

Abstract
Purpose.
Previously, we reported that pioglitazone prevented insulin resistance and cell death in type 2 diabetic retina by reducing TNFα and suppressor of cytokine signaling 3 (SOCS3) levels. Numerous reports suggest prominent vasoprotective effects of insulin growth factor binding protein–3 (IGFBP-3) in diabetic retinopathy. We hypothesized that pioglitazone protects against retinal cell apoptosis by regulating IGFBP-3 levels, in addition to reducing TNFα. The current study explored potential IGFBP-3 regulatory pathways by pioglitazone in retinal endothelial cells cultured in high glucose.
Methods.
Primary human retinal endothelial cells (REC) were grown in normal (5 mM) and high glucose (25 mM) and treated with pioglitazone for 24 hours. Cell lysates were processed for Western blotting and ELISA analysis to evaluate IGFBP-3, TNFα, and cleaved caspase 3 protein levels.
Results.
Our results show that treatment with pioglitazone restored the high glucose–induced decrease in IGFBP-3 levels. This regulation was independent of TNFα actions, as reducing TNFα levels with siRNA did not prevent pioglitazone from increasing IGFBP-3 levels. Pioglitazone required protein kinase A (PKA) and DNA-dependent protein kinase (DNA PK) activity to regulate IGFBP-3, as specific inhibitors for each protein prevented pioglitazone-mediated normalization of IGFBP-3 in high glucose. Insulin growth factor binding protein–3 activity was increased and apoptosis decreased by pioglitazone, which was eliminated when serine site 156 of IGFBP-3 was mutated suggesting a key role of this phosphorylation site in pioglitazone actions.
Conclusions.
Our findings suggest that pioglitazone mediates regulation of IGFBP-3 via activation of PKA/DNA PK pathway in hyperglycemic retinal endothelial cells.
Keywords: pioglitazone, retinal endothelial cells, IGFBP-3, DNA PK
Pioglitazone regulates insulin growth factor binding protein–3 through serine 156, independent of changes in TNFα levels.
Introduction
Diabetic retinopathy is recognized as the leading cause of blindness and visual impairment in the working-age population.1 Retinal microvascular damage caused by hyperglycemia common to both type 1 and type 2 diabetes is a strong factor in diabetic retinopathy complications.2–4 Thiazolidinedione drugs such as rosiglitazone and pioglitazone are ligands for peroxisome proliferator-activated receptor γ (PPARγ) and show promise for treatment of diabetes due to their ability to control systemic glucose levels and insulin resistance.5 Pioglitazone protects against retinal apoptosis in streptozotocin-induced diabetes,5 ischemia/reperfusion,6 and optic nerve crush.7 We recently reported that pioglitazone improved impaired insulin signaling, prevented associated retinal cell death in type 2 diabetic rats, and reduced TNFα and suppressor of cytokine signaling 3 (SOCS3) levels by increasing PPARγ activity. The retinas of type 2 diabetic obese rats had reduced levels of insulin growth factor binding protein-3 (IGFBP-3) protein that was restored with pioglitazone treatment.8 Thus, pioglitazone works in multiple ways to restore normal-glucose levels and prevent retinal damage. However, little is known about pioglitazone actions in retina and there is a need to further elucidate pathways involved in its beneficial effects on diabetic retinopathy. Because we have previously reported that IGFBP-3 levels are reduced in retinal endothelial cells cultured in hyperglycemic conditions and pioglitazone increases IGFBP-3 levels in diabetic rats, we hypothesized that the protective actions of pioglitazone in retina involves IGFBP-3 regulation, in addition to its proinflammatory and insulin-sensitizing properties. We questioned whether pioglitazone regulates IGFBP-3 in retinal endothelial cells cultured in high glucose and which potential pathways may be involved.
Insulin growth factor binding protein–3 stabilizes the insulin growth factors (IGFs) through the formation of IGF/IGFBP complexes. Several reports have suggested protective effects of IGFBP-3 on retinal vasculature.9–11 Insulin growth factor binding protein–3 suppresses apoptosis in diabetic retinopathy both in vivo and in vitro and decreases neovascular tuft formation in murine model of oxygen-induced retinopathy.9,12,13 DNA-dependent protein kinase (DNA PK) modulates IGFBP-3 activity by phosphorylation at sites: Ser156, Ser165, and Thr170, while casein kinase 2 (CK2) modulates IGFBP-3 activity by phosphorylation at sites: Ser111 and Ser113.14,15 Insulin growth factor binding protein–3 is known to inhibit TNFα-induced expression of proinflammatory molecules.16,17 A reciprocal relationship was reported between TNFα and IGFBP-3 in the retina of IGFBP-3 knockout mice and in cultured retinal endothelial cells, where TNFα reduced IGFBP-3, and IGFBP-3 in turn lowered TNFα and TNFα receptor levels.17–19 In those studies, TNFα activated P38 MAPK and CK2, leading to inhibition of IGFBP-3 actions through phosphorylation at sites Ser111 and 113 of IGFBP-3.19
Previous studies have reported that protein kinase A (PKA) increases IGFBP-3 levels through activation of DNA PK–induced phosphorylation of IGFBP-3 on serine 156 in retinal endothelial cells.20 In the present study, we analyzed the actions of pioglitazone on IGFBP-3 in retinal endothelial cells cultured in high glucose and investigated the IGFBP-3 regulatory mechanisms involving PKA, DNA PK, and TNFα. Although TNFα is reported to be decreased by pioglitazone,8 our results demonstrated that pioglitazone actions on IGFBP-3 were independent of TNFα actions. Moreover, pioglitazone required active PKA and DNA PK to increase IGFBP-3 levels and did so through DNA PK–induced phosphorylation of IGFBP-3 on serine 156.
Materials and Methods
Reagents
The IGFBP-3 antibody, β-actin antibody, and protein A/G PLUS-agarose beads were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The Phosphoserine antibody was purchased from EMD Millipore (Temecula, CA, USA). Platelet endothelial cell adhesion molecule (PECAM)-1 antibody was from Cell Signaling (Danvers, MA, USA). Human TNFα siRNA and scrambled siRNA were from Dharmacon RNAi Technologies (Chicago, IL, USA). GenMute transfection reagent was from SignaGen Laborataries (Gaithersburg, MD, USA) and Lipofectamine RNAiMax reagent was from Invitrogen (Carlsbad, CA, USA). Horseradish peroxidase conjugated secondary anti-rabbit antibody was purchased from Promega (Madison, WI, USA). HRP conjugated anti-mouse antibody and SuperSignal WestPico Chemiluminescent Substrate for immunoblot development and signal detection were from ThermoScientific (Rockford, IL, USA). NU7441 and KT5720 were purchased from Tocris Biosciences (Minneapolis, MN, USA). Pioglitazone hydrochloride was purchased from Tocris Biosciences (Minneapolis, MN, USA) and solubilized with dimethyl sulfoxide (DMSO) to 250 mM. The IGFBP-3 NB plasmid DNA was a gift from Maria Grant, MD. The phosphorylation Ser156 site of IGFBP-3 NB plasmid was mutated to alanine as described previously.20
Cell Culture
Primary human retinal endothelial cells were provided by Cell System Corporation (Kirkland, WA, USA) and were grown in M131 medium supplemented with microvascular growth supplements (MVGS), 10 ug/mL gentamycin and 0.25 ug/mL amphotericin B. Cells were maintained at 37°C in a humidified 95% air and 5% CO2 atmosphere. Primary cells only were used within six passages. Cells were maintained in either normal (5 mM) or high glucose (25 mM) for a total of 3 days. Cells were starved for 6 hours before treating with 25 μM pioglitazone and were harvested for protein analysis after 24 hours of pioglitazone treatment. NU7441 (10 uM) and KT5720 (1 uM) were added to the cells for 30 minutes before pioglitazone treatment in order to block the activity of DNA PK and PKA, respectively, prior to the activation of PPARγ. The doses of NU7441 and KT5720 used for inhibiting DNA PK and PKA were selected based on previous reports from our group and others.20–24
Transfection of siRNA and Plasmid DNA
Retinal endothelial cells were transfected with 20 nM TNFα siRNA or scrambled siRNA using Lipofectamine RNAiMAX reagent according to the manufacturer's instructions. Briefly, 60% to 70% confluent cells were used for transfection in a 60-mm culture dish. Sixty picomoles of RNAi was diluted in 500 μL OPTI-MEM medium and separately 15 μL of Lipofectamine RNAiMAX reagent was diluted in 500 μL OPTI-MEM. The two solutions were mixed, incubated for 10 minutes at room temperature, and added to the cells in 60-mm culture dishes. The REC were transfected with TNFα siRNA for 24 hours, followed by transfection of the IGFBP-3 NB plasmid, IGFBP-3 mutant, or control CMV plasmid. Cells were harvested 48 hours later. Plasmids were transfected with GenMute siRNA and DNA transfection reagent according to manufacturer's protocol. Complete medium was added 30 to 60 minutes before transfection to approximately 90% confluent cells. Ten micrograms of the plasmids were mixed with 300 μL transfection buffer and 30 μL GenMute transfection reagent, and incubated for 10 minutes at room temperature. The transfection complexes were added to the REC in 60-mm culture dishes dropwise and replaced with cell growth medium 24 hours after transfection.
Western Blot Analysis
Retinal endothelial cells were rinsed with cold PBS after treatments and scraped in lysis buffer containing protease and phosphatase inhibitors. The lysate was kept on ice for 30 minutes and cleared by centrifugation for 20 minutes at 4°C. Equal amounts of protein from the lysate were separated on the precast tris-glycine gels (Invitrogen) and blotted onto a nitrocellulose membrane. The blots were blocked with TBST (10 mM Tris-HCl buffer, pH 8.0, 150 mM NaCl, 0.1% Tween 20) containing 5% (wt/vol) bovine serum albumin, and then incubated with respective primary antibodies. The antibodies anti-IGFBP3 and anti-phosphoserine were used at 1:500 and β-actin antibody was used at 1:2000 dilutions. Membranes were then washed with TBST and incubated with horseradish peroxidase labeled secondary antibodies. The antigen-antibody complexes were detected using West Pico Chemiluminescence reagent. Mean densitometry of the bands were assessed using Kodak Image Station 4000MM software (Kodak, Carestream Health, Bioimaging, Bend, OR, USA).
ELISA Analysis
An ELISA for cleaved caspase 3 was performed using a cleaved caspase 3 ELISA assay kit (Cell Signaling) according to the manufacturer's instructions to evaluate the levels of active apoptotic markers in the cell lysate. Equal protein was loaded into all wells to allow for comparisons based on optical density. Tumor necrosis factor–α protein concentrations were measured using a TNFα ELISA kit (ThermoFisher, Pittsburgh, PA, USA), based on provided standards. Equal protein concentrations were used in each well to insure that cell numbers did not affect TNFα concentrations.
Immunoprecipitation
The immunoprecipitation of IGFBP-3 was done as described previously.20 Cells were rinsed with PBS and lysed in lysis buffer containing the protease and phosphatase inhibitors for 20 minutes in ice. The lysate was cleared by centrifugation for 20 minutes at 4°C. Equal amount of protein from the lysates were incubated with IGFBP-3 antibody overnight at 4°C with gentle rocking. Protein A/G Plus-Agarose beads were added and incubation was continued for another 2 hours. The beads were then washed with lysis buffer three times and with PBS one time and were heated in Laemmli sample buffer to release the immunocomplexes. The immunoprecipitates were analyzed for IGFBP-3 and phospho–IGFBP-3 by Western blotting.
Apoptosis Analysis
Retinal endothelial cells cultured in normal and high glucose were harvested and washed twice with cold PBS/2% FBS. Cell pellets were resuspended in Annexin V Binding Buffer (BioLegend, San Diego, CA, USA) at a concentration of 5.0 × 106 cells/mL following manufacturer's instructions. Cells were labeled with 5 μL of Annexin V-FITC and 10 μL of Propidium iodide (PI) solution/100 μL of cell suspension. Tubes were vortexed gently and incubated for 15 minutes at room temperature/dark followed by addition of 200 μL of Annexin V Binding Buffer to each sample. Analysis done in BD LSRII Flow Cyometry Analyzer (BD Biosciences, San Jose, CA, USA). Percentage of dead cells defined as Annexin V+PI+. Live cells defined as Annexin VnegPIneg. Experimental analysis performed using FlowJo xV10.0.6 software (Tree Star, Inc., Ashland, OR, USA).
Flow Cytometry Analysis of PECAM Expression
Cells were washed in PBS/2% FBS after harvesting. Cells were labeled with 5 μL anti-human PECAM (aka CD31, clone WM59; BioLegend) directly conjugated to APC-Cy7 to 1.0 × 106 RECs in 95 μL of PBS/2% FBS for 30 minutes/cold/dark. Samples were washed twice and resuspended in 250 μL of PBS/2% FBS before analysis. To determine positivity we used unlabeled sample and mouse IgG1κ isotype control.
Statistics
The experiments were repeated at least three times and the data are presented as mean ± SEM. The data were analyzed by Kruskal-Wallis, followed by Dunn's test. P values less than 0.05 were considered statistically significant. The treatment groups were normalized to the control and represented as fold change. One representative blot is shown for the Western blots.
Results
High Glucose Induced Cell Death in Retinal Endothelial Cells
To investigate whether in vitro high-glucose treatment on primary human REC-induced apoptosis, flow cytometry was used. First, to confirm REC cultures maintain their endothelial cell phenotype through multiple passaging, retinal endothelial cells in normal (5 mM) or high glucose (25 mM) were labeled for PECAM/CD31, a classical endothelial cell marker. Figure 1A confirmed these cells are REC with 85% of cells showing positivity against PECAM-1. Moreover, modulation of glucose levels in the media did change the expression of PECAM-1 as REC grown in both normal and high glucose have similar levels of PECAM-1. Next, we assessed cell death and viability by Annexin V and PI labeling. Briefly, cells cultured in normal and high glucose were labeled for Annexin V and PI simultaneously and analyzed by flow cytometry. Percentage of dead cells is determined by percentage of Annexin V+PI+ cells. As shown in Figure 1B, cells in normal glucose had 4.1% dead cells, whereas high-glucose culture conditions led to 11% dead cells, a 2.7-fold increase in cell death. Total percentage of live cells was 77% in normal-glucose conditions and 72.5% in high glucose. Collectively, REC maintain their PECAM-1 expression in culture and high-glucose culture conditions increased cell death of REC.
Figure 1.

High glucose-induced REC cell death. (A) Flow cytometry analysis of PECAM-1 in REC. Solid histogram shows levels of mouse IgG1κ isotype control and open histogram shows experimental sample results. (B) Annexin V versus PI labeling to determine apoptosis. Normal and high glucose–cultured cells were labeled with Annexin V-FITC and PI prior analysis. Percentage dead cells: percent Annexin V+PI+, percent live cells Annexin VnegPIneg.
Pioglitazone Increases IGFBP-3 in High Glucose Independently of TNFα
One day after plating, the cells were transfected with scrambled siRNA or TNFα siRNA for 24 hours followed by treatment with pioglitazone (25 μM) for the next 24 hours after which cells were harvested for protein analysis. Retinal endothelial cells were maintained in normal (5 mM) and high glucose (25 mM) for 3 days including siRNA transfection and pioglitazone treatment time. Western blot analysis of IGFBP-3 protein levels indicated that high glucose decreased IGFBP-3 levels as compared with normal glucose (Fig. 2A) as had been reported earlier.25 Pioglitazone treatment significantly reversed the decrease in IGFBP-3 levels. Pioglitazone decreased TNFα levels in retinal endothelial cells and Müller cells, as well as in the diabetic retina.8 Additionally, we have previously reported that TNFα decreased IGFBP-3 levels,19 therefore, we wanted to ascertain whether pioglitazone actions on IGFBP-3 were mediated through TNFα. Knockdown of TNFα with siRNA did not eliminate the actions of pioglitazone on IGFBP-3 (Fig. 2A), suggesting that pioglitazone increases IGFBP-3 levels in high glucose via a TNFα-independent mechanism. Tumor necrosis factor–α was knocked down effectively with TNFα siRNA transfection compared to the scrambled siRNA (Fig. 2B).
Figure 2.

Pioglitazone induced IGFBP-3 levels in high-glucose medium in a TNFα independent way. (A) Western blot analysis of IGFBP-3 to β-actin ratio in REC transfected with scrambled and TNFα siRNA for 24 hours followed by treatment with 25 μM pioglitazone for 24 hours in 5 and 25 mM glucose. (B) Bar graph of TNFα levels after TNFα transfection. (A) *P < 0.05 versus untreated NG control. #P < 0.05 versus untreated HG control. N = 4. (B) *P < 0.05 versus untreated NG control. #P < 0.05 versus untreated HG control. $P < 0.05 versus respective scrambled siRNA control. Data are mean ± SEM. N = 4.
Pioglitazone Induced IGFBP-3 Expression Requires PKA Activity
Since PKA has been reported to regulate IGFBP-3 levels,20 we wanted to determine whether pioglitazone uses PKA activity to increase IGFBP-3 levels. Retinal endothelial cells were transfected as before with TNFα siRNA, 1 day after plating cells followed by treatment with the PKA inhibitor, KT5720, for 30 minutes prior to stimulation with pioglitazone for 24 hours. Figure 3 shows that treatment with KT5720 blocked pioglitazone actions to increase IGFBP-3 levels, suggesting that pioglitazone requires PKA activity to induce IGFBP-3. This response was observed in the absence of TNFα indicating that TNFα was not involved in pioglitazone-mediated regulation of IGFBP-3.
Figure 3.
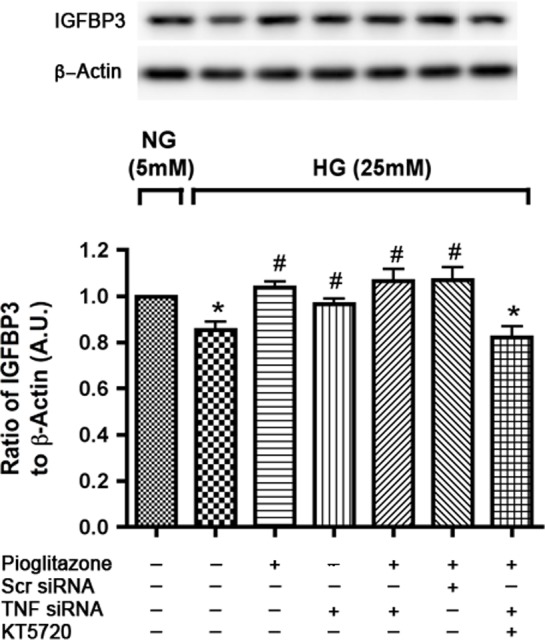
Pioglitazone induced IGFBP-3 expression is through PKA activation in high-ambient glucose. Figure shows bar graph of IGFBP-3 to β-actin levels measured by Western blot in REC cultured in high glucose (25 mM) with TNFα siRNA transfection for 24 hours and then treatment with KT 5720 for 30 minutes followed by 24 hours pioglitazone treatment. Retinal endothelial cells in normal glucose (5 mM) was used as control. *P < 0.05 versus untreated normal glucose control. #P < 0.05 versus untreated high glucose control. Data are mean ± SEM, N = 4.
DNA PK Is Required in Pioglitazone-Induced IGFBP-3 Expression
DNA PK has been reported to play a role in β-adrenergic receptor-PKA induced expression of IGFBP-3 in retinal endothelial cells cultured in high glucose.20 To determine whether this pathway was also involved in pioglitazone actions on IGFBP-3, REC were transfected with TNFα siRNA 1 day after plating and treated with the DNA PK inhibitor, NU7441, for 30 minutes20 followed by 24 hours pioglitazone treatment under high-glucose conditions. Cells were maintained in normal (5 mM) and high glucose (25 mM) medium throughout the experiment. Results show that pioglitazone induces IGFBP-3 levels through DNA PK activation since treatment with NU 7441 prevented pioglitazone to increase IGFBP-3 (Fig. 4).
Figure 4.
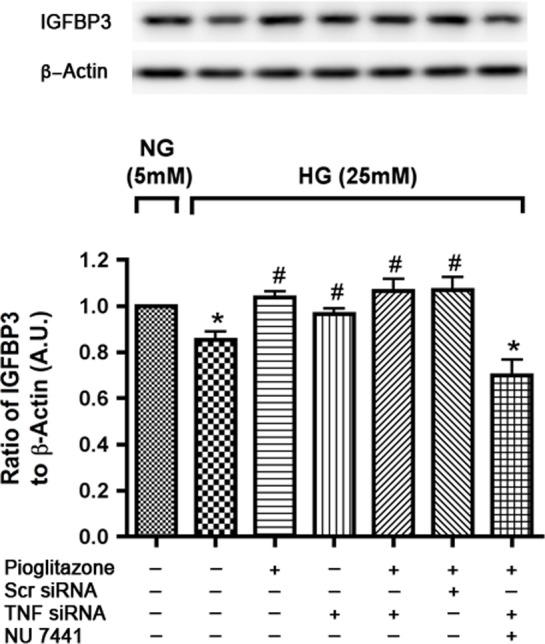
Pioglitazone requires DNA-PK activity to induce IGFBP-3 in high glucose medium. Western blot analysis of IGFBP3 levels in RECs transfected with TNFα siRNA and treated with NU 7441 for 30 minutes followed by pioglitazone treatment for 24 hours in high-glucose medium (25 mM). Retinal endothelial cells in normal glucose (5 mM) were used as control. *P < 0.05 versus untreated normal glucose control. #P < 0.05 versus untreated high glucose control. Data are mean ± SEM, N = 4.
Phosphorylation Site Serine 156 of IGFBP-3 Is Required for Its Pioglitazone-Induced Regulation of IGFBP-3
We have previously shown that serine 156 on IGFBP-3 is key for DNA PK regulation of IGFBP-3.20 Because DNA PK is required for pioglitazone-induced regulation of IGFBP-3, we wanted to determine whether serine 156 on IGFBP-3 is essential for its regulation by pioglitazone. Retinal endothelial cells in high-glucose medium were transfected with TNFα siRNA as before to rule out the involvement of TNFα in pioglitazone-induced IGFBP-3 regulation. Retinal endothelial cells were then transfected with control plasmid, IGFBP-3 NB plasmid, or the IGFBP-3 NB plasmid in which serine at the site 156 is mutated to alanine to prevent phosphorylation. This was followed by treatment with pioglitazone for 24 hours after 1 day of plasmid transfection. Insulin growth factor binding protein–3 was immunoprecipitated and blotted with IGFBP-3 and phosphoserine antibody. Successful transfection is demonstrated in Figure 5 showing high levels of IGFBP-3 after transfection with IGFBP-3 and its mutant plasmid. Pioglitazone increased the ratio of serine phosphorylation of IGFBP-3 to total IGFBP-3 when transfected with IGFBP-3 wild-type and the control plasmid, whereas transfection with the mutant IGFBP-3 plasmid prevented the pioglitazone-induced increase in IGFBP-3 phosphorylation (Fig. 5), demonstrating that this phosphorylation site is critical for IGFBP-3 activation by pioglitazone.
Figure 5.
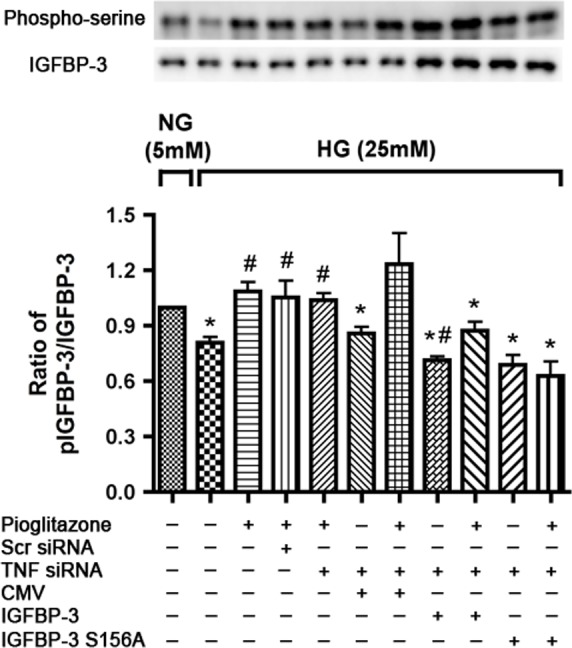
Pioglitazone does not activate IGFBP-3 with transfection of IGFBP-3 S156A plasmid in high glucose medium. Bar graph of ratio of Western blots analysis of p-IGFBP-3 to IGFBP-3 in RECs cultured in high glucose transfected with TNFα siRNA followed by transfection of CMV, IGFBP-3, and IGFBP-3 S156A plasmid next day. Pioglitazone was added for 24 hours after another day. Retinal endothelial cells in normal glucose (5 mM) were used as control. Protein was immunoprecipitated with anti–IGFBP-3 antibody and immunoblotted using anti–IGFBP-3 and antiphosphoserine antibodies. *P < 0.05 versus untreated normal glucose control. #P < 0.05 versus untreated high glucose control. Data are mean ± SEM, N = 4.
Phosphorylation Site Serine 156 of IGFBP-3 Is Involved in Pioglitazone-Induced IGFBP-3 Expression to Inhibit Apoptosis in Hyperglycemic Conditions in RECs
It has recently been demonstrated in our laboratory that pioglitazone reduced apoptosis of REC cultured under hyperglycemic conditions. Insulin growth factor binding protein–3 has also been reported to increase survival of retinal cells in diabetic animals.9 We questioned whether the IGFBP-3 phosphorylation site serine 156 plays a role in antiapoptotic effects of pioglitazone. Cells in high-glucose medium were transfected as described previously with TNFα siRNA followed by transfection with CMV, IGFBP-3, or IGFBP-3 mutant plasmid and subsequently treated with pioglitazone. Transfection with the mutant IGFBP-3 partially prevented the observed decrease in cleaved caspase 3 with pioglitazone treatment when transfected with control and wild-type IGFBP-3 plasmid (Fig. 6). This demonstrates that phosphorylation at this site is involved in pioglitazone-induced IGFBP-3 levels to inhibit apoptosis in high-glucose conditions and pioglitazone actions do not involve TNFα pathway.
Figure 6.
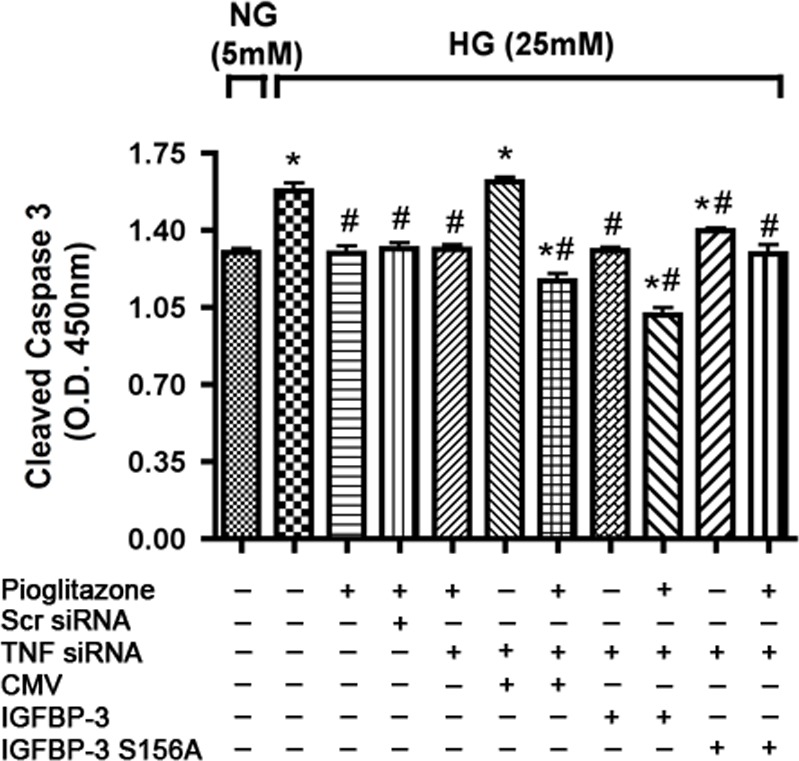
IGFBP-3 S156A plasmid DNA transfection partially reverses inhibition of apoptosis with pioglitazone. Bar graph of cleaved caspase 3 expression of REC in normal (5 mM) and high glucose (25 mM). Retinal endothelial cells in high-glucose medium were transfected with TNFα siRNA for 24 hours followed by transfection with CMV, IGFBP-3, and IGFBP-3 S156A plasmid and subsequent treatment with pioglitazone next day for 24 hours. *P < 0.05 versus untreated normal glucose control. #P < 0.05 versus untreated high glucose control. Data are mean ± SEM, N = 4.
Discussion
In the present study, we show for the first time that pioglitazone, a PPARγ ligand, regulates IGFBP-3 in retinal endothelial cells cultured in high glucose. Insulin growth factor binding protein–3 is reported to suppress retinopathy by protection of the retinal vasculature and restores normal insulin signaling and apoptosis in retina,9,12,18,26,27 thus, making it an ideal candidate for studies related to diabetic retinopathy. Pioglitazone has a protective effect on retinal cell damage associated with retinal ischemia/reperfusion, optic nerve crush, oxygen-induced retinopathy, and diabetic retinopathy.5–7 The activation of PPARγ in the retina by pioglitazone is reported to reduce Müller glial activation, thus protecting retinal ganglion cell death from optic nerve crush.7 Retinal cell damage and activation of glia cells caused by retinal ischemia/reperfusion was also prevented by pioglitazone possibly via NF-κB pathway.6 Downregulation of PPARγ is associated with pathogenesis of oxygen-induced retinopathy and diabetic retinopathy.5,28 Additionally, pioglitazone treatment is reported to improve insulin sensitivity in type 2 diabetic patients.29 We recently reported that pioglitazone restored normal insulin signaling and prevented apoptosis in the type 2 diabetic rat retina, as well as in retinal endothelial and Müller cells exposed to hyperglycemia. Insulin growth factor binding protein–3 protein levels were suppressed in retina of type 2 diabetic BBZDR/Wor rat model8 and in hyperglycemic retinal endothelial cells,13 which was restored with 2 months of pioglitazone treatment to diabetic rats8 and 24 hours of pioglitazone treatment to REC, respectively (Fig. 2). The intent of our study was to understand IGFBP-3 regulation with pioglitazone and dissect the associated mechanisms involved in pioglitazone's action on retinal endothelial cells. Pioglitazone is given to patients at a dose of 30 mg administered once daily.30 The clinical dose used in type 2 diabetic patients is based upon its property to prevent insulin resistance and maintain a constant glycemic state.29,30 Thiazolidinediones drugs are ligand of PPARγ and increase its gene activity. Peroxisome proliferator-activated receptor γ is activated by these ligands at the concentration range similar to the antidiabetic activity. Thiazolidinediones are reported to have antidiabetic actions in preclinical models of insulin resistance and diabetes at a potency, which matches their affinity for PPARγ.31,32 The concentration of pioglitazone used in cell culture studies is usually reflective of the affinity of the ligand for PPARγ in that cell line. Studies have reported use of pioglitazone dose from 2 to 4 0μM effective at activating PPARγ, depending on the cell line used.33,34 We previously reported that 25 μM of pioglitazone was the optimal dose to increase PPARγ activity in retinal endothelial cells. This dose of pioglitazone was also effective in restoring insulin signaling and preventing increased apoptosis with high-glucose treatment in RECs.8 Thus, we used this dose to determine IGFBP-3 regulation by pioglitazone in this study.
Tumor necrosis factor–α plays a role in the pathology of diabetic retinopathy and deletion of the TNFα gene abrogates leukostasis, retinal cell death, and vascular permeability in diabetic mice.35,36 Pioglitazone is reported to prevent TNFα-induced insulin resistance in adipocytes37 and is considered an anti-inflammatory compound.38 It was recently reported that TNFα inhibits IGFBP-3 in human retinal endothelial cells through activation of p38α and casein kinase 2.19 These studies suggest that TNFα could be a key molecule in pioglitazone-mediated IGFBP-3 regulation. However, when TNFα siRNA is applied to REC cultured in high glucose conditions, pioglitazone normalized levels of IGFBP-3 to the same extent as with scrambled siRNA treatment. Thus, we show that pioglitazone does not use TNFα-p38α-casein kinase 2 pathway to regulate IGFBP-3 in REC.
Because pioglitazone did not require TNFα to regulate IGFBP-3, we intended to identify another mechanism by which pioglitazone regulates IGFBP-3 levels in high glucose. Compound 49b, a nonselective β-adrenergic receptors agonist, was reported to increase IGFBP-3 levels through stimulation of DNA PK without activation of casein kinase 2. Additionally, protein kinase A was required for Compound 49b–induced IGFBP-3 regulation.20 The phosphorylation at IGFBP-3 on serine 156 by DNA PK was important for DNA PK-mediated IGFBP-3 regulation.20 Here, we provide evidence that DNA PK plays a role in pioglitazone-induced expression of IGFBP-3. This conclusion is based on our results that show: (1) PKA is required for pioglitazone's actions on IGFBP-3, (2) pioglitazone stimulates IGFBP-3 levels through DNA PK, and (3) serine 156 is the key site of IGFBP-3 phosphorylation activated downstream of pioglitazone to induce IGFBP-3 activation and prevent apoptosis. All these observations were made in the absence of TNFα (using TNF siRNA), thus eliminating the involvement of TNFα in any of the steps in DNA PK–mediated pathway. Our results and those of others suggest that there exists a crosstalk of PKA pathway and PPARγ signaling.39,40 We show that pioglitazone acts on IGFBP-3 via PKA in retinal endothelial cells (Fig. 3). Inversely, the PKA pathway has been reported to interact with PPARγ ligand actions in HEK293 cells.39,41 Protein kinase A can induce PPARγ activity in vitro by phosphorylating it at several domains and stabilizing binding of the ligand PPARγ to DNA.39 In HEK B2 cells, PKA causes inhibition of nuclear exit and activation of DNA PK.42 The PPARγ agonist, rosiglitazone, acts synergistically with cAMP-PKA through cross-talk between the signaling systems in brown adipocytes.40 Taken together, this data suggests that PPARγ signaling interacts with the PKA pathway, leading to an increase in IGFBP-3 levels in REC-cultured hyperglycemic conditions.
Pioglitazone is known to have protective effects in diabetic retinopathy possibly via its anti-inflammatory and antioxidative properties. Our present work suggests that regulatory actions of pioglitazone in retina involve mechanisms other than those involving TNFα. Pioglitazone prevented apoptosis of retinal endothelial cells in high glucose by increasing DNA PK–mediated phosphorylation of IGFBP-3 at serine 156 (Fig. 6). Thus, pioglitazone can act through multiple pathways to confer protection in diabetic retinopathy. Insulin growth factor binding protein–3 might be considered as a novel mediator of pioglitazone's action on pathogenesis underlying diabetic retinopathy, particularly those involving vascular changes. Reduced expression of the endogenous PPARγ in mice aggravates retinal leukostasis and blood– retinal barrier leakage in diabetic mice and PPARγ agonist; rosiglitazone inhibits retinal leukostasis and leakage in diabetic rats.43 Future work investigating the role of IGFBP-3 in pioglitazone-mediated signaling cascades in vascular dysfunction associated with diabetic retinopathy is needed.
Conclusions
This study found that IGFBP-3 levels in retinal endothelial cells cultured in high-ambient glucose were normalized with pioglitazone treatment. Pioglitazone required PKA and DNA PK activity to increase IGFBP-3 levels and did not involve TNFα. Phosphorylation site 156 of IGFBP-3 is key for regulation of IGFBP-3 and associated decrease of cleaved caspase 3 by pioglitazone in hyperglycemic retinal endothelial cells. Taken together, this study provides a mechanism by which pioglitazone regulates IGFBP-3 expression.
Acknowledgments
Supported by grants from the National Eye Institute (R01-EY022330 to JJS; Bethesda, MD, USA); Juvenile Diabetes Research Foundation (2-2011-597 to JJS; Memphis, TN, USA); Oxnard Foundation (JJS; Memphis, TN, USA); Research to Prevent Blindness (James C. Fleming); National Eye Institute Vision Core Grant (PHS 3P30 EY013080 to Dianna Johnson).
Disclosure: S. Thakran, None; Q. Zhang, None; V. Morales-Tirado, None; J.J. Steinle, None
References
- 1. Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet. 2010; 376: 124–136. [DOI] [PubMed] [Google Scholar]
- 2. Jiang Y, Zhang Q, Soderland C, Steinle JJ. TNFalpha and SOCS3 regulate IRS-1 to increase retinal endothelial cell apoptosis. Cell Signalling. 2012; 24: 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steinle JJ. Retinal endothelial cell apoptosis. Apoptosis. 2012; 17: 1258–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001; 158: 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tawfik A, Sanders T, Kahook K, Akeel S, Elmarakby A, Al-Shabrawey M. Suppression of retinal peroxisome proliferator-activated receptor gamma in experimental diabetes and oxygen-induced retinopathy: role of NADPH oxidase. Invest Ophthalmol Vis Sci. 2009; 50: 878–884. [DOI] [PubMed] [Google Scholar]
- 6. Zhang XY, Xiao YQ, Zhang Y, Ye W. Protective effect of pioglitazone on retinal ischemia/reperfusion injury in rats. Invest Ophthalmol Vis Sci. 2013; 54: 3912–3921. [DOI] [PubMed] [Google Scholar]
- 7. Zhu J, Zhang J, Ji M, et al. The role of peroxisome proliferator-activated receptor and effects of its agonist, pioglitazone, on a rat model of optic nerve crush: PPARgamma in retinal neuroprotection. PLoS One. 2013; 8: e68935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jiang Y, Thakran S, Bheemreddy R, et al. Pioglitazone normalizes insulin signaling in the diabetic rat retina through reduction in tumor necrosis factor alpha and suppressor of cytokine signaling 3. J Biol Chem. 2014; 289: 26395–26405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang Y, Zhang Q, Steinle JJ. Intravitreal injection of IGFBP-3 restores normal insulin signaling in diabetic rat retina. PLoS One. 2014; 9: e93788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang Y, Pagadala J, Miller DD, Steinle JJ. Insulin-like growth factor-1 binding protein 3 (IGFBP-3) promotes recovery from trauma-induced expression of inflammatory and apoptotic factors in retina. Cytokine. 2014; 70: 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Q, Jiang Y, Toutounchian JJ, Soderland C, Yates CR, Steinle JJ. Insulin-like growth factor binding protein-3 inhibits monocyte adhesion to retinal endothelial cells in high glucose conditions. Mol Vis. 2013; 19: 796–803. [PMC free article] [PubMed] [Google Scholar]
- 12. Kielczewski JL, Hu P, Shaw LC, et al. Novel protective properties of IGFBP-3 result in enhanced pericyte ensheathment, reduced microglial activation, increased microglial apoptosis, and neuronal protection after ischemic retinal injury. Am J Pathol. 2011; 178: 1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Q, Soderland C, Steinle JJ. Regulation of retinal endothelial cell apoptosis through activation of the IGFBP-3 receptor. Apoptosis. 2013; 18: 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cobb LJ, Liu B, Lee KW, Cohen P. Phosphorylation by DNA-dependent protein kinase is critical for apoptosis induction by insulin-like growth factor binding protein-3. Cancer Res. 2006; 66: 10878–10884. [DOI] [PubMed] [Google Scholar]
- 15. Cobb LJ, Mehta H, Cohen P. Enhancing the apoptotic potential of insulin-like growth factor-binding protein-3 in prostate cancer by modulation of CK2 phosphorylation. Mol Endocrinol. 2009; 23: 1624–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee YC, Jogie-Brahim S, Lee DY, et al. Insulin-like growth factor-binding protein-3 (IGFBP-3) blocks the effects of asthma by negatively regulating NF-kappaB signaling through IGFBP-3R-mediated activation of caspases. J Biol Chem. 2011; 286: 17898–17909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Q, Steinle JJ. IGFBP-3 inhibits TNF-alpha production and TNFR-2 signaling to protect against Retinal Endothelial Cell Apoptosis. Microvasc Res. 2014; 95: 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Q, Jiang Y, Miller MJ, et al. IGFBP-3 and TNF-alpha regulate retinal endothelial cell apoptosis. Invest Ophthalmol Vis Sci. 2013; 54: 5376–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Q, Soderland D, Steinle JJ. TNFalpha Inhibits IGFBP-3 through activation of p38alpha and casein kinase 2 in human retinal endothelial cells. PLoS One. 2014; 9: e103578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Q, Steinle JJ. DNA-PK phosphorylation of IGFBP-3 is required to prevent apoptosis in retinal endothelial cells cultured in high glucose. Invest Ophthalmol Vis Sci. 2013; 54: 3052–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu X, Wang H, Wang P, Chen BP, Wang Y. The Ku-dependent non-homologous end-joining pathway contributes to low-dose radiation-stimulated cell survival. J Cell Physiol. 2011; 226: 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Segal-Raz H, Mass G, Baranes-Bachar K, et al. ATM-mediated phosphorylation of polynucleotide kinase/phosphatase is required for effective DNA double-strand break repair. EMBO Reports. 2011; 12: 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin SL, Johnson-Farley NN, Lubinsky DR, Cowen DS. Coupling of neuronal 5-HT7 receptors to activation of extracellular-regulated kinase through a protein kinase A-independent pathway that can utilize Epac. J Neurochem. 2003; 87: 1076–1085. [DOI] [PubMed] [Google Scholar]
- 24. Steinle JJ, Booz GW, Meininger CJ, Day JN, Granger HJ. Beta 3-adrenergic receptors regulate retinal endothelial cell migration and proliferation. J Biol Chem. 2003; 278: 20681–20686. [DOI] [PubMed] [Google Scholar]
- 25. Zhang Q, Guy K, Pagadala J, et al. Compound 49b prevents diabetes-induced apoptosis through increased IGFBP-3 levels. Invest Ophthalmol Vis Sci. 2012; 53: 3004–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lofqvist C, Chen J, Connor KM, et al. IGFBP3 suppresses retinopathy through suppression of oxygen-induced vessel loss and promotion of vascular regrowth. Proc Natl Acad Sci U S A. 2007; 104: 10589–10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jarajapu YP, Cai J, Yan Y, et al. Protection of blood retinal barrier and systemic vasculature by insulin-like growth factor binding protein-3. PLoS One. 2012; 7: e39398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang F, Gao L, Gong B, et al. Tissue-specific expression of PPAR mRNAs in diabetic rats and divergent effects of cilostazol. Can J Physiol Pharmacol. 2008; 86: 465–471. [DOI] [PubMed] [Google Scholar]
- 29. Yau H, Rivera K, Lomonaco R, Cusi K. The future of thiazolidinedione therapy in the management of type 2 diabetes mellitus. Curr Diab Rep. 2013; 13: 329–341. [DOI] [PubMed] [Google Scholar]
- 30. DeFronzo RA, Tripathy D, Schwenke DC, et al. Pioglitazone for diabetes prevention in impaired glucose tolerance. N Engl J Med. 2011; 364: 1104–1115. [DOI] [PubMed] [Google Scholar]
- 31. Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem. 1995; 270: 12953–12956. [DOI] [PubMed] [Google Scholar]
- 32. Spiegelman BM. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998; 47: 507–514. [DOI] [PubMed] [Google Scholar]
- 33. Wan H, Yuan Y, Qian A, Sun Y, Qiao M. Pioglitazone, a PPARgamma ligand, suppresses NFkappaB activation through inhibition of IkappaB kinase activation in cerulein-treated AR42J cells. Biomed Pharmacother. 2008; 62: 466–472. [DOI] [PubMed] [Google Scholar]
- 34. Verrier E, Wang L, Wadham C, et al. PPARgamma agonists ameliorate endothelial cell activation via inhibition of diacylglycerol-protein kinase C signaling pathway: role of diacylglycerol kinase. Circ Res. 2004; 94: 1515–1522. [DOI] [PubMed] [Google Scholar]
- 35. Joussen AM, Doehmen S, Le ML, et al. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009; 15: 1418–1428. [PMC free article] [PubMed] [Google Scholar]
- 36. Huang H, Gandhi JK, Zhong X, et al. TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Invest Ophthalmol Vis Sci. 2011; 52: 1336–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iwata M, Haruta T, Usui I, et al. Pioglitazone ameliorates tumor necrosis factor-alpha-induced insulin resistance by a mechanism independent of adipogenic activity of peroxisome proliferator–activated receptor-gamma. Diabetes. 2001; 50: 1083–1092. [DOI] [PubMed] [Google Scholar]
- 38. Okada M, Yan SF, Pinsky DJ. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) activation suppresses ischemic induction of Egr-1 and its inflammatory gene targets. FASEB J. 2002; 16: 1861–1868. [DOI] [PubMed] [Google Scholar]
- 39. Lazennec G, Canaple L, Saugy D, Wahli W. Activation of peroxisome proliferator-activated receptors (PPARs) by their ligands and protein kinase A activators. Mol Endocrinol. 2000; 14: 1962–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen HY, Liu Q, Salter AM, Lomax MA. Synergism between cAMP and PPARgamma Signalling in the Initiation of UCP1 Gene Expression in HIB1B Brown Adipocytes. PPAR Res. 2013; 2013: 476049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Subbaramaiah K, Howe LR, Zhou XK, et al. Pioglitazone, a PPARgamma agonist, suppresses CYP19 transcription: evidence for involvement of 15-hydroxyprostaglandin dehydrogenase and BRCA1. Cancer Prev Res. 2012; 5: 1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42. Huston E, Lynch MJ, Mohamed A, et al. EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. Proc Natl Acad Sci U S A. 2008; 105: 12791–12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Muranaka K, Yanagi Y, Tamaki Y, et al. Effects of peroxisome proliferator-activated receptor gamma and its ligand on blood-retinal barrier in a streptozotocin-induced diabetic model. Invest Ophthalmol Vis Sci. 2006; 47: 4547–4552. [DOI] [PubMed] [Google Scholar]