

Background: EmrE is a multidrug transporter.
Results: NMR spectroscopy was used to reveal a structured loop adjoining the substrate-binding and dimerization domains.
Conclusion: The linker structure and composition are conserved across the small multidrug resistance family and necessary for ion-coupled transport.
Significance: Loops in secondary active transporters can play an active role in efflux pump function and require characterization in lipid bilayers.
Keywords: Drug Resistance, Membrane Protein, Nuclear Magnetic Resonance (NMR), Protein Structure, Solid State NMR, EmrE, Membrane Transporter, Secondary Active Transport, Small Multidrug Resistance Family
Abstract
Secondary active transporters undergo large conformational changes to facilitate the efflux of substrates across the lipid bilayer. Among the smallest known transport proteins are members of the small multidrug resistance (SMR) family that are composed of four transmembrane (TM) domains and assemble into dimers. An unanswered question in the SMR field is how the dimerization domain (TM4) is coupled with the substrate-binding chamber (TM1–3). To provide insight for this essential aspect of ion-coupled transport, we carried out a structure-function study on the SMR protein EmrE using solid-state NMR spectroscopy in lipid bilayers and resistance assays in Escherichia coli. The chemical shifts for EmrE were consistent with β-strand secondary structure for the loop connecting TM3 and TM4. Based on these structural results, EmrE mutants were created to ascertain whether a specific loop length and composition were necessary for function. A linker encompassing six extra Gly residues relative to wild-type EmrE failed to give resistance; however, the number of residues in the loop was not the only criterion for a functional efflux pump. Replacement of the central hydrophobic residue with Gly (L83G) also conferred no ethidium resistance phenotype, which supported the conclusion that the structure and length of the loop were both essential for ion-coupled transport. Taken together with a bioinformatics analysis, a structured linker is likely conserved across the SMR family to play an active role in mediating the conformational switch between inward-open and outward-open states necessary for drug efflux. These findings underscore the important role loops can play in mediating efflux.
Introduction
Efflux of antibiotics and antiseptics across the lipid membrane is a mechanism bacteria employ to survive the insults of toxic molecules. Secondary active transport requires coupling with the proton motive force to facilitate drug movement against the concentration gradient that ultimately prevents drugs from interacting with their intended intracellular targets (1, 2). There are five families of efflux pumps associated with multidrug resistance: multi-antimicrobial extrusion proteins (MATE), major facilitator superfamily (MFS),3 ATP-binding cassette (ABC) superfamily, small multidrug resistance (SMR) family, and resistance nodulation division (RND) family (1). The SMRs are membrane protein transporters with 100–140 residues and four transmembrane (TM) domains that confer resistance to a wide variety of toxic compounds, including ethidium, dequalinium, methyl viologen, and acriflavine (3–5). These proteins are the smallest known efflux pumps, which makes them an excellent model system for understanding ion-coupled active transport as well as molecular recognition mechanisms involved in multidrug resistance (6).
EmrE from Escherichia coli, consisting of 110 residues and four transmembrane domains, has been the most extensively studied SMR protein (3). Although a significant number of biochemical and biophysical studies have been conducted, an atomic resolution structure of the backbone and side chains has prevented a complete understanding of ion-coupled transport and the nature of the substrate-binding pocket. The highest resolution structural model is a TPP+-bound crystal structure at 3.8 Å resolution (7), which together with cryo-electron microscopy, NMR, and fluorescence data (8–13) supports the presence of an asymmetric and antiparallel homodimer. Previous experiments have shown that dimer stability is reduced by Cys substitutions at Gly90, Ile94, Gly97, Ile101, and Asn102 that are likely involved in interhelical packing between the monomers (14, 15). In fact, studies of the EmrE homologue Hsmr from Halobacterium salinarum are also consistent with these results showing that mutations at similar positions (G90V, G97V, and V98A) abolish ethidium resistance and destabilize the dimer as assessed by gel electrophoresis (16). Taken together with the structural data (7, 13), it appears that the dimer stability of EmrE is dictated by interhelical contacts between TM4, whereas TM1–3 helices comprise the substrate-binding chamber. Because the dimer stability is a critical aspect of the function of SMR proteins, an unanswered question in the field is whether the loop connecting the dimerization and substrate-binding domains plays an active or a passive role in the ion-coupled transport activity. Indeed, loops have been shown to play important roles in membrane protein function, assembly, stability, and topology (17–27). For example, membrane protein topology has been shown to be driven by the positive-inside rule, where the loops possessing the largest net number of positively charged residues are oriented toward the cytoplasmic side of the bilayer (17–20). In addition, loops can stabilize the structure, as is known for the outer membrane protein OmpA, which enables its function as a bacteriophage receptor (21). Within the MFS family, conserved cytoplasmic loops are necessary for a variety of functions, including proper insertion and assembly (22), transport of lactose with LacY (22, 23), antiport of tetracycline by TetA (24–26), and chemotaxis driven by PcaK (27). However, some loops connecting TM domains seem to possess no specific composition requirement, such as the central linker that connects TM1–6 with TM7–12 in the MFS family (22). These experiments carried out with lactose permease showed that the loop length is conserved and necessary to act as a temporal delay in membrane insertion rather than a specific functional role in the transporter (22).
To unveil the atomic-scale traits of the loop adjoining the dimerization helix with the substrate-binding chamber in EmrE, we carried out a structure-function study using resistance assays and NMR spectroscopy in lipid bilayers. The β-strand character revealed from NMR chemical shifts and the sequence homology analyses suggest structural conservation across the SMR family, which uncovers important clues into the active role this loop plays in the inward-open to outward-open conformational transition in the dimer.
EXPERIMENTAL PROCEDURES
Resistance Assays
Wild-type EmrE in a pMS119EH vector was a generous gift from Prof. Raymond Turner (University of Calgary). Resistance against ethidium bromide was performed with the following nine constructs: wild-type EmrE, Q81G, R82G, L83G, D84G, L83G/D84G, R82G/L83G/D84G (GGG),4 a 6-Gly insert EmrE (6G-RLD), and an EmrE construct lacking a ribosome-binding site (No RBS). All of the mutants were created with an Agilent site-directed mutagenesis kit. Freshly transformed BL21(DE3) cells were grown to an A600 of 1.0 and plated as serial dilutions from 100 to 10−6 onto pH 7.0 LB-agar plates containing 100 μg/ml carbenicillin. The plate with ethidium bromide contained 50 μg/ml. All plates were made in duplicate. The control plates contained carbenicillin without the addition of ethidium to ensure accurate dilutions between the different EmrE mutants.
Expression and Purification of EmrE for NMR Spectroscopy
[U-13C,15N]EmrE and [2-13C,15N]Leu EmrE were expressed and purified as a fusion protein with maltose-binding protein as described previously (8, 9, 28, 29). Reverse-labeled Phe, Leu, and Thr (revFLT) EmrE was expressed as described previously (28) in a minimal medium containing 15NH4Cl (Cambridge Isotope Laboratories) and [13C]glucose (Sigma-Aldrich), supplemented with 100 mg/liter of 15N Thr, 200 mg/liter of natural abundance Leu, and 240 mg/liter of natural abundance Phe. Reverse-labeled Trp, Ile, and Tyr (revWIY) was expressed in a minimal medium containing 15NH4Cl (Cambridge Isotope Laboratories) and [13C]glucose (Sigma-Aldrich), supplemented with 250 mg/liter of natural abundance Trp, Ile, and Tyr.
NMR Sample Preparation
For solution NMR studies, [2H,13C,15N]EmrE back-exchanged to 1H at the amide positions was reconstituted into q = 0.33 DMPC/DHPC bicelles, where q is the ratio between long-chain and short-chain lipids, as described previously (9). For magic-angle spinning (MAS) sample preparation, EmrE in n-dodecyl β-d-maltoside detergent was reconstituted into DMPC or 3:1 DMPC:DMPG liposomes as described previously (9, 28, 29). Briefly, each EmrE sample was reconstituted into liposomes at a lipid:protein ratio of 75:1 (mol:mol) using Bio-Beads (Bio-Rad) at 45-fold excess relative to detergent (w/w). After incubation for 12 h at 4 °C, proteoliposomes were pelleted with ultracentrifugation and resuspended in NMR buffer (20 mm Hepes, 20 mm NaCl, 50 mm DTT, 0.05% NaN3, pH 6.9). Following a second ultracentrifugation step, proteoliposomes were partially dehydrated using lyophilization and rehydrated with water prior to packing between sample spacers in a 3.2-mm thin-walled MAS rotor.
MAS Solid-state NMR Spectroscopy
Data were collected on an Agilent DD2 600-MHz (1H frequency) spectrometer with a triple-resonance BioMAS probe triply tuned to 1H, 13C, and 15N or doubly tuned to 1H and 13C (30). The sample temperature was set to 2, 6, or 9 °C as determined by methanol calibration (31), the spinning rate was set to 12.5 kHz (unless otherwise noted), and the relaxation delay was set to 2 s. In NCA- and NCO-based experiments, the initial 1H-15N cross-polarization utilized a contact time of 0.8–1 ms. The 15N to 13CA or 13CO transfer utilized SPECIFIC-CP (32), where the 15N offset was set to 121 ppm and the 13C offset was set to 58 or 177 ppm, respectively. The SPECIFIC-CP used a tangent adiabatic ramp on the nitrogen channel (33). The Δ/2π and β/2π parameters of the adiabatic cross-polarization were set to 1.2 and 0.3 kHz for the 15N to 13C transfer (34).
The two-dimensional 13C/13C PDSD experiment (35) for [2-13C,15N]Leu EmrE (see Fig. 3C) used a 1H-13C contact time of 450 μs and a mixing period of 800 ms. The indirect dimension was 1562.5 Hz (11.5 ms of maximum evolution time), and the direct dimension was 100 kHz with an acquisition time of 25 ms. The FDR-NCA on revFLT (28) (see Fig. 3E) had a 15N spectral width of 2500 Hz (12 ms of maximum evolution time) and a contact time of 3.55 ms. The two-dimensional 13C/13C double-quantum single-quantum (DQSQ) experiment on revFLT (see Fig. 3B) was run with a spinning rate of 8.333 kHz. The 1H-13C contact time was 0.4 ms. A 480-μs SPC5 element was used for excitation and reconversion (36). The indirect dimension spectral width was set to 16666.7 Hz (6.0 ms of maximum evolution time). For both the FDR-NCA and the DQSQ, the direct dimension was 100 kHz with an acquisition time of 20 ms. The NCACX on U-13C,15N (see Fig. 3A) was collected using the dual acquisition magic angle spinning (DUMAS) sequence in conjunction with a CAN(CO)CA (37). The NCACX had a 15N spectral width of 3125 Hz (9 ms of maximum evolution time), a 15N-13CA contact time of 4.5 ms, and an indirect 13C spectral width of 6250 Hz (4.5 ms of maximum evolution time). The homonuclear 13C transfer used 15 ms of DARR (38, 39). The direct dimension spectral width was 100 kHz with an acquisition time of 20 ms. The CAN(CO)CA (40) on U-13C,15N (see Fig. 3A) used a 1H-13C contact time of 0.4 ms, a 13CA-15N contact time of 4.5 ms, and a 15N-13CO contact time of 5 ms. The homonuclear transfer between 13CO and 13CA was carried out using a 15-ms DARR mixing period (38, 39). The spectral widths and evolution or acquisition times were as follows: indirect 13C spectral width of 6250 Hz (4.5 ms maximum evolution time), 15N spectral width of 3125 Hz (7.4 ms of maximum evolution time), and a direct 13C dimension spectral width of 100 kHz (15 ms of acquisition time). The NCOCX with [U-13C,15N]EmrE (see Fig. 3A) was collected as described previously (28). The CA(N)(CO)CA on [U-13C,15N]EmrE (see Fig. 3F) used a 1H-13CA contact time of 0.75 ms, a 13CA-15N contact time of 4.5 ms, and a 15N-13CO contact time of 4 ms. The 13CO-13CA homonuclear transfer used 25 ms of DARR (38, 39). The indirect 13C dimension was 6250 Hz (4.5 ms of maximum evolution time), and the direct 13C dimension was 100 kHz (20 ms of acquisition time). The NCACX for revWIY (see Fig. 3A) had a 15N-13CA contact time of 4.35 ms. The spectral width and evolution times or acquisition parameters were as follows: 15N spectral width of 3125 Hz (7.7 ms of maximum evolution time), indirect 13C spectral width of 6250 Hz (4.5 ms of maximum evolution time), and a direct 13C dimension of 100 kHz (20 ms of acquisition time).
FIGURE 3.

Spectroscopic characterization of the loop connecting TM3 and TM4 in the drug-free state in DMPC-liposomes. A, the backbone resonances for [U-13C,15N]EmrE were assigned with MAS-SSNMR using a backbone walk with CAN(CO)CA (green), NCACX (red/orange), and NCOCX (black) experiments. Note that the orange NCACX (Asp84) was collected with revWIY-labeled EmrE. B, DQSQ experiment with revFLT shows the arginine residues clustered around 55 ppm. Unlike Arg29 and Arg106, Arg82 is split into two distinct peaks, which results in a weaker signal overall. The assignment for Arg82 is also in good agreement with data collected from solution NMR (Fig. 4). C, similarity of chemical shifts in the TPP+-bound form from solution NMR (blue) are directly transferable to spectra collected in the drug-free form with MAS-SSNMR, which provided the assignment for Leu83 in an 800-ms PDSD experiment on [2-13C,15N]Leu EmrE. D, Gln81, Gly80, and Phe79 are visible in NCACX and NCOCX spectra collected at 750 MHz (1H frequency) with [U-13C,15N]EmrE in 3/1 DMPC/DMPG and are in good agreement with data from solution NMR. E, the FDR-NCA with revFLT served as a spectroscopic filter that highlighted residues i + i relative to the reverse labeled amino acids and allowed for the assignment of Gly80 (28). F, a two-dimensional CA(N)(CO)CA with [U-13C,15N]EmrE was used to assign Gly77 and Phe78.
The NCACX and NCOCX on [U-13C,15N]EmrE in 3:1 DMPC/DMPG liposomes (see Fig. 3D) was collected on a Bruker 750-MHz (1H frequency) spectrometer equipped with an Efree probe (Bruker Biospin). The spinning rate was 13.3 kHz, and the relaxation delay was set to 2.8 s. The 15N dimension spectral width was set to 2660 Hz (7.9 and 8.6 ms of maximum evolution time for the NCACX and NCOCX, respectively). The indirect 13C dimension was 6650 Hz (NCACX, 4.8 ms of maximum evolution time) and 2216.7 Hz (NCOCX, 6.3 ms of maximum evolution time). The NCACX used 75 ms of DARR, and the NCOCX used 25 ms of DARR for the 13C homonuclear transfer (38, 39). The direct dimensions were 100 kHz and 20 ms of acquisition time. The data were processed and analyzed with NMRPipe (41) and Sparky (71).
Solution NMR Spectroscopy
Data were collected on Bruker 600-MHz and 800-MHz spectrometers (1H frequency) equipped with TCI CryoProbes. The three-dimensional HNCA was acquired at 45 °C using a TROSY-HNCA (42–44) at 800 MHz. The experiment had a 15N dimension of 2595 Hz (10 ms) and a 13C dimension of 6037 Hz (9.1 ms). The direct dimension had a spectral width of 11160 Hz and an acquisition time of 46 ms. The relaxation delay was set to 1.2 s. The three-dimensional HNCO was acquired at 45 °C using a TROSY-HNCO (42, 44) at 600 MHz. The experiment had a 15N dimension of 2189 Hz (13.7 ms) and a 13C dimension of 3320 Hz (14.8 ms). The direct dimension had a spectral width of 9014 Hz and an acquisition time of 89 ms. The relaxation delay was set to 1 s. The three-dimensional HN(CA)CO was acquired at 45 °C using a TROSY-HNCACO (42–44) at 600 MHz. The experiment had a 15N dimension of 2067.8 Hz (11.6 ms) and a 13C dimension of 3320 Hz (13.3 ms). The direct dimension had a spectral width of 9014 Hz and an acquisition time of 57 ms. The relaxation delay was set to 1 s.
Sequence Homology Analysis
Search results from UniProt database belonging to the “small multidrug resistance (SMR) protein family” resulted in 161 reviewed sequences. These were reduced in sequence redundancy to less than 90% using the UniRef90 database (45). Multiple sequence alignment was performed with Clustal Omega (46). The sequence logo graph was created using WebLogo (47) and colored using the CINEMA color scheme (48). Amino acids were ranked in terms of hydrophobicity using the hydrophobicity scale described by Monera et al. (49).
RESULTS
Truncated EmrE Lacking Transmembrane-4 Is Prone to Aggregation
Previous work on SMR proteins showed that conserved Gly residues in TM4 play a role in dimer stability (16, 50, 51). These residues in EmrE correspond to Gly90 and Gly97. When mutated to Cys or Trp residues at these positions, EmrE was found to aggregate in vitro (14) and was unable to confer resistance in vivo (15, 52). Because our interests were to determine the structural basis for domain coupling between the proposed dimerization and substrate-binding regions of EmrE, we designed a simple experiment to test whether the helices involved in drug binding were stable in the absence of TM4. To do so, we prepared a construct of EmrE with a stop codon inserted after Arg82 (i.e. no TM4) and purified the truncated protein. The aggregation propensity of this TM1–3 construct was followed using size exclusion chromatography. Fig. 1 shows corresponding chromatograms for truncated and wild-type EmrE in n-dodecyl β-d-maltoside detergent micelles. As observed in the plot, wild-type EmrE eluted as a single peak at ∼12.8 ml, whereas TM1–3 had a large fraction that eluted earlier than the wild-type profile, which was indicative of aggregation and confirmed by gel electrophoresis (data not shown). In addition, the most intense peak shifted to a larger elution volume relative to wild type that is in agreement with destabilization of the dimer quaternary arrangement for the truncated construct. These results are consistent with previous studies that indicate TM4 is necessary for forming a stable EmrE dimer (14–16, 50, 52).
FIGURE 1.
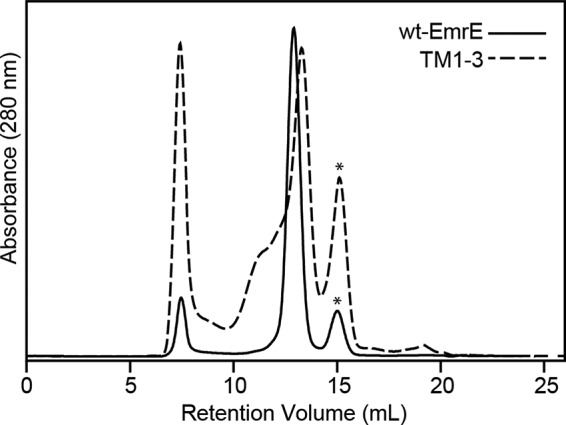
Analysis of the aggregation propensity of TM1–3 by gel filtration chromatography. TM1–3 (dotted line) has a heterogeneous and polydispersed peak in the size-exclusion chromatogram, indicative of the instability of EmrE lacking the TM4 helix. On the other hand, wild-type EmrE (solid line) showed a single, monodispersed peak. The asterisks indicate residual fusion protein partner in the purification.
Sequence Homology in the Small Multidrug Resistance Family
Based on the truncated EmrE results, a question that emerged was whether a structured loop was required to link the TM4 helix with the substrate-binding domain (TM1–3). Using a bioinformatics approach, we carried out a search of the UniProt database for proteins belonging to the “small multidrug resistance (SMR) protein family.” The resulting hits totaled 21,645, which were further narrowed to 161 reviewed proteins in this database and filtered by using the Uniref90 database to group sequences with >90% redundancy together. Multiple sequence alignment (46) of this group allowed for analysis of residue types and loop lengths. Fig. 2A shows the residue propensity at each position relative to wild-type EmrE. Within the TM3-TM4 loop (loop-3), the residue composition showed a preference for a polar or negatively charged residue at position 81, a hydrophobic residue at position 83, and a polar or anionic residue at position 84 (Fig. 2A). With regard to position 83, further analysis showed that ∼80% of the SMRs analyzed contained a large hydrophobic residue (Fig. 2B). Interestingly, position 82 was somewhat variable across the SMR family. To determine whether the length of the linker between TM3 and TM4 was conserved, we used the metric of gaps/sequence, starting five residues before the loop (residue 75 in EmrE) to five residues after the loop (residue 90 in EmrE) based on the crystal structure (Protein Data Bank (PDB) 3B5D). Among the 50 sequences in the Uniref90 set, only one gap was found, which suggested that the length of the loop was highly conserved across the SMR family of transporters.
FIGURE 2.
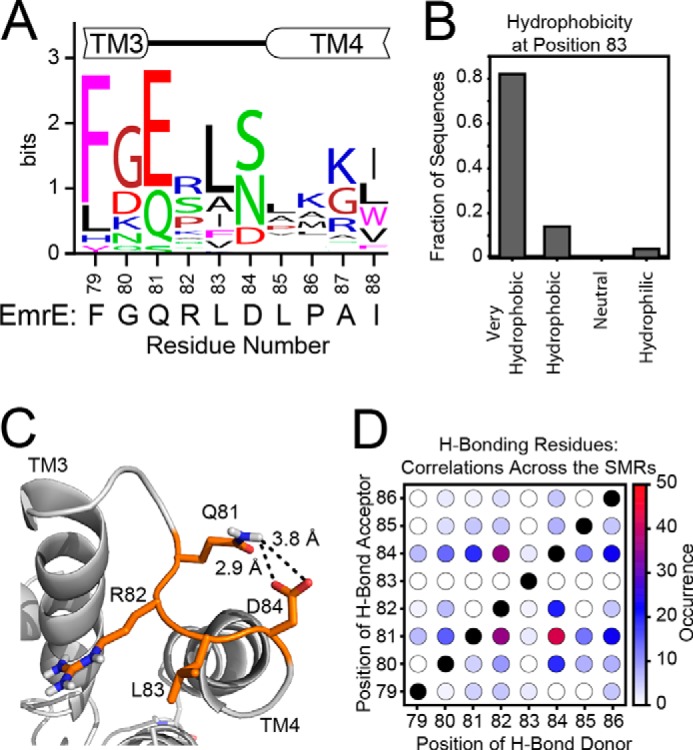
The composition and chemistry of loop-3 is conserved across the SMR family. A, the consensus logo plot for the loop between TM3 and TM4 was generated from the multiple sequence alignment of SMR sequences from the Uniref90 database. The conservation of residue type shows that the loop tends to have a polar or negatively charged residue at position 81, a hydrophobic residue at position 83, and a polar or negatively charged residue at position 84. For comparison, the sequence of EmrE is displayed below the consensus plot. B, a bioinformatics analysis of position 83 reveals that it is occupied by a very hydrophobic residue (Phe, Ile, Trp, Leu, Val, or Met) (49) in more than 80% of sequences analyzed, with another 10% containing a less hydrophobic Ala at this position. C, the 3.8 Å x-ray crystal structure (PDB 3B5D) (7) with the side chains rebuilt shows a possible hydrogen bond between Gln81 and Asp84. D, the occurrences of hydrogen bond acceptors (Asn, Asp, Gln, Glu, His, Ser, Thr, or Tyr) with hydrogen bond donors (Arg, Asn, Gln, His, Lys, Ser, Thr, Trp, or Tyr) were mapped within each of the 50 sequences of the Uniref90 set to determine the possibility of a hydrogen bond within each sequence. The total occurrences across the set were summed and plotted in the heat map. Across the sequences, a hydrogen bond acceptor was frequently found at position 81, and a hydrogen bond donor typically found at position 84.
In addition to the sequence homology analysis, we used the REMO server to rebuild the main chain and side chains of the Cα-only crystal structure coordinates of EmrE bound to TPP+ (53). Inspection of this rebuilt structure revealed the presence of a possible hydrogen bond between the side chains of Gln81 and Asp84 (Fig. 2C) that may participate in stabilizing the structure of the inward-open-facing conformation of the transporter. Note that loop-3 on the outward-open side did not have the same intra-loop hydrogen bond, which was consistent with differential paramagnetic quenching previously reported for the loops in the TPP+-bound form of EmrE (10). To further investigate the hydrogen bond network within loop-3, we carried out an evolutionary analysis to identify structural restraints based solely on homologous primary sequences (54). We surmised that if a hydrogen bond was conserved across the SMR family, both residues involved in the interaction would change in homologues from different species. For example, in Fig. 2, we observed that the most prominent residue at position 81 was a negatively charged amino acid, whereas Gln and Ser comprised the other common side chains, and alternatively, at position 84, the most common residues were polar side chains (Ser and Asn) with the anionic Asp residue at lower abundance. Of the 50 proteins used to comprise this plot, 94% had a hydrogen bond donor (Arg, Asn, Glu, His, Lys, Ser, Thr, Trp, or Tyr) at position 81 or 84 with a hydrogen bond acceptor (Asn, Asp, Gln, Glu, His, Ser, Thr, or Tyr) at the conjugate position. Only two proteins were identified that showed anionic residues at both sites. Interestingly, both of these proteins are members of the paired SMR subfamily (55). A heat map was constructed to further quantify these findings that analyzes all residues surrounding loop-3 in the SMR family (Fig. 2D). As seen in this plot, the only pairwise residues that are highly correlated for hydrogen-bond formation are between 81 and 84. Note that EmrE is atypical of the SMRs analyzed in that it has a hydrogen bond acceptor at position 84 and a hydrogen bond donor at position 81, whereas 84% of the SMRs analyzed had the opposite configuration.
NMR Chemical Shifts Reveal a Structured Loop
To obtain experimental structural insight into the conformation of loop-3 in a native-like environment, we carried out solution and solid-state NMR spectroscopy with isotopically enriched EmrE. It is well known that backbone chemical shifts are highly correlated with the secondary structure of the protein(56, 57). Therefore, to determine the nature of the loop connecting TM3 with TM4, we carried out a series of MAS solid-state NMR (SSNMR) spectroscopy experiments on full-length drug-free EmrE reconstituted into DMPC or 3/1 DMPC/DMPG liposomes (9, 28, 29). We assigned most of the chemical shifts for loop-3 in a de novo fashion using a suite of two-dimensional and three-dimensional DQSQ, PDSD, NCOCX, NCACX, and CAN(CO)CA SSNMR experiments on U-13C,15N, reverse-labeled (28), and selectively labeled samples. Fig. 3A shows the backbone walk from Asp84 to Ala87. The only aspartate residue in EmrE is at position 84, and the unique chemical shift served as an anchor point for these unambiguous assignments. Similarly, we were able to carry out a backbone walk between Phe79 and Gln81 using strip plots that enabled additional structural information on loop-3 (Fig. 3D). Furthermore, we utilized a reverse-labeled Phe-Leu-Thr sample (revFLT) in combination with the FDR-NCA afterglow filter to identify residues located after the reverse-labeled amino acid in the primary sequence (28, 58, 59). Because Gly80 is after Phe79 in EmrE, we observed an N-CA cross-peak for Gly80 in the FDR-NCA experiment shown in Fig. 3E, which is in good agreement with the data collected with solution NMR on TPP+-bound EmrE (described below). From the CA and CO chemical shifts of Gly80, the 15N chemical shift of Gln81 was determined using an NCOCX, which allowed for assignment of Gln81 in the NCACX (Fig. 3D). Additionally, Phe79 could be assigned from the 15N chemical shift of Gly80 in the NCOCX. Lastly, the Gly77-Phe78 pair was assigned from a two-dimensional CA(N)(CO)CA using [U-13C,15N]EmrE with Fig. 3F displaying the direct dimension region for glycine where all peaks arise from Gly-X pairs in the primary sequence. Based on the possible range of chemical shifts for residue types (57), the only unassigned cross-peak in this region was associated with the CA atoms of Gly77 and Phe78.
From the analysis of the three-dimensional sequential experiments, we observed a gap in the assignment between residues Gln81 and Asp84. The asymmetric nature of the dimer previously observed in solution (9, 10) and oriented solid-state NMR (8, 9) provided a possible explanation for the lack of intensity observed for Arg82 and Leu83: peak splitting resulting in reduced intensities and insufficient signal to noise in the three-dimensional experiments. To validate this conclusion, we carried out solution NMR spectroscopy on TPP+-bound EmrE using amide proton back-exchanged 2H,13C,15N-labeled EmrE in DMPC/DHPC (q = 0.33) isotropic bicelles (9). A combination of triple resonance three-dimensional HNCA, HNCO, and HN(CA)CO experiments and previously published work on EmrE (60) was used to assign all residues within the loop (Fig. 4). We found a close similarity between the backbone chemical shifts assigned from MAS with those of the drug-bound form from solution NMR experiments. Therefore, the chemical shifts for Arg82 (Fig. 3B) and Leu83 (Fig. 3C) that were not identified from three-dimensional experiments in MAS could be assigned in two-dimensional spectra using the assignments from solution NMR data. We note that these peaks in two dimensions are weaker than other residues that did not display peak splitting. From the spectra shown in Fig. 3, B and C, we were able to observe two chemical shifts for Arg82 and Leu83 in the MAS spectra.
FIGURE 4.

Spectroscopic characterization of the loop connecting TM3 and TM4 in the TPP+-bound state. A, HNCA strips (blue) from uniformly 13C,15N-labeled, TPP+-bound EmrE in isotropic bicelles. B, HNCO (red) and HN(CA)CO (black) strips from 13C,15N-labeled, TPP+-bound EmrE in isotropic bicelles.
The chemical shift assignment in the drug-free and drug-bound states enabled us to analyze the secondary structure of EmrE in loop-3. Interestingly, a plot of the Cα chemical shift index shown in Fig. 5 indicates a close similarity between drug-free and TPP+-bound EmrE. The narrower line widths from solution NMR enabled both populations to be observed in the spectra; however, these two chemical shifts were generally similar with only slight differences between the two monomers present in the TPP+-bound form (8–10). The chemical shift index is consistent with a structured linker and is in agreement with the chemical shifts observed for loop-3 in the homologous protein Smr from Staphylococcus aureus (61). Specifically, Leu85 and Pro86 have downfield 13Cα chemical shifts characteristic of helix (57), whereas the shifts for Phe79 to Asp84 are consistent with non-helical chemical shifts. This indicates that the TM4 helix begins at Leu85.
FIGURE 5.
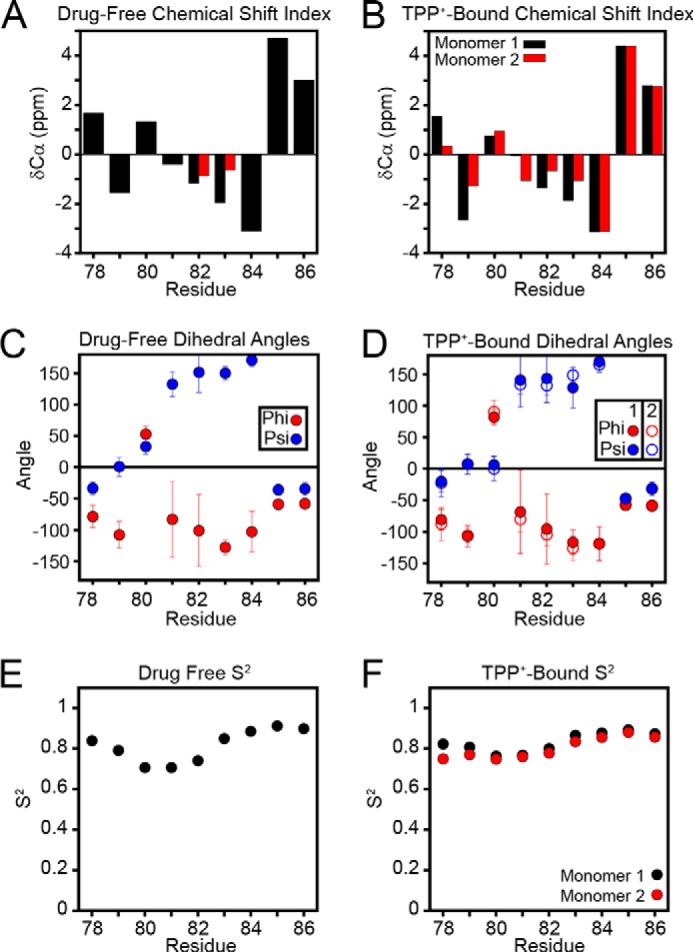
Loop-3 dihedral angles of drug-free and TPP+-bound EmrE. A and B, the chemical shift index (observed - random coil) for CA indicates a β-strand structure in loop-3 for both drug-free EmrE (A) and TPP+-bound EmrE (B) (69, 70). C and D, using the backbone chemical shifts of loop-3, the dihedral angles of this loop were calculated using TALOS+ (62) for both drug-free EmrE (C) and TPP+-bound EmrE (D). Residues 81 through 84 have dihedral angles characteristic of β-strand. E and F, random coil index-predicted order parameters, S2, for loop-3 show that the loop is relatively ordered in both the drug-free (E) and the TPP+-bound (F) states (63, 64).
The assigned chemical shifts for residues 72–87 in the drug-free form of EmrE and residues 77–87 in the TPP+-bound form were used as the input for converting chemical shifts to dihedral angles using the TALOS+ software package (62). The resulting secondary structure plot is shown in Fig. 5, C and D, and supports the presence of similar dihedral angles consistent with a β-strand for residues 81 through 84. A plot of the random coil index-predicted order parameters is shown in Fig. 5, E and F, and indicates a similar dip around the linker region (63, 64). Although these S2 values are slightly lower than those of the TM domain residues, values between 0.7 and 0.8 suggest that the loop is relatively ordered on the picosecond to microsecond timescale (64). Overall, these results are in agreement with the TPP+-bound x-ray structure of EmrE (7) as TM3 helix ends at Gly77 and the loop is composed of residues 78–84. Nevertheless, a complete understanding of the stabilizing forces needed to maintain the structure of loop-3 will require a higher resolution structure of EmrE in the drug-free or drug-bound forms.
Loop-3 Length and Residue Composition Are Required for Resistance
Our atomic-scale characterization of the loop coupled with the bioinformatics analyses suggests that the loop length and residue type are conserved to carry out transport of substrates across the membrane. To further validate this hypothesis, we engineered mutants of EmrE to alter the nature of loop-3 and measured the resistance phenotype relative to the wild-type protein. E. coli was freshly transformed with plasmids containing wild type and mutants of EmrE and added to LB-agar plates in 10-fold serial dilutions to monitor the ethidium resistance phenotype conferred by the transporter. To probe whether the linker length was conserved, we inserted 6 Gly residues between Gln81 and Arg82 (referred to as 6G-RLD). In addition, the importance of the loop composition was analyzed by preparing single-site mutants (Q81G, R82G, L83G, and D84G), a double mutant (L83G/D84G), and a triple mutant of EmrE (R82G/L83G/D84G referred to as GGG mutant). Note that the 6G-RLD construct contained all of the native loop residues, whereas the GGG mutant replaced the wild-type sequence with flexible amino acid residues. The corresponding ethidium resistance assay (Fig. 6B) showed that wild-type EmrE, Q81G, R82G, and D84G did not alter function as indicated by the ability to confer resistance to the host organism up to a dilution factor of 10−5. The fact that the R82G mutant conferred resistance is consistent with the sequence homology analysis that showed variable amino acid substitutions at this position across the SMR family. To the contrary, the L83G, 6G-RLD, and GGG mutants showed no resistance relative to the control (EmrE plasmid containing no ribosome-binding site). These results emphasize the necessity of a central hydrophobic residue within loop-3 for conferring resistance and the conservation of linker length. The absence of a change in resistance phenotype observed for Q81G and D84G mutants suggests that the possible hydrogen bond identified from the crystal structure and bioinformatics analysis would not alone be sufficient to stabilize the structure. To determine whether there was an additive effect of the hydrophobic position with either of the possible hydrogen bond residues, we prepared a double L83G/D84G mutant. We observed only a small difference (<10-fold) between L83G/D84G and L83G, which confirmed that the hydrogen bond is not the primary stabilizing force within loop-3. Instead the hydrophobic position at 83 plays the predominant role in the linker between the substrate-binding and dimerization domains.
FIGURE 6.
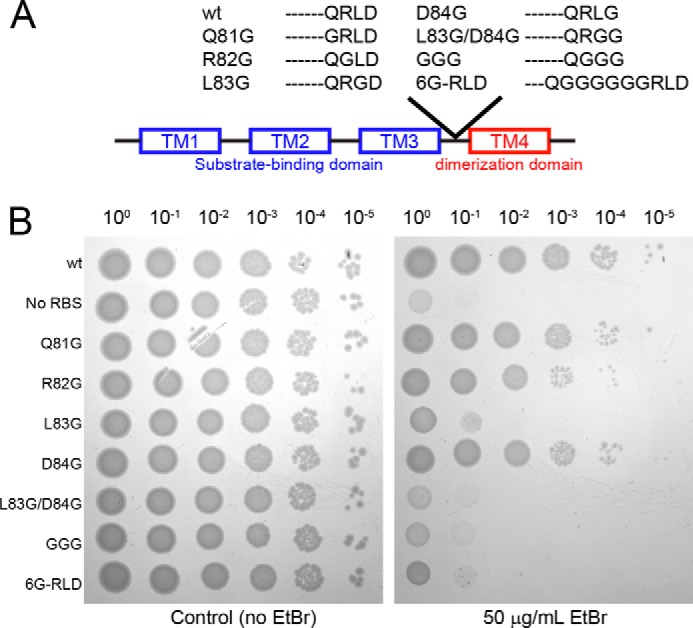
Sensitivity to ethidium resistance with loop-3 mutants. A, a schematic representation of the loop compositions in wild-type (wt) EmrE and the loop-3 mutants. B, right plate, resistance against ethidium bromide was measured using 50 μg/ml ethidium bromide on LB-agar plates with 100 μg/ml carbenicillin. The plate on the left does not contain ethidium bromide and is a control for the dilution factors. All variants of EmrE were in a pMS119EH vector. WT-EmrE, Q81G, R82G, and D84G all show resistance to ethidium, whereas L83G, L83G/D84G, the GGG loop mutant, and 6G-RLD mutant show no resistance, similar to the variant lacking a ribosome-binding site on the transcribed gene (no RBS).
DISCUSSION
EmrE has a unique architecture that consists of two anti-parallel and asymmetric monomers where the helices in each protomer are arranged in a linear fashion (Fig. 7). This assembly enables conformational switching between inward-open and outward-open states that is necessary for transport of substrate or protons across the cellular membrane (i.e. alternating access model (9, 10, 13, 65)). The EmrE dimer requires both protein dynamics within the substrate-binding chamber (TM1–3) and stability of the quaternary arrangement for transport. Based on the structural models (7, 13), evolution appears to have satisfied the latter criterion through the incorporation of TM4 to maintain dimer strength (7, 13, 14, 16, 50). In this work, we provide direct support for this model by showing that an EmrE construct lacking TM4 has a strong tendency to aggregate, which supports the contention that the quaternary architecture stabilizes the tertiary structure. Due to the importance of TM4 as the anchor point of EmrE, we tested the hypothesis that the loop adjoining the substrate-binding region with the dimerization domain played an active role in transport. Our chemical shift data obtained in lipid bilayers and bicelles support this notion and unveiled a key feature of the linker between TM3 and TM4 that consisted of β-strand secondary structure. Interestingly, solution NMR data for loop-3 of the homologous protein Smr from S. aureus were also indicative of a β-strand structure, suggesting a conserved feature in the SMR family (61). To validate our structural work, we carried out resistance assays using engineered mutants of EmrE within loop-3 that showed the linker structure was required for ion-coupled transport and the conferred phenotype. In fact, replacement of the native RLD primary sequence with 3 Gly residues completely abolished resistance. Likewise, the addition of 6-Gly residues within the linker resulted in no conferred phenotype, suggesting that the length of loop-3 was also conserved.
FIGURE 7.
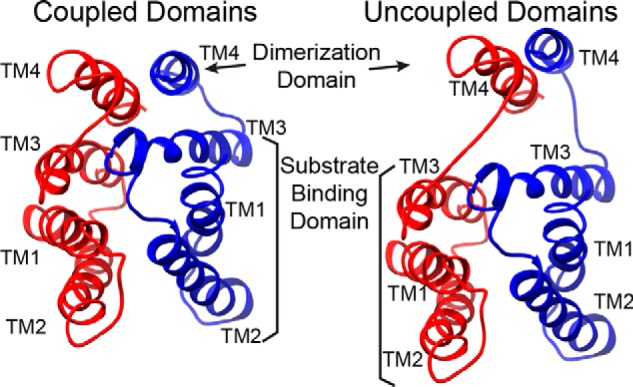
Loop-3 is responsible for coupling substrate binding with dimerization. Model depiction showing that the loop between TM3 and TM4 is responsible for holding the substrate-binding domain (TM1–3) close to the dimerization domain (TM4). When the loop is too flexible or too long, this necessary coupling is lost, and the transporter becomes ineffective. A rebuilt model of the x-ray crystal structure (3B5D) was used to generate the schematic (7).
The conformation of loop-3 highlights the atomic-scale properties necessary for linking the dimerization domain with the substrate-binding region. We propose that the reason for the linker length and composition conservation is due to the need to limit flexibility in the loop with a centrally placed hydrophobic residue. Additionally, a hydrogen bond between Gln81 and Asp84 may play a secondary role in stabilizing the loop conformation. These interpretations are supported by the Cα model of EmrE bound to TPP+ (7), the residue conservation at position 83 (Fig. 2B), a correlated mutational analysis of loop-3 (Fig. 2D), and the lack of resistance provided by the L83G, L83G/D84G, GGG, and 6-Gly mutants. Note that the L83G, L83G/D84G, and GGG mutant disrupted the central hydrophobic residue. To the contrary, the 6 Gly insertions after residue 81 both increased the distance between positions 81 and 84 and introduced greater flexibility into the loop that Leu83 may no longer be able to stabilize. In addition, the 6-Gly mutant data imply that intrahelical contacts between TM3 and TM4 alone are insufficient to couple the dimerization and substrate-binding domains. Indeed, the spatial proximity of TM3 and TM4 is mediated by the presence of a short and structured linker that allows for the functional association between the TM helices (Fig. 7).
Sequence homology analysis also supports the conclusion that the residue composition and length are conserved across the SMR family. In agreement with the multiple sequence alignment for SMR transporters, Arg82 is variable and replacement with Gly resulted in no change in resistance relative to wild-type EmrE. This finding is also interesting in light of the fact that single positively charged side chain substitutions have been shown to influence the single or dual topology nature of EmrE (17). Our results show that a single substitution at Arg82 had no major effect on the dual topology of EmrE. This also indicates that no essential salt bridge exists between the side chains of Arg82 and Asp84 and is consistent with previous mutagenesis studies on EmrE at position 82 (R82C and R82K) that failed to perturb the ethidium resistance phenotype (14, 66–68). The only mutations that altered the phenotype were those that disrupted the central hydrophobic residue.
As a quantitative way to compare our NMR-derived dihedral angles to the TPP+-bound crystal structure (3B5D), we constructed peptides using the experimental values in Fig. 5 and calculated root mean square deviations with the x-ray structure of EmrE. The drug-free state was found to give Cα root mean square deviations of 2.4 Å (monomer A) and 2.3 Å (monomer B), whereas the peptide built from the TPP+-bound dihedral angles had root mean square deviation values of 2.1 Å (monomer A) and 2.2 Å (monomer B). These calculations support the conclusion that the loop-3 structure in lipid bilayers is in general agreement with that in the crystal structure, but further emphasizes the need for a higher resolution structure of the transporter. In addition, a comparison of the dihedral angles between the drug-free and TPP+-bound states shows the same overall trend with only a few subtle differences that may be associated with the helical bending within TM3 at the Gly65-Val66-Gly67 sequence (8, 14). Nevertheless, the primary change upon TPP+ binding is to slow the rate of inward-open to outward-open conformational exchange (9). Although the binding of other substrates may elicit altered structural dynamics (60), it appears that the plastic nature of the drug-free state encodes multidrug recognition and the conferred multidrug resistance phenotypes (9).
In summary, our findings offer a structure-function perspective on the role of a key loop in EmrE that enables transport by holding the dimer together in a specific conformation. The structured loop identified between TM3 and TM4 is likely conserved across the SMR family and plays a critical role in coupling the dimerization and substrate-binding domains necessary to enable ion-coupled transport. Future studies will seek to further unravel an atomic-scale picture of the transport process including the conformational transition accompanying proton and drug efflux that will be compared with other proteins present from different secondary active transport families.
Acknowledgments
The collection of NMR data at the New York Structural Biology Center was made possible by a grant from NYSTAR. We thank Matthew Devany for assistance in using the NMR spectrometer at Hunter College. We thank Prof. Raymond J. Turner at the University of Calgary for providing wild-type EmrE in the pMS119EH vector.
This work was supported, in whole or in part, by National Institutes of Health Grants R01AI108889 and K22AI083745 and start-up funds from New York University (to N. J. T.).
The following mutant designations are used throughout: GGG, R82G/L83G/D84G mutant; 6G-RLD, 6-Gly insert EmrE mutant; No RBS, EmrE construct lacking a ribosome-binding site.
- MFS
- major facilitator superfamily
- SMR
- small multidrug resistance
- TM
- transmembrane
- DMPC
- dimyristoyl phosphatidylcholine
- DHPC
- dihexanoyl phosphatidylcholine
- DMPG
- dimyristoyl phosphatidylglycerol
- MAS
- magic-angle spinning
- FDR
- frequency-selective dipolar recoupling
- DQSQ
- double-quantum single-quantum
- TROSY
- transverse relaxation optimized spectroscopy
- SSNMR
- solid-state NMR
- PDSD
- proton-driven spin diffusion
- rev
- reverse-labeled
- TPP+
- tetraphenylphosphonium
- DARR
- dipolar-assisted rotational resonance.
REFERENCES
- 1. Piddock L. J. (2006) Multidrug-resistance efflux pumps: not just for resistance. Nat. Rev. Microbiol. 4, 629–636 [DOI] [PubMed] [Google Scholar]
- 2. Walsh C. (2000) Molecular mechanisms that confer antibacterial drug resistance. Nature 406, 775–781 [DOI] [PubMed] [Google Scholar]
- 3. Schuldiner S. (2009) EmrE, a model for studying evolution and mechanism of ion-coupled transporters. Biochim. Biophys. Acta 1794, 748–762 [DOI] [PubMed] [Google Scholar]
- 4. Bay D. C., Turner R. J. (2012) Spectroscopic analysis of small multidrug resistance protein EmrE in the presence of various quaternary cation compounds. Biochim. Biophys. Acta 1818, 1318–1331 [DOI] [PubMed] [Google Scholar]
- 5. Bay D. C., Rommens K. L., Turner R. J. (2008) Small multidrug resistance proteins: a multidrug transporter family that continues to grow. Biochim. Biophys. Acta 1778, 1814–1838 [DOI] [PubMed] [Google Scholar]
- 6. Bay D. C., Turner R. J. (2009) Diversity and evolution of the small multidrug resistance protein family. BMC Evol. Biol. 9, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Y.-J., Pornillos O., Lieu S., Ma C., Chen A. P., Chang G. (2007) X-ray structure of EmrE supports dual topology model. Proc. Natl. Acad. Sci. U.S.A. 104, 18999–19004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gayen A., Banigan J. R., Traaseth N. J. (2013) Ligand-induced conformational changes of the multidrug resistance transporter EmrE probed by oriented solid-state NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 52, 10321–10324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cho M. K., Gayen A., Banigan J. R., Leninger M., Traaseth N. J. (2014) Intrinsic conformational plasticity of native EmrE provides a pathway for multidrug resistance. J. Am. Chem. Soc. 136, 8072–8080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morrison E. A., DeKoster G. T., Dutta S., Vafabakhsh R., Clarkson M. W., Bahl A., Kern D., Ha T., Henzler-Wildman K. A. (2012) Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature 481, 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lehner I., Basting D., Meyer B., Haase W., Manolikas T., Kaiser C., Karas M., Glaubitz C. (2008) the key residue for substrate transport (Glu14) in the EmrE dimer is asymmetric. J. Biol. Chem. 283, 3281–3288 [DOI] [PubMed] [Google Scholar]
- 12. Tate C. G., Ubarretxena-Belandia I., Baldwin J. M. (2003) Conformational changes in the multidrug transporter EmrE associated with substrate binding. J. Mol. Biol. 332, 229–242 [DOI] [PubMed] [Google Scholar]
- 13. Fleishman S. J., Harrington S. E., Enosh A., Halperin D., Tate C. G., Ben-Tal N. (2006) Quasi-symmetry in the cryo-EM structure of EmrE provides the key to modeling its transmembrane domain. J. Mol. Biol. 364, 54–67 [DOI] [PubMed] [Google Scholar]
- 14. Amadi S. T., Koteiche H. A., Mishra S., McHaourab H. S. (2010) Structure, dynamics, and substrate-induced conformational changes of the multidrug transporter EmrE in liposomes. J. Biol. Chem. 285, 26710–26718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elbaz Y., Salomon T., Schuldiner S. (2008) Identification of a glycine motif required for packing in EmrE, a multidrug transporter from Escherichia coli. J. Biol. Chem. 283, 12276–12283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poulsen B. E., Rath A., Deber C. M. (2009) The assembly motif of a bacterial small multidrug resistance protein. J. Biol. Chem. 284, 9870–9875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seppälä S., Slusky J. S., Lloris-Garcerá P., Rapp M., von Heijne G. (2010) Control of membrane protein topology by a single C-terminal residue. Science 328, 1698–1700 [DOI] [PubMed] [Google Scholar]
- 18. Kolbusz M. A., Slotboom D. J., Lolkema J. S. (2012) Role of individual positive charges in the membrane orientation and activity of transporters of the small multidrug resistance family. Biochemistry 51, 8867–8876 [DOI] [PubMed] [Google Scholar]
- 19. von Heijne G. (1992) Membrane protein structure prediction: hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225, 487–494 [DOI] [PubMed] [Google Scholar]
- 20. Heijne G. (1986) The distribution of positively charged residues in bacterial inner membrane proteins correlates with the trans-membrane topology. EMBO J. 5, 3021–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koebnik R. (1999) Structural and functional roles of the surface-exposed loops of the β-barrel membrane protein OmpA from Escherichia coli. J. Bacteriol. 181, 3688–3694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weinglass A. B., Kaback H. R. (2000) The central cytoplasmic loop of the major facilitator superfamily of transport proteins governs efficient membrane insertion. Proc. Natl. Acad. Sci. U.S.A. 97, 8938–8943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frillingos S., Gonzalez A., Kaback H. R. (1997) Cysteine-scanning mutagenesis of helix IV and the adjoining loops in the lactose permease of Escherichia coli: Glu126 and Arg144 are essential. Biochemistry 36, 14284–14290 [DOI] [PubMed] [Google Scholar]
- 24. Yamaguchi A., Kimura T., Someya Y., Sawai T. (1993) Metal-tetracycline/H+ antiporter of Escherichia coli encoded by transposon Tn10: the structural resemblance and functional difference in the role of the duplicated sequence motif between hydrophobic segments 2 and 3 and segments 8 and 9. J. Biol. Chem. 268, 6496–6504 [PubMed] [Google Scholar]
- 25. Yamaguchi A., Ono N., Akasaka T., Noumi T., Sawai T. (1990) Metal-tetracycline/H+ antiporter of Escherichia coli encoded by a transposon, Tn10: the role of the conserved dipeptide, Ser65-Asp66, in tetracycline transport. J. Biol. Chem. 265, 15525–15530 [PubMed] [Google Scholar]
- 26. Yamaguchi A., Someya Y., Sawai T. (1992) Metal-tetracycline/H+ antiporter of Escherichia coli encoded by transposon Tn10: the role of a conserved sequence motif, GXXXXRXGRR, in a putative cytoplasmic loop between helices 2 and 3. J. Biol. Chem. 267, 19155–19162 [PubMed] [Google Scholar]
- 27. Ditty J. L., Harwood C. S. (1999) Conserved cytoplasmic loops are important for both the transport and chemotaxis functions of PcaK, a protein from Pseudomonas putida with 12 membrane-spanning regions. J. Bacteriol. 181, 5068–5074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Banigan J. R., Gayen A., Traaseth N. J. (2013) Combination of 15N reverse labeling and afterglow spectroscopy for assigning membrane protein spectra by magic-angle-spinning solid-state NMR: application to the multidrug resistance protein EmrE. J. Biomol. NMR 55, 391–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Banigan J. R., Gayen A., Traaseth N. J. (2014) Correlating lipid bilayer fluidity with sensitivity and resolution of polytopic membrane protein spectra by solid-state NMR spectroscopy. Biochim. Biophys. Acta 10.1016/j.bbamem.2014.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stringer J. A., Bronnimann C. E., Mullen C. G., Zhou D. H., Stellfox S. A., Li Y., Williams E. H., Rienstra C. M. (2005) Reduction of RF-induced sample heating with a scroll coil resonator structure for solid-state NMR probes. J. Magn. Reson. 173, 40–48 [DOI] [PubMed] [Google Scholar]
- 31. Ammann C., Meier P., Merbach A. (1982) A simple multinuclear NMR thermometer. J. Magn. Reson. (1969) 46, 319–321 [Google Scholar]
- 32. Baldus M., Petkova A. T., Herzfeld J., Griffin R. G. (1998) Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol. Phys. 95, 1197–1207 [Google Scholar]
- 33. Baldus M., Geurts D. G., Hediger S., Meier B. H. (1996) Efficient 15N-13C polarization transfer by adiabatic-passage Hartmann-Hahn cross polarization. J. Magn. Reson. A 118, 140–144 [Google Scholar]
- 34. Franks W. T., Kloepper K. D., Wylie B. J., Rienstra C. M. (2007) Four-dimensional heteronuclear correlation experiments for chemical shift assignment of solid proteins. J. Biomol. NMR 39, 107–131 [DOI] [PubMed] [Google Scholar]
- 35. Szeverenyi N. M., Sullivan M. J., Maciel G. E. (1982) Observation of spin exchange by two-dimensional Fourier transform 13C cross polarization-magic-angle spinning. J. Magn. Reson. (1969) 47, 462–475 [Google Scholar]
- 36. Hohwy M., Rienstra C. M., Jaroniec C. P., Griffin R. G. (1999) Fivefold symmetric homonuclear dipolar recoupling in rotating solids: application to double quantum spectroscopy. J. Chem. Phys. 110, 7983–7992 [Google Scholar]
- 37. Gopinath T., Veglia G. (2012) 3D DUMAS: simultaneous acquisition of three-dimensional magic angle spinning solid-state NMR experiments of proteins. J. Magn. Reson. 220, 79–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takegoshi K., Nakamura S., Terao T. (2001) 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 344, 631–637 [Google Scholar]
- 39. Takegoshi K., Nakamura S., Terao T. (2003) 13C-1H dipolar-driven 13C-13C recoupling without 13C rf irradiation in nuclear magnetic resonance of rotating solids. J. Chem. Phys. 118, 2325–2341 [Google Scholar]
- 40. Shi L., Ahmed M. A. M., Zhang W., Whited G., Brown L. S., Ladizhansky V. (2009) Three-dimensional solid-state NMR study of a seven-helical integral membrane proton pump: structural insights. J. Mol. Biol. 386, 1078–1093 [DOI] [PubMed] [Google Scholar]
- 41. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 42. Salzmann M., Pervushin K., Wider G., Senn H., Wüthrich K. (1998) TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc. Natl. Acad. Sci. U.S.A. 95, 13585–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eletsky A., Kienhöfer A., Pervushin K. (2001) TROSY NMR with partially deuterated proteins. J. Biomol. NMR 20, 177–180 [DOI] [PubMed] [Google Scholar]
- 44. Schulte-Herbrüggen T., Sørensen O. W. (2000) Clean TROSY: compensation for relaxation-induced artifacts. J. Magn. Reson. 144, 123–128 [DOI] [PubMed] [Google Scholar]
- 45. Suzek B. E., Huang H., McGarvey P., Mazumder R., Wu C. H. (2007) UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23, 1282–1288 [DOI] [PubMed] [Google Scholar]
- 46. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Crooks G. E., Hon G., Chandonia J.-M., Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Parry-Smith D. J., Payne A. W. R., Michie A. D., Attwood T. K. (1998) CINEMA: a novel Colour INteractive Editor for Multiple Alignments. Gene 221, GC57–GC63 [DOI] [PubMed] [Google Scholar]
- 49. Monera O. D., Sereda T. J., Zhou N. E., Kay C. M., Hodges R. S. (1995) Relationship of sidechain hydrophobicity and α-helical propensity on the stability of the single-stranded amphipathic α-helix. J. Pept. Sci. 1, 319–329 [DOI] [PubMed] [Google Scholar]
- 50. Poulsen B. E., Cunningham F., Lee K. K. Y., Deber C. M. (2011) Modulation of substrate efflux in bacterial small multidrug resistance proteins by mutations at the dimer interface. J. Bacteriol. 193, 5929–5935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Poulsen B. E., Deber C. M. (2012) Drug efflux by a small multidrug resistance protein is inhibited by a transmembrane peptide. Antimicrob. Agents Chemother. 56, 3911–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lloris-Garcerá P., Slusky J. S. G., Seppälä S., Prieß M., Schäfer L. V., von Heijne G. (2013) In vivo Trp scanning of the small multidrug resistance protein EmrE confirms 3D structure models'. J. Mol. Biol. 425, 4642–4651 [DOI] [PubMed] [Google Scholar]
- 53. Li Y., Zhang Y. (2009) REMO: a new protocol to refine full atomic protein models from C-α traces by optimizing hydrogen-bonding networks. Proteins 76, 665–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cuff A. L., Janes R. W., Martin A. C. R. (2006) Analysing the ability to retain sidechain hydrogen-bonds in mutant proteins. Bioinformatics 22, 1464–1470 [DOI] [PubMed] [Google Scholar]
- 55. Jack D. L., Storms M. L., Tchieu J. H., Paulsen I. T., Saier M. H. (2000) A broad-specificity multidrug efflux pump requiring a pair of homologous SMR-type proteins. J. Bacteriol. 182, 2311–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wishart D. S., Sykes B. D., Richards F. M. (1991) Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222, 311–333 [DOI] [PubMed] [Google Scholar]
- 57. Wang Y., Jardetzky O. (2002) Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 11, 852–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Banigan J. R., Traaseth N. J. (2012) Utilizing afterglow magnetization from cross-polarization magic-angle-spinning solid-state NMR spectroscopy to obtain simultaneous heteronuclear multidimensional spectra. J. Phys. Chem. B 116, 7138–7144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Traaseth N. J., Veglia G. (2011) Frequency-selective heteronuclear dephasing and selective carbonyl labeling to deconvolute crowded spectra of membrane proteins by magic angle spinning NMR. J. Magn. Reson. 211, 18–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Morrison E. A., Henzler-Wildman K. A. (2014) Transported substrate determines exchange rate in the multidrug resistance transporter EmrE. J. Biol. Chem. 289, 6825–6836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Poget S. F., Harris R., Cahill S. M., Girvin M. E. (2010) 1H, 13C, 15N backbone NMR assignments of the Staphylococcus aureus small multidrug-resistance pump (Smr) in a functionally active conformation. Biomol. NMR Assign. 4, 139–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Berjanskii M. V., Wishart D. S. (2005) A simple method to predict protein flexibility using secondary chemical shifts. J. Am. Chem. Soc. 127, 14970–14971 [DOI] [PubMed] [Google Scholar]
- 64. Berjanskii M. V., Wishart D. S. (2008) Application of the random coil index to studying protein flexibility. J. Biomol. NMR 40, 31–48 [DOI] [PubMed] [Google Scholar]
- 65. Dutta S., Morrison E. A., Henzler-Wildman K. A. (2014) Blocking dynamics of the SMR transporter EmrE impairs efflux activity. Biophys. J 107, 613–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yerushalmi H., Mordoch S. S., Schuldiner S. (2001) A single carboxyl mutant of the multidrug transporter EmrE is fully functional. J. Biol. Chem. 276, 12744–12748 [DOI] [PubMed] [Google Scholar]
- 67. Yerushalmi H., Schuldiner S. (2000) An essential glutamyl residue in EmrE, a multidrug antiporter from Escherichia coli. J. Biol. Chem. 275, 5264–5269 [DOI] [PubMed] [Google Scholar]
- 68. Mordoch S. S., Granot D., Lebendiker M., Schuldiner S. (1999) Scanning cysteine accessibility of EmrE, an H+-coupled multidrug transporter from Escherichia coli, reveals a hydrophobic pathway for solutes. J. Biol. Chem. 274, 19480–19486 [DOI] [PubMed] [Google Scholar]
- 69. Zhang H., Neal S., Wishart D. (2003) RefDB: A database of uniformly referenced protein chemical shifts. J. Biomol. NMR 25, 173–195 [DOI] [PubMed] [Google Scholar]
- 70. Wishart D. S., Sykes B. D. (1994) Chemical shifts as a tool for structure determination. Methods Enzymol. 239, 363–392 [DOI] [PubMed] [Google Scholar]
- 71. Goddard T. D., Kneller D. G. (2006) SPARKY 3, University of California, San Francisco [Google Scholar]