

Background: Key factors modulating conversion of prion protein into prions remain unclear.
Results: Neutralization of a cluster of lysines within residues 101–110 promoted formation of an N-terminally extended recombinant prion protein amyloid core.
Conclusion: A central lysine cluster strongly modulates folding of prion protein amyloids.
Significance: These findings highlight a key structural factor in the PrPSc-like folding of prion protein.
Keywords: Amyloid, Mutagenesis, Prion, Prion Disease, Protein Folding, Protein Structure
Abstract
The structure of the infectious form of prion protein, PrPSc, remains unclear. Most pure recombinant prion protein (PrP) amyloids generated in vitro are not infectious and lack the extent of the protease-resistant core and solvent exclusion of infectious PrPSc, especially within residues ∼90–160. Polyanionic cofactors can enhance infectivity and PrPSc-like characteristics of such fibrils, but the mechanism of this enhancement is unknown. In considering structural models of PrPSc multimers, we identified an obstacle to tight packing that might be overcome with polyanionic cofactors, namely, electrostatic repulsion between four closely spaced cationic lysines within a central lysine cluster of residues 101–110. For example, in our parallel in-register intermolecular β-sheet model of PrPSc, not only would these lysines be clustered within the 101–110 region of the primary sequence, but they would have intermolecular spacings of only ∼4.8 Å between stacked β-strands. We have now performed molecular dynamics simulations predicting that neutralization of the charges on these lysine residues would allow more stable parallel in-register packing in this region. We also show empirically that substitution of these clustered lysine residues with alanines or asparagines results in recombinant PrP amyloid fibrils with extended proteinase-K resistant β-sheet cores and infrared spectra that are more reminiscent of bona fide PrPSc. These findings indicate that charge neutralization at the central lysine cluster is critical for the folding and tight packing of N-proximal residues within PrP amyloid fibrils. This charge neutralization may be a key aspect of the mechanism by which anionic cofactors promote PrPSc formation.
Introduction
The conversion of normal prion protein (PrPC)3 into its infectious oligomeric/multimeric isoform (PrPSc) is a pathogenic process that underlies all transmissible spongiform encephalopathies or prion diseases. However, the understanding of this process has been hindered to date by the lack of atomic resolution of the three-dimensional structure of PrPSc (1). Thus, relatively coarse biochemical properties are used to characterize PrPSc, such as its detergent insolubility, protease resistance, and secondary structure composition relative to PrPC (2–5). Typically, an N-terminal fragment can be removed from PrPSc by treatment with proteinase K (PK), leaving a ≥19-kDa PK-resistant core of residues ∼90–231, without compromising infectivity (2, 6–8). PrPSc also tends to form linear amyloid fibrils, especially when detached from membranes (9–12). Different conformations of PrPSc are thought to give rise to biologically distinct prion strains that propagate through conformationally faithful seeded or templated polymerization mechanisms (13–17).
Although PrPSc isolated from prion-infected tissue is fully infectious, it is difficult to purify completely and label with site-specific experimental probes. Thus, many researchers have studied PrP conversion and amyloid fibril formation in vitro using recombinant PrP (rPrP) (reviewed in Refs. 1 and 18). However, pure rPrP amyloids tend to be either noninfectious or orders of magnitude less infectious than brain-derived PrPSc (19–21). A major difference between these rPrP amyloids and PrPSc is the folding of the region (residues ∼90–145) that is proximal to the N terminus of the PrPSc PK-resistant core (residues ∼90–231) (22–26). In brain-derived PrPSc, this region is tightly packed and protease-resistant (6, 7, 22, 23, 27), whereas spontaneously generated noninfectious rPrP fibrils are typically more disordered, solvent-accessible, and protease-sensitive within residues ∼90–159 (22, 28). However, more PrPSc-like PK resistance in this region can be enabled by seeding with infectious prions (29, 30) or the addition of cofactors, which may also help to promote the formation of more infectious PrPSc (21, 24, 25, 31).
The mechanism for this effect of anionic cofactors is not clear, but one possibility would be to aid the tight packing of a highly conserved cluster of four lysine residues within amino acids 101–110 (hereafter the central lysine cluster (CLC)) (32). In the absence of cofactors, the close proximity of the positively charged lysine side chains may cause electrostatic repulsion and instability around the CLC within the N-proximal region of the protease-resistant PrP core. In vivo, these cationic charges may be compensated by anionic cofactors, mitigating the electrostatic repulsion of the CLC and allowing residues in the vicinity to pack more tightly as they are in PrPSc.
The potential challenges associated with tight packing of the CLC are particularly exemplified in our recently proposed parallel in-register intermolecular β-sheet (PIRIBS) structure for PrPSc amyloids (32). Within this architecture, the four CLC lysines were not only closely spaced in the primary sequence of the PrPSc subunits, but also only ∼4.8 Å from corresponding lysines in adjacent β-strands. Without charge compensation, electrostatic repulsion might prevent tight packing and destabilize the N-proximal region of the PrP amyloid core (residues 90-∼145). Indeed, molecular dynamics simulations of PrP octamers assembled in a PIRIBS architecture indicated that although the more C-proximal residues ∼160–231 remain relatively stable as the amyloid core of the fibrillar segment, the more N-proximal residues display a greater tendency to be disordered (32). Here we show both in silico and in vitro evidence that neutralization of the cationic CLC side chains allows for the formation of PrP amyloid fibrils with extended, more PrPSc-like PK-resistant amyloid cores and β-sheet secondary structures.
EXPERIMENTAL PROCEDURES
Molecular Dynamics Simulations
Molecular dynamics simulations were performed as previously described (32). A model of a PrPSc fibril comprised of eight monomers of mouse residues 90–231 was constructed using SYBYL v.7.3. Energy minimization was performed using the NAMD program v.2.9 on the Biowulf Linux cluster. After minimization, the positive charges of the lysine side chains within the CLC were neutralized in silico for the “uncharged” model. Molecular dynamics were run on both the charged and uncharged models at a temperature of 310 K for 106 ns using the Biowulf Linux cluster at the National Institutes of Health (Bethesda, MD). Models were analyzed and images were created using VMD and UCSF Chimera.
Mutagenesis
An Agilent Technologies QuikChange kit was used as per the manufacturer's instructions to mutate Lys101, Lys104, Lys106, and Lys110 collectively to alanine or asparagine residues based on the DNA sequence encoding residues 23–231 of the Syrian hamster PrP sequence (accession no. K02234). K4N mutants were made in one QuikChange reaction and K4A mutants with two sequential QuikChange reactions with the following primers: K4N forward, 5′-cacaatcagtggaacaaccccagtaagccaaaaaccaacatgaaccacatggccggc-3′; K4N reverse, 5′-gccggccatgtggttcatgttggtttttggcttactggggttgttccactgattgtg-3′; K4A step 1 forward, 5′-gcttcatgttggtttttggcgcactgggcgcgttccactgattgtgggtgc-3′; K4A step 1 reverse, 5′-gcacccacaatcagtggaacgcgcccagtgcgccaaaaaccaacatgaagc-3′; K4A step 2 forward, 5′-ccggccatgtgcgccatgttggttgctggcgcactgggcg-3′; and K4A step 2 reverse, 5′-cgcccagtgcgccagcaaccaacatggcgcacatggccgg-3′.
Protein Expression and Purification
Recombinant hamster PrPC and K4 mutants were purified as previously described (33). Briefly, Rosetta (DE3) Escherichia coli containing the vector with the PrP sequence (Syrian hamster residues 23–231; accession no. M13899 or the PrP sequence including the lysine mutations described above) were grown in Luria broth medium in the presence of kanamycin and chloramphenicol. Protein expression was induced using the autoinduction system (34, 35) and purified using nickel-nitrilotriacetic acid superflow resin (Qiagen) with an ÄKTA pure chromatography system (GE Healthcare Life Sciences). The purified protein was extensively dialyzed into 10 mm sodium phosphate buffer (pH 5.8) and stored at −80 °C. Protein concentration was determined by measuring absorbance at 280 nm.
Real Time Quaking-induced Conversion (RT-QuIC)
RT-QuIC was carried out as previously described (33). Briefly, WT, K4A, or K4N rPrPC was used as substrate at a concentration of 0.1 mg/ml in RT-QuIC buffer (10 mm phosphate buffer, pH 7.4, 300 mm NaCl, 10 μm thioflavin T (ThT), 10 μm EDTA tetrasodium salt). Samples were loaded into a black 96-well plate with a clear bottom (Nunc). The reactions were treated with a 10−6 dilution of 10% (w/v) hamster normal brain homogenate or 263K scrapie-infected brain homogenate. Reactions that were treated with NBH and became ThT-positive over time are referred to as spontaneous RT-QuIC reactions. To explore seeding activity of the fibrils formed under different conditions, RT-QuIC reactions using WT rPrPC as the substrate were seeded using 10−2 dilutions of the fibrils. All reactions contained a final SDS concentration of 0.002% SDS. The plates were sealed with a film (Nalgene Nunc International sealer) and incubated in BMG Fluostar plate readers at 42 °C with cycles of 1 min of shaking at 700 rpm (double orbital) and 1 min of rest. ThT fluorescence was measured (450 ± 10 nm excitation and 480 ± 10 nm emission; bottom read) every 45 min.
Spontaneous Fibril Formation
Recombinant PrPC stored in sodium phosphate buffer (pH 5.8) was concentrated in 10-kDa spin filters, and the buffer was exchanged with 10 volumes of the following buffers: chaotropic conditions with 1 m guanidine HCl, 3 m urea, 150 mm NaCl in PBS, pH 7.4 (36); and acidic conditions with 20 mm sodium acetate, pH 4.5. The protein was concentrated in the indicated buffer to 0.5–1 mg/ml and incubated at 37 °C with orbital shaking at 600 RPM to initiate fibrillization. Fibril formation was confirmed with ThT fluorescence.
Biochemical Analyses
Reactions containing 0.1 mg/ml rPrP were treated at neutral pH with 10–15 μg/ml PK at 37 °C for 1 h. Protein samples were analyzed using Western blot techniques as previously described (37). Equal volumes of PK-treated reactions were run on 10% Bis-Tris NuPAGE gels (Invitrogen). Proteins were transferred to an Immobilon P membrane (Millipore) using the iBlot Gel Transfer System (Invitrogen). Immunoblotting was carried out using either the α-PrP rabbit polyclonal antibody R20 (1:15,000 dilution, epitope: residues 218–231) or the α-PrP rabbit polyclonal antibody R30 (1:10,000 dilution, epitope: residues 90–104) as described (38, 39).
Transmission Electron Microscopy
For negative stains of amyloid fibrils, Formvar and carbon-coated grids (Ted Pella, Inc., Redding, CA) were subjected to glow discharge, quickly placed onto 10–20-μl droplets of fibril suspensions, and incubated for 60 min at room temperature. The grids were transferred sequentially for 5 min each onto three droplets of deionized water. The grids were then stained negatively with methylamine tungstate (Nanoprobes, Inc., Yaphank, NY) for 15 s, wicked of excess fluid, and air-dried. Negatively stained grids were examined at 80 kV on a model H-7500 transmission electron microscope (Hitachi High Technologies, Dallas, TX) equipped with a model HR-100 digital camera system (Advanced Microscopy Techniques, Woburn, MA).
Fourier Transform Infrared (FTIR) Spectroscopy
FTIR analysis was performed as previously described (33). Briefly, RT-QuIC reactions containing 100 μg of rPrP were treated with 15 μg/ml PK for 1 h at 37 °C to remove any unconverted substrate. PK digestion was stopped using 1 mm Pefabloc SC (Sigma-Aldrich), and samples were pelleted by centrifugation at 20,800 × g at 4 °C for 20 min. The supernatant was removed, and the pellet was resuspended with ultrapure water and centrifuged two times. The pellet was resuspended in nanopure water to a 50% (w/v) slurry. The slurry was dried to a film under a stream of nitrogen on the diamond of a PerkinElmer Spectrum 100 FT-IR spectrometer equipped with a diamond attenuated total reflectance sample unit and an MCT detector. The spectra were background-subtracted and comprised of 32 accumulations (4 cm−1 resolution; 1 cm/s OPD velocity; strong apodization). Second derivative spectra were calculated with nine data points using Spectrum software (PerkinElmer Life Sciences).
RESULTS
Molecular Dynamics Simulations of the Effects of Lysine Cluster Charge Neutralization
We initially tested our hypothesis that electrostatic repulsion between clustered lysines within intermolecular β-sheets can contribute to the instability of the N-proximal region of the PrPSc fibrillar core in silico using a PIRIBS model as an example (32). Starting with an energy-minimized PIRIBS model of an octameric segment of a fibril composed of mouse PrP residues 90–231 (Fig. 1, Energy minimized) (32), we performed molecular dynamics simulations before (Fig. 1, Charged) or after (Fig. 1, Uncharged) artificially neutralizing the positive charges of the 4 lysines within residues 101–110. The simulations were extended for 106 ns to allow for side chain movements and secondary structure changes. As we reported previously, the wild-type model maintained a stable PIRIBS-rich core (residues ∼160–230) but had a higher degree of disorder in the more N-proximal residues, especially around the CLC (Fig. 1, Charged). However, with neutralization of the CLC side chain charges, the N-proximal half of the octamer subunits had less disorder and tighter packing within the fibril as compared with wild-type (Fig. 1, Uncharged). These results provided evidence that charge neutralization of the CLC can promote stabilization of N-proximal residues of PrP 90–231 monomers within a PIRIBS-based fibrillar model, at least in silico. This raises the possibility that charge compensation of the CLC may also be necessary to support the stable packing of the PK-resistant core of bona fide PrPSc.
FIGURE 1.

Removing lysine side chain charges within the CLC in silico improved fibril stability. Starting with an energy-minimized model of a segment of PrPSc amyloid based on a parallel in-register β-sheet assembly of eight mouse PrP 90–231 molecules (Energy minimized) (32), molecular dynamic simulations were performed on either the wild-type octamer with positively charged CLC side chains (Charged) or an octamer in which the CLC side chain charges were neutralized in silico (Uncharged). CLC lysine residues 101, 104, 106, and 110 are depicted in blue, β-strands are in green, and the cysteines forming the native disulfide bond are in yellow. In the energy-minimized octamer of wild-type PrP, the CLC lysine residues were stacked in close intermolecular proximity (Energy minimized). After 106-ns molecular dynamic simulations, the N-proximal region containing the CLC was more disordered in the wild-type (Charged) octamer than in CLC-neutralized (Uncharged) octamer.
Charge Neutralization of the Central Lysine Cluster in Recombinant PrP
To biochemically evaluate the effects of the CLC in recombinant PrP fibril formation, we made rPrP mutants in which Lys101, Lys104, Lys106, and Lys110 of WT hamster rPrP 23–231 were together changed to alanine (K4A) or asparagine (K4N) residues. These mutations replaced the cationic lysine side chains with hydrophobic or neutral polar side chains, respectively. We predicted that the K4N mutant might also allow for the formation of hydrogen bonding between asparagine side chains if, for example, they were stacked in-register in a PIRIBS model as shown in Fig. 1. Such “zippers,” as described by Eisenberg and Jucker (40), might help to further stabilize the region containing these residues within PrP fibrils as has been observed with various peptide amyloids. We investigated the effects of these mutations on both spontaneous and prion-seeded fibril formation as follows.
Spontaneous Fibrillization of Central Lysine Cluster Mutants
We looked initially at the effects of these mutations on spontaneous fibril formation (see “Experimental Procedures”) as a model for sporadic prion diseases. Previous reports have demonstrated that rPrP lacking the N-terminal amino acids 1–89 can spontaneously form fibrils under acidic buffer conditions, even in the absence of chaotropes (41). However, full-length rPrP was reported to be unable to fibrillize under these conditions (42). To investigate whether charge neutralization of the CLC enabled the spontaneous fibrillization of full-length rPrP under the acidic conditions (20 mm sodium acetate, pH 4.5), we compared fibril formation by the WT, K4N, and K4A rPrP molecules. As anticipated, full-length WT rPrP was unable to form fibrils, even after several months (Fig. 2A). In contrast, the K4A and K4N mutants formed fibrils after several weeks. Biochemical analyses showed that in addition to the 10- and 12-kDa PK-resistant species, the K4N and K4A fibrils had an additional ∼17-kDa species, indicative of an extended PK-resistant core (Fig. 2A). This PK-resistant core extended further toward the N terminus, including at least part of the epitope recognized by an antibody directed at residues 90–104 (R30) (Fig. 2A), as is the case for core fragments from PrPSc (39). A similar species with an extended PK-resistant core has been reported for spontaneous rPrP fibrils that were annealed in vitro in brain homogenates (43), which presumably contained cofactors that may act to neutralize the CLC, as we have done artificially in our K4N and K4A mutants, assisting in refolding.
FIGURE 2.
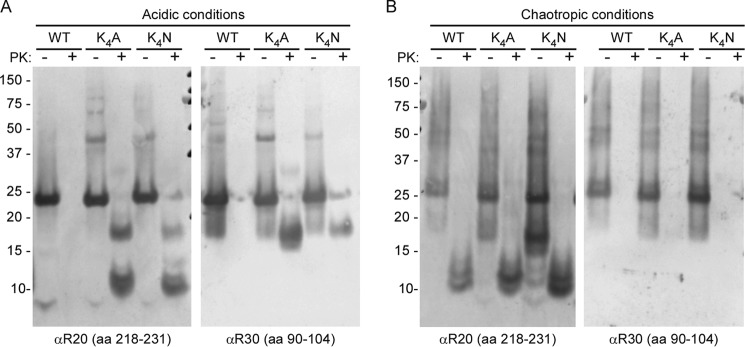
Charge neutralization of the CLC promotes formation of an extended 17-kDa PK-resistant amyloid core. A, WT, K4A, and K4N rPrP 23–231 were incubated under acidic conditions (20 mm sodium acetate, pH 4.5) and then either treated with PK or left untreated prior to Western blot analysis. R20 antiserum to C-terminal residues 218–231 showed 10-, 12-, and 17-kDa PK-resistant core fragments from the K4A and K4N fibrils, whereas no PK-resistant fragments were detectable with WT rPrP. R30 antiserum (residues 90–104) recognized the 17-kDa PK-resistant fragment only with the K4 mutants. B, the WT, K4A, and K4N rPrP molecules all generated 10- and 12-kDa PK-resistant core fragments under chaotropic conditions (1 m guanidine HCl, 3 m urea, 150 mm NaCl in PBS, pH 7.4), but none gave extended 17-kDa fragments. Representative blots are shown of three independent fibril preparations.
In addition to having N-terminally extended PK-resistant amyloid cores, our K4N and K4A fibrils had FTIR spectra indicative of high β-sheet content, with prominent 1625- and 1634-cm−1 bands and a minor 1620-cm−1 band (Fig. 3). The former two bands presumably reflect folding associated with the extended PK-resistant core because the 1634-cm−1 band is minimal or absent in rPrP fibrils lacking a 17-kDa PK-resistant core (Fig. 3, compare acidic to chaotropic). The fibrils formed by K4A and K4N appeared to be highly organized and linear, without any obvious twisting and little bundling (Fig. 4, B and C). Collectively, these results indicated that under acidic conditions and without any neutralization of the lysine side chains, the CLC strongly impeded amyloid fibril formation by WT rPrP.
FIGURE 3.
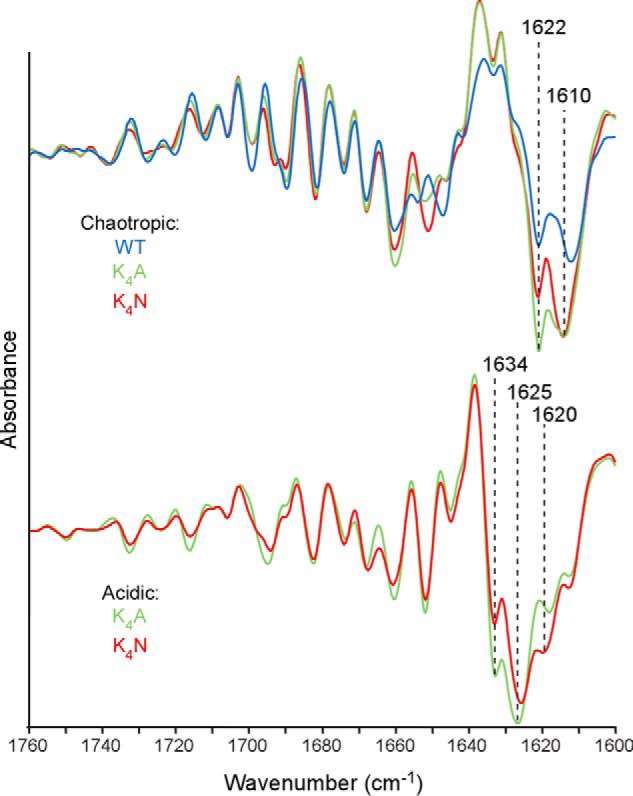
Acidic and chaotropic conditions promote different fibrillar β-sheet structures. Amyloid fibrils of WT, K4A, and K4N rPrP 23–231 molecules formed under the chaotropic conditions (top) described in the legend to Fig. 2 or of the K4A or K4N molecules formed under the acidic conditions (bottom) were treated with PK and analyzed by second derivative FTIR spectroscopy. All of the fibrils had prominent probable β-sheet content as indicated by the strong negative bands in the 1610–1634 cm−1 range. However, whereas the fibrils grown under chaotropic conditions all had predominant bands at 1622 and 1610 cm−1, those bands formed under acidic conditions had strikingly different bands at 1634 and 1625 cm−1. Similar spectral differences were observed in analyses of at least three independent fibril preparations.
FIGURE 4.

Ultrastructure of spontaneously formed fibrils generated under acidic or chaotropic conditions. A, fibrils were analyzed by transmission electron microscopy using methylamine tungstate as a negative stain. No fibrils were visible using WT rPrP and acidic conditions. B and C, highly organized, linear fibrils are visible using K4A (B) and K4N (C) rPrP to form fibrils in acidic buffer conditions. D–F, WT (D), K4A (E), and K4N (F) all formed fibrils characterized by repeating twists and lateral bundling under chaotropic buffer conditions. Scale bar, 100 nm.
Spontaneous WT rPrP fibril formation has also been reported under chaotropic conditions (1 m guanidine HCl, 3 m urea, 150 mm NaCl in PBS, pH 7.4) (36). Under these conditions, the PK-resistant amyloid cores were comprised mainly of 10- and 12-kDa C-proximal fragments. We tested whether the CLC mutations affected the extent of the PK-resistant core in the presence of chaotropes near neutral pH. WT, K4A, and K4N rPrP each formed ThT-positive amyloid fibrils in the chaotropic buffer. Judging from the ThT fluorescence, fibril formation occurred within 24–48 h, i.e. much faster than under acidic conditions tested above (data not shown). In contrast to our findings under the latter conditions, the K4A and K4N mutations did not extend the PK-resistant amyloid core of the fibrils formed under chaotropic conditions. Instead, like WT fibrils, the mutants had PK-resistant cores comprised mainly of 10- and 12-kDa C-proximal fragments (Fig. 2B). FTIR again showed predominant β-sheet content for WT, K4A, and K4N, but each had similar major bands at ∼1610 and 1622 cm−1 that were strikingly distinct from those of fibrils generated under the acidic conditions (Fig. 3). The ultrastructures of the chaotrope-induced WT, K4A, and K4N fibrils were similar to one another, with repeating twists and significant lateral bundling, but distinct from the less bundled acid-induced K4A and K4N fibrils (Fig. 4). These results demonstrated that in the presence of chaotropes the CLC does not appear to modulate or be involved in formation of the PK-resistant fibrillar core.
We then investigated the ability of the spontaneously formed fibrils generated under either chaotropic or acidic conditions to seed new amyloid formation by WT rPrP. The WT, K4A, and K4N fibrils formed under chaotropic conditions each seeded new fibril formation of WT rPrP under the neutral, nonchaotropic conditions of the RT-QuIC assay (10 mm phosphate buffer, pH 7.4, 300 mm NaCl, 10 μm ThT, 10 μm EDTA) (33) (Fig. 5). K4A and K4N fibrils formed under acidic conditions also seeded new WT PrP fibril formation with similar kinetics. Thus, under mild, neutral conditions, efficient seeding of ThT-reactive WT rPrP amyloid fibrils was observed even by the chaotrope-induced fibrils with the 10–12-kDa C-proximal PK-resistant core, providing evidence that the N-proximal extension of the PK-resistant amyloid core is not required for seeding amyloid formation.
FIGURE 5.
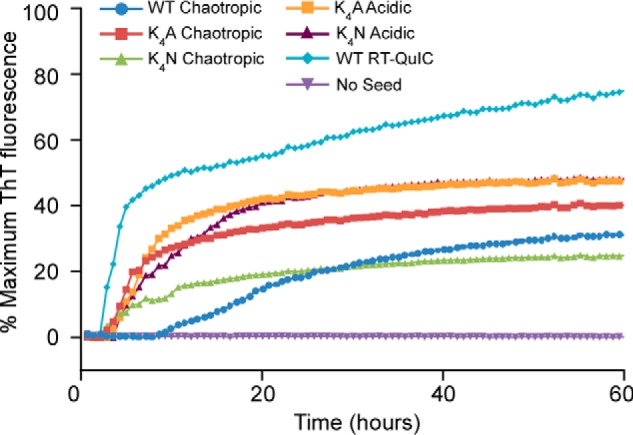
N-terminal extension of the PK-resistant core is not required for seeding further amyloid formation. Amyloid fibrils generated under acidic, chaotropic, or RT-QuIC conditions were used to seed ThT-positive amyloid formation of WT hamster rPrP 23–231 in neutral, nonchaotropic RT-QuIC reactions. Unseeded reactions are shown as a negative control. The data points represent average percentage of maximum ThT fluorescence from four replicate wells as a function of reaction time. All fibrils induced positive enhancements in ThT fluorescence in all replicate wells regardless of whether or not the seed fibril contained an extended (17-kDa) PK-resistant core fragment as indicated in Figs. 2 (A and B) and 6B. Similar results were obtained in three independent experiments.
Prion-seeded Fibrillization of Central Lysine Cluster Mutants
Infectious prion seeds or templates are known to accelerate fibril formation from rPrPC and promote an N-terminal extension of the solvent-excluded, PK-resistant fibrillar core (22, 29, 30). To test the effects of the CLC on prion-seeded fibril formation, scrapie prion (263K)-infected brain homogenate was used to seed the formation of WT, K4A, and K4N rPrP fibrils under the RT-QuIC conditions (Fig. 6A). The prion-seeded WT rPrP fibrils generated C-terminal PK-resistant cores of ∼10 and 12 kDa (Fig. 6B), similar to that of WT fibrils formed spontaneously in reactions treated with NBH (Fig. 8B). However, prion seeding markedly accelerated WT fibril formation (Figs. 6A versus 8A). Prion-seeded K4A and K4N PrP fibrils additionally generated a ∼17-kDa species, composed of residues spanning between the epitopes of the two antisera that detect it, i.e. residues 90–104 and 218–231, indicating an expanded PK-resistant core (Fig. 6B). These data again indicated the importance of charge neutralization of the CLC for more stable folding of the region of PrP residues ∼90–160 within amyloid fibrils under the influence of an infectious PrPSc template.
FIGURE 6.
Prion-templated amyloid formation under neutral, nonchaotropic RT-QuIC conditions. A, 263K scrapie-infected brain homogenate was used to seed fibril formation by WT, K4A, and K4N rPrP 23–231 substrates in RT-QuIC reactions. The data points represent average percentages of maximum ThT fluorescence from four replicate wells as a function of reaction time. Similar results were obtained in at least six independent experiments. B, Western blot analysis of scrapie-seeded, PK-treated RT-QuIC products using antisera to residues 90–104 or 218–231 (R30 and R20, respectively). Extended 17-kDa PK-resistant core fragments reactive with both antisera were always more prominent in the K4A and K4N fibrils than in the WT fibrils in at least six experiments.
FIGURE 8.

Charge neutralization of the CLC allows more rapid generation of spontaneously arising (prion independent) amyloid fibrils by RT-QuIC. A, in unseeded (i.e. treated with normal brain homogenate (NBH)) RT-QuIC reactions, K4A and K4N rPrP 23–231 substrates had shortened lag times to spontaneous fibril formation compared with WT rPrP. The data points represent average percentages of maximum ThT fluorescence from four replicate wells as a function of reaction time. Similar rank orders of relative RT-QuIC kinetics were obtained in six experiments. B, Western blot analysis of unseeded (NBH treated), PK-treated RT-QuIC products using antisera to residues 90–104 or 218–231 (R30 and R20, respectively). Extended 17-kDa PK-resistant core fragments reactive with both antisera were always more prominent in the K4A and K4N fibrils than in the WT fibrils in at least six experiments.
FTIR spectroscopy indicated that prion-seeded WT, K4A, and K4N fibrils each absorbed strongly in the β-sheet region (Fig. 7). The prion-seeded WT PrP fibrils lacked a strong ∼1634-cm−1 probable β-sheet band that is a feature of the 263K scrapie PrPSc seed itself (Fig. 7, inset) (3, 13). However, the K4A and K4N fibrils showed a more pronounced band at ∼1634 cm−1, i.e. close to the ∼1636 cm−1 band of 263K PrPSc (Fig. 7). This, in combination with the PK resistance analyses, provided evidence that in neutral pH and 300 mm NaCl, removal of the charged lysines from the CLC promotes the formation of fibrils with expanded PK-resistant cores and β-sheet structures that are more consistent with those of PrPSc.
FIGURE 7.
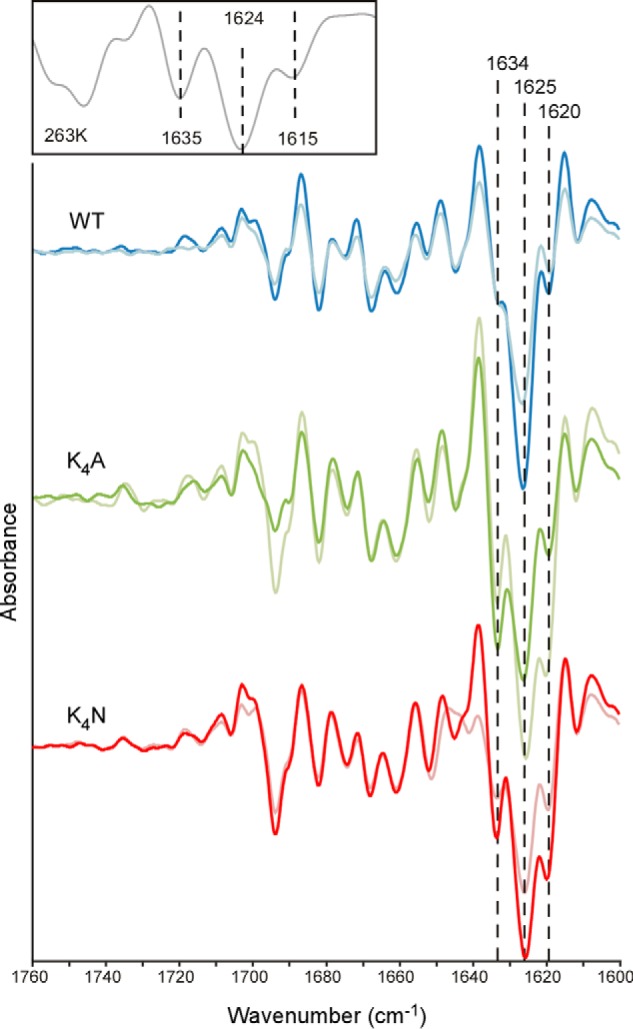
FTIR analysis of prion-templated amyloid formed with WT, K4A and K4N rPrP 23–231 substrates under neutral, nonchaotropic RT-QuIC conditions. Although all of the fibrils had spectra with prominent β-sheet content indicated by the strong negative bands within 1620–1634 cm−1, the K4A and K4N spectra were more similar to the 263K PrPSc spectrum (inset) in having a more prominent 1634 cm−1 band relative to the 1625 cm−1 band that is common to all of the fibril types. Spectra from duplicate RT-QuIC reactions are displayed as representatives from more than six experiments.
In contrast to the acidic conditions, in the neutral, nonchaotropic conditions of RT-QuIC reactions, WT rPrP fibrils were able to form without prion seeding, albeit after lag phases that were much longer than those of seeded reactions (Fig. 8A versus Fig. 6A). These spontaneously arising fibrils had similar PK-resistant cores to the prion-seeded WT rPrP fibrils, giving predominant ∼12-kDa bands that reacted with the C-terminal antiserum on Western blots (Fig. 8). However, FTIR spectra of spontaneously formed WT fibrils had a diminished ∼1634-cm−1 band and a stronger ∼1620-cm−1 β-sheet band, relative to its 1625-cm−1 band, than its prion-seeded counterpart (Fig. 9). K4N and K4A fibrils also formed spontaneously under RT-QuIC conditions and at a faster rate than nontemplated WT rPrP fibrils (Fig. 8). Their biochemical characteristics were similar to their prion-seeded counterparts, including the 17-kDa PK-resistant core (Fig. 8). However, spontaneously formed K4N and K4A fibrils, similar to WT rPrP, had a diminished ∼1634-cm−1 band and an increased 1620-cm−1 band, relative to their 1625-cm−1 bands, when compared with the prion-templated K4N and K4A fibrils (Fig. 9), suggesting that at least some of the tighter N-proximal folding in the prion-seeded K4N and K4A fibrils was promoted by prion templating.
FIGURE 9.
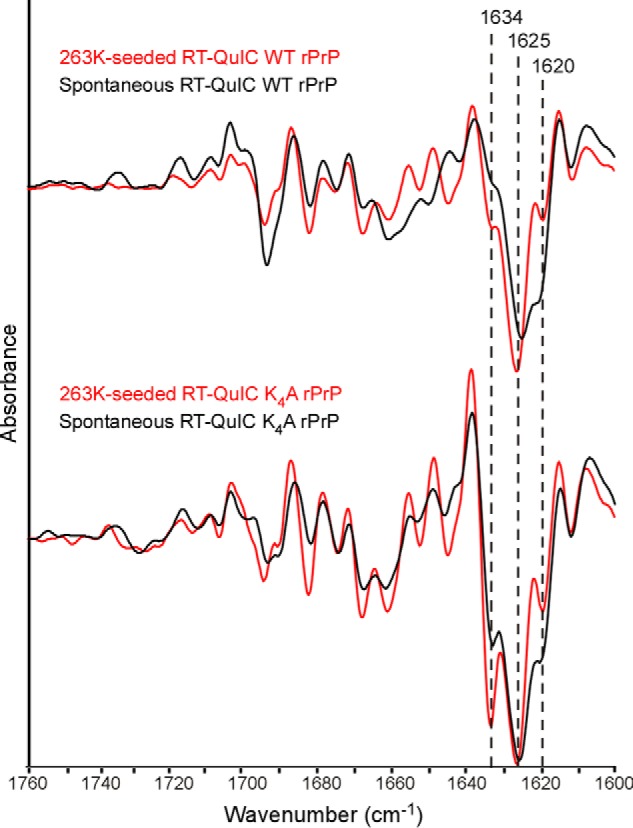
FTIR spectra of scrapie-seeded versus unseeded WT (top) and K4N (bottom) rPrP amyloids formed in RT-QuIC conditions. Unseeded (NBH treated) WT and K4A rPrP amyloid fibrils (black) formed under RT-QuIC conditions and treated with PK had a diminished ∼1634 -cm−1 band and an increased 1620-cm−1 band, relative to their 1625-cm−1 band, when compared with fibrils templated with prion seeds (red). Comparisons of prion-seeded versus unseeded K4N rPrP amyloids showed effects of scrapie seeding on these FTIR bands that were similar to those shown here for the K4A fibrils (data not shown). Similar relative effects of scrapie seeding on the FTIR spectra of WT and K4A rPrP amyloids were seen in at least six experiments. The spectra of the prion-seeded reaction products are the same as those in Fig. 7 and are reproduced here for comparison.
DISCUSSION
The structure of infectious PrPSc fibrils and the manner in which PrP monomers are assembled to give a PK-resistant and solvent-inaccessible amyloid core of residues ∼90–231 remain unclear. However, our present data provide evidence that the CLC is a feature of the PrP sequence that, without charge compensation, inhibits ordered packing within the N-proximal region. We have rationalized this effect using our recently hypothesized PIRIBS model of PrPSc structure as described above. However, any alternative structural model of PrPSc must also explain the empirically observed tight packing of the N-proximal region and its cluster of four cationic lysines within residues 101–110. Such models should also provide explanations for the profound effects of anionic cofactors on the formation of infectious and/or more PK-resistant PrP particles in vitro, whether with crude brain homogenates in PMCA reactions (44) or in biochemically defined reactions containing purified PrPC (26, 45) or rPrPC (24, 31, 46). Building on our previous proposals of the role of endogenous polyanions in PrPSc formation (45), we propose here that anionic cofactors may ion pair with the cationic CLC residues to promote packing of PrP monomers in the N-proximal region of the PK-resistant core. Indeed, several sulfated glycosaminoglycans have been shown to promote PrP conversion (26, 45). Paradoxically, inhibitory effects of glycosaminoglycans and other polyanions have also been reported (47–50). However, because sulfated glycans are known to interact with PrPC (48, 51) as well as PrPSc, it is possible to envision both stimulatory and inhibitory activities of such molecules depending upon experimental context (45). Furthermore, it appears that charge neutralization in the CLC is not all that is required for the formation of fully infectious PrPSc in vitro because inoculation of K4N fibrils into hamsters have so far failed to cause clinical scrapie (data not shown).
The more N-proximal region of the PK-resistant core of PrPSc that contains the CLC has been implicated in the conversion of PrPC to PrPSc. Previous studies have shown that progressive N-terminal deletions up to residue 113 (eliminating the region containing the CLC) have increasingly inhibitory effects on PrPSc formation in cell-free conversion reactions, whereas further deletion to residue 124 had no additional inhibitory effects (52). These findings indicated that the region containing the CLC influences PrPSc formation. Consistent with this conclusion, PrP molecules with K4A CLC mutations like we have studied above were unable to convert to PrPSc when expressed in prion-infected neuroblastoma cells (53). However, a lack of conversion to PrPSc in this cellular setting, which might appear to run counter to our findings that the K4A mutant is more apt to form PrPSc-like fibrils in isolation, may alternatively be due to altered trafficking or subcellular localization of the K4A mutant and not a fundamental inability of K4A to form PrPSc. Nevertheless, these indications of the importance of the CLC region in PrPSc formation are consistent with the possibility that the CLC is critical for the binding of polyanionic cofactors as has been suggested previously (54, 55).
As noted above, experimental conditions that allow WT rPrPC to refold into fibrils with somewhat PrPSc-like extended PK-resistant cores in the absence of cofactors have been described. These conditions include acidic pH in the presence of chaotropes (36), heating in the presence of detergents such as Triton X-100 (43), and prion-templated reactions in the presence of SDS (29, 30). With the latter conditions at least, generation of larger ≥17-kDa PK-resistant fragments is also enhanced by the presence of detergents such as Sarkosyl during the PK digestion (22). However, to the best of our knowledge only the lysine mutations in the CLC that we describe here have allowed in vitro formation of fibrils with an N-terminally extended amyloid core under physiologically compatible conditions in the absence of cofactors, high temperature annealing, or high concentrations of detergents.
In all of the conditions described here, rPrP fibril formation minimally generates amyloid with a stable C-proximal PK-resistant core. Under both the acidic and chaotropic conditions, rPrP amyloid formed with WT or K4 mutants included a 10–12-kDa C-proximal core. Charge neutralization of the CLC allowed further N-terminal extension of this C-proximal amyloid core to 17-kDa under both acidic conditions and neutral RT-QuIC conditions, but not under chaotropic conditions. Previous reports have indicated that fibrils formed from truncated WT rPrP (residues 90–231) under chaotropic or acidic buffer conditions produce similar β-sheet cores comprising residues ∼160–220 (56), indicating that the molecular architecture of the C-proximal core can be formed under divergent conditions. Conversely, as noted above, N-terminal extension of the amyloid core to include the CLC is highly influenced by pH, chaotropes, detergents, high temperature annealing, other cofactors, and/or charge neutralization of the CLC. However, fibrils formed under all of the conditions that we tested had the ∼12-kDa C-proximal PK-resistant core and were able to seed the propagation of further amyloid in the RT-QuIC with similar kinetics. Thus, although the C-proximal amyloid core is sufficient for seeding activity in in vitro fibrillization reactions, infectious prions are associated with major N-terminal extensions of the PK-resistant core. This suggests that infectivity, and possibly strain specificity, are dictated in part by the structure of the more N-proximal regions of the PrPSc PK-resistant core.
Although prion-templated amyloid formation by WT rPrP produced more PrPSc-like FTIR spectra than WT amyloid formed spontaneously (Fig. 9), charge neutralization of the CLC produced spectra with even more apparent PrPSc-like attributes. Thus, mitigating charge within the CLC appeared to allow the amyloid to adopt a more 263K PrPSc-like conformation. However, the differences that remain between the templated K4 mutants and brain-derived PrPSc suggest that other factors are also needed for bona fide PrPSc-like folding. Miller et al. (57) have shown in vitro that sequential addition of select cofactor molecules, including RNA and phospholipids, can induce distinct structural conformers of rPrP. Although such anionic cofactors could compensate the CLC charge and may assist the structural conversion of other parts of PrP molecules, charge neutralization of the CLC alone is not sufficient to fully recapitulate infectious PrPSc structure.
Altogether, our results indicate the critical involvement of the CLC region in modulating the formation, as well as the biochemical and structural aspects of PrP amyloids. The necessity of charge neutralization of the CLC is consistent with the tight packing of the N-proximal region in PrPSc fibrils, whether within the PIRIBS architecture (32) or other potential models for PrPSc structure.
Acknowledgments
We thank Drs. Suzette Priola, Roger Moore, and Matteo Manca for critical evaluation of this manuscript and Anita Mora and Ryan Kissinger for graphics assistance. This study utilized the high performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health (Bethesda, MD).
This work was supported, in whole or in part, by the Intramural Program of the National Institute of Allergy and Infectious Diseases.
- PrP
- prion protein
- rPrP
- recombinant PrP
- PK
- proteinase K
- CLC
- central lysine cluster
- PIRIBS
- parallel in-register intermolecular β-sheet
- RT-QuIC
- real time quaking-induced conversion
- ThT
- thioflavin T
- FTIR
- Fourier transform infrared
- NBH
- normal brain homogenate.
REFERENCES
- 1. Kraus A., Groveman B. R., Caughey B. (2013) Prions and the potential transmissibility of protein misfolding diseases. Annu. Rev. Microbiol. 67, 543–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McKinley M. P., Bolton D. C., Prusiner S. B. (1983) A protease-resistant protein is a structural component of the scrapie prion. Cell 35, 57–62 [DOI] [PubMed] [Google Scholar]
- 3. Caughey B. W., Dong A., Bhat K. S., Ernst D., Hayes S. F., Caughey W. S. (1991) Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry 30, 7672–7680 [DOI] [PubMed] [Google Scholar]
- 4. Safar J., Roller P. P., Gajdusek D. C., Gibbs C. J., Jr. (1993) Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J. Biol. Chem. 268, 20276–20284 [PubMed] [Google Scholar]
- 5. Pan K.-M., Baldwin M., Nguyen J., Gasset M., Serban A., Groth D., Mehlhorn I., Huang Z., Fletterick R. J., Cohen F. E., Prusiner S. B. (1993) Conversion of α-helices into β-sheets features in the formation of the scrapie prion protein. Proc. Natl. Acad. Sci. U.S.A. 90, 10962–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oesch B., Westaway D., Wälchli M., McKinley M. P., Kent S. B., Aebersold R., Barry R. A., Tempst P., Teplow D. B., Hood L. E., Prusiner S. B., Weissmann C. (1985) A cellular gene encodes scrapie PrP 27–30 protein. Cell 40, 735–746 [DOI] [PubMed] [Google Scholar]
- 7. Hope J., Morton L. J., Farquhar C. F., Multhaup G., Beyreuther K., Kimberlin R. H. (1986) The major polypeptide of scrapie-associated fibrils (SAF) has the same size, charge distribution and N-terminal protein sequence as predicted for the normal brain protein (PrP). EMBO J. 5, 2591–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parchi P., Castellani R., Capellari S., Ghetti B., Young K., Chen S. G., Farlow M., Dickson D. W., Sima A. A., Trojanowski J. Q., Petersen R. B., Gambetti P. (1996) Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 39, 767–778 [DOI] [PubMed] [Google Scholar]
- 9. Merz P. A., Somerville R. A., Wisniewski H. M., Iqbal K. (1981) Abnormal fibrils from scrapie-infected brain. Acta Neuropathol. 54, 63–74 [DOI] [PubMed] [Google Scholar]
- 10. Prusiner S. B., McKinley M. P., Bowman K. A., Bolton D. C., Bendheim P. E., Groth D. F., Glenner G. G. (1983) Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 35, 349–358 [DOI] [PubMed] [Google Scholar]
- 11. Diringer H., Gelderblom H., Hilmert H., Ozel M., Edelbluth C., Kimberlin R. H. (1983) Scrapie infectivity, fibrils and low molecular weight protein. Nature 306, 476–478 [DOI] [PubMed] [Google Scholar]
- 12. McKinley M. P., Meyer R. K., Kenaga L., Rahbar F., Cotter R., Serban A., Prusiner S. B. (1991) Scrapie prion rod formation in vitro requires both detergent extraction and limited proteolysis. J. Virol. 65, 1340–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Caughey B., Raymond G. J., Bessen R. A. (1998) Strain-dependent differences in β-sheet conformations of abnormal prion protein. J. Biol. Chem. 273, 32230–32235 [DOI] [PubMed] [Google Scholar]
- 14. Bessen R. A., Kocisko D. A., Raymond G. J., Nandan S., Lansbury P. T., Caughey B. (1995) Nongenetic propagation of strain-specific phenotypes of scrapie prion protein. Nature 375, 698–700 [DOI] [PubMed] [Google Scholar]
- 15. Bessen R. A., Marsh R. F. (1994) Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J. Virol. 68, 7859–7868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Telling G. C., Parchi P., DeArmond S. J., Cortelli P., Montagna P., Gabizon R., Mastrianni J., Lugaresi E., Gambetti P., Prusiner S. B. (1996) Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274, 2079–2082 [DOI] [PubMed] [Google Scholar]
- 17. Castilla J., Morales R., Saá P., Barria M., Gambetti P., Soto C. (2008) Cell-free propagation of prion strains. EMBO J. 27, 2557–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caughey B., Baron G. S., Chesebro B., Jeffrey M. (2009) Getting a grip on prions: oligomers, amyloids, anchors and pathological membrane interactions. Annu. Rev. Biochem. 78, 177–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Legname G., Baskakov I. V., Nguyen H. O., Riesner D., Cohen F. E., DeArmond S. J., Prusiner S. B. (2004) Synthetic mammalian prions. Science 305, 673–676 [DOI] [PubMed] [Google Scholar]
- 20. Kim J. I., Cali I., Surewicz K., Kong Q., Raymond G. J., Atarashi R., Race B., Qing L., Gambetti P., Caughey B., Surewicz W. K. (2010) Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J. Biol. Chem. 285, 14083–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Makarava N., Kovacs G. G., Bocharova O., Savtchenko R., Alexeeva I., Budka H., Rohwer R. G., Baskakov I. V. (2010) Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 119, 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smirnovas V., Kim J. I., Lu X., Atarashi R., Caughey B., Surewicz W. K. (2009) Distinct structures of scrapie prion protein (PrPSc)-seeded versus spontaneous recombinant prion protein fibrils revealed by hydrogen/deuterium exchange. J. Biol. Chem. 284, 24233–24241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smirnovas V., Baron G. S., Offerdahl D. K., Raymond G. J., Caughey B., Surewicz W. K. (2011) Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 18, 504–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deleault N. R., Walsh D. J., Piro J. R., Wang F., Wang X., Ma J., Rees J. R., Supattapone S. (2012) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. U.S.A. 109, E1938–E1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deleault N. R., Harris B. T., Rees J. R., Supattapone S. (2007) Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U.S.A. 104, 9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deleault N. R., Geoghegan J. C., Nishina K., Kascsak R., Williamson R. A., Supattapone S. (2005) Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J. Biol. Chem. 280, 26873–26879 [DOI] [PubMed] [Google Scholar]
- 27. Kocisko D. A., Lansbury P. T., Jr., Caughey B. (1996) Partial unfolding and refolding of scrapie-associated prion protein: evidence for a critical 16-kDa C-terminal domain. Biochemistry 35, 13434–13442 [DOI] [PubMed] [Google Scholar]
- 28. Lu X., Wintrode P. L., Surewicz W. K. (2007) β-Sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc. Natl. Acad. Sci. U.S.A. 104, 1510–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Atarashi R., Wilham J. M., Christensen L., Hughson A. G., Moore R. A., Johnson L. M., Onwubiko H. A., Priola S. A., Caughey B. (2008) Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat. Methods 5, 211–212 [DOI] [PubMed] [Google Scholar]
- 30. Atarashi R., Moore R. A., Sim V. L., Hughson A. G., Dorward D. W., Onwubiko H. A., Priola S. A., Caughey B. (2007) Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat. Methods 4, 645–650 [DOI] [PubMed] [Google Scholar]
- 31. Wang F., Wang X., Yuan C. G., Ma J. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Groveman B. R., Dolan M. A., Taubner L. M., Kraus A., Wickner R. B., Caughey B. (2014) Parallel in-register intermolecular β-sheet architectures for prion-seeded prion protein (PrP) amyloids. J. Biol. Chem. 289, 24129–24142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wilham J. M., Orrú C. D., Bessen R. A., Atarashi R., Sano K., Race B., Meade-White K. D., Taubner L. M., Timmes A., Caughey B. (2010) Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 6, e1001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fox B. G., Blommel P. G. (2009) Autoinduction of protein expression. Curr. Protoc. Protein Sci. 56, 5.23.1–5.23.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 36. Bocharova O. V., Breydo L., Parfenov A. S., Salnikov V. V., Baskakov I. V. (2005) In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J. Mol. Biol. 346, 645–659 [DOI] [PubMed] [Google Scholar]
- 37. Vascellari S., Orrù C. D., Hughson A. G., King D., Barron R., Wilham J. M., Baron G. S., Race B., Pani A., Caughey B. (2012) Prion seeding activities of mouse scrapie strains with divergent PrPSc protease sensitivities and amyloid plaque content using RT-QuIC and eQuIC. PLoS One 7, e48969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Caughey B., Race R. E., Vogel M., Buchmeier M. J., Chesebro B. (1988) In vitro expression in eukaryotic cells of the prion protein gene cloned from scrapie-infected mouse brain. Proc. Natl. Acad. Sci. U.S.A. 85, 4657–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caughey B., Raymond G. J., Ernst D., Race R. E. (1991) N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J. Virol. 65, 6597–6603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eisenberg D., Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cobb N. J., Sönnichsen F. D., McHaourab H., Surewicz W. K. (2007) Molecular architecture of human prion protein amyloid: a parallel, in-register β-structure. Proc. Natl. Acad. Sci. U.S.A. 104, 18946–18951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qi X., Moore R. A., McGuirl M. A. (2012) Dissociation of recombinant prion protein fibrils into short protofilaments: implications for the endocytic pathway and involvement of the N-terminal domain. Biochemistry 51, 4600–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bocharova O. V., Makarava N., Breydo L., Anderson M., Salnikov V. V., Baskakov I. V. (2006) Annealing prion protein amyloid fibrils at high temperature results in extension of a proteinase K-resistant core. J. Biol. Chem. 281, 2373–2379 [DOI] [PubMed] [Google Scholar]
- 44. Deleault N. R., Lucassen R. W., Supattapone S. (2003) RNA molecules stimulate prion protein conversion. Nature 425, 717–720 [DOI] [PubMed] [Google Scholar]
- 45. Wong C., Xiong L.-W., Horiuchi M., Raymond L., Wehrly K., Chesebro B., Caughey B. (2001) Sulfated glycans and elevated temperature stimulate PrPSc dependent cell-free formation of protease-resistant prion protein. EMBO J. 20, 377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deleault N. R., Piro J. R., Walsh D. J., Wang F., Ma J., Geoghegan J. C., Supattapone S. (2012) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. U.S.A. 109, 8546–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Caughey B., Raymond G. J. (1993) Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol. 67, 643–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gabizon R., Meiner Z., Halimi M., Ben-Sasson S. A. (1993) Heparin-like molecules bind differentially to prion proteins and change their intracellular metabolic-fate. J. Cell Physiol. 157, 319–325 [DOI] [PubMed] [Google Scholar]
- 49. Kocisko D. A., Vaillant A., Lee K. S., Arnold K. M., Bertholet N., Race R. E., Olsen E. A., Juteau J. M., Caughey B. (2006) Potent antiscrapie activities of degenerate phosphorothioate oligonucleotides. Antimicrob. Agents Chemother. 50, 1034–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vieira T. C., Cordeiro Y., Caughey B., Silva J. L. (2014) Heparin binding confers prion stability and impairs its aggregation. FASEB J. 28, 2667–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Caughey B., Brown K., Raymond G. J., Katzenstein G. E., Thresher W. (1994) Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and Congo red. J. Virol. 68, 2135–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lawson V. A., Priola S. A., Wehrly K., Chesebro B. (2001) N-terminal truncation of prion protein affects both formation and conformation of abnormal protease-resistant prion protein generated in vitro. J. Biol. Chem. 276, 35265–35271 [DOI] [PubMed] [Google Scholar]
- 53. Abalos G. C., Cruite J. T., Bellon A., Hemmers S., Akagi J., Mastrianni J. A., Williamson R. A., Solforosi L. (2008) Identifying key components of the PrPC-PrPSc replicative interface. J. Biol. Chem. 283, 34021–34028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen S. G., Teplow D. B., Parchi P., Teller J. K., Gambetti P., Autilio-Gambetti L. (1995) Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 270, 19173–19180 [DOI] [PubMed] [Google Scholar]
- 55. González-Iglesias R., Pajares M. A., Ocal C., Espinosa J. C., Oesch B., Gasset M. (2002) Prion protein interaction with glycosaminoglycan occurs with the formation of oligomeric complexes stabilized by Cu(II) bridges. J. Mol. Biol. 319, 527–540 [DOI] [PubMed] [Google Scholar]
- 56. Cobb N. J., Apetri A. C., Surewicz W. K. (2008) Prion protein amyloid formation under native-like conditions involves refolding of the C-terminal α-helical domain. J. Biol. Chem. 283, 34704–34711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miller M. B., Wang D. W., Wang F., Noble G. P., Ma J., Woods V. L., Jr., Li S., Supattapone S. (2013) Cofactor molecules induce structural transformation during infectious prion formation. Structure 21, 2061–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]