

Background: Erythrocytes contribute to nitrite-mediated NO signaling, but the mechanism is unclear.
Results: Deoxyhemoglobin accounts for virtually all NO made from nitrite by erythrocytes with no contributions from other proposed pathways.
Conclusion: Deoxyhemoglobin is the primary erythrocytic nitrite reductase operating under physiological conditions.
Significance: Reduction by deoxyhemoglobin accounts for nitrite-mediated NO signaling in blood mediating vessel tone and platelet function.
Keywords: Erythrocyte, Hemoglobin, Nitric Oxide, Platelet, Redox Signaling, Spectroscopy, Nitrite
Abstract
Nitrite signaling likely occurs through its reduction to nitric oxide (NO). Several reports support a role of erythrocytes and hemoglobin in nitrite reduction, but this remains controversial, and alternative reductive pathways have been proposed. In this work we determined whether the primary human erythrocytic nitrite reductase is hemoglobin as opposed to other erythrocytic proteins that have been suggested to be the major source of nitrite reduction. We employed several different assays to determine NO production from nitrite in erythrocytes including electron paramagnetic resonance detection of nitrosyl hemoglobin, chemiluminescent detection of NO, and inhibition of platelet activation and aggregation. Our studies show that NO is formed by red blood cells and inhibits platelet activation. Nitric oxide formation and signaling can be recapitulated with isolated deoxyhemoglobin. Importantly, there is limited NO production from erythrocytic xanthine oxidoreductase and nitric-oxide synthase. Under certain conditions we find dorzolamide (an inhibitor of carbonic anhydrase) results in diminished nitrite bioactivation, but the role of carbonic anhydrase is abrogated when physiological concentrations of CO2 are present. Importantly, carbon monoxide, which inhibits hemoglobin function as a nitrite reductase, abolishes nitrite bioactivation. Overall our data suggest that deoxyhemoglobin is the primary erythrocytic nitrite reductase operating under physiological conditions and accounts for nitrite-mediated NO signaling in blood.
Introduction
At one time nitrite was thought to be biologically inert (1). However, it is now recognized that nitrite plays important roles in various physiological processes, such as blood pressure control, hypoxic vasodilation, and the inhibition of platelet activation, and its use is being explored for various therapeutic applications (2–4). For example, administration of nitrite by infusion has been shown to increase blood flow, and its physiological administration through dietary nitrate has been shown to decrease blood pressure (5–10). These actions have been associated with production of NO from nitrite reduction in the vasculature (5). Moreover, reduction of nitrite to nitric oxide (NO) has been shown to occur preferentially under hypoxic conditions (5), suggesting a role in hypoxic-NO signaling.
Although the ability of nitrite to modulate blood flow is now widely accepted, its mechanism of bioactivation remains controversial. Deoxygenated (HbFe2+) or partially deoxygenated hemoglobin (Hb) was originally hypothesized to be responsible based on the reaction originally proposed by Brooks (11).
Here, deoxygenated hemoglobin (deoxyHb) reduces nitrite to NO and forms methemoglobin (metHb). The reaction is potentiated in hypoxia and acidosis (5, 12, 13).
A major challenge to the notion that deoxyHb is responsible for nitrite reduction to NO is that once NO is formed in the red blood cell (RBC)5, it will quickly bind to another deoxyHb
or react with oxygenated hemoglobin (oxyHb) to form metHb and nitrate.
Both of the these reactions are extremely fast (3–7 × 107 m−1s−1) (14–18), and the formation of nitrate from NO is irreversible in blood. Thus, based on an analysis of these rate constants, NO formed in the red blood cell is not likely to escape scavenging. Several pathways have been proposed that could potentially lead to the export of NO activity from the red cell (19–27), but none of these proposed mechanisms has been proven, and all have been challenged (28, 29).
Other proposed mechanisms to explain the nitrite role in modulating blood flow include reduction to NO in the vessel by xanthine oxidoreductase (XOR) (9, 30, 31), aldehyde oxidase (30), or smooth muscle myoglobin (32, 33). However, several lines of evidence support a role for the erythrocyte. Experiments performed with aortic ring bioassays showed that nitrite-mediated vasodilation, which increases as the oxygen tension decreases, is increased when red blood cells are present (5). Although this action of red blood cells was challenged in one study (34), subsequent studies clearly demonstrate that NO scavenging by Hb is counterbalanced by nitrite-mediated NO production from deoxyHb (35, 36).
More recently, significant support for a physiological role of the red blood cell in the bioactivation of nitrite and NO signaling comes from studies examining platelet activation by nitrite and red cells (35, 37, 38). NO is known to inhibit platelet activation via soluble guanylate cyclase activation and cGMP signaling (39–42). Whereas nitrite alone has no effect on platelet activation, when nitrite is added in combination with red blood cells, platelet activation is inhibited in association with intracellular platelet cGMP formation (37). The inhibition of platelet activation by nitrite and red blood cells is abrogated by the NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (CPTIO) and promoted by deoxygenation of the red blood cell (35, 37, 38). Importantly, two groups have now shown that nitrite also inhibits platelets systemically in humans; however, this effect only occurs in vitro with exposure of nitrite to whole blood with no effects observed on isolated platelet rich plasma (35, 38). This in vivo effect is potentiated under hypoxia, again consistent with hemoglobin deoxygenation (35). Thus, there is substantial evidence supporting a role of the red blood cell in nitrite bioactivation.
In addition to hemoglobin, XOR, erythrocytic endothelial nitric-oxide synthase (eNOS), and carbonic anhydrase (CA) have been proposed to be responsible for red blood cell-mediated nitrite reduction (9, 31, 43). The resolution of these controversies is central to our understanding of red cell signaling in the vasculature. In the current study we critically evaluate all proposed pathways for nitrite bioactivation by the red blood cell.
EXPERIMENTAL PROCEDURES
Blood was drawn from either normotensive or hypertensive volunteers into sodium heparin tubes under protocols approved by an Internal Review Board at Wake Forest University. Normotensive volunteers had not taken any anti-inflammatory nonsteroidal medication within a week at the time of blood draw. Hypertensive volunteers in this study were taking blood pressure medication. All hypertensive volunteers were required to have systolic blood pressure >130 mm Hg at an office screen visit to be qualified for the study (the average systolic blood pressure was 137.8 ± 4.8, n = 18). RBCs from hypertensive volunteers were used only when it is stated in the text; otherwise, RBCs from normotensive volunteers were employed. Allopurinol was freshly prepared in 0.1 m NaOH and used within 24 h. Immediately before use, allopurinol was diluted either in phosphate-buffered saline (PBS) at pH 7.4, pH 6.8, or pH 6.5. Trusopt, used as an ophthalmic solution (22 mg/ml) of dorzolamide (DZ), was purchased from Merck and diluted in PBS before experiments. L-NAME, sodium nitrite, and ADP were also prepared in PBS either at pH 7.4, pH 6.8, or pH 6.5. DEANONOate was first dissolved in 0.01 m NaOH and then further diluted with PBS just before use in experiments. Sodium nitrite (NaNO2), allopurinol, XOR, sodium dithionite, and ADP were purchased from Sigma. L-NAME and DEANONOate were purchased from Cayman Chemical (Ann Arbor, MI). Monoclonal antibodies against anti-human CD 61 labeled with PerCP fluorescent tag and FITC-labeled GP IIb/IIIa receptor neoepitope (PAC-1) antibodies were purchased from BD Biosciences. Human xanthine oxidase and β actin antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). HRP-labeled secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Normal human and mouse liver tissue lysates were purchased from BioChain (Newark, CA). Premade SDS 4–20% linear gradient gels were purchased from Bio-Rad. Protease inhibitor mixture for mammalian tissues was purchased from Sigma. ECL Prime Western detection kit was purchased from GE Healthcare.
RBCs Solution and Cell-free Hemoglobin Solution Preparation
Immediately after blood draw, whole blood was used in platelet aggregation assays or washed for other assays. Red blood cells were washed by centrifugation (1000 × g for 10 min) in phosphate-buffered saline (PBS) at pH 7.4, pH 6.8, or pH 6.5 at least three times or until the supernatants were clear. After the final wash, red blood cells were resuspended at 0.7 hematocrit (Hct). All the experiments were conducted within 24 h after the blood samples were drawn. Cell-free hemoglobin was prepared as described previously (44). Deoxygenated washed red blood cells were prepared by blowing wet nitrogen across the suspension with gentle rocking at room temperature for 1 h for the platelet activation assay or at least 3 h for other assays. The average deoxygenation percentage of red blood cells was 87.2 ± 7.9% as determined by absorption spectroscopy using a Cary 100 spectrometer equipped with an integrating sphere detector. CO-saturated RBC solutions (RBC-CO) were prepared by exposing washed red blood cells samples to 100% carbon monoxide for at least 3 h with gentle rocking at room temperature. The spectra of CO-bound RBCs were collected using a Cary 100 spectrometer to confirm the saturation of CO. Deoxygenated cell-free hemoglobin was prepared by deoxygenating cell-free hemoglobin using nitrogen, and the average deoxygenation percentage was 96.8 ± 1.5% as determined by absorption spectroscopy using a Cary 50 spectrometer.
Time-resolved Absorption Spectroscopy
Stock solutions of sodium dithionite (400 mm), NADH (200 mm), XOR (1 mm), and nitrite (100 mm) were prepared in deoxygenated HEPES buffer (100 mm, pH 6.5). The allopurinol stock (100 mm) was prepared in 0.1 n NaOH. Samples were prepared in 1-cm quartz cuvettes purged with nitrogen and filled with 4 ml of deoxygenated HEPES buffer. Sodium dithionite (100 μl, 10 mm final concentration), deoxygenated Hb (100 μm final concentration), and NADH (20 μl, 1 mm final concentration) were added to each cuvette. The relative importance of XOR was studied by the addition of 0, 1, or 10 μm XOR in the presence or absence of allopurinol (4 μl, 100 μm final concentration), which is an XOR inhibitor. Nitrite (20 μl, 500 μm final) was added to each cuvette to initiate the reaction. Spectra were taken every 128 s or 180 s at 37 °C in a Cary 100 UV-Vis spectrometer. Half-lives were determined by examination of kinetic fits to basis spectra and determination of when deoxygenated Hb fell to 50% of its initial value. Each experiment was repeated three times.
Nitrite Reduction to NO by Deoxygenated RBCs Measured Using EPR
Washed RBCs (0.7 Hct) from either normotensive or hypertensive volunteers were deoxygenated and mixed with deoxygenated hemoglobin (100 μm). The mixture was equally separated into two aliquots. One aliquot was incubated with inhibitors: either allopurinol (100 μm) for 30 min, l-NG-nitroarginine methyl ester (300 μm) for 30 min, or dorzolamide or Trusopt (750 μm) for 15 min. The other aliquot was incubated with vehicle (PBS solution at the same volume) for 15 or 30 min, respectively, before other procedures. NADH (100 μm) was added to RBCs from hypertensive individuals in some experiments at 37 °C to favor XOR activity. In other experiments 1 mm NADH or xanthine (10 μm) was included. In one set of experiments XOR (10 μm) was also added. Nitrite (100 μm) was added to the mixture and incubated with the mixture at pH 7.4, pH 6.8, or pH 6.5 for 30 min at room temperature or 37 °C. The final hematocrit of the mixture was brought to 0.5 using PBS. During the incubation, a portion of the mixture (0.5 ml) was collected in a tube at 15 and 30 min (or 5 and 30 min for experiments at pH 6.5) and frozen in liquid nitrogen immediately. The rest of the mixture was centrifuged at 3000 × g for 2 min. The supernatant containing cell-free hemoglobin was collected in a tube and frozen at 50 min after the initiation of the reaction. During the experiment, mixtures were either mechanically rocked (at room temperature) or gently rocked manually (at 37 °C). The concentration of the product nitrosyl hemoglobin (HbNO) at each time point was determined by electron paramagnetic resonance (EPR) using a Bruker EMX 10/12 spectrometer as described previously (45).
Nitrite Reduction to NO by CO Saturated RBCs Measured Using EPR
We further explored erythrocytic nitrite reduction using carbon monoxide to block hemoglobin-mediated nitrite reduction using washed RBCs from normotensive volunteers. Nitrite (100 μm) was incubated with the RBC-CO solution for 30 min. To confirm that NO can displace CO from Hb, we added the nitric oxide donor, DEANONOate (100 μm), and examined HbNO formation in CO-saturated and deoxygenated RBCs during a 30-min incubation. Samples were collected and frozen at 15 and 30 min, and the HbNO concentration was examined using EPR.
NO Production Examination from Nitrite and RBCs Measured by a Chemiluminescent Nitric Oxide Analyzer
An in-line nitric oxide analyzer assay was employed using a three-neck round-bottom flask similar to that described previously (12). One of the side necks was connected to incoming argon gas, and the other one was connected to the NaOH trap of a nitric oxide analyzer (Sievers). The flask sat in a water bath at 37 °C throughout the experiment. A nitrite solution (20 ml at 20 mm) at pH 7.4, pH 6.8, or pH 6.5 was degassed in the three-neck round-bottom flask using the neck in the middle for at least 20 min. Washed red blood cells (0.5 Hct) from either normotensive or hypertensive individuals were deoxygenated and incubated with allopurinol (100 μm), L-NAME (300 μm), DZ (750 μm), or vehicle (NaOH or PBS solution at same concentration) at 37 °C and pH 7.4, pH 6.8, or pH 6.5 for 30 min. An aliquot (100 μl) of the washed RBC mixture was added to the deoxygenated nitrite solution, and NO production was detected in the purged gas, which traveled in-line to the analyzer. The released NO was recorded for 20 min. The area below the increased NO signal curve and above the base line was calculated as the amount of NO produced from nitrite reduction. A ratio of the amount of NO produced by RBCs incubated with inhibitors or vehicle, +inhibitor/−inhibitor, was calculated.
Additional experiments were conducted using RBCs from normotensive volunteers. First, instead of deoxygenated RBCs, oxygenated RBCs were incubated for 30 min with allopurinol (100 μm), L-NAME (300 μm), DZ (750 μm), or vehicle and were added to a deoxygenated nitrite solution. Second, deoxygenated RBCs incubated with and without allopurinol (100 μm), L-NAME (300 μm), or DZ (250 μm) were added to a CO-saturated nitrite solution that was prepared by bubbling the nitrite solution using 50% CO balanced with nitrogen for 20 min.
Effects of NO Production from Nitrite, RBCs, and Hemoglobin on Platelet Activation and Aggregation
Inhibition of platelet activation by nitrite in the presence of red blood cells or cell-free hemoglobin was examined and further explored using allopurinol, L-NAME, and DZ. Platelet rich plasma (PRP) was prepared by centrifuging whole blood at 120 × g for 15 min at room temperature. PRP was removed by plastic disposable pipettes without disturbing the buffy coat and was stored in polypropylene tubes for use within 2 h. CO2 was measured in PRP by a carbon dioxide enzymatic kit from BIOO Scientific, Austin, TX according to the manufacturer's recommendations.
All necessary materials were placed inside an air-tight glove bag filled with nitrogen gas. PRP was diluted 1:10 in deoxygenated PBS, pH 7.4, nitrite was added as indicated and washed RBCs were added to 0.2 Hct, and the samples were incubated at 37 °C for 5 min. After 5 min, 20 μm ADP was added and allowed to incubate for 10 min at 37 °C. Samples were then transferred into PAC-1 and CD61 antibodies for 15 min in the dark and diluted 1:50 in 1% formaldehyde to stop the reaction. The procedures were similar for hemoglobin. For hemoglobin experiments, instead of adding RBCs, hemoglobin was added to a concentration of 100 μm and allowed to incubate with platelets and nitrite for 5 min at 37 °C before 2.5 μm ADP was added. The average RBC oxygen saturation was 20%, and the average Hb oxygen saturation was 50% for deoxygenated samples as determined by absorbance spectroscopy. A BD Biosciences FACSCalibur flow cytometer and Cell Quest Pro software were used for data collection and analysis. The activation threshold was set so 99% of baseline platelets were beneath the threshold.
Platelet activation was further explored using allopurinol, L-NAME, or DZ at final concentrations of 100, 100, or 250 μm, respectively. Washed red blood cells (0.5 Hct) were mixed with 15 μl of PRP and nitrite (10 μm) in the presence or absence of allopurinol, L-NAME, or DZ at pH 7.4 or 6.8. The total volume of the assay mixture was 105 μl with a hematocrit of 0.15. After 5 min of incubation at 37 °C, 20 μm ADP (final concentration) was added to each reaction mixture and incubated for another 10 min. Monoclonal antibodies against human CD 61 and PAC-1 were then added, and the samples were further incubated for 15 min in the dark at room temperature. Finally, cells were fixed with 1% buffered formalin and kept in a refrigerator before examination by flow cytometry.
Platelet aggregation was examined using whole blood in the presence or absence of nitrite, allopurinol, or L-NAME and determined by impedance aggregometry using a Multiplate 5.0 Analyzer from diaPharma (West Chester, OH) at Wake Forest University School of Medicine. Multiplate® analysis takes place in a single-use test cell that incorporates dual sensors and a Teflon-coated stirring magnet. Three hundred and fifty microliters of whole blood was incubated in the presence or absence of 10 μm nitrite, 100 μm allopurinol, or 300 μm L-NAME for 5 min at 37 °C. After 5 min of incubation, 300 μl of the reaction mixture was added to the micro cell of the Multiplate 5.0 Analyzer. Aggregation was induced by 8 μm ADP in presence of 0.9% NaCl and 1.5 mm CaCl2 according to the manufacturer's recommendation. Aggregation was induced for 5 min at 37 °C, and platelet aggregation was recorded.
Xanthine Oxidase Fluorometric Assay
A xanthine oxidase fluorometric Assay (Cayman Chemical) was performed following the manufacturer's instructions. This assay was used to confirm that the ability of our allopurinol preparation to inhibit xanthine oxidoreductase. The fluorescence from standard samples with allopurinol (100 μm) was only 24% that from standard samples with control vehicle. Thus, the allopurinol we used for this study effectively inhibits XOR activity.
Xanthine Oxidase Western Blotting
For Western blotting, hemoglobin-free ghost RBCs were prepared from normal human blood as described previously (46) with some modifications. Briefly, RBC ghost cell membranes were prepared in cold hypotonic phosphate buffer, pH 7.4 (20 mosm) with protease inhibitor mixture. Ghost membranes were isolated and washed four times in cold PBS in the presence of protease inhibitor mixture. Proteins were extracted from ghost membranes by modified radioimmune precipitation assay buffer with phosphatase and protease inhibitors. 52 μg of total ghost membrane proteins were loaded onto the SDS-polyacrylamide gel (4–20% linear gradient). Proteins from SDS-PAGE were transferred onto the PVDF membrane and probed for xanthine oxidase. Polyclonal xanthine oxidase antibody at a dilution of 1:500 was added to the membrane and incubated at 4 °C overnight. HRP-labeled second antibody at a dilution of 1:10,000 was used to detect primary antibody. For protein loading control, the same blot was stripped and re-probed with β actin antibody at a dilution of 1:500. In some of the experiments we also used washed human RBCs to look for xanthine oxidase expression in Western blotting. Due to high content of Hb in RBCs, SDS gels were smeared. Thus, we only assayed Hb-free RBC ghost membranes for xanthine oxidase expression. RBCs from eight individual donors were used for ghost membrane preparation and probed for xanthine oxidase expression in Western blotting. All the experiments were repeated three times.
Statistical Analysis
Student's t test was used to test for statistical significance. Results were considered significant when p < 0.05. All values are presented as the mean ± S.D.
RESULTS
Time-resolved Absorption Shows Relative Efficacy of Nitrite Reduction by DeoxyHb Versus XOR
To test the relative efficacy of XOR in reducing nitrite compared with deoxygenated hemoglobin, we performed time-resolved absorption spectroscopy (Fig. 1). In the presence of sodium dithionite, the reaction of Hb and nitrite results in complete conversion of deoxyHb (characterized by a single absorption peak at 555 nm) to HbNO (characterized by two peaks at 540 and 560 nm). The kinetics of the reactions (insets Fig. 1) should increase to the extent XOR simultaneously reduces nitrite to NO. In the absence of XOR (Fig. 1A), the average half-life of the reaction was 9.3 ± 0.8 min. With the addition of 1 μm XOR (Fig. 1B), the half-life decreased to 6.7 ± 3.6 min, and 10 μm XOR (Fig. 1C) decreased the half-life to 2.3 ± 0.7 min. Incubation of allopurinol with 10 μm XOR (Fig. 1D) slowed the reaction down, giving an average half-life of 6.6 ± 0.1 min. These results show that with only 100 μm Hb (∼100 times less than what is in whole blood), ∼1 μm XOR is needed to contribute to NO generation from nitrite; this is greater than the concentration of XOR found in the liver (30).
FIGURE 1.

Effect of XOR on nitrite reduction by Hb. Time resolved absorbance spectra after mixing deoxyhemoglobin (100 μm) and nitrite (500 μm) in the presence of sodium dithionite (10 mm) with NADH (1 mm) (t½ = 9 min) (A), NADH (1 mm) and XOR (1 μm) (t½ = 7.8 min) (B), and NADH (1 mm) and XOR (10 μm) (t½ = 3 min) (C), and NADH (1 mm), XOR (10 μm), and allopurinol (100 μm) (t½ = 6.6 min) (D). Insets show the percentage of deoxyHb (blue) and HbNO (red) as a function of time. Spectra were recorded at 37 °C every 128 s for data shown in panels A, C, and D and every 180 s for data shown in panel B. Samples were all prepared in 100 mm HEPES buffer, pH 6.5.
Allopurinol, a Xanthine Oxidoreductase Inhibitor, Does Not Inhibit NO Production from Red Cells Incubated with Nitrite
The role of xanthine oxidoreductase on erythrocytic nitrite reduction was studied by measuring NO production from nitrite in erythrocytes in the presence or absence of allopurinol using EPR detection of nitrosyl hemoglobin. In the assay nitrite was added to a mixture of deoxygenated RBCs and cell-free Hb (to capture NO released from the red cell) at pH 7.4, 6.8, or 6.5 to monitor production of NO by deoxyHb and/or XOR. Because red cell-encapsulated Hb was present at ∼100-fold excess to cell-free Hb, most Hb-mediated nitrite reduction is expected to be formed inside the red cell (unlike in the case of NO, nitrite does not preferentially react with cell-free Hb as the nitrite/Hb bimolecular rate constant is so small). NO produced from nitrite reduction will bind to deoxyHb to form HbNO. EPR was used to determine the concentration of HbNO formed in the mixture at 15 and 30 min (and at 5 and 30 min for data collected at pH 6.5). The supernatant was also collected at 50 min to assess NO export from the red cell. Allopurinol was used to assess contributions from XOR (Fig. 2 and Table 1). Representative EPR spectra from these experiments conducted with blood drawn from normotensive or hypertensive volunteers and room temperature are shown in Fig. 2, A and B, respectively. Spectra appeared similar in the presence or absence of allopurinol. When RBCs from normotensive volunteers were incubated with allopurinol at pH 6.5 and 37 °C (Fig. 2C), there was no significant difference between the amount of HbNO formed in mixtures with and without allopurinol at 5 min or 30 min in mixtures or 50 min in supernatants. No effect of allopurinol was also observed when RBCs from normotensive volunteers were incubated at pH 7.4 or pH 6.8 at room temperature or 37 °C (Table 1). The addition of xanthine (10 μm) or NADH (1 mm) also did not result in an observable difference when allopurinol was added even though these have been shown to enhance erythrocytic XOR activity (31). When RBCs from hypertensive volunteers were incubated at room temperature and pH 7.4 (Fig. 2D), no statistically significant difference was found between the amounts of HbNO formed with and without allopurinol in mixtures at 15 or 30 min or at 50 min in the supernatants.
FIGURE 2.

Allopurinol (Allo; 100 μm) does not affect HbNO yield detected by EPR. EPR for experiments conducted with blood drawn from normotensive (A, C, and E) and hypertensive volunteers (B, D, and F). A, representative EPR spectra for samples with allopurinol (red) or vehicle (blue) collected from mixtures at 5 (dashed) and 30 min (solid) and from supernatants (SPT) at 50 min (dotted) from normotensive volunteers at pH 6.5 and 37 °C. B, representative EPR spectra for samples using RBCs from hypertensive volunteers at pH 7.4 and room temperature. C, average HbNO concentrations in RBCs from normotensive volunteers at pH 6.5 and 37 °C (n = 3). No significant differences were found for HbNO formed at 5 min (p = 0.8) and 30 min (p = 0.3) in mixtures and 50 min in supernatants (SPT; p = 0.2). D, average HbNO concentrations from experiments when RBCs were drawn from hypertensive volunteers at pH 7.4 and room temperature (n = 6). No significant difference was found for HbNO formed at 15 min (p = 0.4) and 30 min (p = 0.7) in mixtures and 50 min in supernatants (p = 0.5). E, upon the addition of exogenous XOR (10 μm), the addition of allopurinol reduced HbNO formation measured at 30 min after nitrite addition at pH 6.5 and 37 °C using red blood cells from normotensive individuals (p = 0.03, n = 3). No effect of allopurinol was observed at 5 min after nitrite addition in the mixture at 5 min (p = 0.8) or in the supernatants at 50 min (p = 0.6). *, versus −All, p = 0.03, n = 3. F, HbNO concentrations from experiments conducted using red blood cells from hypertensive volunteers with the addition of NADH at pH 7.4 and 37 °C. No significant difference was found between the amounts of HbNO formed in the mixture in the presence or absence of allopurinol (p > 0.6).
TABLE 1.
Nitrite bioactivation by RBCs
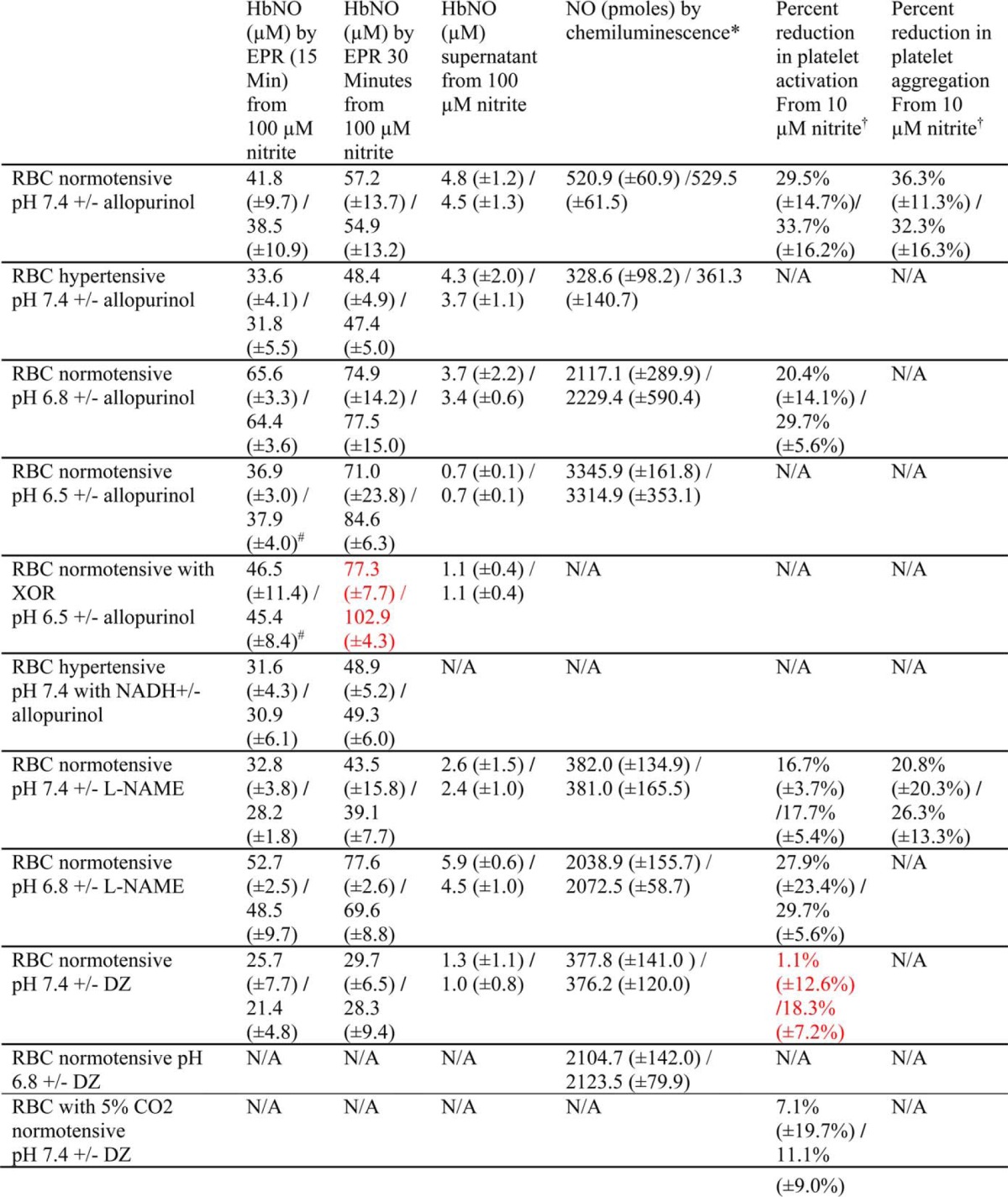
* The amount of NO detected by chemiluminescence is calculated based on a standard curve.
† Percent reduction is calculated by ((+RBCs+intrite)−(RBCs))/RBCs.
# EPR samples were prepared at 5 min.
The red color indicates significance. p < 0.05 comparing + vs. − inhibitor.
N/A; not available.
To further explore a contribution to erythrocytic nitrite reduction by XOR, additional experiments were performed with the addition of 10 μm XOR to the RBCs. In this case, where the experiments were carried out at 37 °C and pH 6.5 for erythrocytes from normotensive individuals, NO production (as measured by HbNO formation) was significantly less when allopurinol was added (Fig. 2E). In addition, experiments with red blood cells from hypertensive volunteers were conducted with the addition of NADH at 37 °C, pH 7.4, and the concentrations of HbNO in the presence or absence of allopurinol were not significantly different (Fig. 2F).
Erythrocytic nitrite reduction by XOR was also determined by chemiluminescent detection of NO in the presence or absence of allopurinol. Deoxygenated RBCs incubated with allopurinol or vehicle were added to a deoxygenated nitrite solution, and NO was produced from nitrite reduction at pH 7.4, 6.8, or 6.5 and 37 °C (Fig. 3 and Table 1). Fig. 3A shows representative raw signals of NO production at pH 7.4 and 37 °C using RBCs from normotensive volunteers. It is apparent that the NO signals produced when RBCs were incubated with allopurinol almost overlaps with the NO signal produced in the absence of allopurinol. Fig. 3B quantified the ratio +Allo/−Allo between NO productions when RBCs were incubated with or without allopurinol for red blood cells from normotensive (0.99 ± 0.1, n = 5) and hypertensive volunteers (0.94 ± 0.1, n = 5) at pH 7.4 and 37 °C. There is no significant difference in NO production by RBCs in the presence or absence of allopurinol using RBCs from either normotensive or hypertensive volunteers. NO production was also examined at pH 6.5 and 37 °C using RBCs from normotensive volunteers (Fig. 3C), and no significant difference was found between RBCs incubated with and without allopurinol. In addition, oxygenated RBCs that had been incubated with allopurinol or vehicle were added to a deoxygenated nitrite solution, and NO production was observed at pH 6.5 (Fig. 3C). The signals from NO production with and without allopurinol overlapped (not shown). Allopurinol had no effect on nitrite reduction when oxygenated RBCs were added to deoxygenated nitrite solution at pH 7.4 or 6.8. Allopurinol also did not have any effect on NO production when experiments were conducted at pH 6.8 (Table 1).
FIGURE 3.

Further studies examining the role of XOR in nitrite reductase activity of XOR using chemiluminescent detection of NO at 37 °C. A, representative signals of chemiluminescence due to NO production from nitrite reduction by adding deoxygenated RBCs to a deoxygenated nitrite solution using RBCs from normotensive volunteers in the presence (+Allo in red) or absence of allopurinol (−Allo in blue) at pH 7.4. Deoxygenated RBCs (0.5 Hct) were added to a nitrite solution (20 ml, 20 mm) after ∼5 min after recording the baseline signal, and NO production was observed for 20 min. B, the ratio of NO production by deoxygenated RBCs (n = 5) in the presence or absence of allopurinol at pH 7.4. No significant difference was found in the presence or absence of allopurinol using RBCs from normotensive (p = 0.7) or hypertensive (p = 0.2) volunteers. C, the ratio of NO production using RBCs at pH 6.5 (n = 3). No significant difference was found between NO production in the presence or absence of allopurinol. The ratio of NO production in the presence or absence of allopurinol from nitrite reduction by adding oxygenated RBCs to a deoxygenated nitrite solution (20 ml, 20 mm) at pH 6.5 was 0.98 (n = 3) and by adding deoxygenated RBCs to a deoxygenated nitrite solution (20 ml, 20 mm) was 1.01 (n = 3). No significant difference was found in the presence or absence of allopurinol for oxygenated (p = 0.8) or deoxygenated (p = 0.8) RBCs. D, Western blot for erythrocytic XOR. Panel A is an immunoblot for XOR, and panel B is an immunoblot for β actin. Lanes were loaded as follows: bovine-purified XOR (lane 1), mouse liver lysate (lane 2), human liver lysate (lane 3), empty (lane 4), human RBC ghost lysates (lanes 5–8). The purified bovine XOR, mouse, and human liver lysates show protein with a molecular mass consistent with the monomer of XOR at 150 kDa, whereas RBC ghost preparations do not.
To examine whether our observed lack of activity of erythrocytic XOR correlated with low levels of this protein on erythrocytes we performed Western blots (Fig. 3D). Whereas substantial detection of the XOR monomer was observed with an XOR positive control as well as with human and rat liver tissue, no XOR was detected from RBC ghost preparations. In all preparations using blood from eight different individuals, no XOR expression was ever observed from red cell ghosts. Thus, overall, our experiments using EPR and time-resolved absorption spectroscopy show that the contribution of XOR to NO production from native erythrocytes is severely limited and that we could only observe an allopurinol-inhibitable signal when exogenous XOR was added at levels substantially higher than that found in the liver.
The role of nitrite reduction by XOR was also determined by the inhibition of platelet activation. We confirmed earlier reports (37) that nitrite inhibits platelet activation in the presence of red blood cells (Fig. 4A). Nitrite had no effect on platelet activation in the absence of red blood cells (data not shown) as established previously (37). These data strongly support the notion that nitrite is reduced by erythrocytes, producing bioavailable NO. As a first step in showing that deoxyHb is the predominant nitrite reductase responsible for this activity, we showed that cell-free deoxyHb can inhibit platelet activation in the presence of nitrite (Fig. 4B). To further explore the mechanism of nitrite-dependent reduction in platelet activation by red cells, we employed the specific inhibitor allopurinol as in our studies using EPR and chemiluminescence at pH 7.4 (Fig. 4C and Table 1) and 6.8 (Fig. 4D and Table 1) under 37 °C. The addition of red blood cells to human PRP and ADP reduces platelet activation at pH 7.4 (p = 0.0001, n = 7) and 6.8 (p = 0.03, n = 4), probably due to ADP uptake by red blood cells. The addition of nitrite (10 μm) further reduces platelet activation at pH 7.4 (p = 0.0005, n = 7) and 6.8 (p = 0.004, n = 4). The addition of allopurinol did not have any additional effect on inhibition of platelet activation at either pH 7.4 or 6.8. In addition, we found that allopurinol had no effect on inhibition of platelet aggregation at pH 7.4 and 37 °C (p = 0.5, Table 1).
FIGURE 4.

Nitrite inhibition of platelet activation at 37 °C. Nitrite was added to PRP and deoxygenated RBC (0.2 Hct) (A) or deoxygenated Hb (100 μm) (B) and allowed to incubate for 5 min before platelets were activated by ADP. A, nitrite together with RBCs (80% deoxygenated) inhibits platelet activation at nitrite concentrations of 1 μm and higher (p < 0.05, n = 6). B, nitrite together with partially deoxygenated cell-free Hb (50% deoxygenated) inhibits platelet activation at nitrite concentrations of 1 μm and higher (p < 0.05, n = 5). C, allopurinol (100 μm) does not affect the inhibition of platelet activation by nitrite (10 μm) and RBCs (0.15 Hct) at pH 7.4 (p = 0.2, n = 7). D, allopurinol (100 μm) does not affect the inhibition of platelet activation by nitrite (10 μm) and RBCs (0.15 Hct) at pH 6.8 (p = 0.2, n = 4). *, p < 0.05.
No Effect on NO Production from Nitrite in the Presence of L-NAME
The effect of eNOS on erythrocytic nitrite reduction was studied by measuring NO production from nitrite in erythrocytes in the presence of L-NAME using EPR detection of nitrosyl hemoglobin, chemiluminescent detection of NO, and inhibition of platelet activation (Table 1). Similarly to the experiments conducted using allopurinol, EPR was used to determine the concentration of HbNO formed at 15 and 30 min in the mixture and 50 min in the supernatant in the presence or absence of L-NAME at pH 7.4 (Table 1) or 6.8 (Table 1) under 37 °C. The results show that the NO synthase inhibitor L-NAME does not affect HbNO yield at pH 7.4 or 6.8. Detection of NO by chemiluminescence was also employed, and the results are shown in Table 1. Deoxygenated RBCs incubated with or without L-NAME were added to a deoxygenated nitrite solution, and NO was produced from nitrite reduction at pH 7.4 or 6.8 and 37 °C. The ratios between NO productions by deoxygenated RBCs in the presence or absence of L-NAME were calculated at pH 7.4 (1.02 ± 0.1, n = 3) and 6.8 (0.98 ± 0.1, n = 3). No significant difference was found due to L-NAME at pH 7.4 (p = 1) or 6.8 (p = 0.7). In addition, oxygenated RBCs incubated with or without L-NAME were added to a deoxygenated nitrite solution at pH 7.4 or 6.8 and 37 °C (data not shown). The ratios between NO productions by oxygenated RBCs in the presence or absence of L-NAME were calculated at pH 7.4 (1.00 ± 0.1, n = 3) and 6.8 (1.01 ± 0.03, n = 3). There was no significant difference due to L-NAME at pH 7.4 (p = 0.9) or 6.8 (p = 0.7). Additionally, we examined the role of eNOS in nitrite-dependent reduction in platelet activation. Similarly to allopurinol, L-NAME did not significantly affect the inhibition of platelet activation by nitrite and RBCs at pH 7.4 (p = 0.8, n = 5, Table 1) or 6.8 (p = 0.9, n = 4, Table 1). In addition, we did not observe a significant effect of L-NAME on nitrite-mediated inhibition of platelet aggregation (Table 1).
Effects of Inhibition of Carbonic Anhydrase on Erythrocytic Nitrite Bioactivation
We tested the effects of inhibition of CA on HbNO formation due to nitrite (100 μm) reduction using EPR at pH 7.4 and 37 °C (Fig. 5A and Table 1). We used two formulations of CA inhibitor dorzolamide (labeled DZ and Trusopt), and neither one had a significant effect on NO production from nitrite as measured by EPR. We also saw no effect of DZ on HbNO formation by RBCs using 10 μm nitrite (p > 0.3, data not shown). Similarly, we detected no effect of DZ on NO production by RBCs using chemiluminescent detection of NO (Fig. 5B and Table 1). However, DZ did significantly blunt nitrite-mediated inhibition of platelet activation when 10 μm nitrite was used (Fig. 5D and Table 1) and trended toward blunting nitrite-mediated inhibition of platelet activation when 1 μm was used (Fig. 5C) but had no effect when 100 μm nitrite was employed (Fig. 5E). Use of DZ did not directly affect platelet inhibition when a NO donor was used (Fig. 5F) and also did not directly react with nitrite as determined by chemiluminescent detection of nitrite in the presence of up to 10 mm DZ (data not shown).
FIGURE 5.

DZ does not affect erythrocytic nitrite reduction. A, two formulations of CA inhibitor dorzolamide (labeled dorzolamide and Trusopt, 750 μm) do not affect HbNO yield from nitrite (100 μm) reduction at pH 7.4 and 37 °C detected by EPR (n = 3). No significant differences were found for HbNO formed at 15 min (p = 0.1) and 30 min (p = 0.7) in mixtures and 50 min in supernatants (p = 0.3). B, the ratio between NO productions by deoxygenated RBCs in the presence or absence of DZ (750 μm) was determined by chemiluminescent detection of NO at pH 7.4 (0.99 ± 0.05, n = 3) and 6.8 (0.99 ± 0.03, n = 3) at 37 °C. No significant difference was found due to DZ at pH 7.4 (p = 0.9) or 6.8 (p = 0.7). C, DZ (250 μm) trended toward affecting the inhibition of platelet activation by nitrite (1 μm) and RBCs (0.15 Hct) at pH 7.4 and 37 °C (p = 0.07, n = 8). * p = 0.01, n = 8. D, DZ (250 μm) affects the inhibition of platelet activation by nitrite (10 μm) and RBCs (0.15 Hct) at pH 7.4 and 37 °C. *, p = 0.01, n = 6. †, p = 0.04, n = 6. E, nitrite (100 μm) enhanced platelet inhibition, and DZ (250 μm) did not affect the inhibition of platelet activation by nitrite and RBCs (0.15 Hct) at pH 7.4 and 37 °C (p = 0.4, n = 10). * p < 0.01, n = 8. F, use of DZ (250 μm) did not directly affect platelet inhibition when a NO donor (2 μm) was used.
Evidence That Hemoglobin Is the Primary Erythrocytic Nitrite Reductase under Physiological Conditions
When red blood cells were partially deoxygenated by 5% CO2, the balance nitrogen instead of pure nitrogen, nitrite (10 μm)-mediated inhibition of platelet activation was no longer significantly affected by DZ (Fig. 6A, Table 1). In addition, 100 μm nitrite strongly inhibited platelet activation when RBCs were deoxygenated with 5% CO2 balance nitrogen, and DZ had no effect (Fig. 6B). Importantly, use of 5% CO2 had no effect on inhibition of platelet activation using NO donors (data not shown). Further evidence that Hb, rather than CA, is primarily responsible for RBC-mediated nitrite bioactivation is shown in Fig. 6C where we confirm that nitrite bioactivation is more potent when the RBCs are deoxygenated as opposed to oxygenated (percent reduction is 10.2% ± 8.5% for deoxyRBCs and 1.7% ± 4.9% for oxyRBCs, p = 0.02).
FIGURE 6.
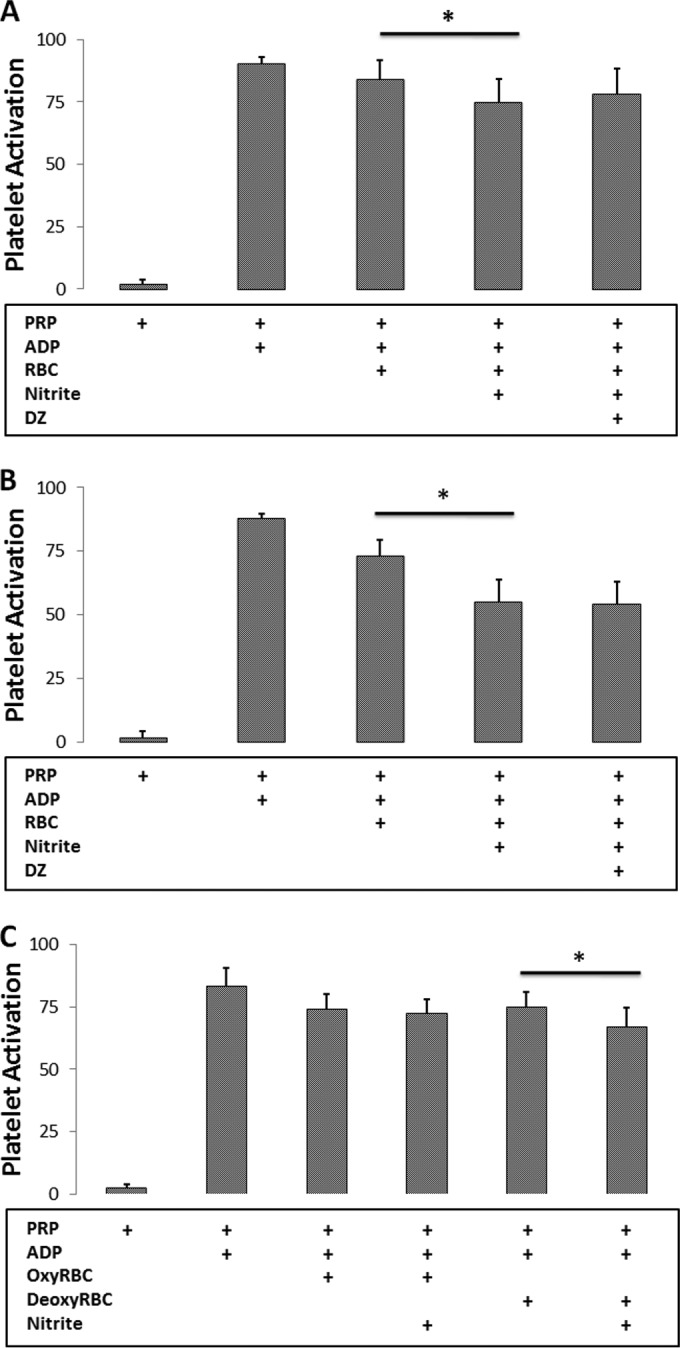
Evidence of hemoglobin as the primary erythrocytic nitrite reductase under physiological conditions. A, DZ (250 μm) no longer affects the platelet activation in the presence of 10 μm nitrite when red blood cells (0.15 Hct) were partially deoxygenated by 5% CO2 balance nitrogen at pH 7.4 and 37 °C. *, p = 0.03, n = 10. B, 100 μm nitrite strongly inhibits platelet activation when RBCs (0.15 Hct) were deoxygenated by 5% CO2 balance nitrogen. DZ (250 μm) no longer had an effect on the platelet activation. *, p < 0.001, n = 7. C, nitrite (10 μm)-dependent platelet inhibition in erythrocytes under oxygenated (percent reduction is 1.7% ± 4.9%) and deoxygenated (percent reduction is 10.2% ± 8.5%) conditions. *, p = 0.005, n = 8.
To further examine whether Hb is primarily responsible for nitrite reduction in the red blood cell, we inhibited its activity using CO. The nitrite reduction activity of any enzymes other than deoxygenated hemoglobin, such as XOR, eNOS, and CA could be elucidated by examining HbNO production using EPR (Fig. 7A). Additionally, the NO donor DEANONOate was added to RBC-CO solution to ensure HbNO could form with NO replacing CO due to hemoglobin's higher affinity for NO compared with CO (47). DEANONOate was also added to deoxygenated RBCs to determine the exact amount of NO released. Fig. 7A shows the representative raw EPR signals of HbNO formed at 15 and 30 min during experiments. The signals of HbNO formed during the incubation of nitrite and RBC-CO at 15 and 30 min are flat. Three repeats of the experiment gave us all zero values for HbNO concentration (data now shown). A lack of HbNO formed during the incubation of RBC-CO and nitrite demonstrate that other enzymes, such as XOR, eNOS, and CA, do not play a big role in erythrocyte-associated nitrite reduction compared with deoxygenated hemoglobin.
FIGURE 7.

NO formation from erythrocytic nitrite reduction in the presence or absence of specific inhibitor is diminished by CO. A, CO diminishes NO formation from erythrocytic nitrite reduction. Representative raw EPR signals of HbNO formed by incubating RBC-CO with nitrite (100 μm), RBC-CO with NO donor (DEANONOate, 100 μm), or deoxygenated RBCs with NO donor (DEANONOate, 100 μm) at pH 7.4 and room temperature. No signals of HbNO formed by incubating RBC-CO with nitrite (indicated by the black arrow). B, representative raw signals of NO production from nitrite reduction by adding deoxygenated RBCs that had incubated with (in red) or without (in blue) allopurinol (100 μm) to a CO saturated nitrite solution at pH 7.4 and 37 °C. Deoxygenated RBCs was added to nitrite solution at ∼2 min, and the signal of NO production was observed for 5 min. C, representative raw signals of NO production from nitrite reduction by adding deoxygenated RBCs that had incubated with (in red) or without (in blue) L-NAME (300 μm) to a CO saturated nitrite solution at pH 7.4 and 37 °C. D, representative raw signals of NO production from nitrite reduction by adding deoxygenated RBCs that had incubated with (in red) or without (in blue) DZ (250 μm) to a CO saturated nitrite solution at pH 7.4 and 37 °C.
CO was also employed in the detection of NO production using chemiluminescence. The signals of NO production were observed when deoxygenated RBCs that had been incubated with or without allopurinol (Fig. 7B), L-NAME (Fig. 7C), or DZ (Fig. 7D), were added to a CO-saturated nitrite solution at pH 7.4 and 37 °C. None of these enzymatic inhibitors has any effect. NO production was greatly attenuated because deoxygenated RBCs bind CO at a very fast rate, and the hemoglobin-mediated nitrite reduction was blocked by CO. The small bumps of signals can be explained by the NO production from nitrite reduction by a small amount of deoxygenated hemoglobin with free heme sites that is present before CO binding. After that, NO production was blocked, and no signal was observed.
We attempted to see if CO would blunt nitrite-mediated inhibition of platelet activation by RBCs, but we found, interestingly, that CO itself blunts NO-mediated inhibition of platelet activation using an NO donor (data not shown), probably because CO displaces NO on the platelet-soluble guanylate cyclase and CO does not activate soluble guanylate cyclase as well as NO.
DISCUSSION
Experiments demonstrating that nitrite only inhibits platelet activation in the presence, but not the absence, of red blood cells and that this inhibition is abrogated when a NO scavenger is employed (37), strongly support a role of the RBC in mediating bioactivation of nitrite. In this work we have explored different pathways that have been previously suggested to be responsible for nitrite bioactivation by RBCs: Hb (5), XOR (9, 31), CA (43), and NOS (31). Our EPR experiments using specific inhibitors of XOR, NOS, and CA clearly show that these do not contribute significantly to NO production in the RBC. Moreover, measurement of HbNO in the supernatant by EPR suggests that they do not contribute to NO export from the RBC either. These results were confirmed in our experiments using chemiluminescence detection of gas-phase NO export. The fact that CO abrogated NO production as measured by EPR supports a primary role of Hb in NO production by the RBC. Inhibitors of XOR and NOS had no effect on nitrite-mediated inhibition of platelet activation by RBCs, as also observed recently (38). However, the CA inhibitor DZ did blunt inhibition of platelet activation under certain conditions but not when 5% CO2 was employed.
Previous work has demonstrated a substantial reduction in nitrite-mediated NO production by RBCs as measured by chemiluminescence when employing NOS and XOR inhibitors at pH 6.8 (31), and XOR was found to be especially important in nitrite bioactivation from RBCs obtained from individuals with hypertension (9). In experiments where we added exogenous XOR, we found that concentrations greater than that found in the liver were necessary to compete with deoxyHb for reduction of nitrite to NO (Figs. 1 and 2). In our studies where we did not add exogenous XOR, we did not observe any effects of XOR or NOS inhibition on RBC-mediated nitrite bioactivation using EPR detection of HbNO, a chemiluminescence assay or inhibition of platelet activation assays. Apparent discrepancies between these results and previous ones using the chemiluminescence assay may be largely due to subtle differences in methods employed, including the fact that our chemiluminescence assay employed 10 mm nitrite, whereas previous measurements employed 10–100 μm nitrite (9, 31). Differences in solution conditions that may have led to different populations of XOR versus xanthine dehydrogenase may also account for some discrepancies. In addition, it is important to point out that whereas we examined RBCs from patients with controlled hypertension who were taking medication, the previous study examined RBCs from drug-naive patients (9). Further work would be useful to elucidate conditions where erythrocytic XOR or NOS contribute to NO production from nitrite by RBCs, but contributions from these enzymes under conditions studied here using various assays were minimal.
We found that inhibition of CA by DZ affected nitrite-mediated inhibition of platelet activation by RBCs only under certain non-physiological conditions, yet DZ had no significant affect on NO production from RBCs. When CO2 was maintained at 5%, DZ no longer had an effect. One possible explanation for these data is that CA forms a species that can become NO in the platelet, but CO2 or bicarbonate competitively inhibits formation of this species. Overall, our results suggest that Hb is the main protein responsible for nitrite bioactivation from RBCs.
Acknowledgment
We acknowledge services provided by the Flow Cytometry Core Laboratory of the Comprehensive Cancer Center, supported in part by NCI, National Institutes of Health Grant P30 CA121291-37.
This work was supported, in whole or in part, by National Institutes of Health Grants HL058091 and HL098032. This work was also supported by the Translational Science Center of Wake Forest University and Hypertension and Vascular Research Center of Wake Forest School of Medicine.
- RCB
- red blood cell
- XOR
- xanthine oxidoreductase
- NOS
- nitric-oxide synthase
- eNOS
- endothelial NOS
- CA
- carbonic anhydrase
- PRP
- platelet rich plasma
- DZ
- dorzolamide
- Hct
- hematocrit
- L-NAME
- NG-nitro-l-arginine methyl ester.
REFERENCES
- 1. Lauer T., Preik M., Rassaf T., Strauer B. E., Deussen A., Feelisch M., Kelm M. (2001) Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc. Natl. Acad. Sci. U.S.A. 98, 12814–12819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gladwin M. T., Schechter A. N., Kim-Shapiro D. B., Patel R. P., Hogg N., Shiva S., Cannon R. O., 3rd, Kelm M., Wink D. A., Espey M. G., Oldfield E. H., Pluta R. M., Freeman B. A., Lancaster J. R., Jr., Feelisch M., Lundberg J. O. (2005) The emerging biology of the nitrite anion. Nat. Chem. Biol. 1, 308–314 [DOI] [PubMed] [Google Scholar]
- 3. Lundberg J. O., Weitzberg E., Gladwin M. T. (2008) The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 7, 156–167 [DOI] [PubMed] [Google Scholar]
- 4. Kevil C. G., Kolluru G. K., Pattillo C. B., Giordano T. (2011) Inorganic nitrite therapy: historical perspective and future directions. Free Radic. Biol. Med. 51, 576–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cosby K., Partovi K. S., Crawford J. H., Patel R. P., Reiter C. D., Martyr S., Yang B. K., Waclawiw M. A., Zalos G., Xu X., Huang K. T., Shields H., Kim-Shapiro D. B., Schechter A. N., Cannon R. O., 3rd, Gladwin M. T. (2003) Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 9, 1498–1505 [DOI] [PubMed] [Google Scholar]
- 6. Modin A., Björne H., Herulf M., Alving K., Weitzberg E., Lundberg J. O. (2001) Nitrite-derived nitric oxide: a possible mediator of “acidic-metabolic” vasodilation. Acta Physiol. Scand. 171, 9–16 [DOI] [PubMed] [Google Scholar]
- 7. Classen H. G., Stein-Hammer C., Thöni H. (1990) Hypothesis: the effect of oral nitrite on blood pressure in the spontaneously hypertensive rat, does dietary nitrate mitigate hypertension after conversion to nitrite. J. Am. Coll. Nutr 9, 500–502 [DOI] [PubMed] [Google Scholar]
- 8. Webb A. J., Patel N., Loukogeorgakis S., Okorie M., Aboud Z., Misra S., Rashid R., Miall P., Deanfield J., Benjamin N., MacAllister R., Hobbs A. J., Ahluwalia A. (2008) Acute blood pressure lowering, vasoprotective, and antiplatelet properties of dietary nitrate via bioconversion to nitrite. Hypertension 51, 784–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghosh S. M., Kapil V., Fuentes-Calvo I., Bubb K. J., Pearl V., Milsom A. B., Khambata R., Maleki-Toyserkani S., Yousuf M., Benjamin N., Webb A. J., Caulfield M. J., Hobbs A. J., Ahluwalia A. (2013) Enhanced vasodilator activity of nitrite in hypertension critical role for erythrocytic xanthine oxidoreductase and translational potential. Hypertension 61, 1091–1102 [DOI] [PubMed] [Google Scholar]
- 10. Larsen F. J., Ekblom B., Sahlin K., Lundberg J. O., Weitzberg E. (2006) Effects of dietary nitrate on blood pressure in healthy volunteers. N. Engl. J. Med. 355, 2792–2793 [DOI] [PubMed] [Google Scholar]
- 11. Brooks J. (1937) The action of nitrite on haemoglobin in the absence of oxygen. Proc. R. Soc. Lond. Ser. B Biol. Sci. 123, 368–382 [Google Scholar]
- 12. Huang Z., Shiva S., Kim-Shapiro D. B., Patel R. P., Ringwood L. A., Irby C. E., Huang K. T., Ho C., Hogg N., Schechter A. N., Gladwin M. T. (2005) Enzymatic function of hemoglobin as a nitrite reductase that produces nitric oxide under allosteric control. J. Clin. Invest. 115, 2099–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang K. T., Keszler A., Patel N., Patel R. P., Gladwin M. T., Kim-Shapiro D. B., Hogg N. (2005) The reaction between nitrite and deoxyhemoglobin: reassessment of the reaction kinetics and stoichiometry. J. Biol. Chem. 280, 31126–31131 [DOI] [PubMed] [Google Scholar]
- 14. Doyle M. P., Hoekstra J. W. (1981) Oxidation of nitrogen-oxides by bound dioxygen in hemoproteins. J. Inorg. Biochem. 14, 351–358 [DOI] [PubMed] [Google Scholar]
- 15. Eich R. F., Li T., Lemon D. D., Doherty D. H., Curry S. R., Aitken J. F., Mathews A. J., Johnson K. A., Smith R. D., Phillips G. N., Jr., Olson J. S. (1996) Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry 35, 6976–6983 [DOI] [PubMed] [Google Scholar]
- 16. Herold S., Exner M., Nauser T. (2001) Kinetic and mechanistic studies of the NO center dot-mediated oxidation of oxymyoglobin and oxyhemoglobin. Biochemistry 40, 3385–3395 [DOI] [PubMed] [Google Scholar]
- 17. Huang K. T., Huang Z., Kim-Shapiro D. B. (2007) Nitric oxide red blood cell membrane permeability at high and low oxygen tension. Nitric Oxide 16, 209–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cassoly R., Gibson Q. (1975) Conformation, co-operativity and ligand-binding in human hemoglobin. J. Mol. Biol. 91, 301–313 [DOI] [PubMed] [Google Scholar]
- 19. Jeffers A., Xu X., Huang K. T., Cho M., Hogg N., Patel R. P., Kim-Shapiro D. B. (2005) Hemoglobin mediated nitrite activation of soluble guanylyl cyclase. Comp. Biochem. Physiol. A. Mol. Integr. Physiol. 142, 130–135 [DOI] [PubMed] [Google Scholar]
- 20. Robinson J. M., Lancaster J. R. (2005) Hemoglobin-mediated, hypoxia-induced vasodilation via nitric oxide: mechanism(s) and physiologic versus pathophysiologic relevance. Am. J. Respir. Cell Mol. Biol. 32, 257–261 [DOI] [PubMed] [Google Scholar]
- 21. Basu S., Grubina R., Huang J., Conradie J., Huang Z., Jeffers A., Jiang A., He X., Azarov I., Seibert R., Mehta A., Patel R., King S. B., Hogg N., Ghosh A., Gladwin M. T., Kim-Shapiro D. B. (2007) Catalytic generation of N2O3 by a concerted nitrite reductase and anhydrase activity of hemoglobin. Nat. Chem. Biol. 3, 785–794 [DOI] [PubMed] [Google Scholar]
- 22. Nagababu E., Ramasamy S., Rifkind J. M. (2006) S-Nitrosohemoglobin: A mechanism for its formation in conjunction with nitrite reduction by deoxyhemoglobin. Nitric Oxide 15, 20–29 [DOI] [PubMed] [Google Scholar]
- 23. Salgado M. T., Nagababu E., Rifkind J. M. (2009) Quantification of intermediates formed during the reduction of nitrite by deoxyhemoglobin. J. Biol. Chem. 284, 12710–12718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Angelo M., Singel D. J., Stamler J. S. (2006) An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc. Natl. Acad. Sci. U.S.A. 103, 8366–8371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Navati M. S., Friedman J. M. (2009) Reactivity of glass-embedded Met hemoglobin derivatives toward external NO; implications for nitrite-mediated production of bioactive NO. J. Am. Chem. Soc. 131, 12273–12279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Navati M. S., Friedman J. M. (2010) Glass matrix-facilitated thermal reduction: a tool for probing reactions of Met hemoglobin with nitrite and nitric oxide. J. Phys. Chem. B 114, 2938–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patel R. P., Hogg N., Kim-Shapiro D. B. (2011) The potential role of the red blood cell in nitrite-dependent regulation of blood flow. Cardiovasc. Res. 89, 507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tu C., Mikulski R., Swenson E. R., Silverman D. N. (2009) Reactions of nitrite with hemoglobin measured by membrane inlet mass spectrometry. Free Radic. Biol. Med. 46, 14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koppenol W. H. (2012) Nitrosation, thiols, and hemoglobin: energetics and kinetics. Inorg. Chem. 51, 5637–5641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li H., Cui H., Kundu T. K., Alzawahra W., Zweier J. L. (2008) Nitric oxide production from nitrite occurs primarily in tissues not in the blood: critical role of xanthine oxidase and aldehyde oxidase. J. Biol. Chem. 283, 17855–17863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Webb A. J., Milsom A. B., Rathod K. S., Chu W. L., Qureshi S., Lovell M. J., Lecomte F. M., Perrett D., Raimondo C., Khoshbin E., Ahmed Z., Uppal R., Benjamin N., Hobbs A. J., Ahluwalia A. (2008) Mechanisms underlying erythrocyte and endothelial nitrite reduction to nitric oxide in hypoxia role for xanthine oxidoreductase and endothelial nitric oxide synthase. Circ. Res. 103, 957–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Totzeck M., Hendgen-Cotta U. B., Luedike P., Berenbrink M., Klare J. P., Steinhoff H.-J., Semmler D., Shiva S., Williams D., Kipar A., Gladwin M. T., Schrader J., Kelm M., Cossins A. R., Rassaf T. (2012) Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation 126, 325–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ormerod J. O., Ashrafian H., Maher A. R., Arif S., Steeples V., Born G. V., Egginton S., Feelisch M., Watkins H., Frenneaux M. P. (2011) The role of vascular myoglobin in nitrite-mediated blood vessel relaxation. Cardiovasc. Res. 89, 560–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dalsgaard T., Simonsen U., Fago A. (2007) Nitrite-dependent vasodilation is facilitated by hypoxia and is independent of known NO-generating nitrite reductase activities. Am. J. Physiol. Heart Circ. Physiol. 292, H3072–H3078 [DOI] [PubMed] [Google Scholar]
- 35. Dautov R. F., Stafford I., Liu S., Cullen H., Madhani M., Chirkov Y. Y., Horowitz J. D. (2014) Hypoxic potentiation of nitrite effects in human vessels and platelets. Nitric Oxide 40, 36–44 [DOI] [PubMed] [Google Scholar]
- 36. Isbell T. S., Gladwin M. T., Patel R. P. (2007) Hemoglobin oxygen fractional saturation regulates nitrite-dependent vasodilation of aortic ring bioassays. Am. J. Physiol. Heart Circ. Physiol. 293, H2565–H2572 [DOI] [PubMed] [Google Scholar]
- 37. Srihirun S., Sriwantana T., Unchern S., Kittikool D., Noulsri E., Pattanapanyasat K., Fucharoen S., Piknova B., Schechter A. N., Sibmooh N. (2012) Platelet inhibition by nitrite is dependent on erythrocytes and deoxygenation. PLOS ONE 7, e30380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Akrawinthawong K., Park J. W., Piknova B., Sibmooh N., Fucharoen S., Schechter A. N. (2014) A flow cytometric analysis of the inhibition of platelet reactivity due to nitrite reduction by deoxygenated erythrocytes. Plos ONE 9, e92435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Loscalzo J. (2001) Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ. Res. 88, 756–762 [DOI] [PubMed] [Google Scholar]
- 40. Wollny T., Iacoviello L., Buczko W., DeGaetano G., Donati M. B. (1997) Prolongation of bleeding time by acute hemolysis in rats: a role for nitric oxide. Am. J. Physiol. Heart Circ. Physiol. 272, H2875–H2884 [DOI] [PubMed] [Google Scholar]
- 41. Azuma H., Ishikawa M., Sekizaki S. (1986) Endothelium-dependent inhibition of platelet aggregation. Br. J. Pharmacol. 88, 411–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schäfer A., Wiesmann F., Neubauer S., Eigenthaler M., Bauersachs J., Channon K. M. (2004) Rapid regulation of platelet activation in vivo by nitric oxide. Circulation 109, 1819–1822 [DOI] [PubMed] [Google Scholar]
- 43. Aamand R., Dalsgaard T., Jensen F. B., Simonsen U., Roepstorff A., Fago A. (2009) Generation of nitric oxide from nitrite by carbonic anhydrase: a possible link between metabolic activity and vasodilation. Am. J. Physiol. Heart Circ. Physiol. 297, H2068–H2074 [DOI] [PubMed] [Google Scholar]
- 44. Huang Z., Louderback J. G., Goyal M., Azizi F., King S. B., Kim-Shapiro D. B. (2001) Nitric oxide binding to oxygenated hemoglobin under physiological conditions. Biochim. Biophys. Acta 1568, 252–260 [DOI] [PubMed] [Google Scholar]
- 45. Kim-Shapiro D. B. (2004) Hemoglobin-nitric oxide cooperativity: is NO the third respiratory ligand? Free Radic. Biol. Med. 36, 402–412 [DOI] [PubMed] [Google Scholar]
- 46. Dodge J. T., Mitchell C., Hanaha D. J. (1963) The preparation and chemical characteristics of hemoglobin-free ghost of human erythrocytes. Arch. Biochem. Biophys. 100, 119–130 [DOI] [PubMed] [Google Scholar]
- 47. Tsai A. L., Berka V., Martin E., Olson J. S. (2012) A “sliding scale rule” for selectivity among NO, CO, and O2 by heme protein sensors. Biochemistry 51, 172–186 [DOI] [PMC free article] [PubMed] [Google Scholar]