

Abstract
ABCA1 mediates the secretion of cellular free cholesterol and phospholipids to an extracellular acceptor, apolipoprotein AI, to form nascent high-density lipoprotein (HDL). Thus, ABCA1 is a key molecule in cholesterol homeostasis. Functional studies of certain Tangier disease mutations demonstrate that ABCA1 has multiple activities, including plasma membrane remodeling and apoAI binding to cell surface, which participate in nascent HDL biogenesis. Recent advances in our understanding of ABCA1 have demonstrated that ABCA1 also mediates unfolding the N terminus of apoAI on the cell surface, followed by lipidation of apoAI and release of nascent HDL. Although ABCA1 mediated cholesterol efflux to apoAI can occur on the plasma membrane, the role of apoAI retroendocytosis during cholesterol efflux may play a role in macrophage foam cells that store cholesterol esters in cytoplasmic lipid droplets.
Keywords: ABCA1, HDL, apoAI, reverse cholesterol transport, atherosclerosis
Numerous epidemiological studies have established an inverse correlation between high-density lipoprotein (HDL) levels and coronary heart disease (CHD)1–3. Although HDL has multiple anti-atherogenic properties4–6, such as anti-inflammatory, anti-oxidant, anti-thrombotic, and anti-apoptotic, the protective effect of HDL is primarily attributed to its ability to remove excess cholesterol from peripheral tissues and deliver it to the liver for biliary excretion, a process called reverse cholesterol transport (RCT)7. The first step in the RCT pathway is the biogenesis of nascent HDL derived from cellular lipids and extracellular lipid-free or lipid-poor apolipoprotein AI (apoAI) in a process mediated by ABCA1; and, this process of cellular cholesterol and phospholipid efflux is the major source of plasma HDL. Genetic modulation of apoAI or ABCA1 in mouse models alters not only HDL biogenesis, but also has effects on atherosclerosis in mouse models8–12. During atherogenesis, the primary cellular pathology is the accumulation of macrophage foam cells in which there is an imbalance between cholesterol influx and efflux. As the lesions progress, the foam cells often die leading to the accumulation of cholesterol rich necrotic plaques in the arterial intima. Therefore, ABCA1 mediated HDL formation is a key mediator regulating macrophage cholesterol homeostasis and this process plays a critically important role in the initiation of early atherosclerotic lesion development. The goal of this review is to summarize research relevant to ABCA1 and its activities in mediating the assembly of cellular lipids and exogenous apoAI to generate nascent HDL.
Plasma HDL is a complex mixture of subspecies
In human plasma, HDL is a heterogeneous collection of lipoprotein particles ranging in diameter from 7 to 12 nm and density from 1.063 to 1.21 g/ml. The nomenclature for HDL subspecies varies depending on the separation technique used13, 14. On the basis of HDL’s buoyant density, ultracentrifugation can separate HDLs into 2 major subfractions, the more buoyant HDL2 (density between 1.063 and 1.125 g/mL) and denser HDL3 (density between 1.125 and 1.21 g/mL). On the basis of size, non-denaturing gradient gel electrophoresis has been used to separate HDL into 5 major subfractions. They are HDL2b, HDL2a, HDL3a, HDL3b, and HDL3c in the order of progressively decreasing size. Non-denaturing 2D gel electrophoresis is probably the best way to separate various apoAI-containing particles into pre-β-1 (corresponding to lipid-poor, or lipid-free apoAI), pre-β-2, α-4, α-3, α-2, and α-1, and pre-α species according to their mass:charge ratio as well as size15. However, it is not correct to think of all of these HDL species and lipid-poor apoAI as static pools of distinct particles, instead, HDL is dynamic with much remodeling, lipolysis, and fusion that can convert smaller particles to larger particles and vice versa. Pre-β-1 particles representing small lipid-free and lipid-poor apoAI are the substrate for ABCA1 leading to the formation of nascent HDL, which in turn is the substrate for lethicin:cholesterol acyltransferase (LCAT) that esterifies free cholesterol into cholesteryl ester, building up the hydrophobic core necessary to generate spherical α-HDL particles. The majority of plasma apoAI-containing particles are spherical particles having α-electrophoretic mobility. Furthermore, this mature HDL can accept additional cellular cholesterol through the activities of cellular ABCG1 and scavenger ester class B type I (SR-BI)16, 17. Finally, the cholesterol ester in HDL is returned to the liver via direct hepatic uptake by SR-BI, or indirectly via transfer to apoB-containing lipoprotein by cholesteryl ester transfer protein (CETP) with subsequent uptake by the liver, where it can be converted to free cholesterol for direct excretion or converted to bile acids for excretion, completing the RCT pathway18.
The proteomics of HDL is very complex, but the overwhelming majority of HDL particles contain apoAI, which is the most abundant apolipoprotein in normal human plasma. Many HDL particles also contain apoAII, the second most abundant protein in HDL, along with other apolipoproteins including apoAV, apoCI, apoCII, apoCIII and apoCIV. Minor subpopulations of HDL particles containing only apoE, were found in human plasma and have been proposed to play a role in RCT19. 188 different proteins have been found on HDL, including the HDL remodeling proteins CETP and LCAT, the HDL oxidizing protein myeloperoxidase, the antioxidant protein paraoxonase1 and numerous other proteins20. Since the largest diameter HDL is around 12 nm, each of these accessory proteins is only on a small fraction of HDL particles. Thus, there are hundreds of different HDL subspecies based on their individual protein composition.
Tangier disease: the role of ABCA1 in HDL biogenesis
In 1961, Fedrickson et al.21 reported a new disease of HDL-deficiency identified in a boy from Tangier Island in the Chesapeake Bay, Virginia. HDL-deficiency, also found in other related subjects from this island, was named as Tangier disease. Individuals with Tangier disease have severe HDL deficiency with less than 5% of normal plasma HDL levels and higher incidence of premature cardiovascular disease, extremely enlarged yellow tonsil, clouding of the cornea, and enlarged spleen and liver22. Cultured fibroblasts from normal subjects exhibit cholesterol efflux to apoAI in a dose dependent fashion, but Tangier disease fibroblasts were deficient in this activity23. In addition, cholesterol efflux to apoAI could be induced in mouse macrophage cell lines by treatment with a cAMP analogue24, although the protein and gene responsible for this efflux activity was not known. It was not until 1999 that the etiology of Tangier disease was identified, by positional cloning, as being due to mutations in the ABCA1 gene25–27. This breakthrough revealed the pivotal role of ABCA1 in generating nascent HDL, but the exact mechanism of HDL assembly by ABCA1 is still unknown and will be discussed below.
ABCA1 is a 2261 amino acid integral membrane protein consisting of 12 transmembrane domains and two ATP binding cassette (ABC) domains (Figure 1). It is a member of a large ABC gene family, with several subfamilies that play diverse roles in transmembrane lipid and ion transport. More than 70 distinct mutations have been identified in the ABCA1 gene from Tangier disease patients, with most mutations residing in either of the two large extracellular domains (ECDs) or in one of the two nucleotide binding domains (NBDs) of ABCA128. There are two intramolecular disulfide bonds between ECD1 and ECD2 that are necessary for apoAI binding and HDL formation.29.
Figure 1.

Domain structure of ABCA1. ABCA1 has 12 membrane spanning domains, two intracellular nucleotide binding domains (NBD), and two large extracellular domains (ECD1 and ECD2). The positions of the Tangier disease mutations in ECD1 and ECD2 that helped solve different activities of ABCA1 are shown, as is a site directed mutation in the first NBD which abolishes ATP hydrolysis and lipid efflux.
The metabolism of HDL initiates with apoAI synthesis in the liver and intestine, but HDL formation requires apoAI interaction with ABCA1. When human ABCA1 is overexpressed in mouse liver and macrophages, plasma HDL and apoAI are increased in the transgenic mice30. Furthermore, the hepatic overexpression of ABCA1, by infusion of adenovirus expressing ABCA1 into mice, induces apoAI-dependent cholesterol efflux from primary hepatocytes and also increases plasma HDL and apoAI levels31, 32. The role of ABCA1 in the formation of HDL particles was further studied in mice with tissue-specific deletion of the ABCA1 gene, showing that the liver is the single most important source of plasma HDL, with ~80% and ~90% reductions in plasma HDL and apoAI in the liver-specific knockout, respectively. Thus, hepatic ABCA1 is critical in maintaining circulating levels of mature HDL particles by direct lipidation of lipid-poor apoAI, slowing apoAI catabolism by the kidney, and prolonging its plasma residence time33. The deletion of intestinal ABCA1 gene leads to an approximately 30% reduction in plasma HDL, and deletion of both hepatic and intestinal ABCA1 results in an approximately 90% decrease in plasma HDL levels34. Monocyte/macrophage ABCA1 contributes to HDL formation, but the contribution to the overall plasma HDL levels is minimal35. However, ABCA1 deficiency in macrophages markedly increases atherosclerosis and foam cell accumulation in hypercholesterolemic apoE deficient mice10.
Activities of ABCA1
Tangier disease mutations and other site directed mutations in ABCA1 have been expressed in ABCA1-GFP fusion protein transfected cells36–38, and here we describe their categorization into four groups. The first group is the maturation defective ABCA1 mutants, with the majority of Tangier disease mutations belonging to this group. ABCA1 is synthesized in the endoplasmic reticulum and is transported to the Golgi and then via vesicles to the plasma membrane. Wild-type (WT) ABCA1 is localized at the plasma membrane and intracellular vesicles and compartments38, 39; however, maturation defective mutants of ABCA1, such as R587W and Q597R in the first large ECD, and S1506L in the second large ECD, have impaired trafficking and are localized only in intracellular compartments, never reaching the plasma membrane36, 37. Because apoAI interacts with ABCA1 on the cell surface, mutants that do not reach the plasma membrane are unable to mediate apoAI binding and lipid efflux.
The second group of ABCA1 mutations consists of the lipid translocation defective mutants, represented by the W590S mutation in the first large ECD of ABCA1. Plasma membrane asymmetry is characterized by the differential distribution of phospholipids across the inner and outer leaflets of the membrane bilayer, which is regulated by several classes of proteins including ABC transporters. The W590S mutant protein is correctly targeted to the plasma membrane, indistinguishable from WT ABCA1. Furthermore, the W590S mutant has normal apoAI binding ability36, 37, 40–42. However, the W590S mutant protein is defective in plasma membrane remodeling. WT ABCA1 mediates the outward translocation (floppase activity) of phosphatidylserine (PS), an anionic phospholipid that is usually sequestered in the inner leaflet of the plasma membrane; however, the W590S mutant protein lacks this activity36, 37, 41, 42, which can be demonstrated by measuring cell surface PS with fluorescently tagged annexin V, a PS specific binding protein. Another method to demonstrate the plasma membrane remodeling activity of ABCA1 is using sodium taurocholate (NaTC) as a weak detergent with extracellular lipid acceptor activity41, WT ABCA1 mediates increased lipid efflux to NaTC, but the W590S mutant protein greatly decreases cholesterol efflux to NaTC. Furthermore, ABCA1 leads to increased plasma membrane fluidity and the release of microparticles in the absence of exogenous acceptors, which leads to modest levels of lipid efflux43. Cholesterol efflux in the absence of acceptors was impaired by the W590S mutation42. Thus, the W590S mutation in the first extracellular domain of ABCA1 is defective in three assays of membrane remodeling, PS translocation, efflux to NaTC, and microparticle efflux in the absence of exogenous acceptors. Despite markedly reduced membrane remodeling activity, the W590S mutant ABCA1 protein has diminished but partial activity to mediate cholesterol efflux to apoAI42.
The third group of ABCA1 mutations consists of the apoAI binding defective mutants, represented by the C1477R mutation in the second large ECD. Expression of WT ABCA1 induces the cellular binding and uptake of apoAI42, 44. In addition, chemical cross-linking experiments show that apoAI directly binds to ABCA112, 45–47. However, most of the apoAI on the cell surface is not thought to be directly bound to ABCA1, but this conclusion is based on several assumptions that are difficult to assess47, 48. Similar to WT ABCA1, the C1477R mutant is expressed in the plasma membrane and competent for PS translocation, efflux to NaTC, and microparticle efflux in the absence of exogenous acceptors; however this mutant has significantly reduced cellular apoAI binding37, 42. Thus, the membrane remodeling and apoAI binding activities of ABCA1 are independent of each other, and appear to be dependent on different extracellular domains. The C1447R mutant also has diminished but partial activity to mediate cholesterol efflux to apoAI42.
The fourth group of ABCA1 mutations consists of the NBD mutants which are correctly localized to the plasma membrane, but disrupt ATP hydrolysis. They are exemplified by the site directed mutations K939M and K1952M49, 50. These mutations are defective in both PS translocation and apoAI binding, and unlike the W590S and C1477R mutations, they are totally defective in mediating cholesterol efflux to apoAI42.
Recently, we generated a fluorescent apoAI indicator of its N-terminal folding state. We used this indicator on cells transfected with different classes of ABCA1 mutations42. We discovered a novel activity of ABCA1, the ability to unfold the N terminus of apoAI on the cell surface42. The W590S ABCA1 mutant promotes apoAI unfolding at the cell surface to the same extent as WT ABCA1, whereas the C1477R and K939M mutants, defective apoAI binding and ATP hydrolysis, respectively, have greatly reduced levels of cell surface apoAI unfolding activity (Table 1). ApoAI has been shown to bind to the K939M-ABCA1 cells and untransfected HEK cells non-specifically. We speculated that this apoAI unfolding activity is a third distinct activity of ABCA1, and this function of ABCA1 to be increased by, but not require, the high affinity apoAI binding. This apoAI unfolding activity of K939M-ABCA1 isoforms could be due to the presence of a separate low affinity apoAI binding site, not distinguishable from the non-specific binding observed in cells lacking ABCA1 expression. This low affinity site on ABCA1 could transiently interact with apoAI and act as a chaperone to mediate apoAI N-terminal unfolding. The presence of the high affinity apoAI binding site in WT and W590S ABCA1 isoforms would promote apoAI proximity to the low affinity binding site and therefore increase apoAI unfolding. In aggregate, it is apparent that for maximal cholesterol efflux to apoAI, ABCA1 must be able to remodel the plasma membrane, bind to apoAI, and mediate the unfolding of its N-terminal domain, and that mutations that disrupt only one activity can still lead to partial efflux to apoAI (Table 1).
Table 1.
Activities of wild-type (WT) ABCA1 and ABCA1 mutants.
| Specific apoAI binding | Lipid translocation | ApoAI unfolding on cell surface | Efflux to apoAI | |
|---|---|---|---|---|
| Control | − | − | − | − |
| WT -ABCA1 | ++ | ++ | ++ | ++ |
| W590S -ABCA1 | ++ | − | ++ | + |
| C1477R -ABCA1 | − | ++ | + | + |
| K939M -ABCA1 | − | − | + | − |
Molday and colleagues have recently been successful in purifying epitope tagged ABCA1 from transfected HEK cells and reconstituting it into unilamellar phospholipid liposomes51, 52. Several exciting findings have come from these studies. First, the ATP hydrolysis activity of ABCA1 could be measured using 32P labeled ATP, and performing this assay before or after detergent solubilization allowed the determination of the directionality of ABCA1 insertion into the membrane. ATP hydrolysis is altered by different phospholipid species, and is decreased by cholesterol in the liposomes. Second, fluorescently labeled fluorescent phospholipid tracers could be reconstituted in the liposomes, and used to follow their translocation after adding exogenous ATP. Since most of the ABCA1 molecules are inside-out (thus the NBD are on the outside of the liposome, while normally they are inside the cell), the net flux of phospholipids in this system is from the outer to inner leaflet of the liposome, and the relative distribution between these leaflets determined after dithionite treatment, a membrane impermeant inactivator of the fluorophore. These elegant studies showed that ABCA1 can translocate PS, phosphatidylcholine, phosphatidylethanolamine, and sphingomylin, consistent with the directionality of ABCA1’s known PS floppase activity. The ATPase-deficient mutants (K939M and K1952M) have no significant phospholipid transport activity52. Furthermore, the Tangier disease mutant W590S has reduced phospholipid flippase activity, around 20% of the WT ABCA152. These ABCA1 reconstituted liposomes did not bind apoAI, and lipid efflux was not assessed.
Models for ABCA1 mediated nascent HDL biogenesis
Nascent HDL biogenesis is essential to maintain cholesterol homeostasis in the body, however, the detailed molecular mechanism by which ABCA1 transforms exogenous apoAI and cellular lipids to assemble nascent HDL is still unknown. Several models have been proposed for this reaction.
The initial models were not detailed in regard to nascent HDL assembly, but only followed the major lipid components of nascent HDL, phospholipid (PL) and free cholesterol (FC). These early models assumed that ABCA1 shuttles lipids onto apoAI, which would accumulate on a growing nascent HDL particle. Fielding et al.53 proposed that ABCA1 mediates the assembly of cellular PL onto exogenous apoAI and that this intermediate in turn could pick up cellular FC in an ABCA1-independent manner. This conclusion was based on the observation that PL efflux from vascular smooth muscle cells to apoAI is less sensitive to vanadate inhibition than FC efflux, and that medium containing apoAI that is conditioned on smooth muscle cells can lead to FC efflux from vascular endothelial cells that do not express ABCA1. However, this was before the discovery that both ABCG1 and SR-BI could mediate FC efflux from cells to HDL in an ABCA1-independent fashion. Wang et al.54 also provided evidence for the two-step pathway by demonstrating that a 30 min pretreatment of ABCA1-expressing cells with 20 mM 2-hydroxypropyl-β-cyclodextrin reduces FC efflux to apoAI without reducing PL efflux, and medium containing apoAI that is conditioned on cyclodextrin-pretreated ABCA1-expressing cells could lead to FC efflux from cells that do not express ABCA1, indicating that cholesterol efflux can be dissociated from phospholipid efflux. Furthermore, photoactive PL could be cross-linked with ABCA1 whereas direct binding of photoactive cholesterol to ABCA1 could not be detected54. However, later research on ABCA7 led the authors to re-assess the two-step pathway. The experiment that included an additional washing step to remove cyclodextrin revealed that conditioned medium with PL/apoAI particles made by ABCA1 could not stimulate passive FC efflux, suggesting that ABCA1 directly mediates both PL and FC efflux to apoAI.55. Furthermore, Smith et al.8 found no evidence to support the two-step pathway of lipid efflux. Instead, a concurrent process model was proposed, in which ABCA1 can directly and concurrently mediate the assembly of PL and FC onto apoAI to generate nascent HDL8. In the RAW264.7 murine macrophage cell line in which ABCA1 expression is inducible by 8-Br-cAMP, FC and PL efflux to apoAI is concurrent both temporally and after treatment with ABCA1 inhibitors. Furthermore, cyclodextrin treatment of RAW264.7 cells partially inhibits 8Br-cAMP-induced efflux of FC and PL to apoAI. They explained some of the observed effects of high-dose cyclodextrin and vanadate as a result of cell damage, as ABCA1-expressing cells are sensitized to both of these treatments, which result in lactate dehydrogenase and PL release by a mechanism independent of extracellular lipid acceptors.
As the PS translocase activity of ABCA1 was being described, Chimini and colleagues37, 49 suggested its role for nascent HDL assembly: 1) ABCA1 mediates the translocation of PS to the plasma membrane outer leaflet; 2) ABCA1 then mediates apoAI binding and the translocation of PL and FC onto apoAI with the released nascent HDL containing cellular PL and FC. Landry et al.56 extended this model showing that the remodeling of the plasma membrane by ABCA1 disrupted lipid rafts to generate loosely packed domains that facilitate apoAI binding to cells. Ueda proposed a four-step model for ABCA1 mediated HDL biogenesis57: first, ATP binding to and hydrolysis by the NBD of ABCA1 leads to conformational changes within ECDs that allow apoAI binding; second, ABCA1 mediates the translocation of phospholipids to the outer leaflet; third, lipids are loaded and accumulate onto the apoAI that is tethered to ABCA1; and fourth, the dissociation of lipid-loaded apoAI from ABCA1 as nascent HDL. In this model, the first apoAI binding step and the second lipid translocation step, both of which are mediated by ABCA1 ATP-hydrolysis, are independent of each other. This model suggests that as apoAI becomes lipid loaded, it loses affinity for ABCA1, facilitating the release of nascent HDL, which is supported by the observation that lipidated apoAI does not interact with ABCA111, 58. Because the W590S mutation impairs the lipid translocation step but not apoAI binding, apoAI can remain bound to the cell in its lipid-free conformation, which has high affinity for interaction with ABCA141, 45.
While the above models assumed ABCA1 mediated assembly of cell lipids incrementally onto a bound apoAI, Rothblat, Phillips and colleagues, even before the discovery of ABCA1, favored a model by which lipid free apoAI, or other amphipathic apolipoproteins or peptides could insert into the plasma membrane and microsolubilize a portion of the membrane to release nascent HDL59, 60. This would generate the concurrent release of both FC and PL. This model has matured, with Phillips proposing a three-step model61. First, there is binding of a small regulatory pool of apoAI to ABCA1, thereby enhancing net phospholipid translocation to the plasma membrane exofacial leaflet; this leads to unequal lateral packing densities in the two leaflets of the phospholipid bilayer. Second, the resultant membrane strain is relieved by bending and by creation of exovesiculated lipid domains. The formation of highly curved membrane surface promotes the binding of apoAI to these domains. Third, this pool of bound apoAI spontaneously solubilizes the exovesiculated domain to create discoidal nascent HDL particles. A key feature of this mechanism is that the lipid translocase activity of ABCA1 leads to high membrane curvature that is required for nascent HDL assembly by apoAI. However, the observation that the W590S mutation is not competent for phospholipid translocation and membrane remodeling but is still able to mediate HDL assembly, albeit at reduced efficiency, questions this model42. As lipid composition of nascent HDL has been shown to be similar to that of the membrane lipid raft, it is possible that the lipids transferred to apoAI are derived from raft like regions of the plasma membrane62.
In a recent study, we identified a new step in the HDL biogenesis, the ABCA1 mediated unfolding the N-terminal domain of apoAI on the cell surface42. The crystal structure of the C-terminal deleted apoAI solved by Mei and Atkinson shows a prominent alpha-helical hairpin loop extending from approximately residues 39-11263. By site directed mutagenesis and covalent modification with self-quenching fluorophore, we showed that residues 39 and 112 are physically pushed further apart on the surface of ABCA1 expressing cells42. We speculated that apoAI’s unfolding would facilitate insertion into the cell membrane, which culminates in the microsoulibilzation of a portion of the membrane and the release of nascent HDL containing cellular phospholipids and cholesterol42.
Putting together all of the observations on ABCA1’s activities, we propose a model for the mechanism of ABCA1 mediated nascent HDL assembly (Figure 2). First, ABCA1 mediates two independent steps, the translocation of phospholipids such as PS from the inner to outer leaflet of the plasma membrane and the cellular binding of apoAI. Next, ABCA1 mediates the partial unfolding of the N-terminus of apoAI. Then, the unfolded apoAI is able to spontaneously insert into the plasma membrane, which resolves in the release of nascent HDL from the cell. We attempted to verify the presence of the intermediate state of apoAI inserted into the cellular membrane, by use of a fluorescent apoAI lipidation indicator, labeled with a lipid sensitive dye42. However, lipidated apoAI was not detected on the surface of ABCA1-expressing cells, implying that the intermediate state with apoAI inserted into the cell lipid bilayer is highly unstable and quickly resolves in the release of nascent HDL42.
Figure 2.
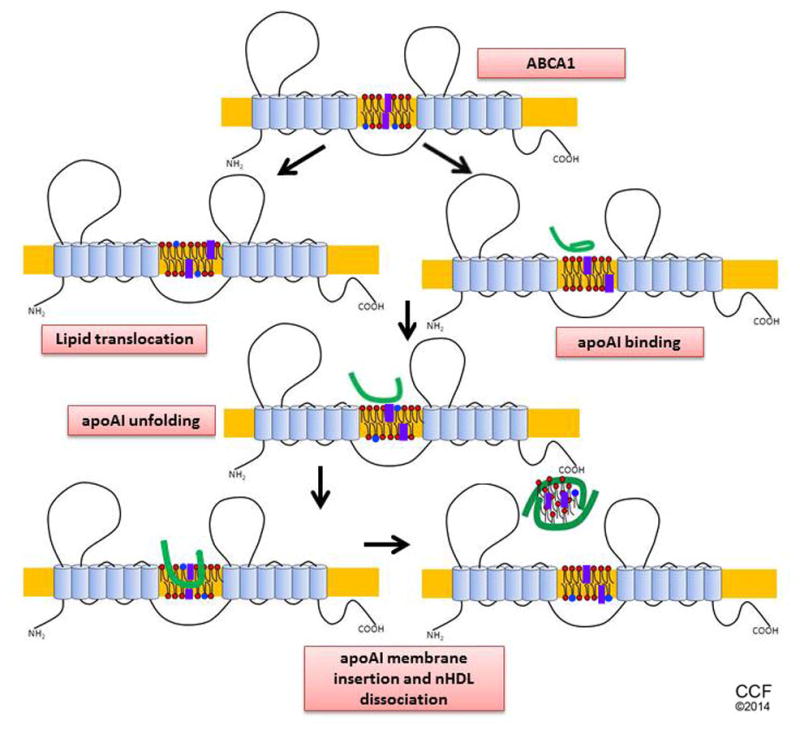
Model for ABCA1 mediated nascent HDL biogenesis. ABCA1 mediates two independent steps, the translocation of phospholipids such as PS from the inner to outer leaflet of the plasma membrane and the cellular binding of apoAI, both of which may be mediated by ATP hydrolysis. ABCA1 then mediates the partial unfolding of the N-terminus of apoAI. The unfolded apoAI is able to spontaneously insert into the plasma membrane, which resolves in the release of nascent HDL from the cell. Denoted are apoAI in green, PC in red, PS in blue, and free cholesterol in purple.
Cellular location of ABCA1 and lipid efflux
ABCA1 resides mainly on the plasma membrane, but also in the intracellular vesicles which transfer ABCA1 to and from the plasma membrane39. Although it is clear that ABCA1 and apoAI can both traffic from the plasma membrane to endosomes39, 44, the role of this cellular uptake in nascent HDL assembly and lipid efflux is controversial44, 64–66. Schmitz and colleagues showed that HDL could be taken up by macrophages cells and then resecreted in a pathway called retroendocytosis67. Smith later showed that apoAI could also be taken up by endocytosis into macrophages, in an ABCA1 dependent manner, and resecreted as nascent HDL, and that blocking endocytosis impairs lipid efflux to apoAI. In addition, ABCA1-mediated lipid efflux has delayed kinetics and is abolished at room temperature, results that are also consistent with a role for endocytosis and vesicular trafficking in this process8. The immunosuppressant cyclosporin A traps ABCA1 on the plasma membrane and also inhibits cholesterol efflux to apoAI, supporting a potential role for ABCA1 endocytic trafficking in ABCA1-mediated lipid efflux68. However, controversy still exists about the mechanism of cyclosporine A inhibition. Ueda and colleagues showed that cyclosporine A inhibits ABCA1 function, including apoAI binding and lipid transport, via direct binding, but it doesn’t abolish cell surface expression of ABCA169. Interestingly, deletion of PEST sequence in ABCA1 leads to impaired internalization of ABCA1, causes higher cholesterol efflux following cell surface labeling, but impaired cholesterol efflux from late endosomes70. Similarly, when ABCA1 and apoAI internalization is blocked, cholesterol efflux from cells that have accumulated cholesterol is decreased, whereas efflux from cells without excess cholesterol is increased66. Thus, retroendocytosis pathway may contribute to HDL formation only when excess cholesterol has accumulated in cells. The retroendocytosis pathway is also supported by data that intracellular pools of cholesterol constituted the major cholesterol source for ABCA1-mediated FC efflux to apoAI71. Although retroendocytosis may play a role in lipid efflux from some cell types, such as cholesterol loaded macrophages, it is most likely that nascent HDL assembly and lipid efflux can also occur on the plasma membrane. In macrophages that were not cholesterol loaded, it has been calculated that the amount of intact apoAI resecreted from the cells is not sufficient to account for the HDL produced by the cholesterol efflux reaction65. Thus, the cellular localization of HDL formation may differ upon whether excess cholesterol has accumulated within cells or not.
Conclusion
ABCA1 is a large membrane protein that is absolutely required for the cellular biogenesis of HDL from apoAI. ABCA1 has many activities, all of which may contribute to the generation of nascent HDL. We present a model in which ABCA1 remodels the plasma membrane and, independently, promotes the cellular binding of apoAI. ABCA1 then leads to the unfolding of a hairpin loop near the N-terminus, which allows apoAI to insert into the cell membrane to microsolubilize cellular lipids that are released as nascent HDL.
Acknowledgments
Illustration by David Schumick, BS, CMI. Reprinted with the permission of the Cleveland Clinic Center for Medical Art & Photography © 2014. All Rights Reserve.
Source of funding
This work was supported by National Institutes of Health grant P01 HL098055.
References
- 1.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The framingham study. Am J Med. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 2.Despres JP, Lemieux I, Dagenais GR, Cantin B, Lamarche B. Hdl-cholesterol as a marker of coronary heart disease risk: The quebec cardiovascular study. Atherosclerosis. 2000;153:263–272. doi: 10.1016/s0021-9150(00)00603-1. [DOI] [PubMed] [Google Scholar]
- 3.Gotto AM., Jr Low high-density lipoprotein cholesterol as a risk factor in coronary heart disease: A working group report. Circulation. 2001;103:2213–2218. doi: 10.1161/01.cir.103.17.2213. [DOI] [PubMed] [Google Scholar]
- 4.Murphy AJ, Woollard KJ, Suhartoyo A, Stirzaker RA, Shaw J, Sviridov D, Chin-Dusting JP. Neutrophil activation is attenuated by high-density lipoprotein and apolipoprotein a-i in in vitro and in vivo models of inflammation. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1333–1341. doi: 10.1161/ATVBAHA.111.226258. [DOI] [PubMed] [Google Scholar]
- 5.Yvan-Charvet L, Pagler TA, Seimon TA, Thorp E, Welch CL, Witztum JL, Tabas I, Tall AR. Abca1 and abcg1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ Res. 2010;106:1861–1869. doi: 10.1161/CIRCRESAHA.110.217281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang SH, Yuan SG, Peng DQ, Zhao SP. Hdl and apoa-i inhibit antigen presentation-mediated t cell activation by disrupting lipid rafts in antigen presenting cells. Atherosclerosis. 2012;225:105–114. doi: 10.1016/j.atherosclerosis.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 7.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. The New England journal of medicine. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith JD, Le Goff W, Settle M, Brubaker G, Waelde C, Horwitz A, Oda MN. Abca1 mediates concurrent cholesterol and phospholipid efflux to apolipoprotein a-i. J Lipid Res. 2004;45:635–644. doi: 10.1194/jlr.M300336-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Westerterp M, Murphy AJ, Wang M, Pagler TA, Vengrenyuk Y, Kappus MS, Gorman DJ, Nagareddy PR, Zhu X, Abramowicz S, Parks JS, Welch C, Fisher EA, Wang N, Yvan-Charvet L, Tall AR. Deficiency of atp-binding cassette transporters a1 and g1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res. 2013;112:1456–1465. doi: 10.1161/CIRCRESAHA.113.301086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aiello RJ, Brees D, Bourassa PA, Royer L, Lindsey S, Coskran T, Haghpassand M, Francone OL. Increased atherosclerosis in hyperlipidemic mice with inactivation of abca1 in macrophages. Arteriosclerosis, thrombosis, and vascular biology. 2002;22:630–637. doi: 10.1161/01.atv.0000014804.35824.da. [DOI] [PubMed] [Google Scholar]
- 11.Wang N, Silver DL, Costet P, Tall AR. Specific binding of apoa-i, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing abc1. J Biol Chem. 2000;275:33053–33058. doi: 10.1074/jbc.M005438200. [DOI] [PubMed] [Google Scholar]
- 12.Oram JF, Lawn RM, Garvin MR, Wade DP. Abca1 is the camp-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem. 2000;275:34508–34511. doi: 10.1074/jbc.M006738200. [DOI] [PubMed] [Google Scholar]
- 13.Asztalos BF, Tani M, Schaefer EJ. Metabolic and functional relevance of hdl subspecies. Curr Opin Lipidol. 2011;22:176–185. doi: 10.1097/MOL.0b013e3283468061. [DOI] [PubMed] [Google Scholar]
- 14.Martin SS, Jones SR, Toth PP. High-density lipoprotein subfractions: Current views and clinical practice applications. Trends Endocrinol Metab. 2014;25:329–336. doi: 10.1016/j.tem.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Asztalos BF, Sloop CH, Wong L, Roheim PS. Two-dimensional electrophoresis of plasma lipoproteins: Recognition of new apo a-i-containing subpopulations. Biochim Biophys Acta. 1993;1169:291–300. doi: 10.1016/0005-2760(93)90253-6. [DOI] [PubMed] [Google Scholar]
- 16.Wang N, Lan D, Chen W, Matsuura F, Tall AR. Atp-binding cassette transporters g1 and g4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci U S A. 2004;101:9774–9779. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. Abcg1 has a critical role in mediating cholesterol efflux to hdl and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Acton S, Rigotti A, Landschulz KT, Xu SZ, Hobbs HH, Krieger M. Identification of scavenger receptor sr-bi as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 19.Krimbou L, Marcil M, Chiba H, Genest J. Structural and functional properties of human plasma high density-sized lipoprotein containing only apoe particles. J Lipid Res. 2003;44:884–892. doi: 10.1194/jlr.M200273-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Toth PP, Barter PJ, Rosenson RS, Boden WE, Chapman MJ, Cuchel M, D’Agostino RB, Davidson MH, Davidson WS, Heinecke JW, Karas RH, Kontush A, Krauss RM, Miller M, Rader DJ. High-density lipoproteins: A consensus statement from the national lipid association. J Clin Lipidol. 2013;7:484–525. doi: 10.1016/j.jacl.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Fredrickson DS, Altrocchi PH, Avioli LV, Goodman DS, Goodman HC. Tangier disease. Ann Intern Med. 1961;55:1016–1031. [Google Scholar]
- 22.Serfatylacrosniere C, Civeira F, Lanzberg A, Isaia P, Berg J, Janus ED, Smith MP, Pritchard PH, Frohlich J, Lees RS, Barnard GF, Ordovas JM, Schaefer EJ. Homozygous tangier-disease and cardiovascular-disease. Atherosclerosis. 1994;107:85–98. doi: 10.1016/0021-9150(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 23.Remaley AT, Schumacher UK, Stonik JA, Farsi BD, Nazih H, Brewer HB., Jr Decreased reverse cholesterol transport from tangier disease fibroblasts. Acceptor specificity and effect of brefeldin on lipid efflux. Arteriosclerosis, thrombosis, and vascular biology. 1997;17:1813–1821. doi: 10.1161/01.atv.17.9.1813. [DOI] [PubMed] [Google Scholar]
- 24.Smith JD, Miyata M, Ginsberg M, Grigaux C, Shmookler E, Plump AS. Cyclic amp induces apolipoprotein e binding activity and promotes cholesterol efflux from a macrophage cell line to apolipoprotein acceptors. J Biol Chem. 1996;271:30647–30655. doi: 10.1074/jbc.271.48.30647. [DOI] [PubMed] [Google Scholar]
- 25.Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denefle P, Assmann G. Tangier disease is caused by mutations in the gene encoding atp-binding cassette transporter 1. Nat Genet. 1999;22:352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 26.Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J, Jr, Hayden MR. Mutations in abc1 in tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 27.Bodzioch M, Orso E, Klucken J, Langmann T, Bottcher A, Diederich W, Drobnik W, Barlage S, Buchler C, Porsch-Ozcurumez M, Kaminski WE, Hahmann HW, Oette K, Rothe G, Aslanidis C, Lackner KJ, Schmitz G. The gene encoding atp-binding cassette transporter 1 is mutated in tangier disease. Nat Genet. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 28.Singaraja RR, Brunham LR, Visscher H, Kastelein JJ, Hayden MR. Efflux and atherosclerosis: The clinical and biochemical impact of variations in the abca1 gene. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:1322–1332. doi: 10.1161/01.ATV.0000078520.89539.77. [DOI] [PubMed] [Google Scholar]
- 29.Hozoji M, Kimura Y, Kioka N, Ueda K. Formation of two intramolecular disulfide bonds is necessary for apoa-i-dependent cholesterol efflux mediated by abca1. J Biol Chem. 2009;284:11293–11300. doi: 10.1074/jbc.M900580200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaisman BL, Lambert G, Amar M, Joyce C, Ito T, Shamburek RD, Cain WJ, Fruchart-Najib J, Neufeld ED, Remaley AT, Brewer HB, Santamarina-Fojo S. Abca1 overexpression leads to hyperalphalipoproteinemia and increased biliary cholesterol excretion in transgenic mice. J Clin Invest. 2001;108:303–309. doi: 10.1172/JCI12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basso F, Freeman L, Knapper CL, Remaley A, Stonik J, Neufeld EB, Tansey T, Amar MJA, Fruchart-Najib J, Duverger N, Santamarina-Fojo S, Brewer HB. Role of the hepatic abca1 transporter in modulating intrahepatic cholesterol and plasma hdl cholesterol concentrations. J Lipid Res. 2003;44:296–302. doi: 10.1194/jlr.M200414-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Wellington CL, Brunham LR, Zhou S, Singaraja RR, Visscher H, Gelfer A, Ross C, James E, Liu GQ, Huber MT, Yang YZ, Parks RJ, Groen A, Fruchart-Najib J, Hayden MR. Alterations of plasma lipids in mice via adenoviral-mediated hepatic overexpression of human abca1. J Lipid Res. 2003;44:1470–1480. doi: 10.1194/jlr.M300110-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Timmins JM, Lee JY, Boudyguina E, Kluckman KD, Brunham LR, Mulya A, Gebre AK, Coutinho JM, Colvin PL, Smith TL, Hayden MR, Maeda N, Parks JS. Targeted inactivation of hepatic abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoa-i. J Clin Invest. 2005;115:1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brunham LR, Kruit JK, Iqbal J, Fievet C, Timmins JM, Pape TD, Coburn BA, Bissada N, Staels B, Groen AK, Hussain MM, Parks JS, Kuipers F, Hayden MR. Intestinal abca1 directly contributes to hdl biogenesis in vivo. J Clin Invest. 2006;116:1052–1062. doi: 10.1172/JCI27352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haghpassand M, Bourassa PA, Francone OL, Aiello RJ. Monocyte/macrophage expression of abca1 has minimal contribution to plasma hdl levels. J Clin Invest. 2001;108:1315–1320. doi: 10.1172/JCI12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singaraja RR, Visscher H, James ER, Chroni A, Coutinho JM, Brunham LR, Kang MH, Zannis VI, Chimini G, Hayden MR. Specific mutations in abca1 have discrete effects on abca1 function and lipid phenotypes both in vivo and in vitro. Circ Res. 2006;99:389–397. doi: 10.1161/01.RES.0000237920.70451.ad. [DOI] [PubMed] [Google Scholar]
- 37.Rigot V, Hamon Y, Chambenoit O, Alibert M, Duverger N, Chimini G. Distinct sites on abca1 control distinct steps required for cellular release of phospholipids. J Lipid Res. 2002;43:2077–2086. doi: 10.1194/jlr.m200279-jlr200. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka AR, Abe-Dohmae S, Ohnishi T, Aoki R, Morinaga G, Okuhira K, Ikeda Y, Kano F, Matsuo M, Kioka N, Amachi T, Murata M, Yokoyama S, Ueda K. Effects of mutations of abca1 in the first extracellular domain on subcellular trafficking and atp binding/hydrolysis. J Biol Chem. 2003;278:8815–8819. doi: 10.1074/jbc.M206885200. [DOI] [PubMed] [Google Scholar]
- 39.Neufeld EB, Remaley AT, Demosky SJ, Stonik JA, Cooney AM, Comly M, Dwyer NK, Zhang M, Blanchette-Mackie J, Santamarina-Fojo S, Brewer HB. Cellular localization and trafficking of the human abca1 transporter. J Biol Chem. 2001;276:27584–27590. doi: 10.1074/jbc.M103264200. [DOI] [PubMed] [Google Scholar]
- 40.Fitzgerald ML, Morris AL, Rhee JS, Andersson LP, Mendez AJ, Freeman MW. Naturally occurring mutations in the largest extracellular loops of abca1 can disrupt its direct interaction with apolipoprotein a-i. J Biol Chem. 2002;277:33178–33187. doi: 10.1074/jbc.M204996200. [DOI] [PubMed] [Google Scholar]
- 41.Nagao K, Zhao Y, Takahashi K, Kimura Y, Ueda K. Sodium taurocholate-dependent lipid efflux by abca1: Effects of w590s mutation on lipid translocation and apolipoprotein a-i dissociation. J Lipid Res. 2009;50:1165–1172. doi: 10.1194/jlr.M800597-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S, Gulshan K, Brubaker G, Hazen SL, Smith JD. Abca1 mediates unfolding of apolipoprotein ai n terminus on the cell surface before lipidation and release of nascent high-density lipoprotein. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1197–1205. doi: 10.1161/ATVBAHA.112.301195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nandi S, Ma L, Denis M, Karwatsky J, Li ZQ, Jiang XC, Zha XH. Abca1-mediated cholesterol efflux generates microparticles in addition to hdl through processes governed by membrane rigidity. J Lipid Res. 2009;50:456–466. doi: 10.1194/jlr.M800345-JLR200. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi Y, Smith JD. Cholesterol efflux to apolipoprotein ai involves endocytosis and resecretion in a calcium-dependent pathway. Proc Natl Acad Sci U S A. 1999;96:11358–11363. doi: 10.1073/pnas.96.20.11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fitzgerald ML, Morris AL, Chroni A, Mendez AJ, Zannis VI, Freeman MW. Abca1 and amphipathic apolipoproteins form high-affinity molecular complexes required for cholesterol efflux. J Lipid Res. 2004;45:287–294. doi: 10.1194/jlr.M300355-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Nagao K, Kimura Y, Ueda K. Lysine residues of abca1 are required for the interaction with apoa-i. Biochim Biophys Acta. 2012;1821:530–535. doi: 10.1016/j.bbalip.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 47.Vedhachalam C, Ghering AB, Davidson WS, Lund-Katz S, Rothblat GH, Phillips MC. Abca1-induced cell surface binding sites for apoa-i. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1603–1609. doi: 10.1161/ATVBAHA.107.145789. [DOI] [PubMed] [Google Scholar]
- 48.Hassan HH, Denis M, Lee DY, Iatan I, Nyholt D, Ruel I, Krimbou L, Genest J. Identification of an abca1-dependent phospholipid-rich plasma membrane apolipoprotein a-i binding site for nascent hdl formation: Implications for current models of hdl biogenesis. J Lipid Res. 2007;48:2428–2442. doi: 10.1194/jlr.M700206-JLR200. [DOI] [PubMed] [Google Scholar]
- 49.Hamon Y, Broccardo C, Chambenoit O, Luciani MF, Toti F, Chaslin S, Freyssinet JM, Devaux PF, McNeish J, Marguet D, Chimini G. Abc1 promotes engulfment of apoptotic cells and transbilayer redistribution of phosphatidylserine. Nat Cell Biol. 2000;2:399–406. doi: 10.1038/35017029. [DOI] [PubMed] [Google Scholar]
- 50.Nagao K, Takahashi K, Azuma Y, Takada M, Kimura Y, Matsuo M, Kioka N, Ueda K. Atp hydrolysis-dependent conformational changes in the extracellular domain of abca1 are associated with apoa-i binding. J Lipid Res. 2012;53:126–136. doi: 10.1194/jlr.M019976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reboul E, Dyka FM, Quazi F, Molday RS. Cholesterol transport via abca1: New insights from solid-phase binding assay. Biochimie. 2013;95:957–961. doi: 10.1016/j.biochi.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 52.Quazi F, Molday RS. Differential phospholipid substrates and directional transport by atp-binding cassette proteins abca1, abca7, and abca4 and disease-causing mutants. J Biol Chem. 2013;288:34414–34426. doi: 10.1074/jbc.M113.508812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fielding PE, Nagao K, Hakamata H, Chimini G, Fielding CJ. A two-step mechanism for free cholesterol and phospholipid efflux from human vascular cells to apolipoprotein a-1. Biochemistry-Us. 2000;39:14113–14120. doi: 10.1021/bi0004192. [DOI] [PubMed] [Google Scholar]
- 54.Wang N, Silver DL, Thiele C, Tall AR. Atp-binding cassette transporter a1 (abca1) functions as a cholesterol efflux regulatory protein. J Biol Chem. 2001;276:23742–23747. doi: 10.1074/jbc.M102348200. [DOI] [PubMed] [Google Scholar]
- 55.Wang N, Lan D, Gerbod-Giannone M, Linsel-Nitschke P, Jehle AW, Chen W, Martinez LO, Tall AR. Atp-binding cassette transporter a7 (abca7) binds apolipoprotein a-i and mediates cellular phospholipid but not cholesterol efflux. J Biol Chem. 2003;278:42906–42912. doi: 10.1074/jbc.M307831200. [DOI] [PubMed] [Google Scholar]
- 56.Landry YD, Denis M, Nandi S, Bell S, Vaughan AM, Zha XH. Atp-binding cassette transporter a1 expression disrupts raft membrane microdomains through its atpase-related functions. J Biol Chem. 2006;281:36091–36101. doi: 10.1074/jbc.M602247200. [DOI] [PubMed] [Google Scholar]
- 57.Nagao K, Kimura Y, Mastuo M, Ueda K. Lipid outward translocation by abc proteins. Febs Lett. 2010;584:2717–2723. doi: 10.1016/j.febslet.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 58.Mulya A, Lee JY, Gebre AK, Thomas MJ, Colvin PL, Parks JS. Minimal lipidation of pre-beta hdl by abca1 results in reduced ability to interact with abca1. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1828–1836. doi: 10.1161/ATVBAHA.107.142455. [DOI] [PubMed] [Google Scholar]
- 59.Gillotte KL, Zaiou M, Lund-Katz S, Anantharamaiah GM, Holvoet P, Dhoest A, Palgunachari MN, Segrest JP, Weisgraber KH, Rothblat GH, Phillips MC. Apolipoprotein-mediated plasma membrane microsolubilization - role of lipid affinity and membrane penetration in the efflux of cellular cholesterol and phospholipid. J Biol Chem. 1999;274:2021–2028. doi: 10.1074/jbc.274.4.2021. [DOI] [PubMed] [Google Scholar]
- 60.Gillotte KL, Davidson WS, Lund-Katz S, Rothblat GH, Phillips MC. Removal of cellular cholesterol by pre-beta-hdl involves plasma membrane microsolubilization. J Lipid Res. 1998;39:1918–1928. [PubMed] [Google Scholar]
- 61.Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC. Mechanism of atp-binding cassette transporter a1-mediated cellular lipid efflux to apolipoprotein a-i and formation of high density lipoprotein particles. J Biol Chem. 2007;282:25123–25130. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- 62.Sorci-Thomas MG, Owen JS, Fulp B, Bhat S, Zhu X, Parks JS, Shah D, Jerome WG, Gerelus M, Zabalawi M, Thomas MJ. Nascent high density lipoproteins formed by abca1 resemble lipid rafts and are structurally organized by three apoa-i monomers. J Lipid Res. 2012;53:1890–1909. doi: 10.1194/jlr.M026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mei XH, Atkinson D. Crystal structure of c-terminal truncated apolipoprotein a-i reveals the assembly of high density lipoprotein (hdl) by dimerization. J Biol Chem. 2011;286:38570–38582. doi: 10.1074/jbc.M111.260422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Denis M, Landry YD, Zha X. Atp-binding cassette a1-mediated lipidation of apolipoprotein a-i occurs at the plasma membrane and not in the endocytic compartments. J Biol Chem. 2008;283:16178–16186. doi: 10.1074/jbc.M709597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Faulkner LE, Panagotopulos SE, Johnson JD, Woollett LA, Hui DY, Witting SR, Maiorano JN, Davidson WS. An analysis of the role of a retroendocytosis pathway in abca1-mediated cholesterol efflux from macrophages. J Lipid Res. 2008;49:1322–1332. doi: 10.1194/jlr.M800048-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Azuma Y, Takada M, Shin HW, Kioka N, Nakayama K, Ueda K. Retroendocytosis pathway of abca1/apoa-i contributes to hdl formation. Genes to cells: devoted to molecular & cellular mechanisms. 2009;14:191–204. doi: 10.1111/j.1365-2443.2008.01261.x. [DOI] [PubMed] [Google Scholar]
- 67.Schmitz G, Robenek H, Lohmann U, Assmann G. Interaction of high density lipoproteins with cholesteryl ester-laden macrophages: Biochemical and morphological characterization of cell surface receptor binding, endocytosis and resecretion of high density lipoproteins by macrophages. Embo J. 1985;4:613–622. doi: 10.1002/j.1460-2075.1985.tb03674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Le Goff W, Peng DQ, Settle M, Brubaker G, Morton RE, Smith JD. Cyclosporin a traps abca1 at the plasma membrane and inhibits abca1-mediated lipid efflux to apolipoprotein a-i. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:2155–2161. doi: 10.1161/01.ATV.0000144811.94581.52. [DOI] [PubMed] [Google Scholar]
- 69.Nagao K, Maeda M, Manucat NB, Ueda K. Cyclosporine a and psc833 inhibit abca1 function via direct binding. Biochim Biophys Acta. 2013;1831:398–406. doi: 10.1016/j.bbalip.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 70.Chen WG, Wang N, Tall AR. A pest deletion mutant of abca1 shows impaired internalization and defective cholesterol efflux from late endosomes. J Biol Chem. 2005;280:29277–29281. doi: 10.1074/jbc.M505566200. [DOI] [PubMed] [Google Scholar]
- 71.Chen W, Sun Y, Welch C, Gorelik A, Leventhal AR, Tabas I, Tall AR. Preferential atp-binding cassette transporter a1-mediated cholesterol efflux from late endosomes/lysosomes. J Biol Chem. 2001;276:43564–43569. doi: 10.1074/jbc.M107938200. [DOI] [PubMed] [Google Scholar]