

Abstract
Dynein light chain LC8 is a small, dimeric, and very highly conserved globular protein that is an integral part of the dynein and myosin molecular motors but appears to have a broader role in multiple protein complexes unrelated to molecular motors. LC8 binds to two families of targets: those having a KXTQT sequence fingerprint and those having a GIQVD fingerprint. All known LC8 binding partners containing these fingerprints share a common binding site on LC8 that raises the question of what determines binding specificity. Here, we present the crystal structure of apo-LC8 at 1.7-Å resolution, which, when compared with the crystal structures of several LC8 complexes, gives insight into the mechanism underlying the binding diversity of LC8. Peptide binding is associated with a shift in quaternary structure that expands the hydrophobic binding surface available to the ligand, in addition to changes in tertiary structure and ordering of LC8 around the binding groove. The observed quaternary shift suggests a mechanism by which binding at one of the two identical sites can influence binding at the other. NMR spectra of titrations with peptides from each fingerprint family show evidence of allosteric interaction between the two binding sites, to a differing degree in the two ligand families. Allosteric interaction between the binding sites may be a mechanism to promote simultaneous binding of ligands from the same family, providing a physiological role for the two fingerprints.
Keywords: dynein light chain, folding scaffold, protein crystallography
Introduction
LC8 (called DYNLL1 in mammals)1 is a highly conserved and widely expressed 10-kDa globular protein identified as an essential component of the dynein2–4 and myosin V5 molecular motors, binding directly to specific sites on the myosin V heavy chain6,7 and the dynein intermediate chain IC74.8,9 Interestingly, as much as 60% of LC8 is not associated with these molecular motors,10 and LC8 has been identified in interactions with a number of nonmotor proteins. Some of these, such as Swallow, a protein essential for RNA localization,11 are associated with active transport along microtubules, leading to the widely held view that LC8 is a dynein cargo adaptor.12,13 But others, such as neuronal nitric oxide synthase,14 are not clearly associated with active transport, leading to the emerging concept that LC8 is primarily a dimerization hub, binding to disordered regions on diverse partners and inducing them to dimerize and form partially ordered structures, often coiled coils.8,15,16 Recently, this was illustrated dramatically for the nuclear pore complex protein Nup159, in which multiple copies of LC8 bind like “beads on a string” and induce formation of a coiled coil nearby.17
LC8 is a dimer containing a pair of central β-sheets composed of four strands from one subunit and one strand crossed over from the other subunit.18 One side of each sheet is flanked by two helices, and the other side forms the dimer interface (Fig. 1a). Two identical binding sites are formed in a groove at the dimer interface, such that both apo- and doubly bound LC8 are symmetric dimers. Ligands bind next to β3 as a sixth strand of the β-sheet.18,19 All known ligands belong to one of two sequence classes, the GIQVD family or the KXTQT family.9,20 Both families share a conserved glutamine residue whose side chain projects out of the binding cleft and forms an N-terminal cap for helix α2.19 As before, we adopted a notation in which the conserved glutamine residue is referred to as Q0 and C-terminal and N-terminal residues are referred to as Q+n and Q−n, respectively. The side chains of residues Q+1, Q−1, and Q−3 project toward the interior of LC8, into a mostly hydrophobic groove lined by the aromatic side chains of residues Phe73, Tyr75, and Tyr77 (Fig. 1b). Hydrophilic residues of strand β1 separate this hydrophobic cavity from the solvent. Interestingly, the peptide binding cleft is about 1 Å wider when bound to the bulkier side chains of a GIQVD ligand versus a KXTQT ligand.19 The width of the cleft in the absence of peptide has not been described, because no crystal structure of apo-LC8 has yet been published and because this was not examined in the NMR structure of apo-LC8.21
Fig.1.
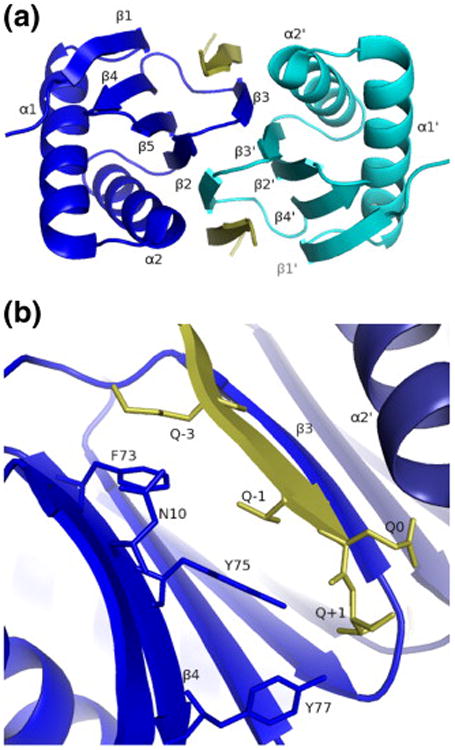
Overview of theLC8–Swa complex:LC8 chain A is shown in blue; LC8 chain B, in dark blue; and Swa peptide, in yellow. (a) View along the 2-fold symmetry axis. (b) Detail of the peptide binding cleft using the Qn notation, where Q0 refers to the conserved glutamine residue of the ligand.
Here, we present the crystal structure of apo-LC8 at 1.7-Å resolution. The structure reveals an unexpected induced-fit structural change and suggests that the binding of ligands to the two sites in dimeric LC8 is not independent. Using NMR spectroscopy, we monitored titrations of LC8 with ligands from both families to provide evidence in solution of an interaction between the binding sites.
Results
Structure solution
The structure of apo-LC8 was solved by rigid-body refinement, using the LC8 chain from the structure of LC8–Swa (peptide consisting of residues 281–297 of Swallow) as the initial model,19 and refined to 1.7-Å resolution, converging at Rcryst=17.9% and Rfree= 21.1% (Table 1). All residues were in the most favored Ramachandran region except for Asn51, which, as in all LC8 structures, has a positive φ angle and is part of a turn that also features a cis-peptide bond between Pro52 and Thr53.
Table 1. Data collection and refinement statistics.
| Apo-LC8 | LC8–Swa | |
|---|---|---|
| Resolution | 100.0–1.70 (1.76–1.70) | 100.0–2.0 (2.11–2.00) |
| Reflections (total/unique) | 525,746/13,961 | 119,020/8784 |
| Completeness | 96.6 (87.0) | 100.0 (100.0) |
| Rmeas a | 6.6 (38.5) | 6.7 (41.7) |
| I/σ | 34.8 (7.5) | 6.3 (1.0) |
| Refinement | ||
| Resolution (Å) | 38.9–1.70 (1.74–1.70) | 35.8–2.0 (2.05–2.00) |
| No. of reflections | 13,151 (843) | 7844 (552) |
| No. of amino acids | ||
| LC8 | 87 | 87 |
| Ligand | – | 10 |
| No. of solvent atoms | 109 | 61 |
| Total no. of atoms | 879 | 869 |
| Average B (all heavy atoms, Å2) | ||
| LC8 | 26.3 | 42.1 |
| Peptide | – | 36.2 |
| Rcryst (%) | 17.9 (19.6) | 21.5 (26.3) |
| Rfree (%) | 21.1 (25.7) | 26.5 (34.7) |
| RMSD bonds (Å) | 0.011 | 0.007 |
| RMSD angles (°) | 1.3 | 0.88 |
| φ, ψ (most favored, %)b | 98.8 | 98.9 |
As defined by Diederichs and Karplus.22
As defined in Lovell et al. 23
The new synchrotron-based structure of LC8–Swa is very similar to the one we reported previously based on home-source X-ray data:19 main-chain atoms align with an RMSD of 0.22 Å. The LC8–Swa structure also agrees well with the LC8–KXTQT motif complex in the recently solved 2.8-Å crystal structure of the ternary complex LC8–TcTex–IC74:24 main-chain atoms align with an RMSD of 0.43 Å. For the comparisons that follow, we used the 2.0-Å LC8– Swa structure as the representative of the KXTQT family because it is the highest-resolution example.
Description of apo-LC8
The apo-LC8 structure has well-defined and continuous density for all but the first three residues of the LC8 main chain, and the peptide binding cleft contains clearly defined bound water molecules, indicating that no complexed LC8 is present in the crystals. The structure also contains one sulfate ion not observed in any of the bound forms. In the following paragraphs, we describe differences between the ligand-bound and apo-LC8 structures in terms of global changes in structure and mobility, local changes in structure and mobility, and the solvent structure in the peptide binding cleft. Also, because the ligand-bound structure is the known reference state, we will describe changes in terms of what happens upon ligand dissociation rather than ligand binding.
Shear movement
Removal of the bound ligand from LC8 is associated with a shear movement25 of the LC8 subunits that decreases the width of the peptide binding cleft (Fig. 2; Table 2). Measured as the distance from 63Cα to 9Cα, the cleft widths are 14.2 Å in LC8–nNOS (a GIQVD ligand), 13.4 Å in LC8–Swa (a KXTQT ligand), and 12.3 Å in apo-LC8. This cleft narrowing reburies 50–100 Å2 of hydrophobic surface exposed by removal of the ligand. The exposed hydrophobic surface area arises mainly from strand β4 (residues Phe73, Tyr75, and Tyr77), with smaller contributions from strand β5 (Ala82 and Leu84). Removing a GIQVD ligand without changing the conformation of the LC8 main chain exposes <200 Å2 to the solvent (Table 2). Removing a KXTQT ligand exposes <150 Å2; in apo-LC8, only <100 Å2 remains exposed.
Fig.2.
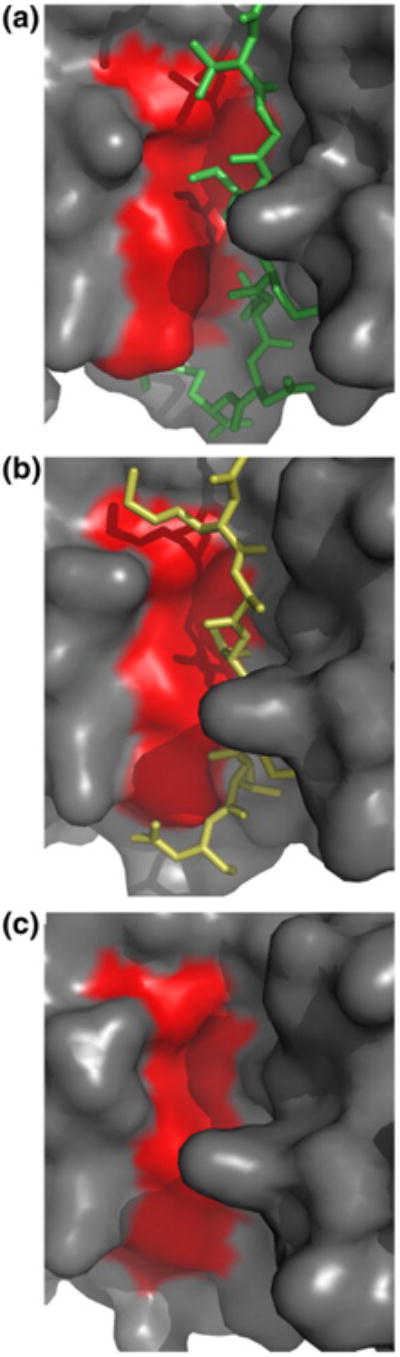
The changing width of the peptide binding cleft. The surface of LC8, looking into the peptide binding groove, is shown for (a) LC8–nNOS, (b) LC8–Swa, and (c) apo-LC8. The surfaces of strands β4 and β5 are shaded red.
Table 2. Induced-fit structural changes in the peptide binding cleft of LC8 due to shear movement.
| Cleft width (Å)a | Cleft surface (Å2)b | PDB entry | |
|---|---|---|---|
| GIQVD ligands | |||
| LC8–nNOS | 14.2 ± 0.2 | 191 | 1cmi18 |
| LC8–nNOS (NMR) | 15.2 ± 0.2 | 182 | 1f9621 |
| KXTQT ligands | |||
| LC8–Swa | 13.4 | 149 | 3e2bc |
| LC8–IC | 13.5 ± 0.1 | 141 | 2pg1d |
| LC8–Bim (NMR) | 13.3 ± 0.5 | 156 | 1f9521 |
| Apo | |||
| Apo-LC8 | 12.3 | 99 | 3bric |
| Apo-LC8 (NMR) | 11.4 ± 0.3 | 104 | 1f3c21 |
Defined as the distance from 63Cα to 9Cα.
Solvent-exposed surface area of strand β4 calculated using the LC8 chain only (i.e., in the absence of ligand).
From this study.
Part of a ternary LC8-IC74-TcTex complex.24
Packing at the dimer interface
Both the interior and the dimer interface of LC8 are well packed, and the packing at the interface is affected only slightly by the shear movement. For LC8–Swa, the average occupied volume (see Materials and Methods) for interior residues (those buried in both monomer and dimer) is within 1% of a reference volume derived from a survey of well defined protein crystal structures.26 The dimer interface, defined as residues that are buried only in the dimer, is even better packed than the interior, with a mean fractional residue volume 2% below the reference volume. A similar pattern is seen for LC8– nNOS (peptide consisting of residues 226–237 of neuronal nitric oxide synthase), in which the ratio of mean observed volume to reference volume is 1.00 at the interface and is 1.03 in the interior. The slightly looser packing in LC8–nNOS may be an artifact of the lower resolution of the structure. Only in apo-LC8 (the highest-resolution structure) is the packing at the interface (mean volume/reference volume = 1.04) less efficient than the packing in the interior (mean volume/reference reference = 1.00); however, the difference in mean occupied volume of 4% is not larger than the per-residue standard deviation observed in a broad sample of well-packed residues.26 Thus, despite the 2-Å displacement of the dimer interface between LC8–nNOS and apo-LC8, packing efficiency changes only very slightly. This is because the interface is very flat and because the side chains make small adjustments in conformation (χ1 and χ2) to avoid collisions, counteracting the movement of the main chain (Fig. 3a). Only one residue (Ile57) converts to a different rotamer to avoid steric clash with its symmetry mate (Fig. 3b).
Fig. 3.
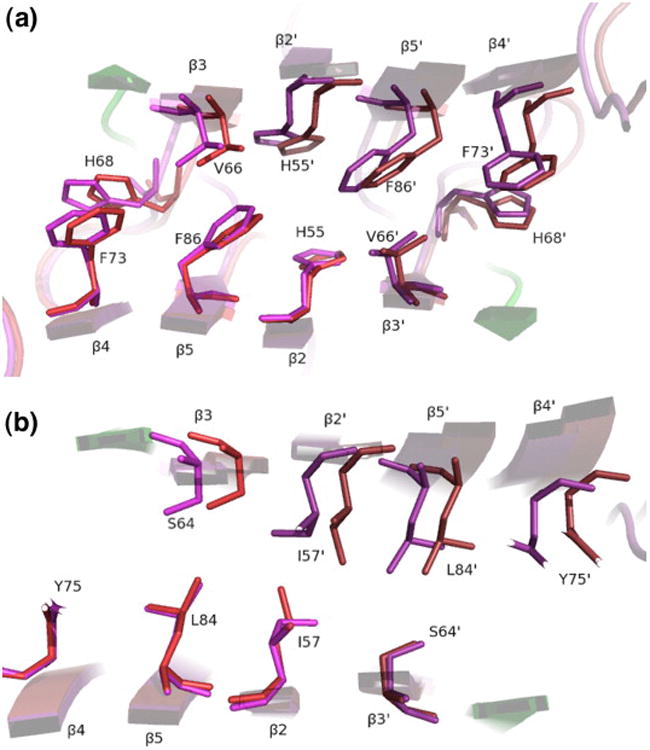
Dimer interface features allowing the shear movement. LC8–nNOS (red/green) is overlaid on apo-LC8 (purple), with alignment based on the lower subunits. The view is along the 2-fold symmetry axis, parallel with the β- strands, in the layer containing (a) Phe86 and (b) Ile57. Note the rotameric interconversion of Ile57. The fluid shear movement of 2 Å is evident in the upper subunits and is facilitated by close residue packing yet lack of side-chain interdigitation.
Local conformational changes and increased heterogeneity upon ligand dissociation
Removal of the ligand is accompanied by a change in conformation of the C-terminus of the protein as the last two residues (Ser88 and Gly89) move away from the dyad axis and into the peptide binding site (Fig. 4a). The β5–β2 interaction is thus shortened by one residue, and hydrogen bonds joining Ser88 O to Ser882 Oγ and Ser88 N to Thr53 O are broken. The space created between β2 and Ser88 is filled by a network of ordered water molecules, making contacts with the side chain of His55 and the backbones of Ser88 and Thr53,all of which are buried in the ligand-bound forms. Residue Ser88 can be phosphorylated in vivo, a potential regulatory mechanism for LC8.27
Fig.4.
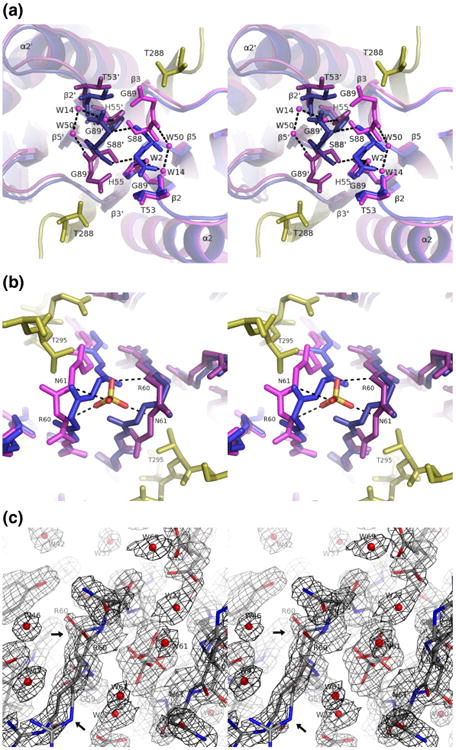
Conformational changes at the exits of the peptide binding groove.(a)Stereoview looking along the 2-fold axis toward the N-terminal exit of the peptide binding groove showing the LC8–Swa complex (blue/yellow) and apo-LC8 (purple), with chain B shaded darker than chain A. Alignment is based on chain A (right side of the figure), excluding the interleaved strand β3. Dashed lines indicate hydrogen bonds. (b) Stereoview along the 2-fold axis toward the C-terminal exit showing the β2–β3 loop. Coloring and alignment are as in (a). In apo-LC8, a sulfate ion sits at the dimer interface, coordinated by the backbone amides of residues 60 and 61. (c) Same view as in (b) showing 2Fo – Fc density for apo-LC8 contoured at 1.0σ. Nitrogen atoms are shown in blue, and oxygen atoms are shown in red. Both alternate conformations are shown overlaid for the backbone in the β2–β3 loop (arrows).
A second rearrangement occurs at the opposite end of the binding groove where the ligand is in contact with the turn connecting β2 and β3 (Fig. 4b). This turn is at the dimer interface, such that in the bound form, the backbone of Asn61 makes hydrogen bonds with the backbone of Asn612. Upon removal of the peptide, this loop becomes less ordered as reflected by the crystallographic B-factors and the appearance of alternate conformations in the backbones of residues Arg60 and Asn61 (Fig. 4c). The two symmetry-related loops move apart, disrupting the interaction between Asn61 and Asn612. A sulfate ion, not seen in any of the peptide-bound forms, occupies the resulting space between the subunits, coordinated in two alternate conformations by the backbone amides of Arg60 and Asn61.
Solvent structure in the ligand binding groove
In apo-LC8, ordered water molecules form hydrogen bonds with all the groove-facing backbone amides and carbonyls of strand β3 (Fig. 5). Some of these water molecules also bind to others to form networks that cover the hydrophobic surface of the groove. One such network covers Phe73 and consists of three water molecules bridging His68 Nδ, Val66 O, Val66 N, and Ser64 Oγ. Another covers Tyr75 and consists of four water molecules bridging Asn10 Nδ to Ser64 N and Ser64 O. Upon ligand binding, the water network over Phe73 is replaced by the side chain of the Q−3 residue.
Fig.5.
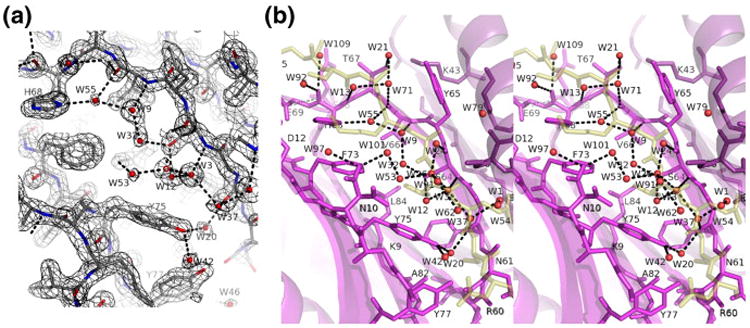
Solvent structure in the apo-LC8 ligand binding groove. (a) 2Fo–Fc density contoured at 1.2σ. Water molecules are shown as red spheres. (b) Ribbon diagram and bound water molecules (red) are shown for apo-LC8 (purple) overlaid with the peptide from LC8–Swa (ghostly yellow). Water 20 (bridging Tyr75 OH and Phe62 O) has an interesting analog in LC8–Swa, where water 1 bridges the same two residues plus Swa Thr293 Oγ and is the only buried water molecule in the structure as well as the most ordered one. An analogous water molecule is missing from LC8–nNOS, due to the different peptide main-chain conformation that leaves insufficient room between the Q−1 and Q+1 residues.
Observation of intermediate states by NMR
To complement the structural information available from crystallography on apo-LC8 and that on doubly occupied LC8, we sought to probe the structure of singly bound forms by using heteronuclear single-quantum coherence (HSQC) spectra to monitor the titration of LC8 with its ligands over a range of 0 equivalent to 1 equivalent. Titrations were performed using peptides derived from two ligands in the KXTQT family [IC (peptide consisting of residues 123–138 of the dynein intermediate chain IC74) and Swa] and one ligand from the GIQVD family (nNOS). As ligand is titrated into LC8, peaks corresponding to apo-LC8 decrease in intensity and peaks corresponding to doubly occupied LC8 appear. Neither peak is appreciably broadened, showing that the free and bound forms are in slow exchange. As noted previously for titration with nNOS,28 at intermediate stages of titration, for some residues, a third, and sometimes fourth, peak grows and then shrinks in intensity during the titration (Fig. 6). Such intermediate peaks representing the singly bound forms can also be observed during titration with Swa and IC, although the peaks occur for fewer residues and tend to be less well resolved from the peaks corresponding to either the apo form or the doubly occupied form. For all three peptides, the intermediate peaks occur only for residues at the dimer interface or at the peptide binding site (Fig. 7).
Fig.6.
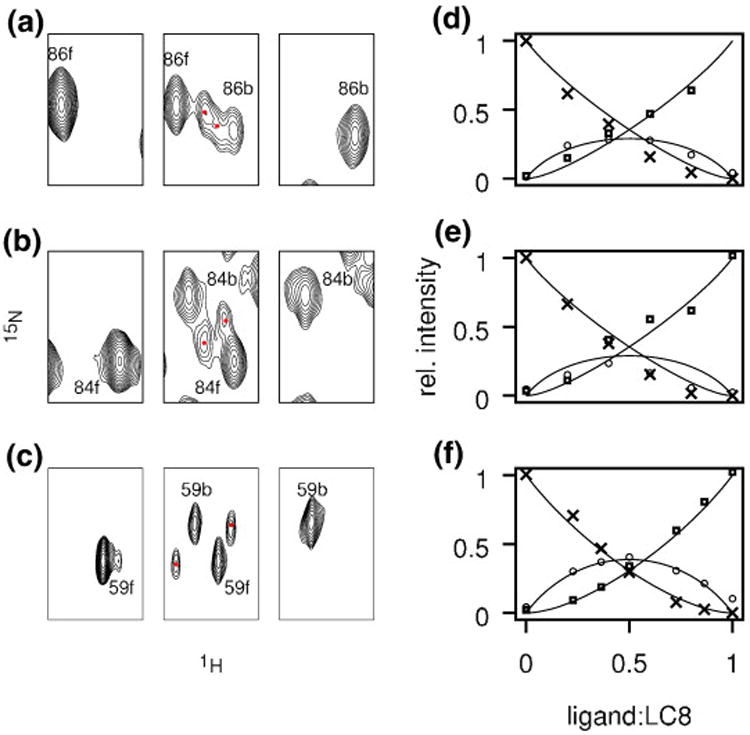
Titration of LC8 with peptide ligands monitored by NMR:(a and d) LC8–IC; (b and e) LC8–Swa; (c and f) LC8– nNOS. (a–c) Excerpts of HSQC spectra with (left to right) 0 equivalent, 0.4 equivalent, and 1 equivalent of ligand added. Peaks for free LC8 (apo) and bound LC8 (doubly occupied) are labeled (f) and (b) respectively. Red dots indicate new peaks arising from singly bound LC8, which are present only in the middle of the titration curve. (d–f) Titration curves for the resonances shown in (a) to (c). Crosses and squares represent relative intensities of peaks corresponding to free and doubly-bound forms, respectively. Circles represent the sum of relative intensities of intermediate peaks. Curves represent populations predicted by the two-site binding model (see Discussion), with K1d/K2d = 6.0 for Swa and IC and K1d/K2d = 2.5 for nNOS.
Fig. 7.
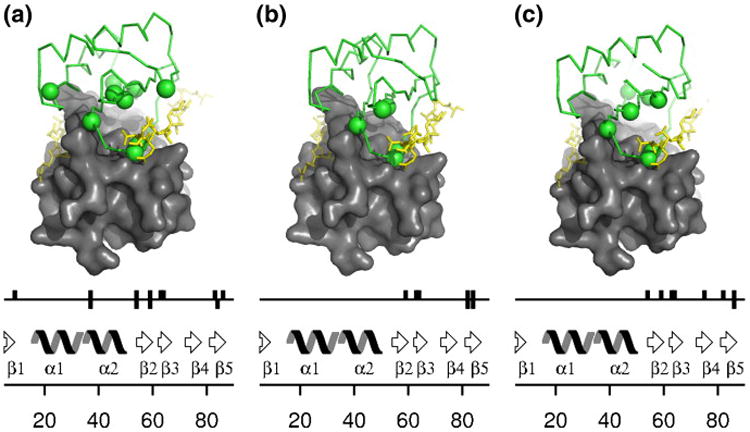
Spatial distribution of residues showing intermediate peaks. (a) nNOS. (b) Swa. (c) IC. The peptide (yellow) shows the location of the occupied binding groove and that of the unoccupied binding groove (faded yellow). On the sequence schematic diagram, upward bars indicate residues for which an intermediate peak is observed near the apo peak and downward bars indicate residues for which an intermediate peak is observed near the doubly bound peak. These same residues are indicated by spheres on the structure.
Discussion
Folding coupled to binding
In protein–protein interactions, the extent to which folding and binding are coupled spans a broad spectrum, from the traditional “lock-and-key” model in which two preorganized interaction surfaces fit together to form a mutually complementary and well packed interface to the other extreme in which one partner has no intrinsic structure and adopts an ordered fold only in the presence of its partner.29 The latter case, which is common among regulatory proteins especially in eukaryotes, is exemplified by the ligands of LC8, which tend to be natively disordered segments of proteins that fold into a β-strand at the recognition site and into a stable coiled coil at a distant site in the presence of LC8.16
LC8, unlike its ligands, has a recognizable binding groove even in the apo form and thus at first glance resembles a protein in the lock-and-key model. However, on closer examination, the binding groove in apo-LC8 is significantly more disordered than LC8 overall, becoming fully ordered only upon formation of the complex: temperature factors decrease, amide protons are better protected from the solvent,19 and fewer Rex terms are required to model 15N relaxation.30 Through steric constraints, the ligand promotes conformational order and homogeneity: the β2–β5 interaction is extended by one residue, and the β2–β3 turn collapses to a single ordered conformation. In the apo-LC8 crystal structure, this turn is partly stabilized by a backbone-coordinated sulfate ion, which very likely arises from the high concentration of ammonium sulfate used in crystallization. In vivo, this sulfate ion may be replaced by a more prevalent ligand, such as phosphate, or may be absent altogether, possibly resulting in an even greater degree of flexibility for this loop than that seen in the crystal structure.
The structural differences between the apo-LC8 and ligand-bound LC8 crystal structures are corroborated by chemical shift perturbations in the NMR spectra of these complexes in solution.19 Such perturbations are sensitive indicators of changes in conformation or contact with a ligand. For LC8, as expected, the largest perturbations occur in strand β3 and the N-terminal of helix α2, the residues lining the peptide binding cleft. However, there are also significant perturbations for the last few residues of strand β5 that are not in contact with the peptide. This is now explained by the conformational change for these residues in apo-LC8 versus bound LC8 (Fig. 4a).
Binding and quaternary structure
The major conformational change seen in ligand-bound versus apo-LC8 is a widening of the binding groove by <1 Å for KXTQT ligands and that by <2 Å for GIQVD ligands. The width of the cleft seems to be determined by the steric contact between the Q+1 residue of the ligand and Tyr77 of LC8 and that between the Q−1 residue and Tyr75. The expansion of the cleft in complexes exposes considerable hydrophobic surface area to the solvent, 50 Å2 for KXTQT ligands and 100 Å2 for GIQVD ligands, which is then covered by the bound ligand. The favorable burial of this hydrophobic surface is presumably what drives the narrowing of the cleft upon release of the ligand. It also implies that the net driving force for ligand binding is less than what would exist for a preformed binding site.
The quaternary shift of LC8 is an example of shear motion at a domain interface.25 Shear motions are sliding movements perpendicular to the interface in which the surfaces remain in close contact, there is no qualitative change in packing (such as a different interdigitation of side chains) and there is little or no change in main-chain conformation, and there are few or no rotameric conversions. Good packing density is maintained by small adjustments in side-chain conformation within a single rotameric well. In LC8, the shear movement seems to be made possible by the rather flat (noninterdigitated) and nonpolar nature of the interface (Fig. 3). The difference between apo-LC8 and LC8–nNOS of 2 Å is apparently the upper limit for shear movement.25 Between these extremes, it appears that the flat interface allows a near-continuum of association geometries, such as the 1-Å shift associated with KXTQT ligands. Fine adjustment of the size of the ligand binding groove through shear motion is one mechanism by which LC8 binds a variety of peptide sequences.
The shear movement provides a rationale for the design of the LC8 dimer interface as adjacent β-sheets with a swapped-over strand. A common model of protein association is that burial of hydrophobic surface contributes a large free energy change but little specificity, and short-range interactions, such as hydrogen bonds and Van der Waals contacts, provide specificity and determine the precise final conformation.31 LC8 dimerization is both moderately tight32 and very specific (the LC8 dimerization interface is not known to bind any other protein). The design of the LC8 dimer interface—two flat β-sheet surfaces with one swapped-over strand—is effective in providing high affinity and specificity while still allowing a large quaternary shear: affinity is provided by the substantial (1000 Å ) hydrophobic surface buried at the flat interface (see Fig. 3), and specificity is provided by the crossed-over β-strand (intersubunit backbone hydrogen bonds).
The shear motion in LC8, albeit a subtle change in conformation (1–2 Å), is supported by several lines of evidence. Our observation was based on a comparison of the crystal structures of apo-LC8, LC8–Swa, and LC8–nNOS (Table 2). Although it was not reported at the time of publication, in retrospect, the same observation can be made by a comparison of the NMR structures of apo-LC8, LC8–Bim, and LC8–nNOS.21 The agreement on this feature between the solution structures and the crystal structures is generally very good, ruling out the possibility that the shear motion is an artifact due to crystal packing effects (a point that is also reinforced by the agreement between KXTQT crystal structures in different space groups). Where there is minor disagreement between the methods (e.g., the 15-Å cleft in the NMR structure of LC8–nNOS versus 14 Å in the crystal structure), we took the crystal structures to be more convincing and accurate, for several reasons. First, the crystal structures are generally of higher quality (75%–80% of residues in the most favored Ramachandran region for the NMR structures versus 98% for the crystal structures) and higher precision (RMSD=0.92 Å for the ensemble of LC8–Bim NMR structures versus 0.18 Å of coordinate error based on Rfree for the crystal structure of LC8–Swa). Also, the NMR structures are based primarily on distance restraints, but their precision (1–2 Å) is as large as the change in quaternary structure we are trying to measure, and in the NMR experiments used, intrasubunit distances are indistinguishable from intersubunit distances. Finally, the NMR structures are determined by a molecular dynamics protocol in which good van der Waals contacts are maintained; thus, in the calculation of the LC8–Bim and LC8–nNOS NMR structures, the peptide binding cleft is forced open by the presence of the ligand, whereas in the calculation of the apo-LC8 structure, the cleft is free to collapse. In contrast, the crystal structures do not suffer from this somewhat circular reasoning: the electron density very clearly demonstrates the change in quaternary structure even at the molecular replacement stage when no ligand is present in the model.
Other pieces of evidence also support the existence of the shear motion in solution. The residues in the turns flanking strand β3 are mobile in apo-LC8 but not in the complex with the KXTQT ligand Bim.30 Since the swapped-over strand β3 is part of the βsheet of the opposite subunit, these residues are precisely the “hinge” that must bend to accommodate the shear movement. Also, a global conformational change coupled to binding is suggested by the observation of intermediate peaks during titration, as described below.
Allostery
LC8 is a symmetric dimer with two identical ligand binding sites. Forming the doubly bound complex (as observed in the crystal structures of LC8– Swa, LC8–IC, and LC8–nNOS) involves two binding steps as shown in Scheme 1.
Scheme 1.

Two-step formation of LC8-ligand complexes, where X is the ligand and the dissociation constants are defined as K1d = [LC8][X]/[LC8 − X] and K2d = [LC8 − X][X]/[X − LC8 − X]. The binding sites and ligands are identical, so [LC8−X]=[X−LC8].
Are the two binding steps independent? The answer depends on the nature of the singly bound intermediate. Three possible models are outlined in Fig. 8. In a two-state model, it is assumed that the effect of ligand binding to LC8 is restricted to small and local conformational changes in the vicinity of the binding site. Binding to one site does not influence binding at the other (i.e., Kd1 = Kd2 ). In a three-state model, binding of the first ligand is linked to a global change in protein conformation (the shear motion) that preorganizes the second binding site. This type of model—originally proposed by Monod, Wyman, and Changeux—matches the behavior of many oligomeric proteins that can undergo a shift in quaternary structure.33 The most general is a four-state model in which binding of the first ligand is linked to a global change in conformation but, unlike in the three-state model, there is no assumption that the conformation in the singly bound state resembles the conformation in the doubly bound state.
Fig. 8.
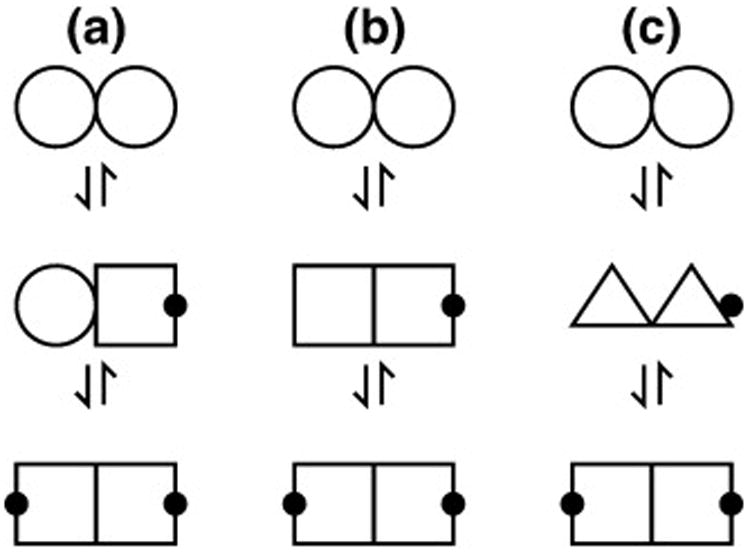
Models for allostery in LC8 binding. Different polygonal shapes represent different conformations of LC8, and a black dot indicates an occupied binding site. (a) Two independent binding sites. Binding at one site does not affect the conformation at the other site. There are only two possible chemical environments, corresponding to the occupied and unoccupied states. (b) Binding coupled to a global conformational change, such as the shear motion. There are three possible chemical environments: the occupied state, the distinct unoccupied state of the free form, and the distinct unoccupied state of the singly bound form. (c) Unique conformation for the singly bound state. There are four possible chemical environments—two corresponding to the same apo and bound states from (a) and (b) and two more from the singly bound state that, lacking symmetry, provides two environments, neither of which is equivalent to the apo or bound state. Only for model (a) are the affinities necessarily equal for the first and second binding steps.
Titration monitored by NMR is a useful method for distinguishing between different allosteric models because the number of unique conformational environments is reflected in the number of distinct chemical shifts that can be observed for each residue.34 Thus, the two-, three-, and four-state models predict two, three, and four distinct peaks per backbone amide, respectively, in 1H–15N HSQC spectra. A two-state model is effectively ruled out for LC8 by the observation of intermediate peaks during titration with all three ligands tested. During titration with KXTQT ligands, residues giving one intermediate peak are most common, and just a few (two in LC8–Swa) give two intermediate peaks. During titration with GIQVD ligands, many more residues give two intermediate peaks. Therefore, binding of KXTQT ligands is largely consistent with the three-state model and binding of GIQVD ligands is most consistent with the four-state model. This difference in behavior may be explained by the difference in size between the two families. In the three-state model, binding the first ligand is linked to the full shear movement, which exposes additional hydrophobic surface area in the other unoccupied binding site. This may be tenable for KXTQT ligands that require a 1-Å shear movement, albeit not for GIQVD ligands that require a 2-Å shear movement. Thus, the global conformational change associated with binding of the first GIQVD ligand may be different from the shear movement observed between the apo and doubly bound crystal structures.
An intriguing consequence of LC8 adopting a three- or four-state model for ligand binding is that the first and second binding constants Kd1 and Kd2 can be different. At the titration midpoint, the relative populations of free, singly bound, and doubly bound LC8 are predicted to be 1:2:1 if independent binding sites are assumed (i.e., Kd1 = Kd2 ). The observed population of the singly bound form is significantly lower than this prediction (Fig. 6), which can be explained by allowing the second association step to be of higher affinity than the first. Conservatively choosing the most similar values that still fit the titration data well results in a Kd1/Kd2 ratio of 2.5 for nNOS and that of 6.0 for IC and Swa.
Overall implications for LC8 function
Several physiological roles for LC8 have been proposed as more binding partners are discovered. LC8 binds to both dynein and putative dynein cargo molecules, leading to the hypothesis that LC8 is a cargo adaptor.11,13,35,36 However, in the known structures of LC8–ligand complexes, both dynein and non-dynein ligands compete for the same two identical binding grooves on LC8.18,19,21 Therefore, models in which LC8 acts as a cargo adaptor are untenable unless LC8 binds one each of two ligands. Arguing against this heterologous binding model is the observation that known LC8 targets tend to be dimeric when bound to LC8 and to have dimerization domains distant from the LC8 binding site.15,24,37,38 Despite the existence of these distant dimerization domains, in cases in which quantitative measurements are available, intrinsic dimerization of the targets in the absence of LC8 is either greatly weakened or entirely absent.7,8,15 This has led to the view that LC8 is a hub protein and that its primary function is to promote dimerization in its partners, both in dynein and in other systems.16
The allosteric behavior of LC8 reported here may discourage heterologous binding and thus further support the hub model. The shear motion of the LC8 dimer interface serves as a mechanism to finely adjust the width of the peptide binding cleft to match the size of the ligand and optimize its affinity. Because the conformational change mainly affects the quaternary structure, it will happen to the same extent at both sites and thus may allow LC8 to discriminate against simultaneously binding a KXTQT ligand and a GIQVD ligand: a 1-Å shifted dimer will bind a GIQVD ligand suboptimally, a 2-Å shifted dimer will bind a KXTQT ligand suboptimally, and a 1.5-Å shifted dimer would bind both ligands suboptimally.
The role of LC8 as a hub further raises the question as to how its diverse collection of targets can share the same interaction site yet bind with high affinity and specificity. The NMR titration experiments reported here have demonstrated that the first binding event influences the second (Kd1/Kd2 = 6 for KXTQT peptide ligands). Such cooperative interactions are a common mechanism to enhance both affinity and specificity in biological systems.39 We propose that allostery in LC8 allows it to act like a switch, preferentially existing either in the apo form or doubly bound to two of the same ligand and disfavoring heterologous states and singly bound states. The distant dimerization domains in various targets can serve to further stabilize doubly bound homodimeric states. With the growing number of known LC8 binding partners, the allosteric interaction between its binding sites, and the structural evidence that the fully occupied form is also the only fully ordered one, we speculate that LC8 present in vivo is not free but is largely bound to homodimers of its many partner proteins.
Materials and Methods
Purification and crystallization
During the structure determination of the LC8–Swa complex that we recently reported,19 the electron density maps from some of the crystals grown from mixtures of LC8 and Swa showed no evidence for the bound peptide. The ligand-free crystals were identical in appearance but had slightly different unit cell dimensions (a = 44.9 ± 0.1 Å versus 44.9±0.1 Å; c=201.6±0.3 Å versus 204.0±0.4 Å).
All crystals were grown at room temperature in hanging drops made by a 1:1 mixture of the reservoir with a stock solution of 1 mM protein and 2 mM peptide in 20 mM Tris– HCl, pH 8.0. The reservoir was composed of 0.2 M potassium sodium tartrate, pH 5.5, 0.1 M sodium citrate, and 2.0 M ammonium sulfate. The crystals were flash frozen in liquid nitrogen following transfer to a cryoprotectant consisting of reservoir solution plus 10% (v/v) glycerol.
X-ray data collection
For apo-LC8, diffraction data were collected on beamline 5.0.3 at Berkeley National Lab's Advanced Light Source (λ = 1.0 Å; Δφ = 1°; high-resolution pass, 120 10-s images; low-resolution pass, 100 3-s images). Data sets were processed using the HKL suite of programs.40 Crystals belong to space group P6122 with a = b = 44.97 Å and c = 202.11 Å, with one molecule in the asymmetric unit and a solvent content of 57%.
Diffraction data for LC8–Swa were collected on Advanced Light Source beamline 8.2.1 (λ = 0.98 Å; Δφ = 1°; 130 4-s images). Images were integrated with MOSFLM,41 and reflections were merged using SCALA.42 Crystals belong to space group P6122 with a=b=44.15 Å and c=203.71 Å, with one molecule in the asymmetric unit and a solvent content of 49%.
Structure determination and refinement
The structure of apo-LC8 was solved by using the LC8 chain from the previously reported LC8–Swa structure [Protein Data Bank (PDB) entry 2p1k19] as an initial model. Before refinement, 10% of the reflection data were set aside for cross-validation. The test set comprised the same reflections used in the LC8–Swa structure plus new randomly selected reflections beyond 2-Å resolution. Molecular replacement at 3.5-Å resolution using MOLREP43 followed by rigid-body refinement using REFMAC44 resulted in Rcryst and Rfree values of 38% and 41%, respectively. The structure was iteratively refined using REFMAC and Coot†, including TLS refinement,45 to final Rcryst and Rfree values of 17.9% and 21.1%, respectively. During refinement, ordered water molecules were added or removed by the criterion of having reasonable hydrogen-bonding partners and a peak in the 2Fo–Fc electron density map of at least 1σ. Water molecules were numbered on the basis of final peak electron density from 1 (the highest) to 109 (the weakest).
The new structure of LC8–Swa was solved similarly. Following rigid-body refinement, Rcryst and Rfree were 40% and 42%, respectively, and there was strong density for the bound peptide. A model for Swa was built into this density. The structure was refined iteratively to Rcryst and Rfree values of 21.5% and 26.5%, respectively.
Per-atom contributions to solvent-accessible surface area were calculated using VOLBL.46 Structure diagrams were produced with PyMOL‡. Occupied volumes of residues were calculated by the Voronoi method47 using the tools available online from the Database of Molecular Movements§. The fractional volume of each residue was defined as the ratio of its calculated volume to a reference volume derived from a set of high-resolution structures selected from the PDB.26 Buried residues were defined as those having less than 5 Å2 of solvent-accessible surface area. Interface residues were defined as residues exposed in the monomer that lose at least 50% of their solvent-accessible surface upon dimerization.
NMR spectroscopy
NMR samples of 15N-labeled LC8 were prepared as described previously.19 1H–15N HSQC spectra were recorded at 303 K with 256 scans per increment (for LC8–Swa and LC8–nNOS) or at 298 K with 64 scans per increment (for LC8–IC) on a 600-MHz Bruker DRX spectrometer. Spectra were processed with NMRPipe.48 Plots of spectra were prepared with burrow-owl.49 For quantitative measurement of peak intensities, peaks were converted to a Gaussian line shape using a Lorentzian-to-Gaussian transform, and then the frequency-domain data were fit by least-squares minimization to Gaussian line shapes using in-house software. Chemical shifts and line widths were constrained to be equal throughout a titration series. Reported intensities were corrected for dilution of the sample due to the addition of peptide stock solution, which was 5 to 10 times the protein concentration.
Theoretical curves for the relative populations of apo, singly bound, and doubly bound forms were derived by solving the following set of equations:
PDB accession codes
The coordinates of apo-LC8 and LC8–Swa have been deposited in the Research Collaboratory for Structural Bioinformatics PDB with accession codes 3bri and 3e2b, respectively.
Acknowledgments
This work was supported by the National Science Foundation through CAREER grant MCB-0417181 (E.J.B.) and the American Heart Association through Award 0720019Z (G.C.B.). We acknowledge the support of the nucleic acid and protein core and the mass spectrometry core of the Oregon State University Environmental Health Sciences Center (National Institute of Environmental Health Sciences, National Institutes of Health, P30 ES00210). We thank Yujuan Song and Jean Yau for protein preparation, Justin Hall for collecting the native diffraction data set for LC8–Swa, and Clare Woodward for critical reading of the manuscript.
Abbreviations used
- nNOS
neuronal nitric oxide synthase
- HSQC
heteronuclear single-quantum coherence
References
- 1.Pfister K, Fisher E, Gibbons I, Hays T, Holzbaur E, McIntosh J, et al. Cytoplasmic dynein nomenclature. J Cell Biol. 2005;171:411–413. doi: 10.1083/jcb.200508078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dick T, Ray K, Salz H, Chia W. Cytoplasmic dynein (ddlc1) mutations cause morphogenetic defects and apoptotic cell death in Drosophila melanogaster. Mol Cell Biol. 1996;16:1966–1977. doi: 10.1128/mcb.16.5.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vallee R, Williams J, Varma D, Barnhart L. Dynein: an ancient motor protein involved in multiple modes of transport. J Neurobiol. 2004;58:189–200. doi: 10.1002/neu.10314. [DOI] [PubMed] [Google Scholar]
- 4.Benashski S, Harrison A, Patel-King R, King S. Dimerization of the highly conserved light chain shared by dynein and myosin V. J Biol Chem. 1997;272:20929–20935. doi: 10.1074/jbc.272.33.20929. [DOI] [PubMed] [Google Scholar]
- 5.Espindola F, Suter D, Partata L, Cao T, Wolenski J, Cheney R, et al. The light chain composition of chicken brain myosin-Va: calmodulin, myosin-II essential light chains, and 8-kDa dynein light chain/PIN. Cell Motil Cytoskeleton. 2001;47:269–281. doi: 10.1002/1097-0169(200012)47:4<269::AID-CM2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 6.Hódi Z, Németh A, Radnai L, Hetényi C, Schlett K, Bodor A, et al. Alternatively spliced exon B of myosin Va is essential for binding the tail associated light chain shared by dynein. Biochemistry. 2006;45:12582–12595. doi: 10.1021/bi060991e. [DOI] [PubMed] [Google Scholar]
- 7.Wagner W, Fodor E, Ginsburg A, Hammer J. The binding of DYNLL2 to myosin Va requires alternatively spliced exon B and stabilizes a portion of the myosin's coiled-coil domain. Biochemistry. 2006;45:11564–11577. doi: 10.1021/bi061142u. [DOI] [PubMed] [Google Scholar]
- 8.Makokha M, Hare M, Li M, Hays T, Barbar E. Interactions of cytoplasmic dynein light chains Tctex-1 and LC8 with the intermediate chain IC74. Biochemistry. 2002;41:4302–4311. doi: 10.1021/bi011970h. [DOI] [PubMed] [Google Scholar]
- 9.Lo K, Naisbitt S, Fan J, Sheng M, Zhang M. The 8-kDa dynein light chain binds to its targets via a conserved (K/R)XTQT motif. J Biol Chem. 2001;276:14059–14066. doi: 10.1074/jbc.M010320200. [DOI] [PubMed] [Google Scholar]
- 10.King S, Barbarese E, Dillman J, Patel-King R, Carson J, Pfister K. Brain cytoplasmic and flagellar outer arm dyneins share a highly conserved Mr 8,000 light chain. J Biol Chem. 1996;271:19358–19366. doi: 10.1074/jbc.271.32.19358. [DOI] [PubMed] [Google Scholar]
- 11.Schnorrer F, Bohmann K, Niesslein-Volhard C. The molecular motor dynein is involved in targeting Swallow and bicoid RNA to the anterior pole of Drosophila oocytes. Nat Cell Biol. 2000;2:185–190. doi: 10.1038/35008601. [DOI] [PubMed] [Google Scholar]
- 12.Naisbitt S, Valtschanoff J, Allison D, Sala C, Kim E, Craig A, et al. Interaction of the postsynaptic density-95/guanylate kinase domain-associated protein complex with a light chain of myosin-v and dynein. J Neurosci. 2000;20:4524–4534. doi: 10.1523/JNEUROSCI.20-12-04524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lo K, Kan H, Chan L, Xu W, Wang K, Wu Z, et al. The 8-kDa dynein light chain binds to p53binding protein 1 and mediates DNA damage-induced p53 nuclear accumulation. J Biol Chem. 2005;280:8172–8179. doi: 10.1074/jbc.M411408200. [DOI] [PubMed] [Google Scholar]
- 14.Jaffrey S, Snyder S. PIN: an associated protein inhibitor of neuronal nitric oxide synthase. Science. 1996;274:774–777. doi: 10.1126/science.274.5288.774. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Hare M, Hays T, Barbar E. Dynein light chain LC8 promotes assembly of the coiled-coil domain of Swallow protein. Biochemistry. 2004;43:4611–4620. doi: 10.1021/bi036328x. [DOI] [PubMed] [Google Scholar]
- 16.Barbar E. Dynein light chain LC8 is a dimerization hub essential in diverse protein networks. Biochemistry. 2008;47:503–508. doi: 10.1021/bi701995m. [DOI] [PubMed] [Google Scholar]
- 17.Stelter P, Kunze R, Flemming D, Hpfner D, Diepholz M, Philippsen P, et al. Molecular basis for the functional interaction of dynein light chain with the nuclear-pore complex. Nat Cell Biol. 2007;9:788–796. doi: 10.1038/ncb1604. [DOI] [PubMed] [Google Scholar]
- 18.Liang J, Jaffrey S, Guo W, Snyder S, Clardy J. Structure of the PIN/LC8 dimer with a bound peptide. Nat Struct Biol. 1999;6:735–740. doi: 10.1038/11501. [DOI] [PubMed] [Google Scholar]
- 19.Benison G, Karplus P, Barbar E. Structure and dynamics of LC8 complexes with KXTQT-motif peptides: Swallow and dynein intermediate chain compete for a common site. J Mol Biol. 2007;371:457–468. doi: 10.1016/j.jmb.2007.05.046. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez-Crespo I, Yelamos B, Roncal F, Albar J, Ortiz de Montellano P, Gavilanes F. Identification of novel cellular proteins that bind to the LC8 dynein light chain using a pepscan technique. FEBS Lett. 2001;503:135–141. doi: 10.1016/s0014-5793(01)02718-1. [DOI] [PubMed] [Google Scholar]
- 21.Fan J, Zhang Q, Tochio H, Li M, Zhang M. Structural basis of diverse sequence-dependent target recognition by the 8 kDa dynein light chain. J Mol Biol. 2001;306:97–108. doi: 10.1006/jmbi.2000.4374. [DOI] [PubMed] [Google Scholar]
- 22.Diederichs K, Karplus P. Improved Rfactors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- 23.Lovell S, Davis I, Arendall W, de Bakker P, Word J, Prisant M, et al. Structure validation by Cαgeometry: φ, ψ Cβ deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 24.Williams J, Roulhac P, Roy A, Vallee R, Fitzgerald M, Hendrickson W. Structural and thermodynamic characterization of a cytoplasmic dynein light chain–intermediate chain complex. Proc Natl Acad Sci USA. 2007;104:10028–10033. doi: 10.1073/pnas.0703614104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerstein M, Lesk A, Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994;33:6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 26.Tsai J, Taylor R, Chothia C, Gerstein M. The packing density in proteins: standard radii and volumes. J Mol Biol. 1999;290:253–266. doi: 10.1006/jmbi.1999.2829. [DOI] [PubMed] [Google Scholar]
- 27.Vadlamudi R, Bagheri-Yarmand R, Yang Z, Balasenthil S, Nguyen D, Sahin A, et al. Dynein light chain 1, a p21-activated kinase 1-interacting substrate, promotes cancerous phenotypes. Cancer Cell. 2004;5:575–585. doi: 10.1016/j.ccr.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 28.Fan J, Zhang Q, Li M, Tochio H, Yamazaki T, Shimizu M, Zhang M. Protein inhibitor of neuronal nitric-oxide synthase, PIN, binds to a 17amino acid residue fragment of the enzyme. J Biol Chem. 1998;273:33472–33481. doi: 10.1074/jbc.273.50.33472. [DOI] [PubMed] [Google Scholar]
- 29.Dyson H, Wright P. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 30.Fan J, Zhang Q, Tochio H, Zhang M. Backbone dynamics of the 8 kDa dynein light chain dimer reveals molecular basis of the protein's functional diversity. J Biomol NMR. 2002;23:103–114. doi: 10.1023/a:1016332918178. [DOI] [PubMed] [Google Scholar]
- 31.Chothia C, Janin J. Principles of protein– protein recognition. Nature. 1975;256:705–708. doi: 10.1038/256705a0. [DOI] [PubMed] [Google Scholar]
- 32.Barbar E, Kleinman B, Imhoff D, Li M, Hays T, Hare M. Dimerization and folding of LC8, a highly conserved light chain of cytoplasmic dynein. Biochemistry. 2001;40:1596–1605. doi: 10.1021/bi002278+. [DOI] [PubMed] [Google Scholar]
- 33.Changeux J, Edelstein S. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 34.Stevens S, Sanker S, Kent C, Zuiderweg E. Delineation of the allosteric mechanism of a cytidylyltransferase exhibiting negative cooperativity. Nat Struct Biol. 2001;8:947–952. doi: 10.1038/nsb1101-947. [DOI] [PubMed] [Google Scholar]
- 35.Navarro C, Puthalakath H, Adams J, Strasser A, Lehmann R. Egalitarian binds dynein light chain to establish oocyte polarity and maintain oocyte fate. Nat Cell Biol. 2004;6:427–435. doi: 10.1038/ncb1122. [DOI] [PubMed] [Google Scholar]
- 36.Lee K, Lee S, Kim B, Chang S, Kim S, Paick J, Rhee K. Dazl can bind to dynein motor complex and may play a role in transport of specific mRNAs. EMBO J. 2006;25:4263–4270. doi: 10.1038/sj.emboj.7601304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu H, Maciejewski M, Takebe S, King S. Solution structure of the TcTex1 dimer reveals a mechanism for dynein–cargo interactions. Structure (Camb) 2005;13:213–223. doi: 10.1016/j.str.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 38.Benison G, Nyarko A, Barbar E. Heteronuclear NMR identifies a nascent helix in intrinsically disordered dynein intermediate chain: implications for folding and dimerization. J Mol Biol. 2006;362:1082–1093. doi: 10.1016/j.jmb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 39.Rao J, Lahiri J, Isaacs L, Weis R, Whitesides G. A trivalent system from vancomycin D-Ala-DAla with higher affinity than avidin–biotin. Science. 1998;280:708–711. doi: 10.1126/science.280.5364.708. [DOI] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 41.Leslie A. Integration of macromolecular diffraction data. Acta Crystallogr, Sect D: Biol Crystallogr. 1999;55:1696–1702. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- 42.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 43.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 44.Murshudov G, Vagin A, Dodson E. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 45.Winn M, Isupov M, Murshudov G. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr Sect D: Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 46.Edelsbrunner H, Facello M, Fu P, Liang J. Measuring proteins and voids in proteins. Proceedings of the 28th Annual Hawaii International Conference on System Sciences 1995;5:256–264. [Google Scholar]
- 47.Richards F. Calculation of molecular volumes and areas for structures of known geometry. Methods Enzymol. 1985;115:440–464. doi: 10.1016/0076-6879(85)15032-9. [DOI] [PubMed] [Google Scholar]
- 48.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe—a multidimensional spectral processing system based on Unix pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 49.Benison G, Berkholz D, Barbar E. Protein assignments without peak lists using higher-order spectra. J Magn Reson. 2007;189:173–181. doi: 10.1016/j.jmr.2007.09.009. [DOI] [PubMed] [Google Scholar]