.2 zymosan beads per cell, whereas those incubated with MDSCs pretreated with control IgG phagocytosed an average of 4.7 zymosan beads per cell (P = 0.0009) (Fig. 9B).
FIG 9.

Loss of suppressive effect of MDSCs pretreated with anti-PD-L1 antibody on AMs. A total of 1 × 105 AMs were cocultured with 5 × 105 MDSCs that were treated with anti-PD-L1 antibody or control IgG. (A) After 16 h of incubation, AMs were isolated, and the PU.1 mRNA levels were determined by real-time RT-PCR. Data are presented as means ± SD from three independent experiments. The level of PU.1 expression in AMs that were not incubated with MDSCs or Gr1BM cells was set as 1, and that in AMs incubated with MDSCs that were pretreated with or without anti-PD-L1 antibody was compared to it. (B) Phagocytosis was assayed, and the number of zymosan beads phagocytosed by AMs incubated with MDSCs pretreated with anti-PD-L1 antibody or control IgG was counted under a confocal microscope.
DISCUSSION
AMs are defective in phagocytosis during PcP, but the mechanisms of this defect are largely unknown. We have recently found that MDSCs accumulate in the lungs during PcP. In this study, we found that AMs from normal mice became defective in phagocytosis when they were incubated with MDSCs from Pneumocystis-infected mice (Fig. 2), suggesting that MDSCs in Pneumocystis-infected lungs can inhibit the phagocytic activity of AMs. We also found that AMs incubated with these MDSCs had a 60% decrease in PU.1 expression compared with those incubated with Gr1BM cells from uninfected mice (Fig. 3). This result is consistent with our previous finding that PU.1 expression in AMs is downregulated during PcP (11). Since PU.1 regulates the expression of many macrophage receptors (11–15), it is conceivable that PU.1 downregulation renders AMs defective in phagocytosis. We also have shown that MDSCs caused PU.1 downregulation in AMs by decreasing histone 3 acetylation (Fig. 4) and increasing CpG methylation (Fig. 5).
In this study, we used biotin-conjugated anti-CD11c antibody and anti-biotin magnetic microbeads to isolate AMs from BAL fluids. Microscopic examination of the isolated cells showed that all of the cells examined had a morphology characteristic of macrophages (Fig. 1), indicating that the great majority of the isolated CD11c+ cells were AMs. This result is consistent with a previous report that greater than 95% of myeloid-derived cells in BAL fluids from normal mice are AMs (55). Since the phagocytosis assay was performed by microscopy, the presence of other non-AM CD11c+ cells (e.g., monocytes or dendritic cells), if any, would not affect the assay. For the investigation of the effect of MDSCs on PU.1 expression in AMs, the presence of these non-AM CD11c+ cells also would not affect the results, because AM is the predominate type of cells that express PU.1 (56).
Results of this study also revealed that AMs upregulated their PD-1 expression when they were cocultured with MDSCs (Fig. 8). Since MDSCs expressed PD-L1 (Fig. 7), this result suggests that MDSCs interact with AMs through PD-1/PD-L1 ligation. Supporting this possibility is the finding that pretreatment of MDSCs from Pneumocystis-infected mice with anti-PD-L1 antibody abrogated their ability to downregulate PU.1 expression and to disable the phagocytic function of AMs (Fig. 9). It remains to be investigated whether PD-1/PD-L1 signaling directly causes epigenetic alterations of the PU.1 gene. However, it has been shown that PD-1 downregulates the expression of Skp2, which is the substrate recognition component of ubiquitin ligase (57), by recruiting histone deacetylase 3 (HDAC3), pro-interleukin-16 (pro-IL-16), and GABPβ1 to inactivate the Skp2 promoter; this Skp2 downregulation results in the suppression of T cell activation (58). It is likely that PD-1/PD-L1 signaling causes PU.1 downregulation by recruiting a histone deacetylase to cause the deacetylation of the PU.1 gene observed in this study.
PD-1 is known to inhibit the phosphorylation of STAT-1, leading to decreased production of IL-12 in monocytes and macrophages (59). Since IL-12 mediates Th1 activation, PD-1 upregulation in these cells may cause defects in adaptive immunity. PD-1 expression also is increased in peritoneal macrophages of mice with sepsis and is correlated with diminished activity of the macrophages to clear bacteria (43). In a dog model of visceral leishmaniasis, PD-1 expression is shown to cause the exhaustion of both CD8+ and CD4+ T cells, as evidenced by their loss of antigen-specific proliferation and gamma interferon (IFN-γ) production, and to impair the phagocytic function of macrophages (60). PD-1 overexpression in T cells is considered a mechanism of immune evasion by a number of microorganisms, such as Helicobacter pylori, Schistosoma mansoni, Mycobacterium tuberculosis, Mycobacterium leprae, LCMV, HIV, and HCV (28, 29, 61–66). Our finding of PD-1 overexpression in AMs during PcP adds Pneumocystis to the list of organisms that use PD-1 expression to disable host defense mechanisms.
Since PD-1 is immunosuppressive, decreasing its levels could be therapeutic. In an SIV model, monoclonal antibody (EH12-1540) against PD-1 was shown to increase the number of virus-specific CD8+ T cells and reduce viral load (67). Treatment of M. tuberculosis-specific IFN-γ-producing T cells in vitro with anti-PD-1 antibody has been shown to prevent them from undergoing apoptosis, and the number of PD-1+ T cells in patients are decreased with effective tuberculosis therapy (68). Similarly, treatment of lymphocytes from sepsis patients with anti-PD-1 or anti-PD-L1 antibodies prevents their apoptosis and increases their production of IL-2 and IFN-γ (69). Since PD-1 expression also impairs immune responses to cancer, a number of clinical trials are in progress using PD-1 MAb, such as BMS-936558 and MK3475, as a supplemental treatment for hematologic malignancies, melanoma, renal cell carcinoma, colorectal cancer, and non-small-cell lung cancer (70). Anti-PD-L1 antibody also has been used to block the interaction between PD-1 and PD-L1, enhancing CD8+ T-cell responses to HIV-1, HSV-1, LCMV, HCV, respiratory syncytial virus, and SIV infections (27, 29, 67, 71–73).
Our discovery of increased PD-1 expression in AMs during PcP provide additional means for the treatment of PcP by blocking the PD-1/PD-L1 signaling pathway with antibodies as described above or suppressing PD-1 expression. PD-1 expression is activated by NFATc1 (nuclear factor of activated T cells c1), AP-1, NF-κB, and Notch (25, 74, 75). Therefore, inactivation of these transcription factors would reduce PD-1 expression. For example, blocking the Notch signaling pathway with DAPT {N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester} leads to the inhibition of PD-1 expression (74). Similarly, inhibition of NFATc1 with the calcineurin inhibitor cyclosporine reduces PD-1 expression (75). PD-1 expression in T cells can be induced by cytokines, such as IL-2, IL-7, IL-15, and IL-21, Toll-like receptor (TLR) signaling, and interferons (76), and is suppressed by T-bet and Blimp-1 (77, 78). Blimp-1 acts by inhibiting NFATc1 production and competing with it for binding to a control region in the PD-1 promoter (77).
Some microbial components can induce PD-1 or PD-L1 expression. For example, the HCV core protein induces PD-1 expression in T cells and monocytes (45, 79). Lipopolysaccharide (LPS) and HIV-1 virions can induce PD-L1 expression in neutrophils (80). PD-1 is induced to express at higher levels by inflammation, as evidenced by the finding that TLR-3 stimulation causes PD-L1 upregulation in dendritic cells, leading to decreased CD4+ T-cell proliferation (81). The activation of TLR-4, TLR-7, and TLR-8 induces PD-L1 expression in neutrophils (80). Our previous finding that TLR-2 mediates inflammatory responses during PcP (82) suggests that PD-1 expression in AMs during PcP is a result of TLR-2 activation. Pneumocystis components that can trigger TLR-2 responses have not been identified. We speculate that Pneumocystis β-glucan plays such a role.
Based on results of this study, we hypothesize the following. During Pneumocystis infection, Pneumocystis components such as β-glucan interact with TLR-2, triggering inflammatory responses and leading to the accumulation of MDSCs. These MDSCs interact with AMs through PD-1/PD-L1 ligation, causing suppressive histone modification and DNA methylation on the PU.1 gene and, as a result, PU.1 downregulation. Thus, the expression of various macrophage receptors is decreased, and AMs become defective in phagocytosis. We also hypothesize that MDSCs use the same mechanism to interact with monocytes, leading to PU.1 downregulation and inhibition of their differentiation into alveolar macrophages. Therefore, the number of AMs is decreased during PcP.
ACKNOWLEDGMENT
This work was supported in part by grant RO3 AI091418 from the National Institutes of Health.
REFERENCES
- 1.Lasbury ME, Lin P, Tschang D, Durant PJ, Lee CH. 2004. Effect of bronchoalveolar lavage fluid from Pneumocystis carinii-infected hosts on phagocytic activity of alveolar macrophages. Infect Immun 72:2140–2147. doi: 10.1128/IAI.72.4.2140-2147.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koziel H, Eichbaum Q, Kruskal BA, Pinkston P, Rogers RA, Armstrong MY, Richards FF, Rose RM, Ezekowitz RA. 1998. Reduced binding and phagocytosis of Pneumocystis carinii by alveolar macrophages from persons infected with HIV-1 correlates with mannose receptor downregulation. J Clin Invest 102:1332–1344. doi: 10.1172/JCI560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fleury J, Escudier E, Pocholle MJ, Carre C, Bernaudin JF. 1985. Cell population obtained by bronchoalveolar lavage in Pneumocystis carinii pneumonitis. Acta Cytol 29:721–726. [PubMed] [Google Scholar]
- 4.Fleury-Feith J, Van Nhieu JT, Picard C, Escudier E, Bernaudin JF. 1989. Bronchoalveolar lavage eosinophilia associated with Pneumocystis carinii pneumonitis in AIDS patients. Comparative study with non-AIDS patients. Chest 95:1198–1201. [DOI] [PubMed] [Google Scholar]
- 5.Young JA, Hopkin JM, Cuthbertson WP. 1984. Pulmonary infiltrates in immunocompromised patients: diagnosis by cytological examination of bronchoalveolar lavage fluid. J Clin Pathol 37:390–397. doi: 10.1136/jcp.37.4.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young JA, Stone JW, McGonigle RJ, Adu D, Michael J. 1986. Diagnosing Pneumocystis carinii pneumonia by cytological examination of bronchoalveolar lavage fluid: report of 15 cases. J Clin Pathol 39:945–949. doi: 10.1136/jcp.39.9.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadaghdar H, Huang ZB, Eden E. 1992. Correlation of bronchoalveolar lavage findings to severity of Pneumocystis carinii pneumonia in AIDS. Evidence for the development of high-permeability pulmonary edema. Chest 102:63–69. [DOI] [PubMed] [Google Scholar]
- 8.Lasbury ME, Durant PJ, Bartlett MS, Smith JW, Lee CH. 2003. Correlation of organism burden and alveolar macrophage counts during infection with Pneumocystis carinii and recovery. Clin Diagn Lab Immunol 10:293–302. doi: 10.1128/CDLI.10.2.293-302.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lasbury ME, Merali S, Durant PJ, Tschang D, Ray CA, Lee CH. 2007. Polyamine-mediated apoptosis of alveolar macrophages during Pneumocystis pneumonia. J Biol Chem 282:11009–11020. doi: 10.1074/jbc.M611686200. [DOI] [PubMed] [Google Scholar]
- 10.Liao CP, Lasbury ME, Wang SH, Zhang C, Durant PJ, Murakami Y, Matsufuji S, Lee CH. 2009. Pneumocystis mediates overexpression of antizyme inhibitor resulting in increased polyamine levels and apoptosis in alveolar macrophages. J Biol Chem 284:8174–8184. doi: 10.1074/jbc.M805787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang C, Wang SH, Liao CP, Shao S, Lasbury ME, Durant PJ, Lee CH. 2010. Downregulation of PU.1 leads to decreased expression of Dectin-1 in alveolar macrophages during Pneumocystis pneumonia. Infect Immun 78:1058–1065. doi: 10.1128/IAI.01141-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eichbaum Q, Heney D, Raveh D, Chung M, Davidson M, Epstein J, Ezekowitz RA. 1997. Murine macrophage mannose receptor promoter is regulated by the transcription factors PU.1 and SP1. Blood 90:4135–4143. [PubMed] [Google Scholar]
- 13.Moulton KS, Semple K, Wu H, Glass CK. 1994. Cell-specific expression of the macrophage scavenger receptor gene is dependent on PU.1 and a composite AP-1/ets motif. Mol Cell Biol 14:4408–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eichbaum QG, Iyer R, Raveh DP, Mathieu C, Ezekowitz RA. 1994. Restriction of interferon gamma responsiveness and basal expression of the myeloid human Fc gamma R1b gene is mediated by a functional PU.1 site and a transcription initiator consensus. J Exp Med 179:1985–1996. doi: 10.1084/jem.179.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panopoulos AD, Bartos D, Zhang L, Watowich SS. 2002. Control of myeloid-specific integrin alpha Mbeta 2 (CD11b/CD18) expression by cytokines is regulated by Stat3-dependent activation of PU.1. J Biol Chem 277:19001–19007. doi: 10.1074/jbc.M112271200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang C, Lei GS, Shao S, Jung HW, Durant PJ, Lee CH. 2012. Accumulation of myeloid-derived suppressor cells in the lungs during Pneumocystis pneumonia. Infect Immun 80:3634–3641. doi: 10.1128/IAI.00668-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabrilovich DI, Nagaraj S. 2009. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. 2011. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol 91:167–181. doi: 10.1189/jlb.0311177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagaraj S, Gabrilovich DI. 2010. Myeloid-derived suppressor cells in human cancer. Cancer J 16:348–353. doi: 10.1097/PPO.0b013e3181eb3358. [DOI] [PubMed] [Google Scholar]
- 20.Green KA, Cook WJ, Green WR. 2013. Myeloid-derived suppressor cells in murine retrovirus-induced AIDS inhibit T- and B-cell responses in vitro that are used to define the immunodeficiency. J Virol 87:2058–2071. doi: 10.1128/JVI.01547-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gantt S, Gervassi A, Jaspan H, Horton H. 2014. The role of myeloid-derived suppressor cells in immune ontogeny. Front Immunol 5:387. doi: 10.3389/fimmu.2014.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGrath MM, Najafian N. 2012. The role of coinhibitory signaling pathways in transplantation and tolerance. Front Immunol 3:47. doi: 10.3389/fimmu.2012.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, Murphy TL, Russell JH, Allison JP, Murphy KM. 2003. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol 4:670–679. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe N, Nakajima H. 2012. Coinhibitory molecules in autoimmune diseases. Clin Dev Immunol 2012:269756. doi: 10.1155/2012/269756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riella LV, Paterson AM, Sharpe AH, Chandraker A. 2012. Role of the PD-1 pathway in the immune response. Am J Transplant 12:2575–2587. doi: 10.1111/j.1600-6143.2012.04224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. 1998. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 28.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 29.Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari C. 2006. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol 80:11398–11403. doi: 10.1128/JVI.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, Hanson HL, Steinberg JP, Masopust D, Wherry EJ, Altman JD, Rouse BT, Freeman GJ, Ahmed R, Grakoui A. 2007. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol 81:2545–2553. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Velu V, Kannanganat S, Ibegbu C, Chennareddi L, Villinger F, Freeman GJ, Ahmed R, Amara RR. 2007. Elevated expression levels of inhibitory receptor programmed death 1 on simian immunodeficiency virus-specific CD8 T cells during chronic infection but not after vaccination. J Virol 81:5819–5828. doi: 10.1128/JVI.00024-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng G, Li S, Wu W, Tan X, Chen Y, Chen Z. 2008. PD-1 upregulation is associated with HBV-specific T cell dysfunction in chronic hepatitis B patients. Mol Immunol 45:963–970. doi: 10.1016/j.molimm.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 33.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, Zarour HM. 2010. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med 207:2175–2186. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. 1999. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11:141–151. doi: 10.1016/S1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 35.Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, Jussif J, Benoit S, Ireland G, Luxenberg D, Askew GR, Milarski KL, Groves C, Brown T, Carito BA, Percival K, Carreno BM, Collins M, Marusic S. 2007. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol 182:124–134. doi: 10.1016/j.jneuroim.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Keir ME, Francisco LM, Sharpe AH. 2007. PD-1 and its ligands in T-cell immunity. Curr Opin Immunol 19:309–314. doi: 10.1016/j.coi.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 37.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. 2001. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 38.Prokunina L, Castillejo-Lopez C, Oberg F, Gunnarsson I, Berg L, Magnusson V, Brookes AJ, Tentler D, Kristjansdottir H, Grondal G, Bolstad AI, Svenungsson E, Lundberg I, Sturfelt G, Jonssen A, Truedsson L, Lima G, Alcocer-Varela J, Jonsson R, Gyllensten UB, Harley JB, Alarcon-Segovia D, Steinsson K, Alarcon-Riquelme ME. 2002. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat Genet 32:666–669. doi: 10.1038/ng1020. [DOI] [PubMed] [Google Scholar]
- 39.Kitazawa Y, Fujino M, Wang Q, Kimura H, Azuma M, Kubo M, Abe R, Li XK. 2007. Involvement of the programmed death-1/programmed death-1 ligand pathway in CD4+CD25+ regulatory T-cell activity to suppress alloimmune responses. Transplantation 83:774–782. doi: 10.1097/01.tp.0000256293.90270.e8. [DOI] [PubMed] [Google Scholar]
- 40.Wang W, Lau R, Yu D, Zhu W, Korman A, Weber J. 2009. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25(Hi) regulatory T cells. Int Immunol 21:1065–1077. doi: 10.1093/intimm/dxp072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang R, Cai Z, Zhang Y, Yutzy WH, Roby KF, Roden RB. 2006. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res 66:6807–6815. doi: 10.1158/0008-5472.CAN-05-3755. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Zeng B, Zhang Z, Zhang Y, Yang R. 2008. B7-H1 on myeloid-derived suppressor cells in immune suppression by a mouse model of ovarian cancer. Clin Immunol 129:471–481. doi: 10.1016/j.clim.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 43.Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, Swan R, Kherouf H, Monneret G, Chung CS, Ayala A. 2009. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U S A 106:6303–6308. doi: 10.1073/pnas.0809422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao D, Xu H, Guo G, Ruan Z, Fei L, Xie Z, Wu Y, Chen Y. 2013. Intrahepatic expression of programmed death-1 and its ligands in patients with HBV-related acute-on-chronic liver failure. Inflammation 36:110–120. doi: 10.1007/s10753-012-9525-7. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Ma CJ, Ni L, Zhang CL, Wu XY, Kumaraguru U, Li CF, Moorman JP, Yao ZQ. 2011. Cross-talk between programmed death-1 and suppressor of cytokine signaling-1 in inhibition of IL-12 production by monocytes/macrophages in hepatitis C virus infection. J Immunol 186:3093–3103. doi: 10.4049/jimmunol.1002006. [DOI] [PubMed] [Google Scholar]
- 46.Cho HY, Choi EK, Lee SW, Jung KO, Seo SK, Choi IW, Park SG, Choi I, Lee SW. 2009. Programmed death-1 receptor negatively regulates LPS-mediated IL-12 production and differentiation of murine macrophage RAW264.7 cells. Immunol Lett 127:39–47. doi: 10.1016/j.imlet.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Cho HY, Lee SW, Seo SK, Choi IW, Choi I, Lee SW. 2008. Interferon-sensitive response element (ISRE) is mainly responsible for IFN-alpha-induced upregulation of programmed death-1 (PD-1) in macrophages. Biochim Biophys Acta 1779:811–819. doi: 10.1016/j.bbagrm.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 48.Serezani CH, Kane S, Collins L, Morato-Marques M, Osterholzer JJ, Peters-Golden M. 2012. Macrophage dectin-1 expression is controlled by leukotriene B4 via a GM-CSF/PU.1 axis. J Immunol 189:906–915. doi: 10.4049/jimmunol.1200257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lasbury ME, Tang X, Durant PJ, Lee CH. 2003. Effect of transcription factor GATA-2 on phagocytic activity of alveolar macrophages from Pneumocystis carinii-infected hosts. Infect Immun 71:4943–4952. doi: 10.1128/IAI.71.9.4943-4952.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reichner JS, Fitzpatrick PA, Wakshull E, Albina JE. 2001. Receptor-mediated phagocytosis of rat macrophages is regulated differentially for opsonized particles and non-opsonized particles containing beta-glucan. Immunology 104:198–206. doi: 10.1046/j.1365-2567.2001.01291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roedel EQ, Cafasso DE, Lee KW, Pierce LM. 2012. Pulmonary toxicity after exposure to military-relevant heavy metal tungsten alloy particles. Toxicol Appl Pharmacol 259:74–86. doi: 10.1016/j.taap.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 52.Polancec DS, Munic Kos V, Banjanac M, Vrancic M, Cuzic S, Belamaric D, Parnham MJ, Polancec D, Erakovic Haber V. 2012. Azithromycin drives in vitro GM-CSF/IL-4-induced differentiation of human blood monocytes toward dendritic-like cells with regulatory properties. J Leukoc Biol 91:229–243. doi: 10.1189/jlb.1210655. [DOI] [PubMed] [Google Scholar]
- 53.Hoogenkamp M, Krysinska H, Ingram R, Huang G, Barlow R, Clarke D, Ebralidze A, Zhang P, Tagoh H, Cockerill PN, Tenen DG, Bonifer C. 2007. The PU.1 locus is differentially regulated at the level of chromatin structure and noncoding transcription by alternate mechanisms at distinct developmental stages of hematopoiesis. Mol Cell Biol 27:7425–7438. doi: 10.1128/MCB.00905-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Gigliotti F, Bhagwat SP, George TC, Wright TW. 2010. Immune modulation with sulfasalazine attenuates immunopathogenesis but enhances macrophage-mediated fungal clearance during Pneumocystis pneumonia. PLoS Pathog 6:e1001058. doi: 10.1371/journal.ppat.1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martin TR, Frevert CW. 2005. Innate immunity in the lungs. Proc Am Thorac Soc 2:403–411. doi: 10.1513/pats.200508-090JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakata K, Kanazawa H, Watanabe M. 2006. Why does the autoantibody against granulocyte-macrophage colony-stimulating factor cause lesions only in the lung? Respirology 11(Suppl):S65–S69. doi: 10.1111/j.1440-1843.2006.00812.x. [DOI] [PubMed] [Google Scholar]
- 57.Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. 2012. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Science Signal 5:ra46. doi: 10.1126/scisignal.2002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Tuzova M, Xiao ZX, Cruikshank WW, Center DM. 2008. Pro-IL-16 recruits histone deacetylase 3 to the Skp2 core promoter through interaction with transcription factor GABP. J Immunol 180:402–408. doi: 10.4049/jimmunol.180.1.402. [DOI] [PubMed] [Google Scholar]
- 59.Ma CJ, Ni L, Zhang Y, Zhang CL, Wu XY, Atia AN, Thayer P, Moorman JP, Yao ZQ. 2011. PD-1 negatively regulates interleukin-12 expression by limiting STAT-1 phosphorylation in monocytes/macrophages during chronic hepatitis C virus infection. Immunology 132:421–431. doi: 10.1111/j.1365-2567.2010.03382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Esch KJ, Juelsgaard R, Martinez PA, Jones DE, Petersen CA. 2013. Programmed death 1-mediated T cell exhaustion during visceral leishmaniasis impairs phagocyte function. J Immunol 191:5542–5550. doi: 10.4049/jimmunol.1301810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, Routy JP, Haddad EK, Sekaly RP. 2006. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med 12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 62.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. 2007. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol 8:239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 63.Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. 2006. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol 176:3000–3009. doi: 10.4049/jimmunol.176.5.3000. [DOI] [PubMed] [Google Scholar]
- 64.Smith P, Walsh CM, Mangan NE, Fallon RE, Sayers JR, McKenzie AN, Fallon PG. 2004. Schistosoma mansoni worms induce anergy of T cells via selective up-regulation of programmed death ligand 1 on macrophages. J Immunol 173:1240–1248. doi: 10.4049/jimmunol.173.2.1240. [DOI] [PubMed] [Google Scholar]
- 65.Jurado JO, Alvarez IB, Pasquinelli V, Martinez GJ, Quiroga MF, Abbate E, Musella RM, Chuluyan HE, Garcia VE. 2008. Programmed death (PD)-1:PD-ligand 1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J Immunol 181:116–125. doi: 10.4049/jimmunol.181.1.116. [DOI] [PubMed] [Google Scholar]
- 66.Alvarez IB, Pasquinelli V, Jurado JO, Abbate E, Musella RM, de la Barrera SS, Garcia VE. 2010. Role played by the programmed death-1-programmed death ligand pathway during innate immunity against Mycobacterium tuberculosis. J Infect Dis 202:524–532. doi: 10.1086/654932. [DOI] [PubMed] [Google Scholar]
- 67.Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, Ahmed R, Amara RR. 2009. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 458:206–210. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh A, Mohan A, Dey AB, Mitra DK. 2013. Inhibiting the programmed death 1 pathway rescues Mycobacterium tuberculosis-specific interferon gamma-producing T cells from apoptosis in patients with pulmonary tuberculosis. J Infect Dis 208:603–615. doi: 10.1093/infdis/jit206. [DOI] [PubMed] [Google Scholar]
- 69.Chang K, Svabek C, Vazquez-Guillamet C, Sato B, Rasche D, Wilson S, Robbins P, Ulbrandt N, Suzich J, Green J, Patera AC, Blair W, Krishnan S, Hotchkiss R. 2014. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care 18:R3. doi: 10.1186/cc13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Topalian SL, Drake CG, Pardoll DM. 2012. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol 24:207–212. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seung E, Dudek TE, Allen TM, Freeman GJ, Luster AD, Tager AM. 2013. PD-1 blockade in chronically HIV-1-infected humanized mice suppresses viral loads. PLoS One 8:e77780. doi: 10.1371/journal.pone.0077780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Telcian AG, Laza-Stanca V, Edwards MR, Harker JA, Wang H, Bartlett NW, Mallia P, Zdrenghea MT, Kebadze T, Coyle AJ, Openshaw PJ, Stanciu LA, Johnston SL. 2011. RSV-induced bronchial epithelial cell PD-L1 expression inhibits CD8+ T cell nonspecific antiviral activity. J Infect Dis 203:85–94. doi: 10.1093/infdis/jiq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Channappanavar R, Twardy BS, Suvas S. 2012. Blocking of PDL-1 interaction enhances primary and secondary CD8 T cell response to herpes simplex virus-1 infection. PLoS One 7:e39757. doi: 10.1371/journal.pone.0039757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mathieu M, Cotta-Grand N, Daudelin JF, Thebault P, Labrecque N. 2013. Notch signaling regulates PD-1 expression during CD8(+) T-cell activation. Immunol Cell Biol 91:82–88. doi: 10.1038/icb.2012.53. [DOI] [PubMed] [Google Scholar]
- 75.Oestreich KJ, Yoon H, Ahmed R, Boss JM. 2008. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol 181:4832–4839. doi: 10.4049/jimmunol.181.7.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O'Shea MA, Fauci AS. 2008. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol 181:6738–6746. doi: 10.4049/jimmunol.181.10.6738. [DOI] [PubMed] [Google Scholar]
- 77.Lu P, Youngblood BA, Austin JW, Mohammed AU, Butler R, Ahmed R, Boss JM. 2014. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J Exp Med 211:515–527. doi: 10.1084/jem.20130208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, Intlekofer AM, Boss JM, Reiner SL, Weinmann AS, Wherry EJ. 2011. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol 12:663–671. doi: 10.1038/ni.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yao ZQ, King E, Prayther D, Yin D, Moorman J. 2007. T cell dysfunction by hepatitis C virus core protein involves PD-1/PDL-1 signaling. Viral Immunol 20:276–287. doi: 10.1089/vim.2006.0096. [DOI] [PubMed] [Google Scholar]
- 80.Bowers NL, Helton ES, Huijbregts RP, Goepfert PA, Heath SL, Hel Z. 2014. Immune suppression by neutrophils in HIV-1 infection: role of PD-L1/PD-1 pathway. PLoS Pathog 10:e1003993. doi: 10.1371/journal.ppat.1003993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Groschel S, Piggott KD, Vaglio A, Ma-Krupa W, Singh K, Goronzy JJ, Weyand CM. 2008. TLR-mediated induction of negative regulatory ligands on dendritic cells. J Mol Med 86:443–455. doi: 10.1007/s00109-008-0310-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang C, Wang SH, Lasbury ME, Tschang D, Liao CP, Durant PJ, Lee CH. 2006. Toll-like receptor 2 mediates alveolar macrophage response to Pneumocystis murina. Infect Immun 74:1857–1864. doi: 10.1128/IAI.74.3.1857-1864.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]