

Abstract
We previously showed that exposure to environmental cigarette smoke (ECS) for 28 days causes extensive downregulation of microRNA expression in the lungs of rats, resulting in the overexpression of multiple genes and proteins. In the present study, we evaluated by microarray the expression of 484 microRNAs in the lungs of either ECS-free or ECS-exposed rats treated with the orally administered chemopreventive agents N-acetylcysteine, oltipraz, indole-3-carbinol, 5,6-benzoflavone, and phenethyl isothiocyanate (as single agents or in combinations). This is the first study of microRNA modulation by chemopreventive agents in nonmalignant tissues. Scatterplot, hierarchical cluster, and principal component analyses of microarray and quantitative PCR data showed that none of the above chemopreventive regimens appreciably affected the baseline microRNA expression, indicating potential safety. On the other hand, all of them attenuated ECS-induced alterations but to a variable extent and with different patterns, indicating potential preventive efficacy. The main ECS-altered functions that were modulated by chemopreventive agents included cell proliferation, apoptosis, differentiation, Ras activation, P53 functions, NF-κB pathway, transforming growth factor–related stress response, and angiogenesis. Some micro-RNAs known to be polymorphic in humans were downregulated by ECS and were protected by chemopreventive agents. This study provides proof-of-concept and validation of technology that we are further refining to screen and prioritize potential agents for continued development and to help elucidate their biological effects and mechanisms. Therefore, microRNA analysis may provide a new tool for predicting at early carcinogenesis stages both the potential safety and efficacy of cancer chemopreventive agents.
Introduction
MicroRNAs (miRNA) provide a major epigenetic mechanism that regulates translation of expressed genes into proteins. These small noncoding RNAs play a role in crucial biological processes, such as cell growth (1), apoptosis (2), development (3), differentiation (4), and endocrine homeostasis (5). MiRNAs have been investigated in a variety of diseases, including diabetes, heart diseases, Alzheimer’s disease, and viral infections (6). The most active area and the starting point for the pathogenetic role of miRNAs is cancer research (7, 8), to such an extent that alterations in miRNA genes have been proposed to be involved in the pathophysiology of many, perhaps, all human cancers (9).
Less attention has been paid to the possible occurrence of miRNA alterations in healthy tissues as a consequence of exposures to environmental and life-style factors, including carcinogens and noxious agents, drugs, and food components. Recently, we provided evidence that exposure of rats to environmental cigarette smoke (ECS) results in extensive downregulation, in the lung, of the expression of several miRNAs involved in a variety of cell functions, such as stress response, apoptosis, proliferation, angiogenesis, and gene transcription (10). These findings were confirmed in a study evaluating miRNA expression in the human airway epithelium of smokers (11) and by our further studies in mice (12). The results of these studies support the view that cigarette smoke mainly works as a tumor promoter by triggering a variety of epigenetic mechanisms (13, 14).
In the present study, we evaluated miRNA expression as a new tool for assessing the ability of chemopreventive agents to modulate physiologic miRNA profiles as well as alterations induced in the lung following exposure of rats to ECS. The investigated chemopreventive agents, all of them administered orally, included dietary agents, such as phenethyl isothiocyanate (PEITC) and indole-3-carbinol (I3C), the synthetic flavone 5,6-benzoflavone (BF), and pharmacologic agents, such as N-acetyl-L-cysteine (NAC) and oltipraz (OPZ). Due to the interest of pursuing a combined chemoprevention strategy, combinations of PEITC with I3C and of NAC with OPZ were also investigated.
Materials and Methods
Animals and treatments
Adult male Sprague-Dawley rats (Harlan Italy), weighing 315 to 320 g, were divided into 16 groups, each composed of eight animals. Eight groups had their whole bodies exposed to ECS for 28 consecutive d, as previously described (15), whereas the remaining rats (Sham exposed) were kept for the same period of time in filtered air. The rats belonging to 14 groups were treated with chemopreventive agents, starting 3 d before exposure to ECS. NAC was given with the drinking water, whereas all other agents were supplemented to the diet. In particular, as detailed in a previous article (15), the experimental groups were as follows: group 1, rats kept in filtered air (Sham); mean diet consumption, 25.0 g/d; body weight gain during the 28 d of the experiment, 17.8%; group 2, rats treated with OPZ (400 mg/kg diet); mean dietary consumption and mean intake of chemopreventive agent, 19.4 g/d and 7.8 mg/rat/d, respectively; body weight gain, 13.2%; group 3, rats treated with PEITC (500 mg/kg diet; 19.2 g/d and 9.6 mg/rat/d); body weight gain, 14.2%; group 4, rats treated with BF (500 mg/kg diet; 20.1 g/d and 10.1 mg/rat/d); body weight gain, 13%; group 5, rats treated with I3C (2,500 mg/kg diet; 25 g/d and 62.5 mg/rat/d); body weight gain, 15.7%; group 6, rats treated with NAC (1,000 mg/kg body weight; 25 g/d and 300 mg/rat/d); body weight gain, 15.1%; group 7, rats treated with a combination of PEITC and I3C (22.5 g/d and 11.3 and 56.3 mg/rat/d, respectively); body weight gain, 15.4%; group 8, rats treated with a combination of OPZ and NAC (22.8 g/d and 9.1 and 300 mg/rat/d, respectively); body weight gain, 16.4%; group 9, ECS-exposed rats. body weight gain, 11.7%; group 10, ECS-exposed rats treated with OPZ (400 mg/kg diet; 18.9 g/d and 7.6 mg/rat/d); body weight gain, 6.3%; group 11, ECS-exposed rats treated with PEITC (500 mg/kg diet; 19.6 g/d and 9.8 mg/rat/d); body weight gain, 6.7%; group 12, ECS-exposed rats treated with BF (500 mg/kg diet; 19.4 g/d and 9.7 mg/rat/d); body weight gain, 6.4%; group 13, ECS-exposed rats treated with I3C (2,500 mg/kg diet; 25 g/d and 62.5 mg/rat/d); body weight gain, 11.8%; group 14, ECS-exposed rats treated with NAC (1,000 mg/kg body weight; 25.0 g/d and 300 mg/rat/d); body weight gain, 13.6%; group 15, ECS-exposed rats treated with a combination of PEITC and I3C (20.9 g/d and 10.5 and 52.3 mg/rat/d, respectively); body weight gain, 7.7%; group 16, ECS-exposed rats treated with a combination of OPZ and NAC (21.8 g/d and 8.7 and 300 mg/rat/d, respectively); body weight gain, 8.6%.
At the end of the experiment, the rats were starved for 24 h, anesthesized deeply with diethyl ether, and killed by cervical dislocation. The lungs were collected, immersed in an RNA-stabilizing buffer, and frozen at −80°C.
The housing and all treatments of animals were in accordance with U.S. NIH and Italian and institutional guidelines.
RNA extraction and miRNA analysis
RNA extraction, quantification, and evaluation of integrity, microarray profiling for 484 rodent miRNAs, and real-time quantitative PCR (qPCR) for miR-let7c were done as previously described (10). The whole list of miRNAs included in the microarray used is available at the Gene Expression Omnibus database (registration number requested).
Analysis of data
Local background was subtracted from raw data, which were then log transformed, normalized, and analyzed by GeneSpring software version 7.2 (Agilent Technologies). Expression data were median centered by using the Gene-Spring normalization option, which normalizes both per gene and per array. Quadruplicate data generated for each miRNA were compared among the various experimental groups by volcano plot analysis, which evaluates both fold variations and statistical significance of differences by ANOVA following Bonferroni multiple testing correction. Global miRNA expression profiles of the various experimental groups were compared by hierarchical cluster analysis and by bidimensional perchloric acid.
Results
Overall microarray evaluation of miRNA expression in the lung of rats treated with chemopreventive agents
As inferred from the scatterplots shown in Fig. 1 (top), the oral administration of either individual chemopreventive agents (BF, NAC, OPZ, or PEITC) or of combined agents (OPZ + NAC or PEITC + I3C) did not appreciably affect miRNA expression profiles in rat lung. In fact, only a negligible number of miRNAs was dysregulated >2-fold following treatment with the chemopreventive regimens tested, compared with Sham-exposed rats. The large majority of the miRNAs falling outside the 2-fold variation interval were poorly expressed in the lung. None of these variations, which are likely to be ascribed to the experimental fluctuations characteristic of low-intensity signals, reached the statistical significance threshold. Note that the sample from rats treated with I3C alone could not be tested due to insufficient amounts of RNA.
Fig. 1.

Scatterplots relating the expression of 484 miRNAs in the lung of rats treated with various chemopreventive regimens to the expression of the same miRNAs in the lung of rats, either Sham-exposed (top) or ECS-exposed (bottom). Central diagonal line in each panel, equivalence of expression between the groups compared on the X and Y scales; two outer diagonal lines in each panel, 2-fold variation intervals.
Overall microarray evaluation of miRNA expression in the lung of rats exposed to ECS and treated with chemopreventive agents
The scatterplots shown in Fig. 1 (bottom) highlight a general trend to modulation of ECS-altered miRNA profiles by all chemopreventive agents tested. In fact, compared with rats exposed to ECS in the absence of chemopreventive treatments, all the agents tended to upregulate miRNA expression, thus counteracting the downregulation induced by ECS alone.
As shown at a glance in Fig. 1, this protective effect varied among the chemopreventive regimens tested. Treatment-related differences in modulating miRNA profiles were well detectable by performing unsupervised hierarchical cluster analysis (Fig. 2). It is evident that Sham and ECS were allocated far away in the dendogram and that, with the exception of I3C, treatment of ECS-exposed rats with all other chemopreventive agents were able to reallocate their miRNA profiles in the same dendogram cluster that included untreated rats (Sham).
Fig. 2.
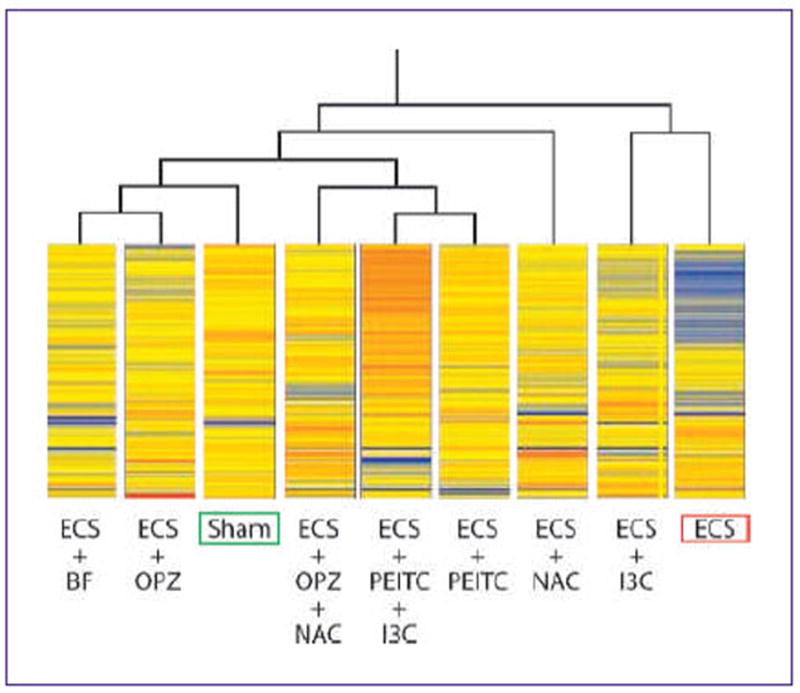
Hierarchical cluster analysis linking the expression profiles of 484 miRNAs among variously treated experimental groups. Each line within each column, the intensity of expression of each miRNA on a color scale, from blue (lowest) to red (highest).
Modulation of the expression of individual miRNAs in the lung of rats exposed to ECS and treated with chemopreventive agents
Table 1 lists the 25 miRNAs whose expression was significantly modulated by chemopreventive agents in rat lung, compared either with unexposed rats (Sham) or ECS-exposed rats. All miRNAs, excepting miR-294, were significantly downregulated by ECS, in the absence of chemopreventive agents (Table 1, first column). Most chemopreventive agents attenuated this effect to a variable extent.
Table 1.
MiRNAs whose expression was significantly modulated by chemopreventive agents in rat lung, compared either with unexposed rats (Sham) or ECS-exposed rats
| miRNA | ECS/Sham | ECS+BF/Sham | ECS+NAC/Sham | ECS+OPZ/Sham | ECS+OPZ+NAC/Sham | ECS+PEITC/Sham | ECS+I3C/Sham | ECS+PEITC+I3C/Sham |
|---|---|---|---|---|---|---|---|---|
| let-7a | 0.24 (0.00–0.77)* | 0.52 (0.27–1.64)† | 0.73 (0.15–1.10)† | 0.85 (0.40–1.19)† | 1.14 (0.82–1.13)‡ | 0.91 (0.69–1.18)† | 0.77 (0.23–1.14) | 1.55 (0.64–2.64)‡ |
| let-7b | 0.24 (0.00–0.85)† | 0.53 (0.00–1.22)‡ | 0.56 (0.00–1.25)†,§ | 1.00 (0.94–1.06)† | 0.90 (0.50–1.31)‡ | 0.84 (0.32–1.35)‡ | 0.60 (0.00–1.28)†,§ | 1.35 (0.40–2.31)† |
| let-7c | 0.22 (0.00–0.80)§ | 0.69 (0.15–1.10)† | 0.72 (0.05–1.02)† | 0.96 (0.71–1.17)‡ | 1.33 (0.67–2.07)‡ | 1.05 (0.85–1.22)‡ | 0.73 (0.27–1.16) | 1.23 (0.70–1.81)‡ |
| let-7f | 0.19 (0.00–0.74)* | 0.38 (0.00–0.85)†,§ | 0.31 (0.00–0.82)†,§ | 0.57 (0.11–1.07)‡ | 1.02 (0.42–1.19)‡ | 0.89 (0.25–1.15)‡ | 0.70 (0.00–0.90) | 1.10 (0.70–1.84)‡ |
| miR-10a | 0.27 (0.00–0.89)* | 1.40 (0.67–2.06)‡ | 0.51 (0.13–1.08)† | 1.02 (0.49–1.20)‡ | 0.85 (0.26–1.15)† | 0.68 (0.41–1.19)† | 0.96 (0.72–1.17)† | 1.80 (0.62–3.26)§,|| |
| miR-26a | 0.32 (0.00–0.99)* | 0.81 (0.30–1.17)† | 1.02 (0.58–1.20)‡ | 1.12 (0.76–1.39)‡ | 1.08 (0.71–1.17)‡ | 1.34 (0.70–1.84)‡ | 1.24 (0.78–1.39)‡ | 1.71 (0.64–2.66)|| |
| miR-30a | 0.31 (0.00–0.96)§ | 0.69 (0.05–1.33) | 0.76 (0.17–1.35) | 0.64 (0.00–1.31) | 0.67 (0.02–1.32) | 0.84 (0.33–1.35) | 0.65 (0.0–1.31) | 1.69 (0.19–3.18)† |
| miR-30c | 0.12 (0.00–0.57)¶ | 0.63 (0.08–1.05)‡ | 0.58 (0.00–1.27)‡,§ | 0.61 (0.00–1.29)‡,§ | 0.76 (0.34–1.18)‡ | 0.60 (0.00–1.28)‡,§ | 0.87 (0.41–1.19)† | 0.95 (0.55–1.20)‡ |
| miR-34b | 0.33 (0.00–0.99)* | 0.76 (0.17–1.35)† | 1.13 (0.59–1.67)‡ | 0.69 (0.05–1.33) | 0.57 (0.00–1.26)†,§ | 0.57 (0.00–1.25)†,§ | 0.99 (0.92–1.07)‡ | 0.81 (0.27–1.35)† |
| miR-34c | 0.32 (0.00–0.97)* | 0.85 (0.36–1.34)† | 1.12 (0.62–1.61)‡ | 0.93 (0.59–1.28)‡ | 0.97 (0.75–1.20)‡ | 0.62 (0.00–1.29) | 0.59 (0.00–1.27) | 2.05 (0.01–4.10)† |
| miR-99b | 0.29 (0.00–0.92)* | 0.68 (0.04–1.33)†,§ | 0.65 (0.00–1.31)†,§ | 0.78 (0.20–1.35)† | 0.61 (0.00–1.28)†,§ | 0.97 (0.73–1.21)‡ | 0.79 (0.21–1.35)† | 1.56 (0.26–2.86)†,§ |
| miR-122a | 0.25 (0.00–0.85)* | 0.52 (0.00–1.21)*,† | 0.61 (0.00–1.29)† | 0.50 (0.00–1.19)†,§ | 0.53 (0.00–1.22)†,§ | 0.59 (0.00–1.27)†,§ | 0.50 (0.00–1.19)†,§ | 0.98 (0.80–1.16)‡ |
| miR-123-prec | 0.32 (0.00–0.97)* | 0.59 (0.00–1.27)*,† | 0.91 (0.52–1.30)† | 1.01 (0.85–1.18)‡ | 1.27 (0.38–2.06)‡ | 1.19 (0.53–1.84)‡ | 0.63 (0.00–1.30)†,§ | 2.07 (0.01–4.13)*,‡ |
| miR-124a-prec | 0.14 (0.00–0.62)* | 0.61 (0.10–1.18) | 0.70 (0.06–1.33)† | 0.63 (0.09–1.18) | 0.80 (0.48–1.25)† | 0.73 (0.12–1.35)† | 0.88 (0.76–1.16)† | 1.19 (0.64–1.78)‡ |
| miR-125a-prec | 0.12 (0.00–0.59)§ | 0.57 (0.00–1.26)†,§ | 0.61 (0.00–1.28)†,§ | 0.69 (0.04–1.33)† | 0.73 (0.02–1.14)† | 0.71 (0.08–1.34)† | 1.01 (0.12–1.19)† | 1.12 (0.65–1.73)‡ |
| miR-125b | 0.30 (0.00–0.69)§ | 0.61 (0.00–1.11)†,§ | 0.76 (0.32–1.25)†,§ | 0.88 (0.35–1.25)‡,§ | 0.76 (0.22–1.23)†,§ | 0.90 (0.52–1.25)‡ | 0.81 (0.35–1.18) | 1.84 (0.43–3.25)‡,§ |
| miR-140s | 0.23 (0.00–0.81)§ | 0.66 (0.07–1.17)§ | 0.90 (0.48–1.32)† | 0.76 (0.17–1.35)† | 0.81 (0.27–1.35)† | 0.89 (0.45–1.33)† | 0.78 (0.36–1.25) | 1.55 (0.27–2.84)‡,§ |
| miR-145-prec | 0.22 (0.00–0.80)¶ | 0.54 (0.00–1.23)† | 0.82 (0.29–1.35)‡ | 0.64 (0.00–1.30)‡ | 0.70 (0.06–1.33)‡ | 0.84 (0.32–1.35)‡ | 0.48 (0.00–1.17)§ | 1.72 (0.18–3.25)‡ |
| miR-146-prec | 0.28 (0.00–0.90)* | 0.89 (0.44–1.33)† | 1.03 (0.80–1.25)‡ | 0.61 (0.00–1.29)†,§ | 1.00 (0.90–1.09)‡ | 1.23 (0.50–1.96)‡ | 0.63 (0.00–1.30)§ | 2.24 (0.50–3.54)*,‡ |
| miR-191-prec | 0.11 (0.00–0.54)* | 0.52 (0.00–1.21)*,† | 0.70 (0.07–1.34)† | 0.62 (0.00–1.29)†,§ | 0.56 (0.00–1.27)†,§ | 0.85 (0.35–1.35)‡ | 0.40 (0.00–1.08)* | 1.43 (0.34–2.51)§,|| |
| miR-192 | 0.24 (0.00–0.84)* | 0.68 (0.03–1.33)§ | 0.70 (0.06–1.33) | 0.59 (0.00–1.27) | 0.96 (0.68–1.23)† | 1.05 (0.74–1.36)† | 0.83 (0.30–1.35) | 1.35 (0.40–2.31)† |
| miR-219-prec | 0.19 (0.00–0.74)* | 0.64 (0.00–1.31) | 0.73 (0.12–1.35) | 0.57 (0.00–1.26) | 0.86 (0.39–1.34) | 0.63 (0.00–1.30) | 0.43 (0.00–1.12)§ | 0.75 (0.15–1.35) |
| miR-222-prec | 0.26 (0.00–0.75)§ | 0.59 (0.00–1.12)† | 0.76 (0.04–1.15)† | 0.84 (0.22–1.23)‡ | 1.09 (0.79–1.31)‡ | 1.23 (0.75–1.39)‡ | 0.53 (0.00–1.07)†,§ | 1.69 (0.52–2.49)‡ |
| miR-223-prec | 0.21 (0.00–0.77)§ | 0.44 (0.00–1.13)‡,§ | 0.50 (0.00–1.19)‡,§ | 0.49 (0.00–1.18)‡,§ | 0.73 (0.12–1.35)‡ | 0.84 (0.34–1.35)‡ | 0.32 (0.00–0.97)* | 1.45 (0.33–2.57)‡ |
| miR-294 | 10.7 (0.00–24.76)§ | 8.76 (0.00–17.78)§ | 9.31 (0.00–21.50)§ | 10.51 (0.00–24.36)§ | 7.67 (0.00–19.97) | 9.34 (0.00–21.66)§ | 5.59 (0.00–10.20) | 7.17 (0.00–18.78) |
NOTE: The reported values are the ratio of miRNA expression in the lung of ECS-exposed rats, either untreated or treated with chemopreventive agents, to that in the lung of Sham-exposed rats. The numbers between brackets are the 95% confidence intervals. Bold values identify the (ECS + chemopreventive agents)/Sham ratios higher than 0.90, which was arbitrarily assumed as demonstrative of a strong protective effect of chemopreventive agents toward ECS-induced miRNA alterations.
Statistical analysis: P < 0.01 compared with Sham.
Statistical analysis: P < 0.05 compared with ECS.
Statistical analysis: P < 0.001 compared with ECS.
Statistical analysis: P < 0.05 compared with Sham.
Statistical analysis: P < 0.01 compared with ECS.
Statistical analysis: P < 0.001 compared with Sham.
In particular, three general situations can be envisaged when comparing the values recorded in ECS-exposed rats receiving a chemopreventive agent with those recorded in ECS-exposed rats in the absence of chemopreventive agents. The first situation, indicating a poor protective effect, was that the chemopreventive agent did not significantly affect ECS-induced miRNA alterations (absence of superscripts *, ‡, or b in Table 1). This situation was observed for 6 of 25 miRNAs in BF-treated rats (24.0%), 4 in NAC-treated rats (16.0%), 6 in OPZ-treated rats (24.0%), 4 in PEITC-treated rats (16.0%), 14 in I3C-treated rats (56.0%), 3 in rats treated with both OPZ and NAC (12.0%), and 2 in rats treated with both PEITC and I3C (8.0%). The second situation, reflecting a moderate protective effect, was that the values recorded in ECS-exposed rats treated with a chemopreventive agent were significantly different from both Sham and ECS (‡ superscripts in Table 1). This situation was observed for 9 of 25 miRNAs in BF-treated rats (36.0%), 7 in NAC-treated rats (28.0%), 6 in OPZ-treated rats (24.0%), 3 in PEITC-treated rats (12.0%), 4 in I3C-treated rats (16.0%), 5 in rats treated with both OPZ and NAC (20.0%), and 7 in rats treated with both PEITC and I3C (28.0%). The third situation, reflecting a strong protective effect, was that the values recorded in ECS-exposed rats treated with a chemopreventive agent were significantly different from ECS only (superscripts §, b, and a in Table 1). This situation was observed for 10 of 25 miRNAs in BF-treated rats (40.0%), 14 in NAC-treated rats (56.0%), 13 in OPZ-treated rats (52.0%), 18 in PEITC-treated rats (72.0%), 7 in I3C-treated rats (28.0%), 17 in rats treated with both OPZ and NAC (68.0%), and 16 in rats treated with both PEITC and I3C (64.0%). Based on these data, it seems that PEITC alone, PEITC + I3C, and OPZ + NAC were the most effective treatments in counteracting miRNA downregulation by ECS. However, it should be noted that, in the case of the combined treatment with PEITC and I3C, miRNA expression often exceeded the baseline situation.
The striking upregulation of miR-294 in ECS-exposed rats was attenuated by treatment with either I3C, NAC + OPZ, or PEITC + I3C. In all three cases, the expression level was no longer significantly different from Sham but did not reach the significance threshold also compared with ECS.
In addition to statistical significance criteria, we assumed an arbitrary threshold value of 0.90 for (ECS + chemopreventive agent)/Sham ratios as a biological indicator of a strong protective effect of a chemopreventive agent toward ECS-induced miRNA alterations (bold values in Table 1).
The biological functions of those ECS-dysregulated miRNAs whose expression was modified by the investigated chemopreventive agents were inferred by selecting genes having a context score of >0.31, as reported in Targetscan,5 Miranda,6 and Sanger Institute7 databases. Table 2 shows an outline of the main functions regulated by miRNAs whose ECS-altered expression was normalized by the investigated chemopreventive agents.
Table 2.
Main functions regulated by those miRNAs whose ECS-altered expression was normalized by chemopreventive agents (bold values in Table 1)
| ECS-altered miRNA | MiRNA-regulated function | Protective chemopreventive agent(s) |
|---|---|---|
| let-7a | Cell proliferation, Ras activation, angiogenesis | PEITC + I3C > OPZ + NAC > PEITC |
| let-7b | Cell proliferation, Ras activation, angiogenesis | PEITC + I3C > OPZ > OPZ + NAC |
| let-7c | Cell proliferation, Ras activation, angiogenesis | OPZ + NAC > PEITC + I3C > PEITC > OPZ |
| let-7f | Cell proliferation, Ras activation, angiogenesis | PEITC + I3C > OPZ + NAC |
| miR-10a | Angiogenesis | PEITC + I3C > BF > OPZ > I3C |
| miR-26a | TGF expression | PEITC + I3C > PEITC > I3C > OPZ > OPZ + NAC |
| miR-30a | Cell adhesion, protein repair, NF-κB activation, EGF activation, cell proliferation | PEITC + I3C |
| miR-30c | Cell adhesion, protein repair, NF-κB activation, EGF activation, cell proliferation | PEITC + I3C |
| miR-34b | P53 effector | NAC > I3C |
| miR-34c | P53 effector | PEITC + I3C > NAC > OPZ + NAC > OPZ |
| miR-99b | Apoptosis | PEITC + I3C > PEITC |
| miR-122a | Stress response | PEITC + I3C |
| miR-123-prec | Angiogenesis | PEITC + I3C > OPZ + NAC > PEITC > OPZ > NAC |
| miR-124a-prec | Stress response | PEITC + I3C |
| miR-125a-prec | Erbb2 activation | PEITC + I3C > I3C |
| miR-125b | Stress response | PEITC + I3C > PEITC |
| miR-140s | P53 effector | PEITC + I3C > NAC |
| miR-145-prec | Protein repair, angiogenesis | PEITC + I3C |
| miR-146-prec | NF-κB activation | PEITC + I3C > PEITC > NAC > OPZ + NAC |
| miR-191-prec | Cell proliferation | PEITC + I3C |
| miR-192 | Ras activation | PEITC + I3C > PEITC > OPZ + NAC |
| miR-219-prec | Elk-1 and Fos activation | None |
| miR-222-prec | Cell proliferation, angiogenesis | PEITC + I3C > PEITC > OPZ + NAC |
| miR-223-prec | Protein repair, Ras activation | PEITC + I3C |
| miR-294 | Gene transcription | None |
Principal component analysis of miRNA expression profiles in rat lung
Principal component analysis (PCA) took into account the data obtained in the 15 experimental groups by analyzing the expression of all 484 miRNAs tested. PCA evaluates the dispersion of data according to the two main components of variance, thus resulting in a bidimensional scattering of the experimental groups, which fall in four quadrants that cluster similar miRNA profiles.
The results of PCA are reported in Fig. 3. It is evident that ECS and Sham fall far away, in two opposite quadrants. All chemopreventive agents, in the absence of exposure to ECS, fall in the same quadrant where Sham is allocated, thus reflecting the poor alterations of miRNA expression exerted by all chemopreventive regimens tested. On the other hand, when given to ECS-exposed rats, all chemopreventive agents tended to depart from ECS and to approach Sham. In the case of the combined treatment with PEITC and I3C, however, PCA allocation was the farthest away from ECS, but at the same time it was far away from Sham, thus reflecting a nonphysiologic modulation of ECS-related miRNA expression.
Fig. 3.
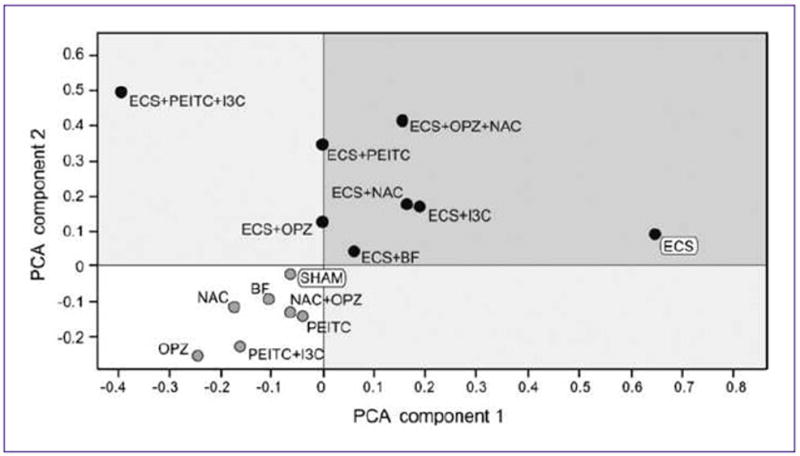
Bidimensional PCA comparing the expression profile of 484 miRNAs among variously treated experimental groups.
Confirmation by qPCR of let-7c expression in rat lung
As shown in Table 1, microarray analyses indicated that the expression of let-7c was markedly downregulated in ECS-exposed rats and was modulated, to a variable extent, by the chemopreventive agents tested. In addition, due to the major relevance of let-7c in pulmonary carcinogenesis, we further analyzed this miRNA by qPCR in the lung of Sham-exposed rats and of ECS-exposed rats, either untreated or treated with the chemopreventive agents. Exposure to ECS caused a downregulation of let-7c expression, as inferred from the increased PCR cycle needed to attain the positivity threshold, compared with Sham (Fig. 4). This effect was attenuated by all but one (BF) chemopreventive agents tested. PEITC and OPZ, when tested individually, had a moderate effect, whereas PEITC + I3C, OPZ + NAC, NAC, and I3C were all very close to the Sham expression level (Fig. 4).
Fig. 4.
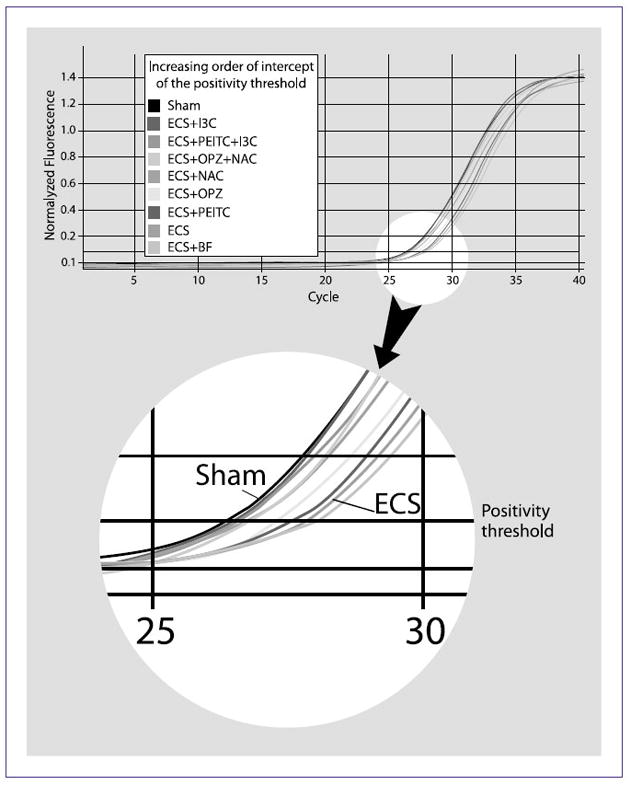
Real-time qPCR amplification curves (top) and positivity threshold (bottom) of let-7c expression in the lung of variously treated rats.
Discussion
Due to the significant difficulties encountered in clinical chemoprevention trials, animal models are very useful to evaluate potential cancer protective effects of dietary and pharmacologic agents, and to prioritize agents for clinical trials. Modulation of early molecular biomarkers, also including those investigated by using “omics” technologies, is of particular interest both for understanding the mechanisms of action of chemopreventive agents and for assessing their safety and efficacy. The following general criteria should be met: (a) as a molecular indicator of safety, an optimal chemopreventive agent should not substantially alter the baseline expression of genes, miR-NAs, and proteins; and (b) as an indicator of efficacy, an optimal chemopreventive agent should be able to counteract the molecular alterations induced by carcinogens and to restore the physiologic situation.
We previously investigated the gene expression profiles of the same chemopreventive agents in the same rat model as those evaluated in the present study (15), producing data that support and complement our present miRNA results. NAC, OPZ, and their combination clustered close to negative controls, thus denoting little alteration of the physiologic situation. At the same time, however, in ECS-exposed lungs, treatments with either OPZ or NAC clustered close to positive controls, thus denoting poor inhibition of ECS-related transcriptome alterations. In contrast, PEITC was the most effective agent in this analysis. The picture from these analyses is complicated by the circumstance that agents appearing to better counteract ECS-related alterations in gene expression also change the physiologic situation (15). In a further study, the lung postmitochondrial fractions of mice treated with Sham, NAC, ECS, or ECS + NAC were analyzed by antibody microarray for the levels of 518 proteins. The oral administration of NAC did not change the basal level of any protein >2-fold and reduced the number of ECS-induced proteins from 50 (9.7%) to 42 (8.1%; ref. 16).
In principle, as supported by the present study, miRNA expression compares favorably to gene and protein expression because the number of existing miRNAs is relatively small, and accordingly, mirnome coverage by microarray analysis is quite broad. In addition, each miRNA targets hundreds of genes (17), thus resulting in an effective global regulation of cell functions. In contrast, only a very small fraction of expressed genes is translated into proteins (18), and the number of detectable proteins is orders of magnitude lower than the number of existing proteins (19). In the interpretation of our data, it should be taken into account that they reflect the net effects of ECS and chemopreventive agents on the lung cell population, which is composed of over 40 cell types involved in a multitude of functions (20). Therefore, we cannot rule out that individual cell types may react in a different way to exogenous stimuli.
The results obtained in the present in vivo study provide evidence, for the first time, that mirnome analysis is a promising tool to investigate cancer chemopreventive agents. None of the chemopreventive regimens tested was found to alter the baseline expression of the 484 analyzed miRNAs to an appreciable extent. On the other hand, the mirnome profiles in the lung of ECS-exposed rats were considerably dysregulated, mainly in the sense of downregulation. These data agree with the results obtained in a previous study (10), where we discussed the correspondence between downregulation of specific miRNAs and upregulation of targeted proteins. In particular, several functions out of those that we found to be dysregulated by ECS at the mirnome level had previously been reported to be altered at both transcriptome and proteome levels. These functions include stress response (16), protein repair (16, 21), RAS activation (22), P53 suppression (21, 23, 24), cell proliferation (16, 23), apoptosis (16, 24, 25), and angiogenesis (16).
The two functions that were most frequently modulated by chemopreventive agents were angiogenesis and transforming growth factor-β (TGF-β)–related response. In fact, all chemopreventive agents counteracted the proangiogenic effect of ECS, which is exerted by downregulating miRNA silencing proangiogenic genes (10). In particular, BF tended to normalize miR-10a; OPZ tended to normalize let-7b, let-7c, miR-10a, and miR-123-prec; PEITC tended to normalize let-7a, let-7c, miR-123-prec, and miR-222; and NAC tended to normalize miR-123-prec. Of the two combinations tested, PEITC + I3C affected several angiogenesis-related miRNAs, including let-7a, let-7b, let-7c, let-7f, miR-10a, miR-123-prec, miR-145-prec, and miR-222-prec, whereas OPZ + NAC tended to attenuate let-7a, let-7b, let-7c, let-7f, miR-123-prec, and miR-222-prec. The antiangiogenic effect is likely to be related to the antioxidant properties of the chemopreventive agents tested. Reactive species, such as hydrogen peroxide and nitric oxide, are involved in the production of factors inducing vessel growth, and their proangiogenic effect is attenuated by factors protecting them from oxidative stress (26, 27). PEITC + I3C, PEITC, I3C, OPZ, and OPZ + NAC, in decreasing rank of activity, counteracted the ECS-downregulated miR-26a silencing of TGF-β. The effect of I3C on miR-26a is consistent with the known ability of this agent to induce p21/p27, which results in an inhibition of cyclin-dependent kinases upregulated during TGF-β–mediated antiproliferative response (28). Modulation of ECS-induced cell proliferation by the combination of OPZ and PEITC occurred through regulation of several miRNAs, including those of the let-7 family, miR-191-prec, and miR-222-prec, which explains the ability of PEITC to slow down the cell cycle through multiple mechanisms, among which the covalent binding to tubulin (29).
In addition, each one of the chemopreventive agents tested specifically modulated other biological functions. The most effectively modulated miRNAs in ECS-exposed rats treated with NAC, an analogue and precursor of reduced glutathione, are involved in NF-κB–mediated stress response (miR-146-prec) and P53 functions (miR-34b, miR-34c, and miR-140s). NAC, besides being a potent scavenger of free radicals, works through multiple mechanisms, also including its ability to inhibit activation and nuclear translocation of NF-κB, to modulate the post-translational increase of P53 expression, and to decrease TGF-β biological activity (30, 31). In particular, attenuation by NAC of ECS-induced miRNA downregulation is consistent with the observed attenuation of ECS-induced proteins or functions in rodent lung. In fact, NAC modulated ECS-altered proteins involved in cell replication (cyclin F), stress response (FOS, IκKα, IκKβ, and IκKγ), and apoptosis (caspase-14; ref. 16), and related functions (30).
In addition to modulation of TGF-β expression (miR-26a), cell proliferation (let-7b and let-7c), and angiogenesis (let-7b, let-7c, miR-10a, and miR-123-prec), OPZ normalized a miRNA involved in P53 function (miR-34c). OPZ is known to interfere with signal transduction pathways that play a central role in cell proliferation (32), to induce wild-type P53 protein and the P53 pathway (33), and to suppress TGF-β (34).
The ECS-downregulated miRNAs affected by PEITC are involved in a variety of functions, including stress response (miR-125b), TGF-β expression (miR-26a), NF-κB activation (miR-146-prec), Ras activation (let-7a, let-7c, and miR-192), cell proliferation (let-7a, let-7c, and miR-222-prec), cell apoptosis (miR-99b), and angiogenesis (let-7a, let-7c, miR-123-prec, and miR-222-prec). These findings are consistent with literature data, showing that PEITC activates the MAPK stress response pathway (35, 36), and is a potent inducer of apoptosis (25). In fact, either alone or in combination with I3C, PEITC was the only agent able to protect miR-99b from ECS-induced downregulation. MiR-192 is involved in H-Ras–mediated oncogenic transformation, which causes elevated generation of reactive oxygen species and renders the cells highly sensitive to PEITC (37).
I3C affected miRNAs involved in P53 functions (miR-34b), TGF-β expression (miR-26a), Erbb2 activation (miR-125a-prec), and angiogenesis (miR-10a). The effect on miR-34b explains at the mirnome level the mechanisms of I3C, which is known to arrest the cell cycle by increasing activated P53 protein in both prostate cancer cells (38) and mammary epithelial cells (39). I3C was the only individual agent that protected miR-125a-prec from ECS downregulation. MiR-125a-prec recognizes as a main target the Erbb2 oncogene, which plays a fundamental role in breast cancer (40) and is also expressed in lung carcinomas (41). I3C exerts antiproliferative and apoptotic effects in breast cancer cells expressing Erbb2 (42). It is noteworthy that I3C was the only chemopreventive agent that had some effect on the ECS-related overexpression of miR-294, which is involved in gene transcription and recognizes transcriptional repressor genes (e.g., zinc finger protein 697; AT-rich interactive domain 4A) as main targets. Accordingly, the increase of miR-294 expression results in a global increase of gene transcription as a response to ECS (10), a mechanism that is attenuated by I3C.
Combined chemoprevention is a very promising strategy in the pharmacologic and dietary prevention of cancer. The combination of NAC and OPZ affected miRNAs that were either individually modulated by both agents (miR-34c and miR-123-prec) or by one of them only (let-7b, let-7c, miR-26a, and miR-146-prec). In addition, this combined chemopreventive regimen affected a series of miRNAs involved in cell proliferation (let-7a, let-7f, and miR-222-prec), Ras activation (miR-192), and angiogenesis (miR-222-prec). Finally, the combination of PEITC and I3C had profound effects on almost all miRNAs downregulated by ECS, which were not affected by the individual agents, thus suggesting that these agents display more than additive effects. Note however that, as previously discussed, the resulting modulation departed from the physiologic situation.
MiRNA genes are frequently located near mouse cancer susceptibility loci (43). We found that chemopreventive agents are effective in protecting from ECS some miRNAs known to be polymorphic in humans (44, 45). This was the case for let-7a (PEITC), miR-125b (PEITC), miR-140s (NAC), and miR-146-prec (NAC, PEITC). Accordingly, it is likely that the efficacy of these chemopreventive agents, when administered to humans, may be influenced by the genetic polymorphisms of these miRNAs. This issue could help to explain the wide interindividual variability observed in the efficacy of these chemopreventive agents when tested in clinical trials (46, 47). It has been predicted that single nucleotide polymorphisms that reside in the miRNA target site may become a gold mine for molecular epidemiology (48). Likewise, the study of polymorphic miRNAs is likely to become an essential tool to develop and qualify chemopreventive agents for clinical trials.
As documented in this study, all of these agents have complex patterns of effects on miRNAs, which become even more complex for their combinations. Objectively comparing and prioritizing between agents for clinical development will require, in part, future studies that can interpret the many complex miRNA patterns associated with agent effects on carcinogenesis. For example, patterns of angiogenesis-related miRNA effects ranged from that of NAC, which modulated only one angiogenesis miRNA (miR-123 prec), to that of PEITC, which modulated several (let-7a, let-7c, miR-222, and miR-123-prec). It will be important to sort out the critical patterns of angiogenesis-related effects (and all other carcinogenesis-related effects) in animal studies of lung cancer development and, ultimately, in early-phase human trials before proceeding to large-scale, definitive clinical testing.
In conclusion, our present study is the first to look at chemopreventive effects on miRNAs and shows the promise of miRNA profiles for predicting the future clinical efficacy and safety of novel chemopreventive agents. This study provides proof-of-concept and technology validation, and we are refining the technology to both screen and prioritize potential agents for continued drug development and to provide biological and mechanistic insights into specific promising agents. Objective prioritization of agents will require sophisticated statistical modeling. A valuable contribution of our future work will be to look at correlations between miRNA results and tumor results in the same model. Our future studies also will examine miRNA polymorphisms—for example, in smokers who develop or do not develop lung cancer—for profiles that may identify individuals who are sensitive (or resistant) to specific agents and thus advance the evolution of personalized prevention.
Acknowledgments
We thank Dr. Ilaria Righi for the bibliographical search and assistance in preparation of the manuscript.
Grant Support
NIH National Cancer Institute (contract N01-CN-53301).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005;33:1290–7. doi: 10.1093/nar/gki200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu P, Guo M, Hay BA. MicroRNAs and the regulation of cell death. Trends Genet. 2004;20:617–24. doi: 10.1016/j.tig.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Karp X, Ambros V. Developmental biology. Encountering microRNAs in cell fate signaling. Science. 2005;310:1288–9. doi: 10.1126/science.1121566. [DOI] [PubMed] [Google Scholar]
- 4.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–6. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 5.Poy MN, Eliasson L, Krutzfeldt J, et al. A pancreatic-islet specific miRNA regulates insulin secretion. Nature. 2004;432:226–30. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 6.Couzin J. MicroRNAs make big impression in disease after disease. Science. 2008;319:1782–4. doi: 10.1126/science.319.5871.1782. [DOI] [PubMed] [Google Scholar]
- 7.Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR-15 and miR-16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM. MicroRNA expression and function in cancer. Trends Mol Med. 2006;12:580–7. doi: 10.1016/j.molmed.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Calin GA, Croce CM. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 2006;66:7390–4. doi: 10.1158/0008-5472.CAN-06-0800. [DOI] [PubMed] [Google Scholar]
- 10.Izzotti A, Calin GA, Arrigo P, Steele VE, Croce CM, De Flora S. Down-regulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009;23:806–12. doi: 10.1096/fj.08-121384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schembri F, Sridhar S, Perdomo C, et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc Natl Acad Sci U S A. 2009;106:2319–24. doi: 10.1073/pnas.0806383106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Izzotti A, Calin GA, Steele VE, Croce CM, De Flora S. Relationships of microRNA expression in mouse lung with age and exposure to cigarette smoke and light. FASEB J. 2009;23:3243–50. doi: 10.1096/fj.09-135251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trosko JE, Upham BL. The emperor wears no clothes in the field of carcinogenic risk assessment: ignored concepts in cancer risk assessment. Mutagenesis. 2005;20:81–92. doi: 10.1093/mutage/gei017. [DOI] [PubMed] [Google Scholar]
- 14.Upham BL, Trosko JE. Oxidative-dependent integration of signal transduction with intercellular gap junctional communication in the control of gene expression. Antioxid Redox Signal. 2009;11:1–11. doi: 10.1089/ars.2008.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Izzotti A, Bagnasco M, Cartiglia C, et al. Modulation of multigene expression and proteome profiles by chemopreventive agents. Mutat Res. 2005;591:212–23. doi: 10.1016/j.mrfmmm.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 16.Izzotti A, Bagnasco M, Cartiglia C, et al. Chemoprevention of genome, transcriptome, and proteome alterations induced by cigarette smoke in rat lung. Eur J Cancer. 2005;41:1864–74. doi: 10.1016/j.ejca.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 17.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 18.Zong Q, Schummer M, Hood L, Morris DR. Messenger RNA translation state: the second dimension of high-throughput expression screening. Proc Natl Acad Sci U S A. 1999;96:10632–6. doi: 10.1073/pnas.96.19.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melton L. Proteomics in multiplex. Nature. 2004;429:101–7. doi: 10.1038/429101a. [DOI] [PubMed] [Google Scholar]
- 20.Pinkerton KE, Joad JP. Influence of air pollution on respiratory health during perinatal development. Clin Exp Pharmacol Physiol. 2006;33:269–72. doi: 10.1111/j.1440-1681.2006.04357.x. [DOI] [PubMed] [Google Scholar]
- 21.Izzotti A, Cartiglia C, Longobardi M, et al. Alterations of gene expression in skin and lung of mice exposed to light and cigarette smoke. FASEB J. 2004;18:1559–61. doi: 10.1096/fj.04-1877fje. (Full text published online, 2 August 2004) [DOI] [PubMed] [Google Scholar]
- 22.Yao R, Wang Y, Lubet RA, et al. K-Ras mutations in lung tumors from p53 mutant mice exposed to cigarette smoke. Exp Lung Res. 2005;31:271–81. doi: 10.1080/0190214059090386. [DOI] [PubMed] [Google Scholar]
- 23.De Flora S, Balansky RM, D’Agostini F, et al. Molecular alterations and lung tumors in p53 mutant mice exposed to cigarette smoke. Cancer Res. 2003;63:793–800. [PubMed] [Google Scholar]
- 24.Izzotti A, Cartiglia C, Longobardi M, et al. Gene expression in the lung of p53 mutant mice exposed to cigarette smoke. Cancer Res. 2004;64:8566–72. doi: 10.1158/0008-5472.CAN-04-1420. [DOI] [PubMed] [Google Scholar]
- 25.D’Agostini F, Izzotti A, Balansky RM, Bennicelli C, De Flora S. Modulation of apoptosis by chemopreventive agents. Mutat Res. 2005;591:173–86. doi: 10.1016/j.mrfmmm.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 26.Gerald D, Berra E, Frapart YM, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–94. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 27.Zhao F, Nozawa H, Daikonnya A, Kondo K, Kitanaka S. Inhibitors of nitric oxide production from hops (Humulus lupulus L. ) Biol Pharm Bull. 2003;26:61–5. doi: 10.1248/bpb.26.61. [DOI] [PubMed] [Google Scholar]
- 28.Chinni SR, Li Y, Upadhyay S, Koppolu PK, Sarkar FH. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. 2001;20:2927–36. doi: 10.1038/sj.onc.1204365. [DOI] [PubMed] [Google Scholar]
- 29.Mi L, Xiao Z, Hood BL, et al. Covalent binding to tubulin by isothiocyanates. A mechanism of cell growth arrest and apoptosis. J Biol Chem. 2008;283:22136–46. doi: 10.1074/jbc.M802330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Flora S, Izzotti A, D’Agostini F, Balansky RM. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis. 2001;22:999–1013. doi: 10.1093/carcin/22.7.999. [DOI] [PubMed] [Google Scholar]
- 31.De Flora S, Izzotti A, Albini A, D’Agostini F, Bagnasco M, Balansky RM. Antigenotoxic and cancer preventive mechanisms of N-acetyl-L-cysteine. In: Kelloff GJ, Hawk ET, Sigman CC, editors. Cancer chemoprevention. Vol. 1. New York: The Humana Press Inc; 2004. pp. 37–67. [Google Scholar]
- 32.Madhukar BV, Dimitrov NV, Meyer-Leece C, Contreras ML, Crowell J. Inhibition of mitogen-activated protein kinase activity of human lymphocytes after oral administration of Oltipraz. Mol Cancer Ther. 2002;1:1125–8. [PubMed] [Google Scholar]
- 33.Chi WJ, Doong SL, Lin-Shiau SY, Boone CW, Kelloff GJ, Lin JK. Oltipraz, a novel inhibitor of hepatitis B virus transcription through elevation of p53 protein. Carcinogenesis. 1998;19:2133–8. doi: 10.1093/carcin/19.12.2133. [DOI] [PubMed] [Google Scholar]
- 34.Kang KW, Choi SH, Ha JR, Kim CW, Kim SG. Inhibition of dimethylnitrosamine-induced liver fibrosis by [5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione] (oltipraz) in rats: suppression of transforming growth factor-β1 and tumor necrosis factor-α expression. Chem Biol Interact. 2002;139:61–77. doi: 10.1016/s0009-2797(01)00286-1. [DOI] [PubMed] [Google Scholar]
- 35.Hu R, Kim BR, Chen C, Hebbar V, Kong AN. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cells. Carcinogenesis. 2003;24:1361–7. doi: 10.1093/carcin/bgg092. [DOI] [PubMed] [Google Scholar]
- 36.Kong AN, Owuor E, Yu R, et al. Induction of xenobiotic enzymes by the MAP kinase pathway and the antioxidant or electrophile response element (ARE/EpRE) Drug Metab Rev. 2001;33:255–71. doi: 10.1081/dmr-120000652. [DOI] [PubMed] [Google Scholar]
- 37.Trachootham D, Zhou Y, Zhang H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–52. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Hsu JC, Dev A, Wing A, Brew CT, Bjeldanes LF, Firestone GL. Indole-3-carbinol mediated cell cycle arrest of LNCaP human prostate cancer cells requires the induced production of activated p53 tumor suppressor protein. Biochem Pharmacol. 2006;72:1714–23. doi: 10.1016/j.bcp.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 39.Brew CT, Aronchik I, Hsu JC, et al. Indole-3-carbinol activates the ATM signaling pathway independent of DNA damage to stabilize p53 and induce G1 arrest of human mammary epithelial cells. Int J Cancer. 2006;118:857–68. doi: 10.1002/ijc.21445. [DOI] [PubMed] [Google Scholar]
- 40.Xiang B, Chatti K, Qiu H, et al. Brk is coamplified with ErbB2 to promote proliferation in breast cancer. Proc Natl Acad Sci U S A. 2008;105:12463–8. doi: 10.1073/pnas.0805009105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.López-Malpartida AV, Ludeña MD, Varela G, Pichel JG. Differential ErbB receptor expression and intracellular signaling activity in lung adenocarcinomas and squamous cell carcinomas. Lung Cancer. 2009;65:25–33. doi: 10.1016/j.lungcan.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 42.Rahman KM, Aranha O, Glazyrin A, Chinni SR, Sarkar FH. Translocation of Bax to mitochondria induces apoptotic cell death in indole-3-carbinol (I3C) treated breast cancer cells. Oncogene. 2000;19:5764–71. doi: 10.1038/sj.onc.1203959. [DOI] [PubMed] [Google Scholar]
- 43.Sevignani C, Calin GA, Nnadi SC, et al. MicroRNA genes are frequently located near mouse cancer susceptibility loci. Proc Natl Acad Sci U S A. 2007;104:8017–22. doi: 10.1073/pnas.0702177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saunders MA, Liang H, Li WH. Human polymorphism at microRNAs and microRNA target sites. Proc Natl Acad Sci U S A. 2007;104:3300–5. doi: 10.1073/pnas.0611347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duan R, Pak C, Jin P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum Mol Genet. 2007;16:1124–31. doi: 10.1093/hmg/ddm062. [DOI] [PubMed] [Google Scholar]
- 46.Van Schooten FJ, Nia AB, De Flora S, et al. Effects of oral N-acetylcysteine: a multi-biomarker study in smokers. Cancer Epidemiol Biomarkers Prev. 2002;11:167–75. [PubMed] [Google Scholar]
- 47.Seow A, Vainio H, Yu MC. Effect of glutathione-S-transferase polymorphisms on the cancer preventive potential of isothiocyanates: an epidemiological perspective. Mutat Res. 2005;592:58–67. doi: 10.1016/j.mrfmmm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 48.Chen K, Song F, Calin GA, Wei Q, Hao X, Zhang W. Polymorphisms in microRNA targets: a gold mine for molecular epidemiology. Carcinogenesis. 2008;29:1306–11. doi: 10.1093/carcin/bgn116. [DOI] [PubMed] [Google Scholar]