

Background: Crystallography and mutagenesis indicate that Asp309 is required for substrate binding and catalysis.
Results: Substrate binding in aromatase is pH-dependent. Such a dependence is missing in D309N mutant.
Conclusion: The apparent pKa for Asp309 is 8.2, and the residue is protonated at physiological pH.
Significance: The assigned pKa indicates the role of Asp309 in proton delivery for aromatization reaction.
Keywords: Aspartate (Aspartic Acid), Cytochrome P450, Enzyme Catalysis, Heme, Steroid, Aromatase, Estrogen, pKa
Abstract
Aromatase (CYP19A1), the enzyme that converts androgens to estrogens, is of significant mechanistic and therapeutic interest. Crystal structures and computational studies of this enzyme shed light on the critical role of Asp309 in substrate binding and catalysis. These studies predicted an elevated pKa for Asp309 and proposed that protonation of this residue was required for function. In this study, UV-visible absorption, circular dichroism, resonance Raman spectroscopy, and enzyme kinetics were used to study the impact of pH on aromatase structure and androstenedione binding. Spectroscopic studies demonstrate that androstenedione binding is pH-dependent, whereas, in contrast, the D309N mutant retains its ability to bind to androstenedione across the entire pH range studied. Neither pH nor mutation perturbed the secondary structure or heme environment. The origin of the observed pH dependence was further narrowed to the protonation equilibria of Asp309 with a parallel set of spectroscopic studies using exemestane and anastrozole. Because exemestane interacts with Asp309 based on its co-crystal structure with the enzyme, its binding is pH-dependent. Aromatase binding to anastrozole is pH-independent, consistent with the hypothesis that this ligand exploits a distinct set of interactions in the active site. In summary, we assign the apparent pKa of 8.2 observed for androstenedione binding to the side chain of Asp309. To our knowledge, this work represents the first experimental assignment of a pKa value to a residue in a cytochrome P450. This value is in agreement with theoretical calculations (7.7–8.1) despite the reliance of the computational methods on the conformational snapshots provided by crystal structures.
Introduction
Human aromatase (CYP19A1) is the cytochrome P450 that catalyzes the terminal step of estrogen biosyntheses, converting androstenedione, testosterone, and 16α-hydroxytestosterone to estrone, 17β-estradiol, and 17β,16α-estriol, respectively (1–3). This enzyme has drawn considerable interest for its ability to construct an aromatic ring and for its success as a breast cancer therapeutic target (4, 5). The reaction occurs in three distributive steps, each requiring one equivalent of molecular oxygen and NADPH (6). The first two steps are accepted to be hydroxylations of the steroid C19 methyl group, whereas the final step relies on a debated mechanism that deformylates the C19 aldehyde and aromatizes the steroid A-ring (Scheme 1A) (7). Although the ferric peroxo species has been generally accepted to mediate the third catalytic step, recent studies by different groups strongly support the involvement of Compound I in the third lyase step (8, 9). Despite decades of investigation, additional mechanistic studies are warranted to understand the mechanisms of this complex, pharmacologically relevant enzyme.
SCHEME 1.

A, summary of aromatase reaction with the conversion of androstenedione to estrone via 19-oxoandrostenedione. The reversible protonation of Asp309 is illustrated for the final step. B, chemical structure of anastrozole. C, chemical structure of exemestane.
Recently, crystal structures of CYP19A1 have suggested that a protonated aspartic acid residue in the “acid-alcohol” pair (Asp309-Thr310) is critical for substrate binding, orientation, and catalysis (10) (Fig. 1). Theoretical calculations based on one crystal structure (11) and site-directed mutagenesis studies (12) supported the structural data. Furthermore, the crystal structures in complex with androstenedione revealed a close contact (2.7 Å) between the 3-keto oxygen atom of androstenedione and the δ-O atom of Asp309 (10, 12). Similar distances between these atoms in the crystal structures with exemestane and other steroidal inhibitors have likewise been observed (13). An explanation for these close contacts is that Asp309 participates in a hydrogen bond to the 3-keto oxygen atom (Fig. 1). To permit this hydrogen-bonding configuration, the Asp309 side chain must be protonated, and its pKa must be elevated relative to that measured for the free amino acid in aqueous solution. To the extent that it contributes to the acid-alcohol pair present in the active sites of many P450 enzymes, precise tuning of this residue's pKa near physiological pH could optimize the reversible protonation required to drive the canonical P450 catalytic cycle. Computational prediction using an approach based on the Poisson Boltzmann equation and the crystal structure of human placental aromatase also estimated a pKa of 7.7 for Asp309 (11). Furthermore, subsequent hybrid quantum mechanics/molecular mechanics studies of the third catalytic step revealed a mechanism in which the Asp309 proton is cyclically relayed between the side chain and the substrate to circumvent accumulation of charge and stabilize intermediates in the deformylation/aromatization reaction (11). Although inferences from crystallography suggest protonation of Asp309, and theory highlights the potential value of this proton for catalysis, additional experimental evidence is required to support an elevated pKa for Asp309 in aromatase.
FIGURE 1.
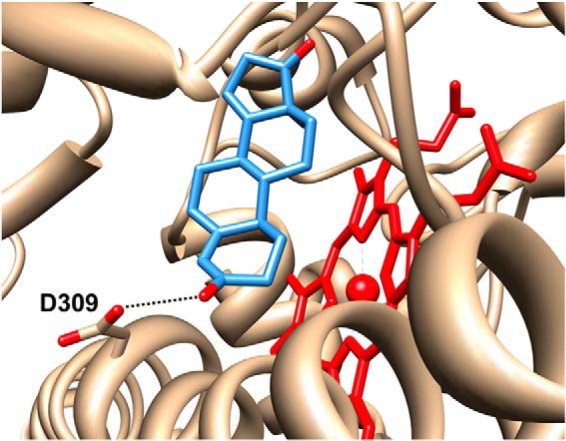
Active site of human aromatase (Protein Data Bank entry 4KQ8). The 3-keto oxygen atom of androstenedione (blue) is within hydrogen-bonding distance (2.9 Å) of the Asp309 side chain.
In a previous work, using an N-terminally truncated form of human aromatase expressed in Escherichia coli (rArom-WT)3 and sharing the same structural and functional features of the full-length wild-type enzyme, we found that the mutation of Asp309 to a non-titratable asparagine residue abolishes aromatase catalytic activity (12). In the present work, we describe the pH dependence of aromatase ligand binding. Exploiting the reliable spin state transition that occurs upon binding of androstenedione in the active site, we measured the enzyme's ability to retain androstenedione using UV-visible and resonance Raman (rR) spectroscopy. Taken together with spectroscopic experiments using the D309N mutant, the steroidal inhibitor exemestane, and the azole inhibitor anastrozole, the results lead us to assign an elevated pKa to Asp309 in aromatase, supporting a critical role for this residue. Additionally, in order to investigate potential physiological implications, possible variations in the catalytic parameters were also investigated in the pH regime where the enzyme is catalytically active in the physiological environment of the cell, which ranges from 6.5 to 7.4.
EXPERIMENTAL PROCEDURES
Computational pKa predictions
pKa values of the ionizable residues in the available crystal structures of rArom, including Asp309, were calculated using the empirical pKa predictor PROPKA3 (14).
Expression and Purification of rArom and the D309N Mutant
rArom-WT was expressed and purified as described previously (15). The enzyme was purified in the absence of ligand, with 10 μm androstenedione, 10 μm exemestane, or 1 μm anastrozole. The D309N mutant was purified in the presence of 500 μm androstenedione to improve yields of this protein. The P450 content of purified aromatase samples was measured using reduced CO difference spectra. The CO binding assay was performed at 30 °C by monitoring the absorbance at 450 nm after complete reduction of rArom with 12.5 mm sodium dithionite and bubbling with CO. Enzyme concentrations were determined using the differences in absorbance at 450 and 490 nm (A450–490) in [Fe2+CO] − [Fe2+] difference spectra and an extinction coefficient at 450 nm for the Fe2+CO complex of 91,000 m−1 cm−1 (16). All UV-visible absorption measurements were made with an Agilent 8453E UV-visible spectrophotometer.
UV-visible Absorption and Ligand Titrations
Androstenedione binding in the active site of aromatase results in a reliable shift of the heme iron from a low (S = ½) to high (S = ) spin electronic configuration. Typical of P450 enzymes, this spin shift results from displacement of the distal water ligand by the substrate (17). In aromatase, this is evidenced by a shift in the Soret band from 418 nm (low spin) to 394 nm (high spin) as androstenedione is titrated into the enzyme (15). The pH dependence of the shift in the Soret band was evaluated using two approaches. First, rArom (1 μm) purified in the absence of added ligands was incubated with 10 μm androstenedione for 5 min at 25 °C in 0.1 m potassium phosphate buffer for pH 6.5–8.0 and 0.1 m Tris buffer for pH 8.5 and 9.0. Second, the experiment was performed using rArom co-purified with 10 μm androstenedione and incubated for 5 min at 25 °C at different pH. All buffers used in the UV-visible absorption titrations also contained 20% glycerol, 0.1% Tween 20, and 1 mm β-mercaptoethanol. The percentage of enzyme in the high spin state (HS) was calculated as follows. Because there is no detectable high spin enzyme at pH 10 and no detectable low spin enzyme at pH 6.5, the spectral difference in the absorbance at 394 nm and at 418 nm between these two spectra (A394pH 6.5 − A394pH 10) − (A418pH 6.5 − A418pH 10) is defined as (ΔA394 − ΔA418)max and set to 100% HS. The percentages of high spin enzyme (%HS) at the remaining pH values were calculated as 100(ΔA394 − ΔA418)/(ΔA394 − ΔA418)max. This transformation was necessary to compare different set of experiments with slightly varying protein concentrations. pKa values were determined by fitting the percentages of high spin enzyme and pH values to the equation,
using SigmaPlot version 10.0. ymax and ymin are the asymptotic values of the sigmoidal fit (18).
Dissociation constants for androstenedione were determined by monitoring the shift in the Soret band from 418 to 394 nm as the concentration was titrated from 0.1–10 μm androstenedione into 0.5–1 μm rArom in 0.1 m potassium phosphate buffer for pH 6.5–8.0 and 0.1 m Tris buffer for pH 8.5 and 9.0. After each ligand addition, equilibrium conditions were reached after 3 min at 25 °C, and the spectra were recorded. The dissociation constants Kd were calculated using the equation,
where [S]free is [S]total − [E·S] and [E·S] = ΔA394–418 [E]total/ΔA394–418max.
Because the addition of the steroidal inhibitor exemestane causes the same low to high spin transition as the substrate, the pH dependence for exemestane binding was studied in the same experimental conditions as for androstenedione, and the Kd values were calculated on the basis of Equation 2.
The pH dependence for anastrozole binding was likewise tested over the aforementioned pH range by monitoring the red shift of the Soret band from 418 to 422 nm that results from replacement of the distal water ligand with the nitrogen heterocycle of the inhibitor (19). Titrations with increasing amounts of anastrozole (0.05–1 μm) were also performed to determine the pH dependence of the Kd values for this ligand. 0.1 m potassium phosphate buffer was used for pH 6.5–8.0 and 0.1 m Tris buffer for pH 8.5 and 9.0. Also in this case, after each ligand addition, the sample was equilibrated for 3 min at 25 °C before recording the spectrum. The dissociation constants Kd were calculated using the equation,
where [I]free is [I]total − [E·I] and [E·I] = ΔA422–418 [I]total/ΔA422–418max.
Circular Dichroism Spectroscopy
Far-UV (200–250 nm) CD spectra were collected using 1 μm rArom-WT in a 0.1-cm path length cell. All CD spectra were recorded at room temperature on a Jasco-815 instrument (Jasco Instruments, Inc., Easton, MD).
Resonance Raman Spectroscopy
rR spectra were obtained following excitation using the 406.7-nm line of a Coherent Innova 302C krypton ion laser. Laser powers at the sample were 35 milliwatts. Spectra were collected using an f/9.7 single grating monochromator (Acton SP2750, Princeton Instruments) at a 100 μm slit width using 1800-groove/mm gratings, and imaged using a 1340 × 400 pixel back-illuminated CCD camera with UV-optimized coatings (PyLoN 400BR eXcelon, Princeton Instruments). Reported spectra are the mean of three 20-min scans and are unsmoothed. Reference calibrations were performed with respect to a mercury vapor lamp. The nonlinear fluorescence background of rR spectra were removed using asymmetric least squares (20) in MATLAB. The concentrations of rArom and D309N were 10–15 μm in 0.1 m potassium phosphate (pH 6.5–8.0) buffer containing 5% glycerol and containing either 10 μm androstenedione or 1 μm anastrozole. 0.1 m Bicine buffer was used for measurements at pH 8.5 and 9.0. To determine the relative Raman cross-sections of the ν3 band arising from the high and low spin states of rArom, 0.2 m potassium sulfate was added as an internal standard to samples that were essentially high or low spin rArom. The ν3 bands of the heme and the 981 cm−1 band of the sulfate ion were fit to Gaussian line shapes using nonlinear least squares. Their corresponding intensities were determined by numerical integration of the peak areas. Following normalization of the high and low spin ν3 band intensities to the sulfate intensities, the high to low spin intensity ratio (IHS/ILS) was determined to be 0.68. This value was used to compute the relative high and low spin populations of rArom and D309N, which in this work are expressed as the percentage of the enzyme in the high spin state. Bands contributing to the propionates, vinyl, and 1620–1650 cm−1 regions were deconvoluted using two- and three-Gaussian fits in MATLAB.
Substrate Turnover
Aromatase activity assay was carried out by the water release method (21). The protein (30 nm) was incubated with 30 nm human cytochrome P450 reductase (Invitrogen) and the substrate 1-β-3H-androstenedione (PerkinElmer Life Sciences) for 5 min at room temperature. The reaction was then initiated by the addition of 1 mm NADPH and incubated for 10 min at 37 °C. The reaction was terminated with the addition of trichloroacetic acid (30%, v/v). The precipitated material was pelleted by centrifugation, and the resulting supernatant was subject to solid-phase extraction using a Strata X solid-phase extraction column (Phenomenex) to remove the unreacted 1-β-3H-androstenedione. The eluted aqueous phase containing 3H2O was counted with a Tri-Carb 2100TR liquid scintillation analyzer. (Packard Bioscience).
RESULTS
Empirical pKa Predictions
The PROPKA3 method was used to estimate the pKa value of Asp309 in the available crystal structures of aromatase. PROPKA is an empirical method that has been parameterized with a set of 85 experimentally determined pKa values of aspartate and glutamate residues in proteins. Utilizing a test set of 201 Asp and Glu pKa values, the root mean square deviation error of this approach was shown to be 0.79 pH units. The previously estimated pKa for Asp309 in the crystal structure of full-length human placental aromatase bound to androstenedione was predicted to be 7.7 (11). Additional crystal structures of the placental and recombinant aromatase with androstenedione and steroidal inhibitors have since become available, and they all suggest the presence of a hydrogen bond between the 3-keto oxygen atom androstenedione and the δ-O atom of Asp309. Accordingly, PROPKA3 also predicts pKa values for Asp309 in the range 7.7–7.9 in these structures (Table 1). The PROPKA analysis predicted a slightly higher pKa of 8.1 for the truncated form of aromatase used in this work. This variation is not of concern because these calculations are performed using crystal structures, which represent snapshots of the protein conformation and do not reflect the solution conformational heterogeneity of the enzyme. Furthermore, minor differences in the crystallization conditions, packing effects, and ligand-induced perturbations of the protein conformation induce minor differences in the electrostatic environment of the ionizable residues, thereby resulting in slightly different pKa predictions. Nonetheless, the PROPKA3 method consistently predicts a substantially elevated pKa for Asp309 across available crystal structures, and the fluctuations in these predictions are well within the S.D. value reported for the computational method.
TABLE 1.
Predicted pKa values of Asp309 in available aromatase crystal structures
UV-visible Titrations with Androstenedione
The pH dependence of the spin shift that is reliant on the presence of androstenedione in the active site was investigated by UV-visible absorption spectroscopy using enzyme purified with and without saturating amounts of androstenedione. The UV-visible spectra of rArom-WT at different pH values are illustrated in Fig. 2A. This pH range was selected because the sodium dithionite reduced form of the enzyme effectively binds CO and has a Soret band at 450 nm in the UV-visible absorption spectrum, consistent with properly folded, functional enzyme. At pH lower than 5.5 and higher than 10, the Soret band is found at 420–422 nm for rArom-WT in the absence of ligand, and the protein is not able to bind CO and show the typical peak at 450 nm, even in the presence of androstenedione. In general, at pH values greater than 7.0, there is a partial high to low spin shift of the Soret band from 394 to 418 nm, reflecting the displacement of androstenedione. When the pH is changed in the reverse direction, from 10 to 6.5, the opposite effect is observed, with the Soret band shifting from 418 to 394 nm. An isosbestic point at 407 nm is observed in both titrations. Incubating rArom-WT purified in the absence of ligand with saturating amounts of androstenedione, it was not possible to obtain a complete low to high spin transition at pH higher than 7.0. Control experiments using potassium phosphate buffers set at different concentrations to maintain consistent the ionic strengths at different pH (e.g. 125 mm KPi, pH 7.0, and 100 mm KPi, pH 7.4) and using different buffers for the same pH values (e.g. 100 mm Tris and 100 mm Bicine for pH 8.5) were performed in order to exclude possible spin transition effects due to ionic strength and/or buffer composition. The pH dependence of the high and low spin content evident in the UV-visible spectra was analyzed as described under “Experimental Procedures,” and the results, expressed as the percentage of high spin enzyme, are illustrated in Fig. 2C. In each case, the data are fitted using non-linear least squares to reveal an apparent pKa value for the spin transition. Using rArom-WT purified with and without androstenedione, the apparent pKa values are 8.2 ± 0.3 and 8.1 ± 0.2, respectively. A global fit analysis of the raw experimental data (changes at 394 and 418 nm as a function of pH; Fig. 2A, inset), taking into account all sets of experiments, was also performed, leading to pKa values of 8.2 ± 0.4 and 8.1 ± 0.3 for the experiments where substrate displacement and binding were monitored, respectively.
FIGURE 2.

A, UV-visible spectra of rArom-WT in complex with 10 μm androstenedione at pH 6.5 (solid black line), 7.0 (dashed gray line), 7.4 (dotted black line), 8.0 (solid dark gray line), 8.5 (dashed black line), and 10.0 (light gray solid line). Inset, plot of the absorbance values at 394 nm (white squares) and 418 nm (black squares) as a function of pH. B, UV-visible spectra of rArom-D309N in complex with 10 μm androstenedione at pH 6.5 (solid black line), 7 (solid gray line), and 9.0 (dashed black line). Spectra were collected using 1 μm rArom in a 1-cm path length cell. C, titration curves resulting from spectral transitions with varying pH observed for rArom-WT purified in the absence of ligand incubated with 10 μm androstenedione (white squares fitted by the dashed line), for rArom-WT co-purified with 10 μm androstenedione (black circles fitted by the solid line), and rArom-D309N co-purified with 500 μm androstenedione (black triangles). D, far-UV CD spectra of rArom-WT at pH 6.5 (solid black line), 7 (solid gray line), and 10 (dashed black line).
The pH dependence of the Kd values for binding was determined by titrating 0.1–10 μm androstenedione into rArom-WT purified in the absence of substrate. Kd values for androstenedione are compiled in Table 2. The Kd value is increased 3-fold, from 0.4 to 1.2 μm, by increasing the pH from 6.5 to 7.4. At pH 8.0, it was not possible to induce a complete shift of the Soret band from 418 to 394 nm despite the presence of an apparently saturating amount of androstenedione (10 μm). At higher pH values, no shift of the Soret band to 394 nm was observed. An increase in the Kd values is observed as the pH increases, showing that the binding affinity of rArom-WT for the substrate is higher at lower pH (Table 2).
TABLE 2.
Dissociation constants (Kd) of rArom-WT for the substrate androstenedione and the inhibitor anastrozole obtained at different pH
| pH |
Kd |
||
|---|---|---|---|
| Androstenedione | Exemestane | Anastrozole | |
| μm | |||
| 6.5 | 0.4 ± 0.1 | 0.25 ± 0.13 | 0.13 ± 0.03 |
| 7.0 | 0.9 ± 0.1 | 0.42 ± 0.17 | 0.19 ± 0.03 |
| 7.4 | 1.2 ± 0.1 | 0.61 ± 0.04 | 0.29 ± 0.03 |
| 8.0 | 2.6 ± 0.2 | 2.8 ± 0.2 | 0.23 ± 0.02 |
| 8.5 | No spin shift | No spin shift | 0.08 ± 0.01 |
| 9.0 | No spin shift | No spin shift | 0.13 ± 0.02 |
To assess the importance of reversible protonation of Asp309 for androstenedione binding, the pH dependence of the androstenedione-induced spin shift was similarly investigated in the D309N mutant (Fig. 2B). This mutant was successfully expressed and purified with yields comparable with those of rArom-WT in the presence of androstenedione. However, attempts to purify this mutant in the absence of ligand resulted in heme-free protein. This behavior precluded experiments titrating androstenedione into ligand-free D309N and subsequent Kd determinations. Over the pH range from 6.5 to 10.0, the dithionite-reduced D309N mutant could bind to CO, yielding a Soret band at 450 nm in the UV-visible absorption spectrum, identical to that of rArom-WT. Conversely, the D309N mutant remained predominately in the high spin state over this pH range (higher than 95%) (Fig. 2, B and C). The lack of pH dependence in substrate binding in the D309N mutant indicates that the protonation-deprotonation equilibrium of Asp309 is an important contributor to the pH dependence of the androstenedione-induced spin transition.
Circular Dichroism Spectroscopy
Far-UV CD spectroscopy was used to determine whether pH variations induce more global changes in rArom-WT structure that might explain the pH dependence of ligand binding. The CD spectra of ligand-free rArom-WT did not show any significant pH-dependent changes in the secondary structure of the protein (Fig. 2D). Furthermore, the consistent molar ellipticity at 222 nm of −17,000 degrees cm2 dmol−1 corresponds to the presence of 50% of helical structure (22), in keeping with the value calculated for the crystal structure of this protein and with previous CD studies on other human cytochromes P450 (23, 24).
UV-visible Titrations with Exemestane and Anastrozole
To further probe the role of the Asp309 protonation state in ligand interactions, the pH dependence of rArom-WT for binding exemestane, a structural analog of androstenedione, and the 1,2,4-triazole inhibitor anastrozole were evaluated (Scheme 1, B and C). Crystallography suggests that exemestane participates in a similar hydrogen-bonded interaction with Asp309 (13). By contrast, anastrozole lacks a functional group to exploit this hydrogen bond. The latter notion is supported by hyperfine sublevel correlation spectroscopy demonstrating that a nitrogen nucleus from the triazole ring coordinates directly with the heme iron, orienting the remaining azole nitrogens out of reach of a Asp309 hydrogen bond (19). As expected, exemestane binding to rArom-WT is pH-dependent (Fig. 3A). Analysis of the pH-dependent shift from 418 to 394 nm using enzyme purified with and without exemestane resulted in apparent pKa values of 8.3 ± 0.1 and 8.4 ± 0.2, respectively (Fig. 3B). Similar to androstenedione, there is an increase in exemestane's Kd as the pH is changed from 6.5 to 8.0 (Table 2). The UV-visible spectra of the anastrozole·rArom-WT complexes were identical across the pH range, each characterized by the distinct, red-shifted Soret band at 422 nm (Fig. 3C). Finally, the Kd values for anastrozole vary from 0.08 μm at pH 8.5 to 0.29 μm at pH 7.4 with no apparent correlation between pH and Kd (Table 2).
FIGURE 3.

A, UV-visible spectra of rArom-WT in complex with 10 μm exemestane at pH 6.5 (solid black line), 8.0 (dotted black line), 8.5 (solid gray line), and 9.0 (dashed black line). Spectra were collected using 1.2 μm rArom in a 1-cm path length cell. B, titration curves resulting from spectral transitions with varying pH observed for rArom-WT purified in the absence of ligand incubated with 10 μm exemestane (black circles fitted by the solid line) and for rArom-WT co-purified with 10 μm exemestane (white squares fitted by the dashed line). C, UV-visible spectra of rArom-WT (1.5 μm) in complex with 1 μm anastrozole at pH 6.5 (solid black line), 8.0 (dotted black line), 8.5 (solid gray line), and 9.0 (dashed black line). Error bars, S.D.
Resonance Raman Spectroscopy of Androstenedione Complexes
rR spectra were measured to investigate the changes in heme electronic structure resulting from the pH-induced decrease in the high spin population of rArom-WT. The high frequency regions of the rR spectra of rArom-WT co-purified in the presence of androstenedione are illustrated in Fig. 4A. The high frequency region of the spectra contains bands that are sensitive to the oxidation (ν4), and spin state (ν3) as well as those that are sensitive to the spin state and coordination environment (ν2). The oxidation state vibrations, ν4, occur at 1374–1376 cm−1 in the spectra of rArom-WT at all pH values. These values are as expected for a P450 enzyme in the ferric oxidation state. Conversely, intensities of those bands that depend on the spin state vary substantially over this pH range. At pH 6.5, the predominant ν3 band occurs at 1488 cm−1, consistent with essentially the entire enzyme population in the high spin state. As the pH is increased in 0.5-unit increments, the intensity of ν3 centered at 1488 cm−1 decreases, whereas the intensity of this band increases at 1504 cm−1, reflecting accumulation of the low spin state of the enzyme. The incremental pH increase likewise shifts ν2 from 1571 cm−1 to 1588 cm−1, also telling of the spin state shift. The high to low spin intensity ratio (R = IHS/ILS) for ν3 was used to calculate the relative populations of the high and low spin state at each pH value. Because the high spin ν3 peak area overlaps with the glycerol band at 1470 cm−1, the isolated ν3 intensity was determined by fitting the overlapping peaks with two Gaussian functions. The intensity of the high spin component was then determined by numerical integration of the peak centered at 1488 cm−1. The corresponding ν3 band arising from the low spin configuration was well separated from the high spin component and could be fit to a single Gaussian function. Calculation of the percentage of the enzyme populations in the high spin configuration (%HS) were computed using the equation, %HS = 100 × IHS/(R × ILS + IHS). Nonlinear least-squares fitting (Fig. 5) of the %HS and pH values to the equation, %HS = 100/1 + 1(pH-pKa), results in an apparent pKa for the high to low spin transition of 8.2 ± 0.1 (95% confidence interval, r2 = 0.97). This value corroborates the value determined by the UV-visible spectroscopy titration experiments.
FIGURE 4.

rR spectra of rArom-WT·androstenedione and rArom-D309N·androstenedione complexes at pH 6.5–9.0 with 406.7-nm laser excitation. The high frequency (A) and low frequency (B) regions of the rR spectra of rArom-WT and the high frequency (C) and low frequency (D) regions of the rArom-D309N mutant in the presence of 10 μm androstenedione are illustrated. Spectra in the high and low frequency regions were normalized to ν4 and ν7, respectively. Proteins were purified in the presence of androstenedione as described under “Experimental Procedures.” Protein concentrations were 10–15 μm.
FIGURE 5.

pH dependence of the high spin content of rArom-WT·androstenedione (solid circles), rArom-WT·exemestane (open squares), and rArom-D309N (open circles). Intensities for the high and low spin positions of ν3 were determined by numerical integration of their corresponding Gaussian line shapes. Error bars, S.D.
The low frequency regions of the rR spectra for rArom-WT co-purified in the presence of androstenedione are illustrated in Fig. 4B. The low frequency region contains the heme skeletal stretching modes as well as bending vibrations of the peripheral vinyl and propionate moieties. As is typical in the rR spectra of P450 enzymes, the heme stretching modes ν7 and ν8, centered at 677–678 and 345 cm−1 are resistant to pH titration in the presence of androstenedione. In rArom-WT, the dominant propionate bending frequency (δprop) appears at 378 cm−1, and the peripheral vinyl bending modes (δvinyl) appear at 417 cm−1. Fitting of the δvinyl envelopes generally resulted in two components centered at 413–415 and 422–427 cm−1, with the blue-shifted extremes dominating at higher pH values. These assignments are made based on the complete vibrational assignments of the heme in myoglobin and cytochrome c (25, 26). Changes in the chemical environments surrounding these functional groups are reflected in shifts of the corresponding frequencies in the rR spectrum. The δpropionates frequency positions provide insight into the hydrogen bonding and/or ionic interactions surrounding the heme propionates that anchor the cofactor into the active site. The low frequency regions of the rR spectra of rArom-WT at different pH values are nearly identical, indicating that pH titration does not influence the enzyme environment surrounding the heme periphery or change the hydrogen-bonding and/or ionic interactions anchoring the propionates into the aromatase active site. These spectra confirm that pH does not influence the immediate environment of the heme macrocycle.
As in the UV-visible experiments, rR spectra of the D309N mutant were likewise collected in the presence of 10 μm androstenedione (Fig. 4, C and D). The D309N mutation conservatively replaces the Asp309 side chain with a nontitratable functional group capable of contributing a hydrogen bond to the 3-keto oxygen atom over the entire pH range evaluated. Indeed, rR spectra in the high frequency region are identical from pH 6.5 to 9.0, and the enzyme populations are essentially entirely high spin (Fig. 4C). Calculation of the percentage of enzyme in the high spin state using the previously described Gaussian line shape analysis confirmed that the D309N mutant remained greater than 90% high spin over the pH range (Fig. 5). Furthermore, features of the low frequency region, most notably the δprop and δvinyl are likewise similar to that observed for rArom-WT (Fig. 4D). The constitutive protonation of the asparagine side chain putatively maintains the binding of androstenedione over the functional pH range, and the identical positions and pH resistance of the vibrational markers reporting the heme environment indicate that neither the mutation nor pH perturbs the heme-androstenedione interaction.
Resonance Raman Spectroscopy of Exemestane and Anastrozole Complexes
The rR spectra of rArom-WT in the presence of 10 μm exemestane and 20 μm anastrozole were measured to monitor the pH-dependent changes in heme electronic structure and environment in the presence of these ligands. rR spectra of rArom-WT with exemestane mirror those with androstendione. In the high frequency region, both ν3 and ν2 shift from their high to low spin positions in a pH-dependent manner (Fig. 6A). Line shape analyses of the ν3 bands result in an apparent pKa of 8.4 ± 0.1 (95% confidence interval, r2 = 0.94) (Fig. 5). This value is also in close agreement with the values determined in UV-visible experiments. The rR spectra of the low frequency region also mirror those of the androstendione complex and display the same features across the pH range, illustrating that pH does affect the immediate heme environment in the exemestane complex. Spectra of the rArom-WT·anastrozole complexes are nearly superimposable, although, in contrast to androstenedione, they reveal that aromatase remains entirely in the low spin state, with ν3 and ν2 bands occurring at 1504 and 1588 cm−1, respectively (Fig. 6C). Likewise, the positions of ν8, δprop, and δvinyl also remain fixed, indicating that pH also apparently does not affect the environments of the heme peripheral substituents in the anastrozole·rArom-WT complex (Fig. 6D).
FIGURE 6.

rR spectra of rArom-WT·exemestane and rArom-WT·anastrozole complexes at pH 6.5–9.0 with 406.7-nm laser excitation. The high frequency (A) and low frequency (B) regions of the rR spectra of rArom-WT in the presence of 10 μm exemestane and the high frequency (C) and low frequency (D) regions of the rArom-WT in the presence of 20 μm anastrozole are illustrated. Spectra in the high and low frequency regions were normalized to ν4 and ν7, respectively. Proteins were purified in the presence of exemestane and anastrozole as described under “Experimental Procedures.” Protein concentrations were 10–15 μm.
rArom-WT Enzyme Kinetics
Aromatase activity as a function of pH was previously studied using a microsomal system and reported to be pH-dependent with an optimum at pH 7.4 (27). Following establishment of the elevated pKa for Asp309, we assessed the role of pH in regulating aromatase catalytic parameters in the physiological pH 6.5–7.4. Androstenedione is capable of saturating the enzyme and consistently induces a high spin transition of the enzyme in this pH range. Intracellular pH can vary by ±0.9 pH units, as in neuronal firing. The catalytic activity of rArom-WT purified in the ligand-free form was assayed at pH 6.5, 7.0, and 7.4 by measuring the rArom-WT-catalyzed release of 3H2O from 1β-3H-androstenedione. At these pH values, the reaction rate increased hyperbolically with increasing substrate concentration. The experimental data were fitted to the Michaelis-Menten equation to obtain the values of Km and Vmax reported in Table 3. The Km values increase ∼1.5-fold from pH 6.5 to 7.4, showing the same increasing trend as for the Kd values determined in UV-visible titration experiments. Furthermore, the values of Km for the conversion of androstenedione into estrone are lower than the values of Kd for androstenedione binding, in line with previous results (6) and consistent with an ordered sequential mechanism.
TABLE 3.
Catalytic parameters (Km and Vmax) of rArom-WT for the substrate androstenedione obtained at three different pH values
| pH | Km | Vmax |
|---|---|---|
| nm | nmol product/min/mg protein | |
| 6.5 | 221 ± 20 | 17.8 ± 0.8 |
| 7.0 | 266 ± 25 | 23.5 ± 1.1 |
| 7.4 | 334 ± 42 | 19.8 ± 3.0 |
DISCUSSION
Ionizable residues in proteins are of profound importance for enzyme catalysis, structural stability, and protein-ligand interactions. The pKa values of ionizable residues depend strongly on their electrostatic environment. Thus, their degree of solvation by water and ions as well as the nature of neighboring residues, especially their charge states, influence these values. Forsyth et al. (28) surveyed 200 protein carboxyl amino acid pKa values determined by NMR and identified empirical relationships between descriptors of the aspartate or glutamate milieu and their pKa values. Relevant environmental factors included the solvent-accessible surface area, electrostatic potential, and the number of hydrogen bonds involved. The mean pKa value of aspartates among these proteins was 3.4 ± 1.0 units. Aspartate residues having pKa values greater than 5.5 were predominately buried in enzyme active sites with solvent-accessible surface areas of less than 20 Å2. These residues' environments were characterized by a low electrostatic potential and few if any hydrogen bonds (28).
Many cytochrome P450 enzymes carry adjacent, conserved amino acids in their active sites known as the “acid-alcohol” pair. The Asp251-Thr252 pair in CYP101 has undergone extensive mutagenesis and biophysical studies to confirm that these residues are critical to delivering protons to reduced oxygen intermediates and drive the canonical P450 catalytic cycle (29–31). The proton delivery mechanism that has been generally accepted for P450s requires that the acidic residue is first protonated, and this proton is relayed to the ferric peroxo and ferric hydroperoxo intermediates in the catalytic cycle via the hydroxyl oxygen of the alcohol residue. Insomuch that a corresponding acid-alcohol pair is conserved in aromatase, it is reasonable that an analogous proton delivery mechanism is operative. However, in crystal structures with androstenedione and inhibitors based on this scaffold, these ligands intervene in the proton delivery network through an apparent hydrogen bond to Asp309. Additionally, the 13-Å2 solvent-accessible surface area of this residue is in line with other protein aspartates known to have elevated pKa values. Both of these features are consistent with Asp309 protonation at physiological pH. In accord with these observations, computational prediction using PROPKA3, an approach that carefully accounts for desolvation and electrostatic contributions, estimates the Asp309 pKa to range from 7.7 to 8.1 among the available crystal structures. These estimates are 4.3–4.7 units higher than the average pKa values reviewed by Forsyth et al. (28). In view of the inferred importance of Asp309 in proton delivery, ancillary observations in crystallography, and strong computational support for protonation of this residue, a complementary experimental approach was used to investigate the pKa value of this residue as a validation of the theoretical predictions.
In many proteins, the direct measurement of the pKa values of multiple ionizable side chains can be simultaneously measured using NMR. Such measurements in the active site of P450 enzymes are complicated by the presence of the paramagnetic ferric ion. Attempts to silence this effect, such as generation of the diamagnetic carbonmonoxy-ferrous complex, often fail because these species are not stable for the time required at ambient temperature to collect the chemical shift data. In this work, site-directed mutagenesis, UV-visible absorption, rR spectroscopy, and enzyme kinetics were used to probe the role of Asp309 in maintaining androstenedione in a binding orientation that induces the high to low spin transition of the heme iron. Collectively, these approaches indeed measure a clear effect of Asp309 protonation on ligand binding and enzymatic activity and hence provide an indirect measurement of the pKa. The integrated approach affords exclusion of structural perturbations, other than the protonation/deprotonation of Asp309, as predominant contributors to the observed pH dependence of androstenedione binding to aromatase.
These results demonstrate that the binding of androstenedione is pH-dependent, and the apparent pKa for the spin transition is ∼8.2. The Km for androstenedione was shown to increase, going up to pH 7.4, and the same trend is observed for both Km and Kd values determined spectrophotometrically. Similar trends are observed for exemestane; however, the predicted pKa is slightly higher than that obtained with androstenedione. This observation is probably attributable to additional hydrophobic contacts between exemestane and the active site that contribute to its higher binding affinity. Conversely, the D309N mutant binds to androstenedione and remains greater than 90% high spin over the investigated pH range. The results suggest that whereas a protonated residue in position 309 is requisite for positioning androstenedione in the active site, reversible protonation is necessary for catalytic activity. The observations are both consistent with the putative proton delivery function of this residue and the recently proposed mechanism for the deformylation reaction (10). Both the low frequency region of the rR spectra and circular dichroism studies narrow the pH dependence to the protonation state of Asp309. The environments of the heme vinyl groups and the hydrogen bonding environments of the propionates are independent of pH in both rArom-WT and the D309N mutant. Furthermore, circular dichroism studies do not reveal substantial changes in the secondary structure of the enzyme while varying the pH. Anastrozole, a triazole inhibitor of aromatase that does not require interaction with Asp309, binds to aromatase in a pH-independent manner. Both the measured Kd values and low frequency rR spectra are the same across the pH range. The strong evidence supporting the necessity for reversible protonation of Asp309 for substrate binding and catalysis and the opposing evidence that the observed pH dependence does not result from global structural perturbations of the enzyme led us to assign a pKa of 8.2 to Asp309 in the active site of aromatase.
In addition to affording aromatase mechanistic flexibility, the elevated pKa of this residue may have physiological significance. Aromatase is expressed in extragonadal tissues, including the brain, where intracellular pH changes are detected, for example, in both neurons and glial cells in response to electrical stimulation or membrane ligands (32–38). Kinetic parameters were evaluated over the pH range in which androstenedione demonstrated induction of the low to high spin transition (pH 6.5–7.4). This pH range is chosen also on the basis of previous reports showing that in vertebrate neurons, different depolarizing stimuli, such as application of excitatory amino acids and glutamate, also induce pH acidification followed by alkalinization, altering the pH by 0.9 pH units (37, 38).
In conclusion, we have applied an integrated approach to investigate the possibility of and extraordinarily elevated pKa for Asp309 of the conserved acid-alcohol pair in the active site of human aromatase. Taken together with previous investigations, these results support that protonation of this residue is necessary for androstenedione binding, whereas reversible protonation is necessary for catalytic turnover. The agreement between the empirical pKa prediction based on the rArom-WT crystal structure (Protein Data Bank entry 4KQ8) and the experimental measurements is remarkable despite the approximations made in the computational approach, including ignorance of protein dynamics. This study underscores the value of such methods to support interpretation of experimental results. Although the importance of Asp309 protonation has been illustrated in molecular dynamics and quantum mechanics/molecular mechanics studies, the physiological impact of such fine regulation remains unclear. This phenomenon may be relevant in the brain, where rapid changes in estrogen levels might be correlated with small alterations of pH during neurotransmission. To our knowledge, this work represents the first experimental assignment of a pKa value in the active site of a cytochrome P450 enzyme.
Acknowledgments
We thank Prof. Sebastiano Colombatto for the access to his radioactive laboratory for the aromatase activity assay.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM092827 (to J. C. H.) and R01GM086893 (to D. G.). This work was also supported by an Italian Ministry of Education, Universities, and Research FIRB grant (Project RBFR12FI27_004) (to G. D.) and United States Office of Naval Research Grant N000141210773 (to J. C. H.).
- rArom
- recombinant aromatase
- rR
- resonance Raman
- HS
- high spin state
- Bicine
- N,N-bis(2-hydroxyethyl)glycine.
REFERENCES
- 1. Thompson E. A., Jr., Siiteri P. K. (1974) The involvement of human placental microsomal cytochrome P-450 in aromatization. J. Biol. Chem. 249, 5373–5378 [PubMed] [Google Scholar]
- 2. Thompson E. A., Jr., Siiteri P. K. (1974) Utilization of oxygen and reduced nicotinamide adenine dinucleotide phosphate by human placental microsomes during aromatization of androstenedione. J. Biol. Chem. 249, 5364–5372 [PubMed] [Google Scholar]
- 3. Simpson E. R., Mahendroo M. S., Means G. D., Kilgore M. W., Hinshelwood M. M., Graham-Lorence S., Amarneh B., Ito Y., Fisher C. R., Michael M. D. (1994) Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr. Rev. 15, 342–355 [DOI] [PubMed] [Google Scholar]
- 4. Brueggemeier R. W., Hackett J. C., Diaz-Cruz E. S. (2005) Aromatase inhibitors in the treatment of breast cancer. Endocr. Rev. 26, 331–345 [DOI] [PubMed] [Google Scholar]
- 5. Santen R. J., Brodie H., Simpson E. R., Siiteri P. K., Brodie A. (2009) History of aromatase: saga of an important biological mediator and therapeutic target. Endocr. Rev. 30, 343–375 [DOI] [PubMed] [Google Scholar]
- 6. Sohl C. D., Guengerich F. P. (2010) Kinetic analysis of the three-step steroid aromatase reaction of human cytochrome P450 19A1. J. Biol. Chem. 285, 17734–17743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Akhtar M., Calder M. R., Corina D. L., Wright J. N. (1982) Mechanistic studies on C-19 demethylation in oestrogen biosynthesis. Biochem. J. 201, 569–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoshimoto F. K., Guengerich F. P. (2014) Mechanism of the third oxidative step in the conversion of androgens to estrogens by cytochrome P450 19A1 steroid aromatase. J. Am. Chem. Soc. 136, 15016–15025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khatri Y., Luthra A., Duggal R., Sligar S. G. (2014) Kinetic solvent isotope effect in steady-state turnover by CYP19A1 suggests involvement of Compound 1 for both hydroxylation and aromatization steps. FEBS Lett. 588, 3117–3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghosh D., Griswold J., Erman M., Pangborn W. (2009) Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 457, 219–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sen K., Hackett J. C. (2012) Coupled electron transfer and proton hopping in the final step of CYP19-catalyzed androgen aromatization. Biochemistry 51, 3039–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lo J., Di Nardo G., Griswold J., Egbuta C., Jiang W., Gilardi G., Ghosh D. (2013) Structural basis for the functional roles of critical residues in human cytochrome P450 aromatase. Biochemistry 52, 5821–5829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghosh D., Lo J., Morton D., Valette D., Xi J., Griswold J., Hubbell S., Egbuta C., Jiang W., An J., Davies H. M. L. (2012) Novel aromatase inhibitors by structure-guided design. J. Med. Chem. 55, 8464–8476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Olsson M. H. M., Sřndergaard C. R., Rostkowski M., Jensen J. H. (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 [DOI] [PubMed] [Google Scholar]
- 15. Di Nardo G., Breitner M., Sadeghi S. J., Castrignanň S., Mei G., Di Venere A., Nicolai E., Allegra P., Gilardi G. (2013) Dynamics and flexibility of human aromatase probed by FTIR and time resolved fluorescence spectroscopy. PLoS One 8, e82118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Omura T., Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J. Biol. Chem. 239, 2370–2378 [PubMed] [Google Scholar]
- 17. Schenkman J. B., Remmer H., Estabrook R. W. (1967) Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 3, 113–123 [PubMed] [Google Scholar]
- 18. Silverstein T. P. (2012) Fitting imidazole 1H NMR titration data to the Henderson-Hasselbalch equation. J. Chem. Educ. 89, 1474–1475 [Google Scholar]
- 19. Maurelli S., Chiesa M., Giamello E., Di Nardo G., Ferrero V. E. V., Gilardi G., Van Doorslaer S. (2011) Direct spectroscopic evidence for binding of anastrozole to the iron heme of human aromatase. Peering into the mechanism of aromatase inhibition. Chem. Commun. (Camb.) 47, 10737–10739 [DOI] [PubMed] [Google Scholar]
- 20. Eilers P. H. C., Boelens H. F. M. (2005) Baseline correction with asymmetric least squares smoothing. Leiden University Medical Centre Report [Google Scholar]
- 21. Lephart E. D., Simpson E. R. (1991) Assay of aromatase activity. Methods Enzymol. 206, 477–483 [DOI] [PubMed] [Google Scholar]
- 22. Chen Y. H., Yang J. T., Chau K. H. (1974) Determination of the helix and β form of proteins in aqueous solution by circular dichroism. Biochemistry 13, 3350–3359 [DOI] [PubMed] [Google Scholar]
- 23. Yun C. H., Song M., Ahn T., Kim H. (1996) Conformational change of cytochrome P450 1A2 induced by sodium chloride. J. Biol. Chem. 271, 31312–31316 [DOI] [PubMed] [Google Scholar]
- 24. Yun C. H., Ahn T., Guengerich F. P. (1998) Conformational change and activation of cytochrome P450 2B1 induced by salt and phospholipids. Arch. Biochem. Biophys. 356, 229–238 [DOI] [PubMed] [Google Scholar]
- 25. Smulevich G., Mauro J. M., Fishel L. A., English A. M., Kraut J., Spiro T. G. (1988) Heme pocket interactions in cytochrome c peroxidase studied by site-directed mutagenesis and resonance Raman spectroscopy. Biochemistry 27, 5477–5485 [DOI] [PubMed] [Google Scholar]
- 26. Hu S., Kincaid J. R. (1991) Resonance Raman characterization of nitric oxide adducts of cytochrome P450cam: the effect of substrate structure on the iron-ligand vibrations. J. Am. Chem. Soc. 113, 2843–2850 [Google Scholar]
- 27. Osawa Y., Higashiyama T., Shimizu Y., Yarborough C. (1993) Multiple functions of aromatase and the active site structure: aromatase is the placental estrogen 2-hydroxylase. J. Steroid Biochem. Mol. Biol. 44, 469–480 [DOI] [PubMed] [Google Scholar]
- 28. Forsyth W. R., Antosiewicz J. M., Robertson A. D. (2002) Empirical relationships between protein structure and carboxyl pKa values in proteins. Proteins 48, 388–403 [DOI] [PubMed] [Google Scholar]
- 29. Denisov I. G., Mak P. J., Makris T. M., Sligar S. G., Kincaid J. R. (2008) Resonance Raman characterization of the peroxo and hydroperoxo intermediates in cytochrome P450. J. Phys. Chem. A 112, 13172–13179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davydov R., Makris T. M., Kofman V., Werst D. E., Sligar S. G., Hoffman B. M. (2001) Hydroxylation of camphor by reduced oxy-cytochrome P450cam: mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J. Am. Chem. Soc. 123, 1403–1415 [DOI] [PubMed] [Google Scholar]
- 31. Denisov I. G., Makris T. M., Sligar S. G., Schlichting I. (2005) Structure and chemistry of cytochrome P450. Chem. Rev. 105, 2253–2277 [DOI] [PubMed] [Google Scholar]
- 32. Chesler M. (2003) Regulation and modulation of pH in the brain. Physiol. Rev. 83, 1183–1221 [DOI] [PubMed] [Google Scholar]
- 33. Takahashi K. I., Copenhagen D. R. (1996) Modulation of neuronal function by intracellular pH. Neurosci. Res. 24, 109–116 [DOI] [PubMed] [Google Scholar]
- 34. Schwiening C. J., Kennedy H. J., Thomas R. C. (1993) Calcium-hydrogen exchange by the plasma membrane Ca-ATPase of voltage-clamped snail neurons. Proc. Biol. Sci. 253, 285–289 [DOI] [PubMed] [Google Scholar]
- 35. Hartley Z., Dubinsky J. (1993) Changes in intracellular pH associated with glutamate excitotoxicity. J. Neurosci. 13, 4690–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Irwin R. P., Lin S. Z., Long R. T., Paul S. M. (1994) N-methyl-d-aspartate induces a rapid, reversible, and calcium-dependent intracellular acidosis in cultured fetal rat hippocampal neurons. J. Neurosci. 14, 1352–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang G. J., Randall R. D., Thayer S. A. (1994) Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2+ loads. J. Neurophysiol. 72, 2563–2569 [DOI] [PubMed] [Google Scholar]
- 38. Zhan R. Z., Fujiwara N., Tanaka E., Shimoji K. (1998) Intracellular acidification induced by membrane depolarization in rat hippocampal slices: roles of intracellular Ca2+ and glycolysis. Brain Res. 780, 86–94 [DOI] [PubMed] [Google Scholar]