

Background: Mutant-selective IDH1 inhibitors are potential cancer therapeutics, but the mechanistic basis for their selectivity is not yet well understood.
Results: Inhibitor binding modes and kinetic mechanisms were characterized.
Conclusion: The inhibitors selectively inhibit mutant IDH1 by interacting with a magnesium-binding residue.
Significance: Targeting metal-binding residues with drug-like small molecules is a feasible strategy for IDH1 inhibition.
Keywords: Anticancer Drug, Drug Action, Drug Discovery, Enzyme Inhibitor, Metalloenzyme, IDH1 Inhibitor, Cancer Metabolism, Mechanism of Inhibition
Abstract
Cancer-associated point mutations in isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) confer a neomorphic enzymatic activity: the reduction of α-ketoglutarate to d-2-hydroxyglutaric acid, which is proposed to act as an oncogenic metabolite by inducing hypermethylation of histones and DNA. Although selective inhibitors of mutant IDH1 and IDH2 have been identified and are currently under investigation as potential cancer therapeutics, the mechanistic basis for their selectivity is not yet well understood. A high throughput screen for selective inhibitors of IDH1 bearing the oncogenic mutation R132H identified compound 1, a bis-imidazole phenol that inhibits d-2-hydroxyglutaric acid production in cells. We investigated the mode of inhibition of compound 1 and a previously published IDH1 mutant inhibitor with a different chemical scaffold. Steady-state kinetics and biophysical studies show that both of these compounds selectively inhibit mutant IDH1 by binding to an allosteric site and that inhibition is competitive with respect to Mg2+. A crystal structure of compound 1 complexed with R132H IDH1 indicates that the inhibitor binds at the dimer interface and makes direct contact with a residue involved in binding of the catalytically essential divalent cation. These results show that targeting a divalent cation binding residue can enable selective inhibition of mutant IDH1 and suggest that differences in magnesium binding between wild-type and mutant enzymes may contribute to the inhibitors' selectivity for the mutant enzyme.
Introduction
Metabolic enzymes have become the focus of intensive efforts in basic cancer biology and the discovery of new targets for cancer drug therapy (1, 2). Isocitrate dehydrogenase, which catalyzes the conversion of isocitrate to α-ketoglutarate (αKG),2 is one of the metabolic enzymes most strongly associated with cancer risk and clinical outcome. IDH1 and IDH2, the isoforms that have been implicated in cancer, are NADP+-dependent enzymes localized to cytosol/peroxisome and mitochondria, respectively (3–5). IDH1 and IDH2 are the only metabolic enzymes yet identified in which a few single-point mutations are highly associated with cancer risk (the classic genetic signature of an oncogene). Mutation of arginine 132 in IDH1 is common in gliomas (up to 70% of secondary gliomas (6)), with IDH2 mutations represented at much lower frequency (7). In cytogenetically normal acute myeloid anemia, mutated IDH1 is found in 10.9% of patients (8), whereas mutated IDH2 (more frequently at Arg140 and less frequently at Arg172 (9)) is found in 12.1% (10).
Metabolic profiling of cancer cells expressing the R132H mutant IDH1 and in vitro characterization of the enzymatic activity of this IDH1 mutant led to the surprising discovery that the oncogenic mutation, in addition to causing a loss of normal enzymatic function (7, 11), also enabled a neomorphic enzymatic activity: the NADPH-dependent reduction of αKG to d-2-hydroxyglutarate (2HG) (12). The mutations associated with the neomorphic activity are also associated with other changes in catalytic active site function: Km values for both isocitrate and Mg2+ in the residual isocitrate dehydrogenase reaction of the mutant enzyme are much higher than the corresponding values for the wild-type enzyme (∼300-fold higher in the case of Mg2+) (12). The observation of the neomorphic activity, together with the oncogene-like genetics of the IDH mutations, led to the hypothesis that 2HG acts as an oncometabolite. Subsequent in vitro experiments demonstrated that 2HG is an inhibitor of histone demethylases and TET family 5-methylcytosine hydroxylases at the high (∼10 mm) concentrations observed in tumors (13–15), suggesting that 2HG induces dysregulation of methylation, with possible oncogenic effects. 2HG has also been proposed to promote oncogenic transformation by activating EGLN, an α-ketoglutarate-dependent prolyl hydroxylase involved in the hypoxia-inducible factor signaling pathway (16).
In vivo experiments using small molecule inhibitors of mutant IDH1 and IDH2 also support a role for 2HG in maintenance of undifferentiated tumor phenotypes and the potential clinical utility of mutant IDH inhibitors (17, 18). The IDH2 allosteric inhibitor AGI-6780 relieves the differentiation block in TF-1 erythroleukemia cells expressing an IDH2 mutant enzyme, and it stimulates the differentiation of primary acute myeloid anemia blasts (18). AGI-5198, an IDH1 inhibitor that has been reported to inhibit competitively with respect to αKG and noncompetitively with respect to NADPH (19), acts on IDH1 mutant glioma cells in vivo to inhibit 2HG accumulation (17) and to reverse histone methylation and induce the expression of genes associated with astrocytic differentiation (20). The IDH2 inhibitor AG-221, developed by Agios Pharmaceuticals, is currently in clinical trials as a therapy for hematological malignancies (ClinicalTrials.gov NCT 01915 498).
Because of the potential of IDH1/2 inhibitors as anticancer therapeutics, a number of groups have investigated the structural and mechanistic aspects of IDH inhibition. Kinetic and/or structural studies suggest that some inhibitors may bind at the αKG/isocitrate binding site (17, 19, 21), whereas others, such as the IDH2 inhibitor AGI-6780, bind allosterically to the interface between the two protomers of the IDH dimer (18). Based on the structure of AGI-6780-bound IDH2 (18), this inhibitor has been hypothesized to prevent catalysis by locking IDH2 in an “open,” precatalytic, inactive conformation that is similar to that observed in the absence of αKG or isocitrate (22, 23) and distinct from the “closed” conformation observed when the αKG/isocitrate site is occupied (12, 24).
Because of the importance of wild-type IDH in primary metabolism, achieving selective inhibition of the mutant enzyme over the wild-type is a critical issue in designing IDH inhibitors for therapeutic use. The IDH inhibitors reported to date achieve moderate to high mutant selectivity, but the structural/mechanistic basis for this selectivity remains an important unsolved question. In the case of allosteric IDH inhibitors in particular, the dimer interface is well separated from the residues mutated in cancer, suggesting that direct interactions between the mutated residue and allosteric inhibitors do not account for mutant selectivity.
The current study demonstrates that small molecules from two unrelated structural classes act as selective allosteric inhibitors of mutant IDH1 and act by the unusual mechanism of competing with the catalytically essential magnesium ion. Inhibitor binding thereby prevents the assembly of a catalytically competent magnesium binding site. Competitive binding with magnesium may contribute to the mutant selectivity of the inhibitor, because Mg2+ is much more effective at saturating its binding site in the wild-type enzyme than the mutant enzyme. These results also suggest that catalytically essential divalent cation binding residues in other enzymes may also be targetable using drug-like small molecules.
EXPERIMENTAL PROCEDURES
High Throughput Screening
High throughput screening assays were performed in 1536-well format (Corning 2728). Screening compounds (10.67 μm) were preincubated with homodimeric R132H IDH1 and NADPH at 37 °C for 30 min, followed by addition of αKG and incubation at 37 °C for 1.5 h. Final reaction conditions were the following: 30 nm R132H IDH1, 100 μm NADPH, 250 μm αKG, 50 mm Tris-HCl, pH 7.4, 20 mm MgCl2, 0.01% Tween 20, and 1% DMSO. The AmpliteTM detection reagent (AAT Bioquest; 5 μl) was added to terminate the IDH1 reaction, followed by incubation at room temperature for 30 min. The fluorescence intensity was measured using an Envision (PerkinElmer Life Sciences) plate reader with excitation and emission values of 540 and 590 nm.
General Methods for Protein Purification
Protein purity was estimated to be >95% based on SDS-PAGE. The identity of the purified proteins was confirmed by mass spectrometry. Following purification, proteins were quick-frozen in liquid nitrogen prior to storage at −80 °C.
Expression/Purification of Homodimeric Mutant IDH1
Mutant human IDH1 genes (R132H or R132C, based on the wild-type sequence NM_005896) bearing an N-terminal Strep tag followed by a TEV cleavage sequence were synthesized with codon usage optimized for Escherichia coli expression (DNA 2.0). The genes were subcloned into pET28a to produce the expression constructs. Expression was in E. coli BL21 DE(3), with induction using isopropyl β-d-thiogalactopyranoside (1 mm). Cleared cell lysates were applied to a StrepTactin affinity column (30 ml; GE Healthcare), which was washed with Buffer A (50 mm Tris, pH 7.4, 200 mm NaCl, 10% glycerol, 5 mm DTT; 150 ml). The Strep-tagged IDH1 mutants were eluted with 2.5 mm desthiobiotin in Buffer A. Fractions containing IDH1 were pooled and subjected to digestion with His-tagged TEV protease (Invitrogen; 0.3 units of TEV/μg of protein to be cleaved) overnight at 4 °C while dialyzing against Buffer A. The cleaved proteins were separated from TEV protease via application to a nickel-NTA column (Qiagen; 0.25 ml) and elution with a step gradient of imidazole in Buffer A (50, 100, 250, and 500 mm, in that sequence). Following a second StrepTactin affinity purification to remove residual uncleaved protein, the proteins were concentrated to 3–4 mg/ml.
Expression/Purification of Homodimeric Wild-type IDH1
The human IDH1 gene (NM_005896) bearing an N-terminal hexahistidine tag followed by a TEV cleavage sequence was synthesized and subcloned into pET28. Expression and purification were carried out as for the mutant homodimer, with the following exceptions. The cleared cell lysate was applied to a nickel-NTA column (Qiagen; 5 ml), and proteins were eluted using elution with a step gradient of imidazole in buffer A (0, 50, 100, 250, and 500 mm, in that sequence). After TEV cleavage of the IDH1 eluted from the nickel-NTA column, the cleaved protein was separated from TEV protease via reapplication to a nickel-NTA column and elution with Buffer A in the absence of imidazole.
Expression/Purification of Heterodimeric R132H/Wild-type IDH1
Wild-type IDH1 bearing an N-terminal His tag followed by a TEV cleavage site and R132H IDH1 bearing an N-terminal Strep tag followed by a TEV cleavage site were subcloned into pRSF-Duet (11), enabling bacterial expression of both genes on the same plasmid. Expression and cell lysis were carried out as for the homodimeric IDH constructs, and purification was carried out using a tandem affinity approach (Strep and His tags) based on the separations used for the homomeric constructs. Purification on a StrepTactin column was used to separate wt/wt dimers from mt/wt heterodimers and mt/mt homodimers. Fractions containing heterodimers and mt/mt homodimers were pooled and purified using nickel-NTA chromatography to separate mt/mt homodimers from mt/wt heterodimers. Fractions containing mt/wt heterodimers were pooled and subjected to TEV cleavage, and the cleaved protein was applied to a second nickel-NTA column (to remove TEV and uncleaved wt IDH1), with elution carried out as for the first column. A second StrepTactin chromatography step was carried out to remove proteins containing an uncleaved mutant protomer. The cleaved mt/wt heterodimeric protein was concentrated to ∼1 mg/ml.
Expression and Purification of IDH2
The following construct was prepared, based on the construct previously employed for expression of the porcine enzyme (25): an N-terminal His tag, followed by the sequence coding for GSGSGS as a linker, followed by the maltose-binding protein sequence (based on AF031088.1), followed by the thrombin cleavage sequence, followed by wild-type or mutant (R172K or R140Q) IDH2 (based on NM_002168.2, coding for the mature polypeptide, amino acid residues 40–452). For expression, E. coli BL21 DE(3) was transformed with the expression construct. Cleared cell lysate was applied to an amylose affinity column (New England Biolabs; 3 ml), and the column was washed with buffer B (30 mm triethanolamine, pH 7.4, 150 mm Na2SO4, 2 mm MnSO4, 10% glycerol; 15 ml), followed by elution with buffer B containing 10 mm maltose (20 ml). Fractions containing IDH2 were pooled, applied to a Superdex 200 column (120 ml), and eluted with buffer B. IDH2-containing fractions were pooled and subjected to cleavage with biotinylated thrombin (Novagen) overnight at 4 °C. Biotinylated thrombin was removed via binding to a streptavidin column, and a second amylose affinity chromatography step was carried out to remove uncleaved IDH2. The purified protein was dialyzed against Buffer C (30 mm triethanolamine, pH 7.4, 150 mm Na2SO4, 10% glycerol) overnight at 4 °C to remove MnSO4. The protein was concentrated to ∼0.2 mg/ml.
Synthesis of Compound 1 and Compound 2
Compound 1 was synthesized according to the following modified published procedure (26). Briefly, a 100-ml round-bottomed flask was charged with 4-(2,4,4-trimethylpentan-2-yl)phenol (2.06 g, 10 mmol), 1H-imidazole (1.02 g, 15.00 mmol), and paraformaldehyde (600.52 mg, 20.00 mmol) in ethanol (10 ml) to give a pale yellow solution which was heated to ∼60 °C. After 16 h, the reaction mixture was concentrated in vacuo to give an oil that was then heated at ∼130 °C. The reaction mixture was dry loaded onto a silica gel column and eluted with DCM/MeOH (gradient from 100/0 to 90/10) to give an off-white solid (with a higher retardation factor (Rf)) on TLC, consistent with 2-((1H-imidazol-1-yl)methyl)-4-(2,4,4-trimethylpentan-2-yl)phenol by NMR and LC-MS, and a pale yellow gum (lower Rf), consistent with a mixture of 2,6-bis((1H-imidazol-1-yl)methyl)-4-(2,4,4-trimethylpentan-2-yl)phenol and 2-((1H-imidazol-1-yl)methyl)-6-(hydroxymethyl)-4-(2,4,4-trimethylpentan-2-yl)phenol by LC-MS and NMR. The latter mixture was further purified by silica gel column (EtOAc/MeOH: gradient from 100/0 to 95/5) to give 1.02 g (28% yield) of a pale yellow semi-solid, consistent with the desired product, 2,6-bis((1H-imidazol-1-yl)methyl)-4-(2,4,4-trimethylpentan-2-yl)phenol. 1H NMR (d4-MeOH, 400 MHz) δ (ppm): 7.68 (s, 2H), 7.07 (s, 4H), 6.95 (s, 2H), 5.24 (s, 4H), 2.01 (s, 1H), 1.64 (s, 2H), 1.26 (s, 6H), 0.61 (s, 9H). MS: ES+ (M+H)+ = 367.5 m/z; ES− (M-H)− = 365.5 m/z.
Synthesis of Compound 2 and Chiral purification of Enantiomer 1 and Enantiomer 2
In a 100-ml round-bottomed flask, ortho-tolualdehyde (238.43 μl, 2 mmol) and 3-fluoroaniline (194.19 μl, 2.00 mmol) were mixed in 5 ml of MeOH. After stirring for 45 min at room temperature, benzoimidazol-1-yl-acetic acid (391.49 mg, 2.00 mmol) was added. After a further 15 min stirring at room temperature, cyclohexyl isocyanide (250.33 μl, 2.00 mmol) was added. The reaction mixture was stirred overnight at room temperature. A solid precipitated and was isolated by filtering through a sintered glass funnel. The solid was rinsed with cold MeOH, then dried under vacuum to afford compound 2 (2-(2-(1H-benzo[d]imidazol-1-yl)-N-(3-fluorophenyl)acetamido)-N-cyclohexyl-2-(o-tolyl)acetamide as a white powder (815 mg, % 81% yield). 1H NMR (300 MHz, DMSO-d6) d: 8.05 (s, 1H), 8.00–7.98 (d, 1H, J = 8.1), 7.65–7.61 (d, 1H, J = 9.3), 7.39–7.36 (d, 1H, J = 8.7), 7.27–6.87 (m, 7H), 6.78–6.75 (d, 1H, J = 7.5), 6.20 (s, 1H), 5.04–4.69 (m, 2H), 3.61–3.59 (m, 1H), 2.40 (s, 3H), 1.77–1.50 (m, 5H), 1.26–0.94 (m, 5H). MS: ES+ (M + H)+ = 499.5 m/z; ES− (M − H)− = 497.5 m/z.
The enantiomerically pure compounds were isolated by separation of the racemate (100 mg) using a chiral HPLC column (Chiralcel, OD 10 μm 3 × 25 cm column, UV 254 nm), eluting with 80% heptane, 20% EtOH, 0.1% TEA. The flow rate was increased from 40 to 80 ml/min after elution of the first product peak. The two desired enantiomers (36.4 and 34.8 mg) were isolated as white powders. Assignment of stereochemistry was based on biological activity: within this chemotype, compounds with (S) configuration have been reported to be the active species against R132H IDH1 (17, 27). Therefore, enantiomer 1 (active against R132H IDH1; with a longer retention time on the chiral column) is assigned with the (S) configuration, whereas enantiomer 2 (inactive; with a shorter retention time) is assigned with the (R) configuration. 1H NMR and LC-MS analyses were comparable to the racemic mixture.
Synthesis of 13C5-2HG
A six-step synthesis starting from [13C5]l-glutamic acid was conducted to obtain 13C5-2HG. First, tetrafluoroboric acid-diethyl ether complex (5.4 ml, 39.4 mmol) was slowly added to a suspension of l-[13C5]glutamic acid (3.0 g, 19.7 mmol) and sodium sulfate (3.0 g, 21.1 mmol) in benzyl alcohol (35 ml) at room temperature and under argon (28). After 20 h of stirring, the reaction mixture was diluted with tetrahydrofuran (100 ml) and was filtered. Triethylamine (6.1 ml, 43.7 mmol) was added, and the solution was concentrated to an oil. The addition of ethyl acetate (200 ml) caused precipitation, thus affording 4.97 g of a white solid after filtration. This solid was dissolved into water (160 ml) and acetic acid (40 ml). The solution was cooled to 0 °C, and sodium nitrite (2.76 g, 40.0 mmol) was added over 20 min as a solution in water (40 ml) (29, 30). The mixture was stirred for 2.5 h under argon, after which time the reaction was cooled to 0 °C and was quenched by addition of 2 m methylamine in tetrahydrofuran (20 ml, 40.0 mmol). The mixture was stirred for 15 min, and the volatiles were evaporated under vacuum. The remaining aqueous solution was extracted with ethyl acetate (4 × 80 ml), and the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated by rotary evaporation to a lower volume (evaporation to dryness causes lactonization). Column chromatography (SiO2, DCM:MeOH:AcOH, 95:5:1) afforded the desired product containing acetic acid as a contaminant (attempts to remove acetic acid increased lactone formation).
The product was rapidly used as such in the next step by dissolving it into anhydrous N,N-dimethylformamide (20 ml). Sodium bicarbonate (3.5 g, 41.7 mmol) and iodomethane (6.1 ml, 98.0 mmol) were added, and the reaction was stirred for 48 h at room temperature and under argon (30). Ethyl acetate (100 ml) was added, and the salts were filtered off. Water (50 ml) was added followed by saturated aqueous thiosulfate (50 ml). The mixture was extracted with ethyl acetate (3 × 80 ml). The organic layer was then washed with brine (2 × 100 ml) and was dried over sodium sulfate, filtered, and concentrated under vacuum. Column chromatography (SiO2, n-Pent: EtOAc, 8:2) afforded [13C5]-(S)-2-hydroxypentanedioic 5-benzyl ester 1-methyl ester as a clear oil (2.7 g, 53% from [13C5]-l-glutamic acid). 1H NMR (200 MHz, CDCl3) δ (ppm): 7.5–7.3 (m, 5H), (d, 3JC-H = 3.2 Hz, 2H), 4.26 (dm, (d, JC-H = 143.4 Hz, 1H), 3.79 (d, 3JC-H = 3.8 Hz, 3H), and 3.10–1.50 (4m, 4H).
[13C5](S)-2-Hydroxypentanedioic 5-benzyl ester 1-methyl ester (2.5 g, 9.7 mmol) was dissolved into anhydrous tetrahydrofuran (80 ml). Triphenylphosphine (3.1 g; 11.8 mmol) and 4-nitrobenzoic acid (4.9 g, 29.3 mmol) were added, and the mixture was cooled to −5 °C. Diisopropyl azodicarboxylate (2.4 ml, 12.2 mmol) was added dropwise, and the reaction was allowed to stir overnight at room temperature and under argon. The solvent was evaporated, and the remaining material was dissolved into ethyl acetate (100 ml). This solution was washed with brine (80 ml) and dried over sodium sulfate, filtered, and evaporated. Column chromatography (SiO2, Pent:EtOAc, 9:1 then 8:2) afforded [13C5](R)-2-(4-nitrobenzoyloxy)pentanedioic acid 5-benzyl ester 1-methyl ester as a clear, yellowish oil (3.5 g, 8.61 mmol, 88.6%). 1H NMR (200 MHz, CDCl3) δ (ppm): 8.3–8.2 (m, 4H), 7.4–7.3 (m, 5H), 5.35 (dm, JC-H = 152.8 Hz, 1H), 5.13 (d, 3JC-H = 3.2 Hz, 2H), 3.78 (d, 3JC-H = 3.8 Hz, 3H), 2.58 (dm, JC-H = 128.6 Hz, 2H), and 2.4 (dm, JC-H = 129 Hz, 2H). The product was analyzed by chiral HPLC ee = 96.3%: Chiralcel OD-H 250 × 4.6-mm 5-μm column, UV 254 nm, elution with heptane: EtOH, 80:20 at 1 ml/min and at 40 °C. Rf Major = 14.56 min and Rf minor = 12.55 min.
Potassium carbonate (5.0 g, 36.2 mmol) was added to a solution of [13C5](R)-2-(4-nitrobenzoyloxy)pentanedioic acid 5-benzyl ester 1-methyl ester (3.0 g, 7.38 mmol) in methanol (40 ml) at 0 °C. After 30 min of stirring, the mixture was diluted with dichloromethane (50 ml), and water (20 ml) followed by saturated aqueous ammonium chloride (40 ml) were added. The aqueous layer was extracted with dichloromethane (3 × 80 ml). The combined organic extracts were dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. Column chromatography (SiO2, Pent: EtOAc, 8:2) afforded 890 mg of the desired product containing 20 mol % of the corresponding lactone (as observed by 1H NMR). Based on the 1H NMR spectrum, the 890-mg batch was a mixture of [13C5](R)-hydroxyglutaric acid dimethyl ester (4.22 mmol) and [13C5](R)-5-oxotetrahydrofuran-2-carboxylic acid methyl ester (0.844 mmol), thus giving a combined yield of 68.5%. 1H NMR (600 MHz, CDCl3) δ (ppm): 4.94 (dm, JC-H = 158.6 Hz, 1Hminor), 4.24 (dm, JC-H = 147.6 Hz, 1HMajor), 3.81 (d, 3JC-H = 3.9 Hz, 3Hminor), 3.79 (d, 3JC-H = 3.8 Hz, 3HMajor), 3.67 (d, 3JC-H = 3.8 Hz, 3HMajor), 2.86–2.90 (m, 1HMajor, OH), 2.77–2.48 (2m, 4Hminor and 2.5HMajor), 2.11–1.98 (m, 1HMajor), and 1.87–1.76 (m, 0.5HMajor).
The mixture of [13C5](R)-hydroxyglutaric acid dimethyl ester and [13C5](R)-5-oxotetrahydrofuran-2-carboxylic acid methyl ester (890 mg, 5.06 mmol) was dissolved into 1 m sodium hydroxide (10.7 ml, 10.7 mmol) and stirred for 48 h at room temperature. After this time, the reaction mixture was freeze-dried, giving the desired 13C5-2HG (disodium salt) as a white powder (1.07 g, >100%). 1H NMR (600 MHz, D2O) δ (ppm): 3.98 (dm, JC-H = 145.6 Hz, 1H), 2.20 (dm, JC-H = 125.4 Hz, 2H), 1.95 (dm, JC-H = 135.5 Hz, 1H) and 1.78 (dm, JC-H = 130.2 Hz, 1H); 13C NMR (150 MHz, D2O) δ (ppm): 170.8 (dd, 2JC-C = 51.6 Hz and 3JC-C = 3.7 Hz), 169.1 (dd, 2JC-C = 54.5 Hz and 3JC-C = 3.0 Hz), 60.0 (ddd, 2JC-C = 54.5 Hz, 3JC-C = 36.6 Hz and 4JC-C = 3.7 Hz) and 21.4 (ddd, 2JC-C = 51.6 Hz, 3JC-C = 35.6 Hz and 4JC-C = 3.0 Hz) and 18.9 (2JC-C = 36.6 Hz, 3JC-C = 35.8 Hz); [α] D25 = +7.4° (c = 1 H2O @ 25 °C); MS: ES− (M-1) = 152 m/z; water content: 7.7% (Karl Fischer analysis); sodium content: 23.3% (ion chromatography).
Measurement of Reductive Activity of Mutant IDH1 or IDH2 (Conversion of αKG to 2HG) by LC-MS/MS
The conversion of αKG to 2HG by mutant IDH1 or IDH2 was monitored by quantifying 2HG using LC-MS/MS. Assays were conducted in a 30-μl volume in a 384-well microplate (Corning 3824) in a buffer containing 50 mm HEPES, 150 mm NaCl, 20 mm MgCl2, 1 mm DTT, and 0.005% Tween 20, pH 7.4. For compound IC50 determinations, substrates were used at concentrations equivalent to 1–2-fold of their Km values (Table 1). Inhibitors were incubated with enzyme and NADPH for 30 min, followed by initiation of the IDH reaction by addition of the substrate αKG. For the time-dependent inhibition study, compound 1 or 2 was preincubated with 10 nm R132H IDH1 for the time indicated in Fig. 1 before the reductive reaction was initiated. For steady-state kinetic analysis of inhibition, the enzyme was also preincubated with the inhibitor for 30 min. Reactions were carried out for 15–20 min at room temperature, followed by quenching with 6% formic acid and 16 μm 13C5-2HG (10 μl). The quenched reaction mixture was diluted 4-fold with acetonitrile and subjected to LC-MS/MS analysis.
TABLE 1.
Biochemical assay conditions for determining compound inhibitory potency using mutant or wild-type IDH
| Enzyme | IDH1 |
IDH2 |
||||||
|---|---|---|---|---|---|---|---|---|
| R132H/R132H | R132C/R132C | R132H/WT | R132H/WT | WT/WT | R140Q/R140Q | R172K/R172K | WT/WT | |
| Reaction direction | Reductive | Reductive | Reductive | Oxidative | Oxidative | Reductive | Reductive | Oxidative |
| Enzyme (nm) | 10 | 4 | 1 | 0.3 | 0.3 | 1.5 | 1.5 | 0.3 |
| NADPH (reductive) or NADP+ (oxidative) (μm) | 2 | 0.8 | 1 | 33 | 50 | 2 | 2 | 10 |
| αKG (reductive) or isocitrate (oxidative), μm | 600 | 150 | 350 | 41 | 25 | 5900 | 4000 | 15 |
FIGURE 1.
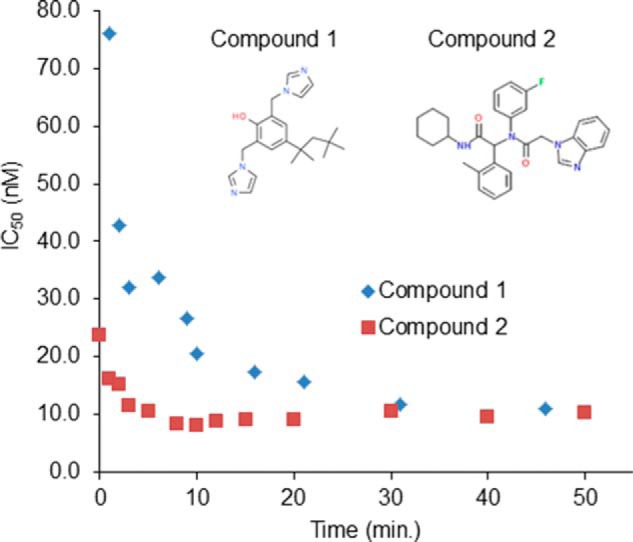
Time-dependent inhibition of R132H IDH1 by compounds 1 and 2. Compounds were preincubated with the enzyme for the time indicated, and IC50 values were determined as described under “Experimental Procedures.”
LC-MS/MS analysis was carried out on a TSQ Vantage triple stage quadrupole mass spectrometer (Thermo Scientific) operated in electrospray negative ion mode. The mass spectrometer was coupled to a 1200 HPLC system (Agilent). Samples were injected using a PAL HTC autosampler (LEAP Technologies). Separation of 2HG from other reaction components was achieved on a weak anion exchange column (20 mm × 2.1 mm, 5 μm; Thermo Biobasic) at a flow rate of 1.0 ml/min. Mobile phase B (0.2% NH4OH in acetonitrile) was held at 90% for 15 s, before ramping mobile phase A (25 mm NH4HCO3 in water) from 10 to 75% over 30 s. Following this, mobile phase B was held at 90% for 18 s. The MRM transitions for 2HG and 13C5-2HG (internal standard) were m/z 147 → 128 and m/z 152 → 134, respectively. Instrument parameters were: spray voltage, 2500 V; vaporizer temperature, 300 °C; sheath gas pressure, 60 liter/h; auxiliary gas pressure, 10 liter/h; capillary temperature, 350 °C; and collision energy, 30 V. Mass spectrometry data were acquired and processed using Xcalibur (Thermo Scientific). The peak area of 2HG was normalized using the peak area of the internal standard 13C5-2HG.
Initial velocity data were fitted to the Michaelis-Menten equation using GraphPad Prism 6 (GraphPad Software) to obtain Vmax and Km. For mechanism of inhibition studies, steady-state kinetics data were globally fitted to various inhibition models (competitive, noncompetitive, and uncompetitive (31)) using SigmaPlot 12.3 (Systat Software, Inc.). Inhibition modality was determined based on the goodness of fits (AICc values) of alternative kinetic models to the data.
Measurement of Oxidative Activity of IDH1 and IDH2 (Conversion of Isocitrate to αKG)
The oxidative conversion of isocitrate to αKG was monitored indirectly in kinetic mode via the concomitant reduction of NADP+ to NADPH, with fluorescence detection of NADPH. Formation of NADPH was monitored using fluorescence measurements (excitation, 340 nm; emission, 460 nm) on an EnVision spectrophotometer (PerkinElmer Life Sciences). Reactions were conducted at room temperature in 384-well plates (Corning 3820), in a final volume of 30 μl. The reaction mixture contained 50 mm HEPES, pH 7.4, 150 mm NaCl, 20 mm MgCl2, 0.005% Tween 20, and 1 mm DTT with 0.3–1 nm enzyme. For determination of kinetic parameters, one substrate concentration was fixed at a saturating value (typically >15-fold Km), whereas the concentration of the other substrate was varied. For IC50 determinations, isocitrate and NADP+ concentrations were fixed at a value of ∼1–2 Km for the enzyme in question (Table 1).
Testing for Reversibility of Inhibition
The reversibility of inhibition by compounds 1 and 2 was determined by preincubating R132H IDH1 and each compound in the presence of NADPH (5 μm R132H IDH1, 10 μm compound, 1 mm NADPH) at room temperature for 1 h, followed by a 500-fold dilution with a solution containing substrate αKG (final concentrations: 10 nm R132H IDH1, 20 nm compound 1 or compound 2, 0.6 mm αKG, 2 μm NADPH in a buffer containing 50 mm HEPES, 150 mm NaCl, 20 mm MgCl2, 1 mm DTT, and 0.005% Tween 20, pH 7.4). The experiment was conducted in a 384-well plate, and the production of 2HG at nine time points in a reaction course of 0–30 min was quantified by the LC-MS/MS assay described above.
Measurement of Reductive Activity of R132H IDH1 as a Function of Mg2+ and Inhibitor Concentration Using a Fluorescence Intensity Assay
The initial velocity of the homodimeric R132H IDH1 reductive reaction was measured using varying concentrations of inhibitors and MgCl2. The reactions were conducted in a 30-μl volume, and reaction mixtures contained 10 nm R132H IDH1, 5 μm NADPH, 4 mm αKG, 50 mm HEPES, pH 7.4, 150 mm NaCl, 0.005% Tween 20, and 1 mm DTT. The concentrations of inhibitors and MgCl2 evaluated are as indicated in Fig. 6. Decrease of NADPH fluorescence was monitored (excitation/emission, 340/460 nm) for 30 min at room temperature. The steady-state kinetic data were globally fitted to various inhibition models (competitive, noncompetitive, and uncompetitive (31)) using SigmaPlot 12.3. Inhibition modality was determined based on the goodness of fits (AICc values) of alternative kinetic models to the data.
FIGURE 6.

Both compounds 1 and 2 inhibit R132H IDH1 competitively with respect to Mg2+. Alternative inhibition models were fitted to steady-state inhibition data and are illustrated with Lineweaver-Burk plots. The fits to the competitive model (left panel) are the best, as judged by comparison of AICc. The unit of inhibitor concentrations shown in the insets is nanomolar.
Determination of Ligand/Enzyme Interaction Using Surface Plasmon Resonance (SPR)
Avi-tagged homodimeric IDH1 mutants R132H and R132C were purified using the same procedure as the non-Avi-tagged variants and biotinylated using biotin ligase (Avidity). Wild-type IDH1 was chemically biotinylated to a low level (no more than 1:1 modification stoichiometry by intact protein mass spectrometry) with EZ-Link Sulfo-NHS-LC-LC-Biotin (Pierce) and immobilized to streptavidin sensor chips (GE Healthcare) according to the manufacturer's instructions. SPR experiments were performed on a Biacore T200 instrument (GE Healthcare) using a running buffer containing 50 mm Tris, pH 7.4, 150 mm NaCl, 10 mm MgCl2, 1 mm tris(2-carboxyethyl)phosphine, 0.005% surfactant P20 (GE HealthCare), 3% DMSO. Biotinylated mutants R132H and R132C and WT enzymes were immobilized onto the different flow cells on a streptavidin sensor chip (GE Healthcare) preconditioned with 40 mm NaOH and 1 m NaCl. To obtain NADPH-free enzyme (NADPH was co-purified with R132H IDH1), 10 mm αKG in the running buffer was injected over a sensor chip immobilized with R132H for 1 min, followed by washing of the chip with the running buffer (an independent experiment showed that co-purified NADPH was completely removed by treating the enzyme with αKG followed by dialysis, as indicated by an fluorescence emission spectrum showing that excitation of protein tryptophan at 290 nm was no longer associated with fluorescence emission at 426 nm because of NADPH fluorescence). Instrument parameters were: flow rate = 50 μl/min; contact (association) time = 75 s; and dissociation time = 75 s. For studying the interaction of inhibitors with enzymes bound to NADPH, 100 μm of NADPH was included in the running buffer.
Thermodynamic studies were conducted in the temperature range of 5–40 °C, taking measurements at 5 °C intervals. Thermodynamic parameters were obtained by fitting equilibrium binding constants (KD) determined at different temperatures to the van't Hoff equation (BiaEvaluation software v 1.0; GE Healthcare). All other binding studies were conducted at room temperature. Competitive binding experiments were carried out with one inhibitor present at saturating concentration in the running buffer and the other inhibitor presented to the bound enzyme at varying concentrations from a sample plate. SPR data were analyzed using BiaEvaluation software (GE Healthcare).
Determination of the Interaction between Ligand and Enzyme by Differential Scanning Fluorimetry
Inhibitors (0.2 mm) were combined with R132H IDH1 (0.5 mg/ml) and 10× SYPRO Orange (S6650; Invitrogen) in a buffer containing 100 mm HEPES (pH 7.0) and 100 mm NaCl. The thermal stability of the protein in the reaction mixture (10 μl) was measured using a CFX 96 RT-PCR instrument (Bio-Rad), employing a temperature gradient from 20 °C to 95 °C at an increment of 1 °C/min. Melting temperature values (Tm) were determined from the melting curves either using the instrument software or by fitting the initial fluorescence increase to a Boltzmann function (Y = Bottom + (Top − Bottom)/(1 + exp ((V50 − X)/Slope)), where Y is the fluorescence signal, and X is the logarithm of the temperature in Kelvin).
Construction of an R132H IDH1-expressing Cell Line and Measurement of the Effects of IDH1 Inhibitors on 2HG Production
A cell line with inducible IDH1 mutant expression was generated by stably transfecting Flp-In-T-Rex-293 cells (Invitrogen) with the plasmid DNA encoding R132H IDH1, followed by ZeocinTM selection. For cell-based 2HG measurements, 50,000 IDH1 mutant-expressing cells were plated in 95 μl/well of growth media (DMEM, Invitrogen; 10% Tet-free FBS, Clontech) in the presence or absence of doxycycline (Clontech, 1 μg/ml) and allowed to adhere overnight. Compounds were added the following day (3-fold dilution from 50 μm for compound 1 and 3-fold dilution from 10 μm for compound 2; final DMSO, 0.5%), and the cells were incubated for an additional 24 h. An aliquot (20 μl) of the conditioned medium was then harvested and analyzed for 2HG using the LC-MS/MS assay described above for recombinant IDH proteins. Cell viability was assessed using the CellTiterGlo kit (Promega) according to the manufacturer's instructions.
Crystallization, Structure Determination, and Structure Refinement for Compound 1 in Complex with Homodimeric R132H IDH1
R132H IDH1 protein for structural and differential scanning fluorimetry studies was expressed and purified according to the method of Dang et al. (12). For crystallization in the absence of inhibitor, R132H IDH1 was concentrated to 12 mg/ml and was incubated overnight in the presence of NADPH (5 mm). Crystals were obtained using the hanging drop vapor diffusion technique, using 22% PEG5000MME, 100 mm Tris, pH 6.5, and 220 mm ammonium sulfate. 25% glycerol was added as a cryo-protecting agent prior to freezing.
For co-crystallization of R132H IDH1 and compound 1, the protein was concentrated to 8 mg/ml in 50 mm Tris, pH 7.5, 200 mm NaCl, 5 mm DTT, and 10% glycerol and incubated with 1 mm compound 1 overnight. Crystals were grown by the hanging drop method at 19 °C in 26% PEG8K, 100 mm Tris, pH 8.0, and 200 mm malonate. 15% glycerol was added prior to freezing.
For the R132H IDH1/NADPH structure, the diffraction data were collected at the APS synchrotron to a resolution of 2.07 Å. The structure was determined via molecular replacement, using the published wild-type 1T09 structure (22) as the model. For the R132H IDH1/compound 1 structure, diffraction data were collected at the Soleil synchrotron to a resolution of 1.88 Å. The structure was determined via molecular replacement, using the published 3MAR structure (23) as the model. In both cases, there are two molecules of R132H IDH1 in the asymmetric unit, each one associated with a NADPH molecule. Subsequent refinement was carried out to an Rfree of 22.4% and 21.1% respectively using Buster (BUSTER-TNT 2.11.5; GlobalPhasing Ltd., Cambridge, UK) and COOT (32). Crystallization statistics are listed in Table 2.
TABLE 2.
Data collection and refinement statistics for R132H IDH1/NADPH in complex with compound 1
The data in parentheses correspond to the highest resolution shells. To calculate Rfree, 5% of the reflections were excluded from the refinement. Rsym is defined as Rsym = Σhkl Σi|Ii(hkl) − <I(hkl)>|/Σhkl Σi Ii(hkl). A final quality check was done with MolProbity (37). The atomic coordinates have been deposited in the Protein Data Bank under codes 4UMX for the compound 1-bound structure and 4UMY for the compound-unbound structure.
| R132H IDH1/NADPH | R132H IDH1/NADPH/compound 1 | |
|---|---|---|
| Data collection | ||
| Space group | P43212 | P43212 |
| Cell dimensions | ||
| a, b, c (Å) | 82.68, 82.68, 300.37 | 81.41, 81.41, 305.92 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50.0-2.07 (2.14-2.07) | 305.9-1.88 (1.98-1.88) |
| Rsym or Rmerge | 6.5 (59.3) | 10.8 (66.3) |
| I/σI | 31.3 (4.4) | 17.8 (3.3) |
| Completeness (%) | 99.4 (99.1) | 100 (100) |
| Redundancy | 9.7 (9.9) | 14.0 (9.8) |
| Refinement | ||
| Resolution (Å) | 2.07 | 1.88 |
| No. of reflections | 64186 | 84917 |
| Rwork/Rfree | 22.4/20.0 | 21.1/18.3 |
| No. of atoms | ||
| Protein | 6130 | 6479 |
| Ligand/ion | 122 | 129 |
| Water | 286 | 766 |
| B-factors | ||
| Protein | 52.0 | 31.9 |
| Ligand/ion | 56.0 | 34.7 |
| Water | 54.8 | 41.3 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.010 | 0.010 |
| Bond angles (°) | 1.04 | 1.02 |
RESULTS
A high throughput screening campaign against homodimeric R132H IDH1 identified a potent inhibitor, bis-imidazole phenol (IC50 13 ± 5 nm), referred to here as compound 1 (Fig. 1). Its inhibition of R132H IDH1 is time-dependent, as shown by dependence of IC50 values on the preincubation time (Fig. 1), but fully reversible (data not shown). Compound 1 is selective for IDH1 mutant enzymes over wild-type IDH1 and mutant or wild-type IDH2 (Table 3). It induces a substantial thermal stabilization of R132H IDH1 (+2.4 °C) as measured using differential scanning fluorimetry, consistent with high affinity binding to the target. This inhibitor is also active against mutant IDH1 in cells; it dose-dependently inhibits 2HG production from an engineered cell line that expresses R132H IDH1 and produces 2HG at a high level (a ∼30-fold increase compared with WT) (Fig. 2). Compound 1 does not affect cell viability at concentrations up to 25 μm (determined by luminescence cell viability assay; data not shown). The potent and selective inhibitory activity of compound 1 against IDH1 mutant enzymes and its promising cellular activity prompted us to investigate the mechanistic and structural details of its interactions with mutant IDH1.
TABLE 3.
IC50 of compound 1 and compound 2 against wild-type and mutant IDH1 and IDH2
H/H, R132H/R132H; C/C, R132C/R132C; H/W, R132H/WT; W/W, WT/WT; K/K, R172K/R172K; Q/Q, R140Q/R140Q; red., reductive reaction of IDH (converting αKG to 2 HG); oxi., oxidative reaction of IDH (converting isocitrate to αKG); Cpd 1, compound 1; Cpd 2, compound 2.
| IC50 |
||||||||
|---|---|---|---|---|---|---|---|---|
| IDH1 |
IDH2 |
|||||||
| H/H (red.) | C/C (red.) | H/W (red.) | H/W (oxi.) | W/W (oxi.) | K/K (red.) | Q/Q (red.) | W/W (oxi.) | |
| μm | μm | |||||||
| Cpd 1 | 0.011 | 0.259 | 0.048 | 0.180 | >10 | >10 | >10 | >10 |
| Cpd 2 | 0.042 | 0.004 | 0.080 | 0.143 | 1.998 | >10 | >10 | >10 |
FIGURE 2.

Compounds 1 and 2 inhibit 2HG production in R132H IDH1-expressing HEK-293 cells. Cells were treated with compound or DMSO (final DMSO concentration, 0.5%) for 24 h before 20 μl of cell culture supernatant was harvested for 2HG quantitation by LC-MS/MS. Percentage of 2HG inhibition: %I = 100 × (1 − (amount of 2HG in compound treated sample/amount of 2HG in DMSO-treated sample)).
We used steady-state enzyme kinetics to investigate whether compound 1 might compete with R132H IDH1 substrates. In parallel with compound 1, we also characterized compound 2 (Fig. 1), a published inhibitor representative of a series with a scaffold prepared using the Ugi reaction (27). The active enantiomer of this inhibitor (enantiomer 1) is 2–3-fold more potent than its racemic mixture against purified enzyme or R132H IDH1 expression cells (see notes in Fig. 4A). The inactive enantiomer of compound 2 (enantiomer 2) does not inhibit either the purified enzyme or R132H IDH1-expressing cells at concentrations up to 10 μm. Compound 2 is comparably potent to compound 1 against the purified enzyme (Table 3) and potently inhibits cellular 2HG production (Fig. 2). For both compound 1 and compound 2, enzyme and inhibitor were preincubated for 30 min before initiating the enzymatic reaction so that the time-dependent pre-equilibration of enzyme and inhibitor (Fig. 1) would not contribute to the observed kinetics. We evaluated the effect of substrate (αKG and NADPH) concentrations (four sets: 2× Km or 100× Km for NADPH and 1× Km or 20× Km for αKG) on IC50 values of the inhibitors against R132H IDH1. These IC50 values are independent of substrate concentration (IC50 range: 0.006–0.011 μm for compound 1 and 0.037–0.073 μm for compound 2), suggesting that both inhibitors inhibit noncompetitively. To test this hypothesis, we conducted a steady-state kinetic study in which the initial velocities were measured while varying the concentrations of inhibitor and substrates. The data were fitted to competitive, uncompetitive, and noncompetitive kinetic models (Fig. 3), with the best fits coming from the noncompetitive inhibition model (data for compound 2 is not shown).
FIGURE 4.

A, compounds 1 and 2 bind to both free and NADPH-bound R132H IDH1 enzyme forms and bind selectively to one enantiomer of compound 2. SPR binding isotherms were measured for compound 1 (top panel), compound 2 (middle panel), and the two enantiomers of compound 2 (bottom panel). Saturable binding of only the active enantiomer (enantiomer 1) but not the inactive enantiomer (enantiomer 2) indicates that inhibitor binding to R132H IDH1 immobilized on SPR sensor chip is due to a specific interaction. The data were fitted to a steady-state affinity model. For the purpose of comparison, biochemical IC50 and cellular IC50 (2HG inhibition) of each compound are shown on the top of each SPR binding isotherm. B, immobilization of R132H IDH1 onto the SPR chip surface does not compromise binding of its native ligands NADPH and NADP+. Binding and dissociation time traces (left panels) are fitted to a 1:1 kinetic binding model (original data and fits are shown in colors and in black, respectively), and binding isotherms obtained from steady-state response levels (right panels); response units were determined at 4 s before the end of each compound injection) are independently fitted to the Langmuir equation. The KD values calculated based on kon and koff from the fits of the binding kinetics are consistent with the values obtained by fitting the steady-state binding data.
FIGURE 3.

Compound 1 inhibits R132H IDH1 noncompetitively with respect to NADPH and αKG, respectively. Alternative inhibition models were fitted to steady-state inhibition data for compound 1. Inhibition data with respect to NADPH are illustrated with Michaelis-Menten plots (top row), whereas data with respect to αKG are illustrated with Lineweaver-Burk plots (bottom row). The fits to the noncompetitive model (far-right panel) are the best, as judged by comparison of AICc. The unit of inhibitor concentrations shown in the insets is micromolar.
As an additional test for noncompetitive inhibition, we used SPR to characterize inhibitor binding to apo- and NADPH-bound R132H IDH1. Steady-state kinetic studies on the order of substrate binding indicated that mutant forms of IDH1 follow an ordered, sequential kinetic mechanism (19, 33); therefore, both apo- and NADPH-bound forms of the enzyme will be populated during the catalytic cycle and may bind inhibitors. In SPR experiments, both compounds bind to apo- and NADPH-bound enzyme forms with similar affinities (Fig. 4A), as predicted for noncompetitive inhibition; reversible NADPH and NADP+ (a substrate and a product of the reductive reaction catalyzed by R132H IDH1) binding to apo R132H IDH1 on the SPR chip is directly observed as well (Fig. 4B), indicating that the protein at the chip surface is in a native-like state in both apo- and NADPH-bound forms. SPR experiments also indicate that this binding is also observed for the R132C mutant, but not for wt IDH1 (data not shown). These results thus suggest that the binding observed in the SPR experiments is related to enzyme inhibition; both binding and inhibition are selective for the mutant enzymes. Taken together, these data suggest that compounds 1 and 2 bind to allosteric site(s). Notably, compounds structurally very similar to compound 2 (designated as inhibitor 1 and ML309) have previously been reported to be competitive inhibitors with respect to αKG (17, 19).
As a direct test of the hypothesis that compound 1 binds to an allosteric site, we solved crystal structures of the NADPH-bound form of R132H IDH1 in the absence and presence of compound 1. The structure of R132H IDH1/NADPH in the absence of inhibitor was determined to 2.13 Å. Similar to the published 3MAR structure (3.4 Å) (23), this structure displays an open conformation with the metal-binding regulatory segment unfolded (i.e. the metal binding site is not assembled). The structure of R132H IDH1/NADPH in complex with compound 1 was determined to 1.9 Å resolution. Like the unbound enzyme, the inhibitor-bound enzyme adopts an open conformation. Compared with R132H IDH1/NADPH, the compound 1-bound enzyme exhibits pronounced movement of one protomer with respect to the other (Fig. 5A). The compound 1-bound enzyme also exhibits additional density at the interface between the protomers. This density (Fig. 5B) corresponds in part to an ordering of residues 276–286 (the metal-binding regulatory segment) of the A protomer, whereas the equivalent residues of the B protomer remain disordered. Compound 1 binds to the A protomer near the dimer interface (Fig. 5C), consistent with an allosteric mode of inhibition. Residues 278–286 are found as a loop and a small helix, with Asp279 making a hydrogen bond with the hydroxyl of compound 1 and Val281 stacking against its phenyl ring. Surprisingly, no other productive interactions are observed between the protein and the rest of the ligand. In the compound 1-bound structure, residues 132–141 (including Tyr139, a residue believed to play a vital role in catalysis (23)) are found far from the catalytic active site in both protomers (similar to what is observed in other αKG-free structures). This structural arrangement differs from the conformation adopted in the presence of substrate, where these residues make up part of the active site, and the hydroxyl group of Tyr139 is oriented toward the substrate binding site (23) (Fig. 5D).
FIGURE 5.

X-ray structure of compound 1 with homodimeric R132H IDH1. A, superimposition of structures of R132H IDH1/NADP+ dimer (blue/violet) and R132H IDH1/NADP+/compound 1 dimer (cyan/green), with the A protomers aligned. A large movement of the B protomer can be seen upon binding of compound 1 (black arrow). B, electron density maps of NADPH and compound 1. C, close-up of the dimer interface in the R132H IDH1/NADPH/compound 1 structure (A protomer in cyan and B protomer in green). D, the conformation of the clasp domain (circled) and segment 1 in the compound 1-bound structure and 3INM. Segment 1 (residue 132–141) in the compound 1-bound structure is disordered (green and cyan for protomer A and protomer B, respectively). Tyr139, a residue believed to play a vital role in catalysis, is no longer oriented toward the active site as it is in the substrate-bound structure (magenta).
The direct interaction of compound 1 with Asp279 (with Asp275 and Asp252′, one of the three aspartate residues that chelate the catalytically essential divalent ion (12)) in the structure suggests that compound 1 may compete with Mg2+ for interaction with Asp279. To investigate this possibility, we conducted a steady-state kinetic study in which initial velocities were measured as a function of compound 1 and Mg2+ concentrations. Kinetic data in the absence of inhibitor enabled the measurement of a Km value for Mg2+ in the reductive reaction of R132H IDH1 (6250 μm, compared with a value of 19 μm measured for the oxidative reaction of the WT enzyme). Fitting the inhibition data to competitive, uncompetitive, and noncompetitive inhibition models shows that the competitive model provides the best fit (Fig. 6), confirming that binding of compound 1 to the enzyme disrupts its ability to bind the catalytically essential divalent ion. In addition, compound 1 does not bind detectably to R132H IDH1 in the presence of NADH, αKG, and magnesium (data not shown), conditions that mimic turnover conditions (NADH binds to the enzyme but does not support catalysis). This observation suggests that compound 1 binding prevents the enzyme from achieving the catalytically competent conformation that magnesium binding supports. It is important to note that the inhibitor is present at concentrations several orders of magnitude below the concentration of Mg2+, precluding direct chelation of Mg2+ as a possible mechanism of inhibition.
We have been unsuccessful in obtaining a crystal structure for compound 2, leaving an open question of whether compound 2 and compound 1 share a common allosteric binding site. To address this question, we conducted a competitive binding study of compound 2 with compound 1 using SPR. We found that in the presence of a saturating concentration of one inhibitor, the binding of the other inhibitor is weakened (i.e. it no longer reaches binding saturation in the concentration range tested), as would be expected if the two compounds bind to the enzyme by competing for the same binding pocket at the dimer interface. Consistent with this conclusion, enantiomer 1 of compound 2, like compound 1, inhibits R132H IDH1 competitively with respect to Mg2+ (Fig. 6); this result indicates that compound 2 also prevents the enzyme from binding to divalent ion, possibly via an interaction with Asp279. Together, these studies suggest that compounds 1 and 2 may share a common allosteric binding site.
As a complementary method for comparing the binding modes of compound 1 and compound 2, we measured the temperature dependence of binding for these compounds using SPR. Although compounds 1 and 2 differ structurally, their thermodynamic binding profiles are similar: entropy of binding makes a favorable contribution (−TΔS: −53 and −75 KJ/mol for compounds 1 and 2, respectively) to the overall binding free energy (ΔG: −34 KJ/mol for both compounds 1 and 2), whereas enthalpy makes an unfavorable contribution (ΔH: 18 and 41 KJ/mol for compounds 1 and 2, respectively). These results suggest that the binding of both inhibitors is mostly driven by increased hydrophobic interactions in the complex and is consistent with a similar mode of binding for the two inhibitors.
The observation of an allosteric binding site at the dimer interface raises the question of how binding of a ligand at this site will affect the heterodimeric mutant/wt IDH1 variant, proposed to be the key species mediating 2HG production in cellular contexts (34). Consistent with previously published results (35), the wt and mutant subunits in a dimer appear to operate independently of each other. Although all homodimeric IDH mutants showed very little enzymatic activity in converting isocitrate to αKG (oxidative reaction) compared with their activity in converting αKG to 2HG (neomorphic reductive reaction) (data not shown), the heterodimer R132H/WT IDH1 possesses comparable catalytic efficiency for both reactions (kcat/Km = 0.77 μm−1 s−1 for the oxidative reaction; kcat/Km = 1.02 μm−1 s−1 for the neomorphic reductive reaction). Compounds 1 and 2 inhibit both the neomorphic reductive activity of the heterodimer R132H/WT IDH1 and the oxidative activity of the enzyme with less than 2- and 4-fold differences in potency, respectively (Table 3), suggesting that binding of the inhibitors locks both subunits of the heterodimer in inactive conformations. This observation is thus consistent with the hypothesis that binding of compound 1 prevents binding of the catalytic metal ion in both subunits of an IDH1 dimer.
DISCUSSION
Steady-state enzyme kinetic studies support an allosteric mode of IDH1 inhibition for both compounds 1 and 2 (here defining an allosteric site as a site distinct from αKG and NADPH binding sites). Inhibition of R132H IDH1 by these compounds is noncompetitive with respect to both substrates of the reaction (αKG and NADPH) (Fig. 3) but competitive with respect to Mg2+ (Fig. 6). One possibility consistent with the steady-state kinetic data is that the inhibitor binds competitively with respect to magnesium in one or more of the apo-, NADPH-bound, and αKG/NADPH-bound forms. This mechanism (Fig. 7) predicts the observed noncompetitive/mixed inhibition with respect to NADPH and αKG; if the inhibitor could not bind to either apo-, NADPH-, and/or NADPH/αKG-bound forms, competitive or uncompetitive inhibition with respect to at least one of the substrates would be expected. This mechanism also predicts the observed competitive inhibition with respect to Mg2+, because both the inhibitor and magnesium ion compete for the enzyme/αKG/NADPH complex, but only the magnesium-bound complex is catalytically competent. The steady-state kinetic studies therefore suggest that compounds 1 and 2 bind to a site on the enzyme that is distinct from the substrate binding sites and whose occupancy prevents simultaneous binding by magnesium.
FIGURE 7.
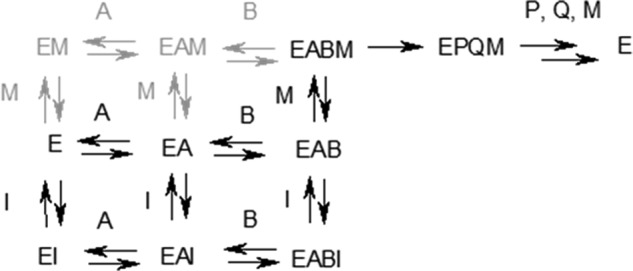
Proposed kinetic mechanism for inhibition of the reductive reaction of R132H IDH1. A, NADPH; B, αKG; M, Mg2+; I, compound 1 or compound 2; P, 2HG; Q, NADP+. Species in black have either been detected directly via SPR or are required to accommodate steady-state kinetics results; the involvement of species in gray is neither required nor ruled out by experimental results to date.
Biophysical studies also support the hypothesis that compounds 1 and 2 each bind to the same discrete allosteric site. The observation of saturable and approximately equipotent binding to apo- and NADPH-bound enzyme (Fig. 4A, top and middle panels) demonstrates that inhibitor binding occurs outside of the NADPH-binding site. In addition, preincubation with compound 2 prevents binding of compound 1 and vice versa. The simplest interpretation for this result is that the two inhibitors bind to the same physical site on the enzyme.
One complication in the interpretation of the biophysical studies is the discrepancy between the dissociation constant observed in SPR experiments (∼10−7–10−6 m; Fig. 4A) and the kinetic inhibition constants (∼10−8 m; Table 3). This discrepancy notwithstanding, the conformation of the enzyme required for co-factor binding appears to be well preserved after the protein is immobilized onto SPR sensor chip: binding of NADPH or NADP+ (the product of the reductive reaction) is well fitted to a 1:1 binding model (Fig. 4B), and the KD value for NADPH measured by SPR agrees well with a value previously obtained with the protein in free solution (33). The binding of inhibitor to the immobilized protein is due to a specific interaction, as demonstrated by the saturable binding of only the active enantiomer of compound 2 (enantiomer 1) but not its inactive enantiomer (enantiomer 2) (Fig. 4A, bottom panel). The time dependence of inhibitory potency (Fig. 1) may contribute to the differences between dissociation constants and inhibition constants. For both of the inhibitors, the highest inhibitory potency requires preincubation with the enzyme on the time scale of minutes (half-times of 2–3 min). However, in the SPR experiment, dissociation is measured after an association phase of just over 1 min, which may bias the measurement toward the observation of lower affinity binding modes. Hindered solvent access to the dimer interface of surface-bound IDH1 compared with IDH1 in solution may also be a contributing factor if it prevents the binding reaction from reaching equilibrium on the time scale of the experiment. These factors may also contribute to the complexity of the observed SPR sensorgrams, which are not well fitted to a 1:1 kinetic model (data not shown). Finally, differential affinity for magnesium ion among the apo- and/or substrate/product-bound forms of the enzyme may also contribute to discrepancies between dissociation constants and IC50 values. Although the dissociation constants are measured for an individual enzyme form in the presence of competing magnesium ion, the IC50 reflects the binding of inhibitors to multiple distinct enzyme forms for which the inhibitor may compete more or less effectively with magnesium.
The structure solved from co-crystals of R132H IDH1 and compound 1 indicates that compound 1 binds at the interface between the two protomers, in a site that does not overlap with the substrate binding sites but does include residues that would ordinarily bind magnesium. In the presence of compound 1, the regulatory helix of the A protomer is partially unfolded and oriented so that Asp279 (which, along with Asp275, is a Mg2+-chelating residue found in this helix) does not face the active site. Compound 1 actually interacts directly with Asp279, forming a hydrogen bond. Helix 251–261 in protomer B, which contains Asp252′ (the third Mg2+ chelating residue (12)), is also displaced as a result of compound binding. As a result of these conformational changes, the network of residues that orient Mg2+ for catalysis in the active site is completely disrupted. One surprising aspect of the structure is that compound 1 is bound in a shallow pocket and makes few contacts with the protein, in contrast to what might be expected for a potent (IC50 ∼10−8 m) inhibitor. These contacts may be strong enough to account for the observed binding, or compound 1 may adopt a subtly different binding pose in solution.
The disruption of Mg2+ binding sites in both protomers may help explain the observation that binding of compound 1 (and presumably compound 2) at the dimer interface inhibits both active sites of the dimer (as shown by their activity against mutant/wt heteromers), even though the structure shows direct engagement only with protomer A. Another factor that may contribute to inhibition of catalytic activity in both protomers is displacement of residues 132–141 (referred to as segment 1 in the 3MAR structure (23)) from the active site. These residues are disordered in the substrate-free/NADPH-bound structure but ordered and located near the catalytic active site in substrate-bound structures (23); they therefore appear to constitute an important functional element of the active site. In the presence of compound 1, Tyr139 (which appears to be required for efficient catalysis (23)) is no longer oriented toward the active site in either protomer (Fig. 5D). In protomer A, the regulatory segment that interacts directly with the inhibitor also blocks access of segment 1 to the active site, possibly explaining the reorientation of Tyr139 in this protomer. Structural coupling between the two subunits may explain why Tyr139 is also excluded from the active site in protomer B. The clasp domain in the compound 1-bound structure (which interlocks the two subunits and is proximal to Segment 1) is displaced compared with its position in the substrate-bound structure (3INM), which may constrain segment 1 in protomer B to adopt a conformation similar to that in protomer A.
Although the structural consequences of inhibitor binding to the divalent cation binding residue Asp279 are complex, the direct contact between the inhibitor and this residue appears to be the primary driver for inhibitory activity. This unusual mechanism of action may contribute to the observed selectivity of the inhibitors for mutant IDH1 over the wild-type. The markedly higher (>300-fold) Km for magnesium ion observed for the R132H IDH1 reductive reaction compared with that observed for the oxidative reaction of the wild-type enzyme indicates that Mg2+ will be a much more effective competitor for the wild-type divalent cation site than for the corresponding mutant site. In a cellular environment, the free magnesium ion concentration is likely to be ∼10−4–10−3 m (36), much greater than the Km for the wild-type oxidative reaction but much less than the Km for the mutant reductive reaction. Under such conditions, inhibition of wild-type homodimers would be expected to be negligible, whereas inhibition of mutant-containing dimers would be expected to be close to maximally potent. Although the IDH2 allosteric inhibitor AGI-6780 almost certainly has a different mechanism of action from the IDH1 inhibitors described here (calcium ions are observed in the divalent cation binding site when AGI-6780 is bound to IDH2 (18)), it may also prevent the divalent cation binding site from achieving a catalytically competent conformation: AGI-6780 forms a hydrogen bond with Gln316 in the metal binding regulatory segment (18), which may prevent it from undergoing any substantial conformational change during the catalytic reaction (23).
The inhibitory mechanism presented here suggests that the disorganization of the active site induced by oncogenic IDH mutations not only favors their unusual neomorphic activity but also imposes a neomorphic vulnerability to pharmacological inhibitors directed toward the metal-binding regulatory segment. Structural plasticity caused by the mutations may allow this segment to adopt new, catalytically inactive conformations that are stabilized by drug-like small molecules in competition with the catalytically active conformation stabilized by Mg2+. Inhibition by binding of drug-like molecules to divalent cation sites may also be a feasible strategy for other metalloenzyme targets. Divalent cation sites with sufficient flexibility (either intrinsic, as for natively unfolded metalloproteins, or caused by a pathogenic mutation) may be able to adopt conformations accommodating small molecule ligands that differ substantially in structure and charge from the divalent metal ions that bind these sites physiologically.
Acknowledgments
We thank our colleagues Dr. Oleg Schmidt-Kittler and Dr. Carlos Garcia-Echeverria for helpful discussions.
The atomic coordinates and structure factors (codes 4UMX and 4UMY) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- αKG
- α-ketoglutarate
- 2HG
- d-2-hydroxyglutaric acid
- IDH
- isocitrate dehydrogenase
- NTA
- nitrilotriacetic acid
- SPR
- surface plasmon resonance.
REFERENCES
- 1. Tennant D. A., Durán R. V., Gottlieb E. (2010) Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 10, 267–277 [DOI] [PubMed] [Google Scholar]
- 2. Levine A. J., Puzio-Kuter A. M. (2010) The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330, 1340–1344 [DOI] [PubMed] [Google Scholar]
- 3. Reitman Z. J., Yan H. (2010) Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 102, 932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dang L., Jin S., Su S. M. (2010) IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med. 16, 387–397 [DOI] [PubMed] [Google Scholar]
- 5. Geisbrecht B. V., Gould S. J. (1999) The human PICD gene encodes a cytoplasmic and peroxisomal NADP+-dependent isocitrate dehydrogenase. J. Biol. Chem. 274, 30527–30533 [DOI] [PubMed] [Google Scholar]
- 6. Parsons D. W., Jones S., Zhang X., Lin J. C., Leary R. J., Angenendt P., Mankoo P., Carter H., Siu I. M., Gallia G. L., Olivi A., McLendon R., Rasheed B. A., Keir S., Nikolskaya T., Nikolsky Y., Busam D. A., Tekleab H., Diaz L. A., Jr., Hartigan J., Smith D. R., Strausberg R. L., Marie S. K., Shinjo S. M., Yan H., Riggins G. J., Bigner D. D., Karchin R., Papadopoulos N., Parmigiani G., Vogelstein B., Velculescu V. E., Kinzler K. W. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan H., Parsons D. W., Jin G., McLendon R., Rasheed B. A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G. J., Friedman H., Friedman A., Reardon D., Herndon J., Kinzler K. W., Velculescu V. E., Vogelstein B., Bigner D. D. (2009) IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wagner K., Damm F., Göhring G., Görlich K., Heuser M., Schäfer I., Ottmann O., Lübbert M., Heit W., Kanz L., Schlimok G., Raghavachar A. A., Fiedler W., Kirchner H. H., Brugger W., Zucknick M., Schlegelberger B., Heil G., Ganser A., Krauter J. (2010) Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J. Clin. Oncol. 28, 2356–2364 [DOI] [PubMed] [Google Scholar]
- 9. Paschka P., Schlenk R. F., Gaidzik V. I., Habdank M., Krönke J., Bullinger L., Späth D., Kayser S., Zucknick M., Götze K., Horst H. A., Germing U., Döhner H., Döhner K. (2010) IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 28, 3636–3643 [DOI] [PubMed] [Google Scholar]
- 10. Thol F., Damm F., Wagner K., Göhring G., Schlegelberger B., Hoelzer D., Lübbert M., Heit W., Kanz L., Schlimok G., Raghavachar A., Fiedler W., Kirchner H., Heil G., Heuser M., Krauter J., Ganser A. (2010) Prognostic impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood 116, 614–616 [DOI] [PubMed] [Google Scholar]
- 11. Zhao S., Lin Y., Xu W., Jiang W., Zha Z., Wang P., Yu W., Li Z., Gong L., Peng Y., Ding J., Lei Q., Guan K. L., Xiong Y. (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 324, 261–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dang L., White D. W., Gross S., Bennett B. D., Bittinger M. A., Driggers E. M., Fantin V. R., Jang H. G., Jin S., Keenan M. C., Marks K. M., Prins R. M., Ward P. S., Yen K. E., Liau L. M., Rabinowitz J. D., Cantley L. C., Thompson C. B., Vander Heiden M. G., Su S. M. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Figueroa M. E., Abdel-Wahab O., Lu C., Ward P. S., Patel J., Shih A., Li Y., Bhagwat N., Vasanthakumar A., Fernandez H. F., Tallman M. S., Sun Z., Wolniak K., Peeters J. K., Liu W., Choe S. E., Fantin V. R., Paietta E., Löwenberg B., Licht J. D., Godley L. A., Delwel R., Valk P. J., Thompson C. B., Levine R. L., Melnick A. (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S. H., Ito S., Yang C., Xiao M. T., Liu L. X., Jiang W. Q., Liu J., Zhang J. Y., Wang B., Frye S., Zhang Y., Xu Y. H., Lei Q. Y., Guan K. L., Zhao S. M., Xiong Y. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chowdhury R., Yeoh K. K., Tian Y. M., Hillringhaus L., Bagg E. A., Rose N. R., Leung I. K., Li X. S., Woon E. C., Yang M., McDonough M. A., King O. N., Clifton I. J., Klose R. J., Claridge T. D., Ratcliffe P. J., Schofield C. J., Kawamura A. (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koivunen P., Lee S., Duncan C. G., Lopez G., Lu G., Ramkissoon S., Losman J. A., Joensuu P., Bergmann U., Gross S., Travins J., Weiss S., Looper R., Ligon K. L., Verhaak R. G., Yan H., Kaelin W. G., Jr. (2012) Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Popovici-Muller J., Saunders J. O., Salituro F. G., Travins J. M., Yan S., Zhao F., Gross S., Dang L., Yen K. E., Yang H., Straley K. S., Jin S., Kunii K., Fantin V. R., Zhang S., Pan Q., Shi D., Biller S. A., Su S. M. (2012) Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med. Chem. Lett. 3, 850–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang F., Travins J., DeLaBarre B., Penard-Lacronique V., Schalm S., Hansen E., Straley K., Kernytsky A., Liu W., Gliser C., Yang H., Gross S., Artin E., Saada V., Mylonas E., Quivoron C., Popovici-Muller J., Saunders J. O., Salituro F. G., Yan S., Murray S., Wei W., Gao Y., Dang L., Dorsch M., Agresta S., Schenkein D. P., Biller S. A., Su S. M., de Botton S., Yen K. E. (2013) Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626 [DOI] [PubMed] [Google Scholar]
- 19. Davis M. I., Gross S., Shen M., Straley K. S., Pragani R., Lea W. A., Popovici-Muller J., DeLaBarre B., Artin E., Thorne N., Auld D. S., Li Z., Dang L., Boxer M. B., Simeonov A. (2014) Biochemical, cellular and biophysical characterization of a potent inhibitor of mutant isocitrate dehydrogenase IDH1. J. Biol. Chem. 289, 13717–13725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rohle D., Popovici-Muller J., Palaskas N., Turcan S., Grommes C., Campos C., Tsoi J., Clark O., Oldrini B., Komisopoulou E., Kunii K., Pedraza A., Schalm S., Silverman L., Miller A., Wang F., Yang H., Chen Y., Kernytsky A., Rosenblum M. K., Liu W., Biller S. A., Su S. M., Brennan C. W., Chan T. A., Graeber T. G., Yen K. E., Mellinghoff I. K. (2013) An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zheng B., Yao Y., Liu Z., Deng L., Anglin J. L., Jiang H., Prasad B. V., Song Y. (2013) Crystallographic investigation and selective inhibition of mutant isocitrate dehydrogenase. ACS Med. Chem. Lett. 4, 542–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu X., Zhao J., Xu Z., Peng B., Huang Q., Arnold E., Ding J. (2004) Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 279, 33946–33957 [DOI] [PubMed] [Google Scholar]
- 23. Yang B., Zhong C., Peng Y., Lai Z., Ding J. (2010) Molecular mechanisms of “off-on switch” of activities of human IDH1 by tumor-associated mutation R132H. Cell Res. 20, 1188–1200 [DOI] [PubMed] [Google Scholar]
- 24. Ceccarelli C., Grodsky N. B., Ariyaratne N., Colman R. F., Bahnson B. J. (2002) Crystal structure of porcine mitochondrial NADP+-dependent isocitrate dehydrogenase complexed with Mn2+ and isocitrate. Insights into the enzyme mechanism. J. Biol. Chem., 277, 43454–43462 [DOI] [PubMed] [Google Scholar]
- 25. Soundar S., Jennings G. T., McAlister-Henn L., Colman R. F. (1996) Expression of pig heart mitochondrial NADP-dependent isocitrate dehydrogenase in Escherichia coli. Protein Expr. Purif. 8, 305–312 [DOI] [PubMed] [Google Scholar]
- 26. Yan J.-M., Zhang Z.-J., Yuan D.-Q., Xie R.-G., Zhao H.-M. (1994) Direct imidazolylmethylation of phenols. Synthetic Commun. 24, 47–52 [Google Scholar]
- 27. Popovici-Muller J., Salituro F. G., Saunders J. O., Travins J. M., Yan S. (2012) Therapeutically active compositions and their method of use. WO 2012/009678 [Google Scholar]
- 28. Albert R., Danklmaier J., Hönig H., Kandolf H. (1987) A simple and convenient synthesis of β-aspartates and γ-glutamates. Synthesis 1987, 635–637 [Google Scholar]
- 29. Deechongkit S., You S.-L., Kelly J. W. (2004) Synthesis of all nineteen appropriately protected chiral α-hydroxy acid equivalents of the alpha-amino acids for boc solid-phase depsi-peptide synthesis. Org. Lett. 6, 497–500 [DOI] [PubMed] [Google Scholar]
- 30. Liu R. C., Huang W., Ma J. Y., Wei B. G., Lin G. Q. (2009) BF3. Et2O catalyzed diastereoselective nucleophilic reactions of 3-silyloxypiperidine N,O-acetal with silyl enol ether and application to the asymmetric synthesis of (+)-febrifugine. Tetrahedron Lett. 50, 4046–4049 [Google Scholar]
- 31. Segel I. H. (1975) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme System, John Wiley & Sons, Inc., New York [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rendina A. R., Pietrak B., Smallwood A., Zhao H., Qi H., Quinn C., Adams N. D., Concha N., Duraiswami C., Thrall S. H., Sweitzer S., Schwartz B. (2013) Mutant IDH1 enhances the production of 2-hydroxyglutarate due to its kinetic mechanism. Biochemistry 52, 4563–4577 [DOI] [PubMed] [Google Scholar]
- 34. Jin G., Reitman Z. J., Duncan C. G., Spasojevic I., Gooden D. M., Rasheed B. A., Yang R., Lopez G. Y., He Y., McLendon R. E., Bigner D. D., Yan H. (2013) Disruption of wild-type IDH1 suppresses d-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 73, 496–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pietrak B., Zhao H., Qi H., Quinn C., Gao E., Boyer J. G., Concha N., Brown K., Duraiswami C., Wooster R., Sweitzer S., Schwartz B. (2011) A tale of two subunits: how the neomorphic R132H IDH1 mutation enhances production of αHG. Biochemistry 50, 4804–4812 [DOI] [PubMed] [Google Scholar]
- 36. Grubbs R. D. (2002) Intracellular magnesium and magnesium buffering. Biometals 15, 251–259 [DOI] [PubMed] [Google Scholar]
- 37. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]