

Background: Aggregation and fibrillation of amyloid β peptides (Aβ) in lipid bilayers lead to membrane disruption.
Results: Small vesicles accelerated Aβ fibrillation, whereas large vesicles promoted amorphous aggregation.
Conclusion: Moderate membrane-curvature-dependent interaction is an important factor for Aβ aggregation.
Significance: Two types of membrane binding, one leading to amyloid nucleation and the other to non-productive binding, may be common to various amyloidogenic proteins.
Keywords: Amyloid, Amyloid-β (AB), Lipid, Membrane, Protein Aggregation, Amyloid Fibrils, Supersaturation
Abstract
The deposition of amyloid β (Aβ) peptides is a pathological hallmark of Alzheimer disease. Aβ peptides were previously considered to interact specifically with ganglioside-containing membranes. Several studies have suggested that Aβ peptides also bind to phosphatidylcholine membranes, which lead to deformation of membranes and fibrillation of Aβ. Moreover, the role of membrane curvature, one type of deformation produced by binding of proteins to a membrane, in the binding and fibrillation of Aβ remains unclear. To clearly understand the relationship between the binding, consequent membrane deformation, and fibrillation of Aβ, we examined the amyloid fibrillation of Aβ-(1–40) in the presence of liposomes of various sizes. Membrane curvature increased with a decrease in the size of the liposomes. We used liposomes made of 1,2-dioleoyl-sn-glycero-3-phosphocholine to eliminate electrostatic effects. The results obtained showed that liposomes of smaller sizes (≤50 nm) significantly accelerated the nucleation step, thereby shortening the lag time of fibrillation. On the other hand, liposomes of larger sizes decreased the amount of fibrils but did not notably affect the lag time. The morphologies of fibrils, which were monitored by total internal reflection fluorescence microscopy, atomic force microscopy, and transmission electron microscopy, revealed that the length of Aβ-(1–40) fibrils became shorter and the amount of amorphous aggregates became larger as liposomes increased in size. These results suggest that the curvature of membranes coupled with an increase in water-accessible hydrophobic regions is important for binding and concentrating Aβ monomers, leading to amyloid nucleation. Furthermore, amyloid fibrillation on membranes may compete with non-productive binding to produce amorphous aggregates.
Introduction
Alzheimer disease is one of the most deleterious neurological diseases. The main cause of this disease has been attributed to the deposition of amyloid β (Aβ)3 peptides in the brain (1–3); therefore, a large number of studies have examined the mechanism underlying the fibrillation of Aβ peptides in vivo and in vitro. Aβ peptides are produced by the sequential cleavage of the amyloid precursor protein with β and γ secretases. Although Aβ peptides are initially disordered in solution, they fold to form an oligomeric nucleus, possibly with a large amount of cross-β structures. Elongation then occurs, resulting in the formation of mature amyloid fibrils (3–5). The structures and toxicities of oligomers, which are directly responsible for cytotoxicity, were the recent targets of studies on Alzheimer disease as well as other amyloidosis (6). In a previous study using NMR, it was reported that one of the initial structures of oligomers was a non-β-sheet structure containing 3–10 helix in solution (7).
Aβ peptide concentrations in vivo were shown to be a few nm (∼5 nm) (8–11), whereas fibrillation in vitro requires an order of magnitude higher concentration (3, 12). To clarify the reason for this difference, many studies proposed that Aβ peptides might interact with lipid membranes, which decreases diffusion dimensions and increases the local concentration of Aβ peptides (13–15). Various studies showed that specific membrane components (e.g. cholesterol, sphingomyelin, ganglioside, etc.) interact specifically with Aβ peptides, and this interaction has an important role in Aβ fibrillation (13, 16–20); however, other studies challenged this proposal (21, 22). Recently, a two-step mechanism of membrane disruption by Aβ peptides has been proposed in which (i) Aβ oligomers bind to the membrane to form ion-permeable pores, and (ii) the process of Aβ fibrillation causes membrane disruption mainly due to the interaction of Aβ fibrils with gangliosides (20, 23).
Amyloid fibrillation of various proteins, other than Aβ peptides, in lipid membranes has been extensively studied. Lipid membranes are known to be involved in the fibrillation of α-synuclein and human islet amyloid polypeptide, which are associated with Parkinson disease and type 2 diabetes, respectively (24–27). Various kinds of lipids that affect amyloid fibrillation have also been studied, and the findings obtained revealed that electrostatic interactions between proteins and membranes facilitated the binding of proteins to the membranes as well as their fibrillation (25, 28, 29). The size or radius of liposomes may also be an important factor for determining these interactions; α-synuclein binds to small unilamellar vesicles, and islet amyloid polypeptide interacts with high curvature membranes (30–32).
However, the role of membrane properties on the fibrillation of Aβ peptides has not yet been elucidated in detail. Previous studies using giant unilamellar vesicles of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) suggested that Aβ peptide binding caused membrane deformation (33, 34). A simulation study revealed that the accumulation of Aβ peptides deformed membranes to produce highly curved membranes (35).
Thus, the deformation of membranes may be one of the key factors that accelerate fibrillation. Moreover, the presence of Aβ peptides in the synapses in vivo caused a dysfunction in synaptic vesicles with a diameter of ∼40 nm (36). Because the binding of Aβ peptides to a membrane and the subsequent deformation are a linked phenomenon, the presence of deformed membranes may accelerate the binding and fibrillation of Aβ. Membrane curvature, one possible type of membrane deformation, can be controlled by changing the radius of liposomes; therefore, studies are needed to verify the relationship between membrane deformation, binding, and fibrillation.
We herein examined the effects of liposomes of various diameters on the amyloid fibrillation of Aβ-(1–40) to determine whether the fibrillation depends on the membrane curvature and consequent increase in water-accessible hydrophobic regions. We chose DOPC and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) as the liposome components because Aβ binding was reported to bend phosphocholine membranes (35), and phosphocholine, a zwitterionic lipid, allowed us to focus on the effects of membrane curvature while minimizing the effects of electrostatic interactions (16, 37). We previously observed that although Aβ-(1–40) is relatively easy to keep in monomeric states, Aβ-(1–42) forms nuclei for spontaneous fibrillation much faster than Aβ-(1–40) (38). Consequent explosive fibrillation of Aβ-(1–42) resulted in a large number of short fibrils, minimizing the interaction with the surface. Thus, to address the effects of membranes, we chose Aβ-(1–40), although it will be important to compare Aβ-(1–40) and Aβ-(1–42) to obtain further insight into the interactions of Aβ with membranes.
The results of the present study revealed that membrane curvature is an important factor for determining the amyloid fibrillation of Aβ-(1–40). Liposomes of larger sizes decreased the length of amyloid fibrils, and this was accompanied by an increase in the amount of amorphous aggregates. These results suggest that amyloid fibrillation on membranes competes with amorphous aggregation, thereby providing an insight into the mechanism underlying synaptic vesicle impairments.
EXPERIMENTAL PROCEDURES
Materials
Lyophilized Aβ-(1–40) purchased from Peptide Institute Inc. (Osaka, Japan) was dissolved in a 0.05% (w/w) ammonia solution at a concentration of 100 μm and stored at −80 °C. Phospholipids DOPC, POPC, 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rho-DOPE) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). Thioflavin T (ThT) and 8-anilino-1-naphthalenesulfonate (ANS) were obtained from Wako Pure Chemical Industries Ltd. (Osaka, Japan) and Sigma, respectively. All other reagents were obtained from Nacalai Tesque (Kyoto, Japan).
Preparation of Liposomes
Liposomes were prepared as previously described (39). Briefly, lipids (∼10 mm) were stored in methanol stock solutions. Lipid films were prepared by drying appropriate amounts of the stock solution in a stream of dry nitrogen followed by desiccation for at least 1 h to ensure removal of the organic solvent. Appropriate buffer solutions were added and vortexed to rehydrate the lipid films. After 10 freeze-thaw cycles, lipid suspensions were extruded 20 times through 50-, 100-, or 200-nm polycarbonate membranes (Whatman, Clifton, NJ) using a miniextruder (Avanti Polar Lipids, Inc., Alabaster, AL) just before use. Liposomes with a diameter of 30 nm were prepared by an ethanol injection method with slight modifications (40). Appropriate lipids (∼100 mm) in methanol were diluted to 10% (v/v). After vortexing, the lipid suspension in the tubes were set on a water bath-type ultrasonic transmitter with a temperature controller (ELESTEIN SP070-PG-M, Elekon Sci. Inc., Chiba, Japan) that applied repetitive ultrasonication pulses to samples from three directions (41). The temperature was set to 37 °C, and ultrasonic pulses were applied with repeats of 1 min of ultrasonication and 9 min of quiescence for 3 h. After ultrasonication, this suspension was centrifuged to precipitate multilamellar liposomes or/and aggregations of lipids followed by the collection of the supernatant. As this supernatant may contain methanol, it was dialyzed against an appropriate buffer twice. After dialysis, the concentration of lipids was quantified. Liposome sizes were measured by dynamic light scattering using DynaPro Titan (Wyatt Technology Co., Goleta, CA). Liposomes containing the fluorescence lipid (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl (Rho-DOPE)) (0.1% mol) or DOPS were prepared using the same procedure as described above.
Analytical Ultracentrifugation
The sedimentation velocity data were obtained by a Beckman-Coulter Optima XL-A analytical ultracentrifuge (Beckman Coulter, Miami, FL). The samples were first centrifuged at 3000 rpm (700 g) for 5 min to stabilize the absorbance and temperature. Next, centrifugation was performed at 60,000 rpm (250,000 × g) and 4 °C. Absorbance data at 220 nm were collected at an interval of 20 min at the radial increment of 0.003 cm in the continuous scanning mode. The absorbance data were analyzed by using sedfit to obtain the sedimentation coefficients. The parameters of solvent conditions (partial specific volume, density and viscosity) were calculated by using sednterp.
Total Internal Reflection Fluorescence Microscopy (TIRFM), Atomic Force Microscopy (AFM), and Transmission Electron Microscopy (TEM)
The prism-type TIRFM system was used to observe individual amyloid fibrils. The details of this system were described previously (42, 43). Briefly, an aliquot (14 μl) of sample solution was deposited on a quartz glass slide, and a fibril image was obtained with TIRFM. We used an argon laser (532 nm) and a helium-cadmium laser (442 nm) to excite ThT and rhodamine, respectively. AFM images were obtained using a Digital Instruments Nanoscope IIIa scanning microscope (Veeco Instruments Inc., Plainview, NY) as reported previously (44). TEM images were obtained using a HITACHI H-7650 transmission microscope (Hitachi, Tokyo, Japan) at 20 °C with a voltage of 80 kV and magnification of 30,000. The sample solution (5 μl) was spotted onto a collodion-coated copper grid (Nisshin EM Co., Tokyo, Japan). After 1 min, the remaining solution was removed with filter paper, and 5 μl 2% (w/w) uranyl acetate was spotted onto collodion-coated copper grids. After 1 min, the remaining solution was removed in the same manner.
Amyloid Fibrillation and ThT Fluorescence Assay
The formation of Aβ-(1–40) fibrils was examined at 10 μm in 10 mm sodium phosphate buffer (pH 7.5) containing 5 μm ThT, 100 mm NaCl, and DOPC or POPC liposomes at various concentrations. These solutions (200 μl) were added to the 96-well microplates (Greiner-Bio-One, Tokyo, Japan), and a seal was affixed (PowerSeal CRISTAl VIEW, Greiner-Bio-One). The plates were set on a microplate reader (SH-9000, Corona Electric Co., Ibaraki, Japan), and amyloid fibrillation was monitored by ThT fluorescence. It is noted that, although the morphology of Aβ fibrils is likely to depend on agitation of solution (45), we used the fibrillation conditions without agitation. The fluorescence intensity of ThT at 490 nm was measured with an excitation wavelength of 450 nm at 37 °C. The lag time of fibrillation was defined as the time at which ThT fluorescence reached 10% of the maximum.
ANS Assay to Evaluate Hydrophobicity
ANS fluorescence was measured using the Hitachi fluorescence spectrophotometer F4500 with the excitation wavelengths at 350 nm. The fluorescence of ANS was monitored by adding 2 μm ANS to the solution. The concentrations of liposomes and amyloid fibrils were 2 μm.
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) of Aβ-(1–40) monomers to the two different sizes of DOPC liposomes (30 nm and 100 nm) in 10 mm phosphate buffer (pH 7.5) containing 100 mm NaCl was performed by using a VP-ITC instrument (GE Healthcare) at 37 °C. The concentrations of Aβ-(1–40) in the syringe and liposomes in the cell were 100 μm and 5 mm, respectively. The reference power for the base line was set to 10 μcal/s, and the initial delay was 1800 s. Titration of Aβ-(1–40) was comprised of 23 injections with the spacing time of 900 s or 300 s and the filter period of 2 s. The injection volume was 12 μl for each, and the stirring speed was 242 rpm.
The heat of dilution (and mixing) of Aβ-(1–40) titrated to the same buffer was also measured. The apparent values of the change in enthalpy (appKa) and the association constant (appKa) were obtained by the fit of the binding isotherms to the one-site binding model incorporated in Origin 7.1. By using the relationships appΔGbind = −RTln(appKa) = appKa − TappΔSbind, the apparent thermodynamic values of the change in Gibbs free energy (appΔGbind) and entropy (appΔSbind) were further calculated. The apparent dissociation constant (appKd) was obtained based on the relation of appKa = 1/appKa. It should be noted that appKa values are more susceptible to the fit than appKa values.
RESULTS
Preparation of Liposomes and Aβ Solution
To investigate the effects of the liposome size on the fibrillation of Aβ-(1–40), we prepared liposomes of four sizes (∼200, 100, 50, and 30 nm) and analyzed the kinetics of fibrillation monitored by ThT fluorescence. We first confirmed the size of DOPC liposomes with TEM (Fig. 1, A–D) and dynamic light scattering (Fig. 1, E–H). The results obtained indicated that we could prepare liposome samples of distinct sizes with relatively small distributions. We call the respective preparations 30-, 50-, 100-, and 200-nm liposomes.
FIGURE 1.

Preparation of liposomes of various sizes and Aβ monomers. A–D, TEM images of liposomes of various diameters: 30 (A), 50 (B), 100 (C), and 200 nm (D). The scale bar represents 100 nm. E–H, distribution of liposome diameters determined by TEM and dynamic light scattering (DLS). The liposome diameters prepared were 30 (E), 50 (F), 100 (G), and 200 (H) nm. I, sedimentation boundary profiles of Aβ monomers. A sedimentation pattern was recorded at 60,000 rpm and 4 °C by monitoring the absorbance at 220 nm, and fitted traces at intervals of 20 min are shown. J, sedimentation coefficient distributions derived from sedimentation boundary profiles of Aβ monomers.
To check whether Aβ-(1–40) samples before the fibrillation experiments were monomeric, we conducted a sedimentation velocity measurement (Fig. 1, I and J). The sedimentation coefficient distribution for Aβ-(1–40) solution revealed a sharp peak at ∼0.3 S. Using this value, we calculated the molecular weight to be ∼4700, consistent with that of Aβ-(1–40) monomer, 4300.
The absence of oligomeric aggregates was further confirmed by native-PAGE followed by Western blotting with antibodies to Aβ-(1–40) monomers (data not shown). In addition, we observed the kinetics of Aβ-(1–40) fibrillation with samples with and without ultracentrifugation to remove possible aggregates, where we also checked the effects of 30- and 100-nm liposomes. We did not observe significant differences between Aβ-(1–40) samples with and without ultracentrifugation (data not shown). Therefore, we used Aβ-(1–40) preparations without ultracentrifugation assuming that we can address fibrillation of Aβ-(1–40) monomers.
Dependence of Fibrillation on the Liposome Concentration
We monitored the fibrillation of 10 μm Aβ-(1–40) in 10 mm phosphate buffer (pH 7.5) containing 100 mm NaCl and 5 μm ThT at 37 °C using amyloid-specific ThT fluorescence (46). It has been reported that Aβ-(1–40) fibrils with 2-fold symmetry with twisted morphology predominate when fibrils are grown in vitro with agitation of the Aβ-(1–40) solution, whereas fibrils with 3-fold symmetry with straight ribbon morphology predominate when fibrils are grown without agitation (45). Throughout the paper, we prepared the fibrils without agitation because agitation might affect the morphologies of liposomes.
Spontaneous fibrillation without agitation occurred with a lag time of 12 h (Fig. 2A). The straight and long morphology of mature fibrils was confirmed by TIRFM, AFM, and TEM (see below). These results were consistent with the findings of our previous studies (38, 47).
FIGURE 2.

Effects of 30 and 100 nm DOPC liposomes on amyloid fibrillation of Aβ-(1–40) at 37 °C without shaking. A and D, kinetics monitored by ThT fluorescence in the absence and presence of 30 nm (A) or 100 nm (D) DOPC liposomes at various concentrations at 37 °C. The concentrations of the liposomes are described in each figure. Amyloid fibrillation at the respective liposome concentrations was monitored at three wells, and the representative kinetics is shown. The insets show the average lag times. B, C, E, and F, dependences of the relative lag time (B and E) and maximal ThT amplitude (C and F) on the 30-nm (B and C) or 100-nm (E and F) DOPC liposome concentrations. Dotted lines are the values obtained in the absence of liposomes. The error bars and gray zones indicate the S.D. among three experiments.
To examine the effects of liposomes on the fibrillation of 10 μm Aβ-(1–40), we added 30- or 100-nm liposomes at 10, 50, 100, 250, 500, 750, and 1000 μm (Fig. 2, A and D). We observed a significant acceleration in fibrillation in the presence of 30-nm liposomes at 10 μm as revealed by a decrease in the lag time from ∼12 h to ∼7.6 h (Fig. 2A, inset): the relative lag time in 30-nm liposomes at 10 μm was 0.6 (Fig. 2B). Further increases in the liposome concentration slightly decreased the lag time. We compared the amount of fibrils by the ThT intensities at the end of the measurements (i.e. final ThT intensities at about 40 h). When the ThT intensity decreased after the maximum, we used the maximal ThT intensity. The final ThT intensity was similar to that in the absence of liposomes at all the liposome concentrations examined (Fig. 2C). In contrast, the presence of 100-nm liposomes at various concentrations slightly decreased the lag time (Fig. 2, D, inset, and E). The final ThT intensity in the presence of 100-nm liposomes was independent of the liposome concentration within an acceptable level of error (Fig. 2F).
These results suggested that 10 μm liposomes were sufficient to examine the effects of liposomes on the fibrillation of 10 μm Aβ-(1–40). The binding equilibrium between Aβ-(1–40) and liposomes that affected amyloid fibrillation may have been established even at low concentrations of liposomes. Thus, we used 10 μm DOPC liposomes to examine the effects of liposomes of various sizes on the fibrillation of Aβ-(1–40).
Dependence of Fibrillation on the Liposome Size
The kinetics of Aβ-(1–40) fibrillation was examined in the presence of 10 μm DOPC liposomes of various diameters (i.e. 30, 50, 100, and 200 nm) (Fig. 3A). The size of DOPC liposomes notably affected both the lag time and final ThT intensity (Fig. 3, C and D). The lag time was the shortest for the 30-nm liposomes and increased with the size of liposomes, returning to the value in the absence of liposomes at 100- or 200-nm liposomes (Fig. 3, A, inset, and C). On the other hand, the maximal ThT intensities observed in the presence of 30- or 50-nm liposomes were similar to that in the absence of liposomes and were larger than those in the presence of 100- or 200-nm liposomes (Fig. 3D). The slope of the elongation process after the lag time was slightly less steep in the presence of 100- or 200-nm DOPC liposomes (Fig. 3A). These results suggested that small liposomes accelerated fibrillation without changing the final amount of fibrils, whereas the larger liposomes decreased the amount of fibrils without an apparent decrease in the fibrillation rate.
FIGURE 3.

Effects of liposomes of various sizes on Aβ amyloid fibrillation. A and B, kinetics monitored by ThT fluorescence in the absence (black) or presence of DOPC (A) or POPC (B) liposomes of 30 (pink), 50 (yellow), 100 (green), or 200 nm (blue) at 37 °C without shaking. Liposome and Aβ concentrations were 10 μm. The insets show the average lag times. C and D, dependences of the relative lag time (C) and maximal ThT amplitude (D) on the liposome size. Dotted lines are the values obtained in the absence of liposomes. The error bars and gray zones indicate the S.D. among three experiments.
We performed the same experiments with POPC liposomes at 10 μm (Fig. 3B). Both the lag time and final ThT intensity revealed similar dependences on the size of liposomes to those observed for DOPC liposomes (Fig. 3, B, inset, C, and D), although the final ThT intensities of POPC liposomes were smaller than those of DOPC liposomes. The smaller final ThT intensities of POPC liposomes might be caused by the difference of the membrane surface properties due to the difference of acyl chains or of the temperature of gel-liquid crystalline phase transition; transition temperatures are −17 and −2 °C for DOPC and POPC membranes, respectively. Although there were some differences between DOPC and POPC, we could observe a similar effect of liposome size on the kinetics of Aβ-(1–40) fibrillation. These results suggested that the size-dependent effects of liposomes may be common to various liposomes. We then attempted to address why 30- and 50-nm liposomes significantly promoted the nucleation reaction, whereas larger liposomes reduced the maximal ThT value without notable effects on the lag time.
Hydrophobic Surface of Membranes Exposed to a Solvent
To clarify the mechanism underlying the promotion of nucleation by small liposomes, we assumed that Aβ-(1–40) interacted preferentially with the parts of membranes where water molecules could penetrate. The penetration depth of water representing water-accessible hydrophobic regions may depend on the size of the liposomes and may be larger (deeper) for small size liposomes. Smaller liposomes (≤50 nm) may have more of these regions than larger liposomes. A previous study reported that the insertion of the C terminus of Aβ peptides that contained a series of hydrophobic amino acids into membranes was crucial to the structural conversion of monomeric Aβ peptides to amyloid fibrils (48). Moreover, the interface between the head groups and acyl chains of lipids was shown to be responsible for the binding and conformational conversion of Aβ peptides (49, 50). Hence, a more detailed characterization of the relationship between the kinetics of Aβ fibrillation and hydrophobicity of liposomes will be important.
To examine this hypothesis, we assessed the water-accessible hydrophobic surface of liposomes by measuring ANS fluorescence at 2 μm in the presence of various sizes of DOPC liposomes at 2 μm (Fig. 4A). ANS is a hydrophobic dye, the fluorescence of which increases markedly in a hydrophobic environment, and is often used to detect water-accessible hydrophobic regions in the intermediates of protein folding (51). Using this assay, we speculated that we may be able to detect the exposure of the hydrophobic groups (i.e. acyl chains) of membranes.
FIGURE 4.

Exposed hydrophobicity of liposomes and its effects on the fibrillation of Aβ. A, fluorescence spectra of 2 μm ANS in the absence and presence of 2 μm DOPC liposomes and 2 μm Aβ fibrils. Black dotted line, ANS alone; black solid line, Aβ fibrils; blue dotted line, liposomes of 200 nm in diameter; blue solid line, Aβ fibrils and DOPC liposomes of 200 nm in diameter; magenta dotted line, liposomes of 30 nm in diameter; magenta solid line, Aβ fibrils and liposomes of 30 nm in diameter. B, ANS fluorescence (20 μm) at the maximal wavelength in the presence of 2 μm liposomes at various diameters in the presence (red) and absence (orange) of 2 μm Aβ fibrils. Blue circles indicate the difference between the presence and absence of Aβ fibrils. C, the relationship between the distance, d, between two head groups of lipids, and the diameter of DOPC liposomes. D and E, dependences of ANS fluorescence at 500 nm (D) and lag time (E) on d (distance). Dashed lines indicate the ANS fluorescence (B) or lag time (E) in the absence of liposomes. The error bars indicate the S.D. among three experiments. All experiments are conducted at 37 °C.
The fluorescence intensity of ANS was low at a maximum of 520 nm in the absence of liposomes (Fig. 4A). The fluorescence of ANS increased after the addition of 30-nm liposomes at 2 μm, with a blue shift observed in the maximum to 500 nm that suggested the partial burial of the dye in a hydrophobic environment. The increase observed in ANS fluorescence and the blue shift was less for larger liposomes. (Fig. 4, A and B). These results indicated that the water-accessible hydrophobic regions (or solvent penetration) depended on the size of liposomes; smaller liposomes exposed more hydrophobic regions to the solvent than larger liposomes.
To address the dependence of ANS fluorescence on the liposome size, we calculated the distance between two head groups of lipids, d. As shown in Fig. 4C, we assumed a simplified model in which the inner leaflet was ignored, and the lipid of the outer leaflet was represented by a rectangle. According to this model, the inside circumference was π(R-2l), and the number of rectangles was represented by n = π(R-2l)/A0.5, where R, l, and A are the diameter of a liposome, the height of a lipid, and occupied area for each lipid molecule, respectively. Consequently, d was calculated by d = πR/n − A0.5. The estimated value of d was plotted against R (Fig. 4C), which showed that d was inversely proportional to R. The dependence of d on the diameter of liposomes was consistent with that of the ANS fluorescence of DOPC liposomes with a linear relationship between the ANS fluorescence and d, thereby arguing the critical role of membrane curvature (Fig. 4D). Importantly, the lag time of amyloid fibrillation showed a linear relationship with d, which indicated that membrane curvature and consequently water-accessible hydrophobic surfaces were critical for determining the lag time (Fig. 4E).
On the other hand, the fluorescence of ANS increased in the presence of 2 μm Aβ-(1–40) fibrils, with a slight blue shift observed in the maximum to 510 nm (Fig. 4A). Thus, the fluorescence of ANS may also be useful for monitoring conformational changes in Aβ-(1–40) as well as those of lipids. A further blue shift in the fluorescence of ANS to 480 nm occurred after the addition of 30-nm liposomes at 2 μm, and this was accompanied by an increase in the intensity of fluorescence. The subtracted fluorescence spectrum suggested that the binding of Aβ-(1–40) fibrils to membranes led to the additional binding of ANS (or a more hydrophobic environment on the prebound ANS molecules). The extent of the blue shift and increase in fluorescence intensity were less after the addition of 100-nm liposomes at 2 μm, suggesting that additional ANS binding may be less for larger liposomes.
Fibril Morphology
We examined the morphologies of fibrils prepared in the presence of DOPC liposomes with various diameters using TIRFM, AFM, and TEM (Fig. 5). We used liposomes in which rhodamine-labeled lipids were included for TIRFM so that we could also monitor the location of the liposomes. Liposomes were deformed by interaction with surfaces; therefore, the exact morphology of liposomes could not be detected by our measurements. The three methodologies covered a wide range of dimensions from nm to μm.
FIGURE 5.
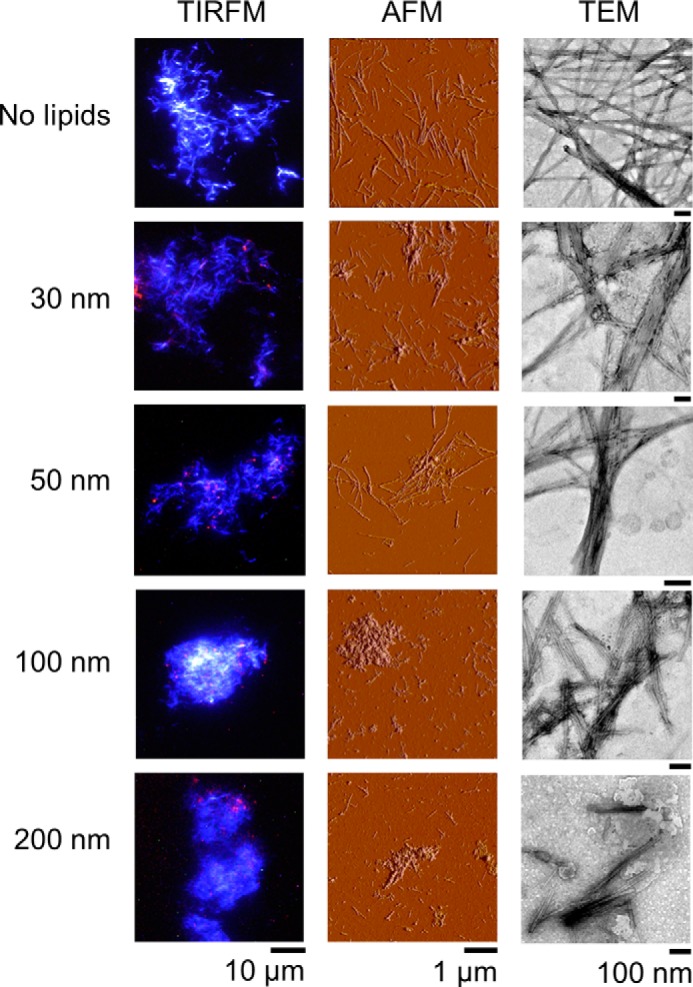
TIRFM, AFM, and TEM images of fibrils under DOPC liposomes of various sizes. The concentrations of Aβ and liposomes were 10 μm. In TIRFM images, fibrils and liposomes were monitored separately by ThT (blue) and rhodamine (red), respectively. The scale bars of TIRFM, AFM, and TEM are 10 μm, 1 μm, and 100 nm, respectively.
In the absence of liposomes, Aβ-(1–40) fibrils were straight with a length up to several μm. They were well dispersed without clustering. Fibrils were still long and dispersed in the presence of 30- or 50-nm liposomes, although some clusters and fibril clumps were observed. In the TIRFM images, the locations of liposomes revealed by rhodamine fluorescence always overlapped with amyloid fibrils, which indicated that the liposomes were the sites of amyloid nucleation. The sizes of liposomes were markedly smaller than those of the fibrils.
AFM images indicated that fibrils became shorter and amorphous aggregates accumulated, with an increase in the size of liposomes, as was evident from AFM images obtained in the presence of 100- and 200-nm liposomes. TEM images in the presence of 100- and 200-nm liposomes revealed that fibrils associated with the ruptured liposomes. TIRFM images in the presence of 100- and 200-nm liposomes showed the significant clustering of fibrils. TEM images in the presence of large liposomes showed thick and short fibrils. Taken together, the morphologies of Aβ-(1–40) fibrils observed by TIRFM, AFM, and TEM depended on the size of the coexisting liposomes, which indicated that fibrils became shorter and clustered with an increase in the size of the liposomes, and the amount of amorphous aggregates increased.
Binding of Aβ-(1–40) to Liposomes Characterized by ITC
To examine the interactions between Aβ-(1–40) and DOPC liposomes, we performed ITC measurements (Fig. 6). Liposomes with the sizes of 30 nm and 100 nm were chosen to address the effects of high and low curvatures, respectively, on Aβ-(1–40) fibrillation. Consecutive titrations of Aβ-(1–40) to 30-nm liposomes in the ITC cell showed the positive ITC peaks with gradual decreases in the intensity (Fig. 6A). The heats of dilution and mixing of Aβ-(1–40) to buffer were much smaller than those of Aβ-(1–40) titrations to liposomes (Fig. 6A, inset). Thus, endothermic reactions indicated intermolecular interactions between Aβ-(1–40) and 30-nm liposomes.
FIGURE 6.

Isothermal titration calorimetry of DOPC liposomes with Aβ-(1–40). A and B, ITC thermograms (upper panel) and binding isotherms (lower panel) on titrating 30-nm (A) or 100-nm (B) liposomes with Aβ-(1–40) are shown. ITC thermograms were obtained after the base-line correction. The spacing time between the first and second titrations was 900 s in A. The inset in A below the thermogram of Aβ titration indicates the small endothermic heat of dilution and mixing on Aβ-(1–40) to buffer. The same scale of heat was used for comparison. The concentrations of liposomes were expressed by the molar ratio in the binding isotherms of those of lipid monomers based on the study by Otzen and co-workers (53). The solid lines in the lower panels indicate the fitted curves.
On the other hand, ITC thermogram of Aβ-(1–40) titrations to 100-nm liposomes showed exothermic peaks that decreased gradually in amplitude (Fig. 6B). The extents of heat were larger than those observed when Aβ-(1–40) was titrated to 30-nm liposomes (Fig. 6A). These data suggested that although Aβ-(1–40) was bound to both of 30- and 100-nm liposomes, the detailed mechanisms were distinct.
To obtain further insights into interactions between Aβ-(1–40) and liposomes, we evaluated apparent thermodynamic parameters based on analyses of binding isotherms. The values of appKd of Aβ-(1–40) for 30-nm and 100-nm liposomes were 67.1 μm and 36.4 μm, respectively. These values were smaller than those of α-synuclein binding to the liposomes, which mimicked presynaptic membranes (appKd = 261 μm), or the liposomes, which consisted of 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (appKd = 93 μm) (52). Accordingly, appΔGbind (−6.3 kcal/mol) of Aβ to 100-nm liposome was smaller than that of Aβ to the 30-nm liposomes (−5.9 kcal/mol). These indicated that apparent binding affinity of Aβ-(1–40) to DOPC liposomes was slightly stronger than those of α-synuclein to model liposomes and that affinity of Aβ to 100-nm liposome was slightly stronger than that to 30-nm liposome.
Interestingly, the sign and amplitude of appKa and appΔSbind were different depending on the size of liposomes. The Aβ-30-nm liposome binding was driven by the positive entropy change (T appΔSbind = 14.3 kcal/mol) to overcome the unfavorable endothermic reaction (appKa = 8.4 kcal/mol). On the contrary, the Aβ-100-nm liposome binding was favored by the negative enthalpy change (appKa = −62.0 kcal/mol) to overcome the entropy loss (T appΔSbind = −55.7 kcal/mol). The large value of appKa in the Aβ-100-nm liposome binding may imply the complex mechanism in comparison with the Aβ-30-nm liposome binding. Similar complicated ITC results were observed for the α-synuclein binding to liposomes consist of 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (53).
Moreover, biphasic patterns of each ITC peak in titration of Aβ to 100-nm liposomes suggested apparent two processes; the fast process with a sharp and intense peak was followed by the slow process with a broad and low intensity peak (Fig. 6B). These indicated that binding of Aβ to 100-nm liposomes was not a simple intermolecular interaction, although further studies are required to clarify the details of the liposome size-dependent binding of Aβ.
As can be seen in the ITC thermograms (Fig. 6), the binding reactions between Aβ-(1–40) and liposomes did not saturate, which brought uncertainty of the fitting. Thus, the error values of Ka were large. Calculation of other thermodynamic parameters by using Ka with the large error further increased uncertainty. To obtain a saturated curve, which can decrease errors, it was required to increase the concentrations of Aβ-(1–40) in the syringe. However, the concentration of Aβ-(1–40) (100 μm) used was already high, and it was not practical to further increase the Aβ-(1–40) concentration due to the formation of nonspecific aggregations. Although decreasing temperature to delay nonspecific aggregation might be an alternative possibility at higher Aβ-(1–40) concentrations, there were concerns about changing the binding mode and/or aggregation pathway compared with those at 37 °C. Thus, we conclude that although the errors were large, the ITC results clearly showed distinct bindings of Aβ-(1–40) to the two kinds of liposomes and suggested that the binding mode and aggregation pathway of Aβ-(1–40) depend on the degree of membrane curvature.
Effects of Anionic Phospholipids on the Fibrillation
To address the role of electrostatic interactions between Aβ-(1–40) and membranes in fibrillation, we examined the effects of liposomes composed of DOPC and DOPS at a ratio of DOPC:DOPS = 1:1 (i.e. 50% DOPS liposomes). DOPS is one of anionic phospholipids reported to accelerate fibrillation of Aβ-(1–40), and it has been suggested that phosphatidylserine groups of DOPS liposomes interact with positive-charged Lys residue at the C terminus of Aβ-(1–40) (37). However, the dependence on the size of liposomes has not been examined. We monitored the fibrillation Aβ-(1–40) in the presence of 50% DOPS liposomes of various sizes (Fig. 7A). Only the 30-nm liposomes with 50% DOPS shortened the lag time (Fig. 7B).
FIGURE 7.

Effects of DOPS on the DOPC-dependent fibrillation of Aβ-(1–40). A and B, the kinetics of Aβ-(1–40) fibrillation in the presence of 50% DOPS liposomes of 30 (pink), 50 (yellow), 100 (green), or 200 (blue) nm in diameter or without lipids (black) (A) and the dependence of relative lag time on the liposome size (B). The inset in A represents the average lag time at various liposome sizes. C and D, the kinetics of Aβ(1–40) in the presence of 100% DOPC (pink), 50% DOPS (purple), and 100% DOPS (blue) liposomes of 30 nm in diameter (C) and the dependence of relative lag time on the DOPC content (D). The inset in C represents the average lag times at different contents of DOPS.
We then prepared the 30-nm liposomes composed of 100% DOPC, 50% DOPC and 50% DOPS, or 100% DOPS and monitored the fibrillation of Aβ-(1–40) in the presence of these liposomes (Fig. 7, C and D). The liposomes containing 50% DOPS promoted the nucleation reaction as well as 100% DOPC liposomes. However, the degree of promotion of DOPS liposomes was small.
Thus, although the DOPS liposomes were reported to accelerate the Aβ-(1–40) fibrillation (37), we saw no clear acceleration under our conditions. On the contrary, we observed slightly adverse effects to suppress the accelerating effects produced by small DOPC liposomes. It is possible that, under our experimental conditions, the electrostatic interaction between the negative-charged head group of PS and positive-charged amino acid residues of Aβ-(1–40) tightly trapped the Aβ-(1–40) molecules on liposomes so that further nucleation became difficult.
DISCUSSION
We herein showed that DOPC liposomes markedly accelerated the fibrillation of Aβ-(1–40) and that these effects depended on the size of liposomes even at the same total concentration of phospholipids; smaller liposomes more strongly accelerated fibrillation. Acceleration was suppressed by an increase in the liposome size. The amount of fibrils decreased, and that of amorphous aggregates increased.
To understand these distinct effects of liposomes on fibrillation, we assumed two types of interactions between Aβ-(1–40) and liposomes; one was productive binding leading to nucleation and fibrillation, and the other was non-productive binding that formed amorphous aggregates (Fig. 8, A and B). An analysis of the water-accessible hydrophobicity of liposomes probed by ANS binding and estimation of curvature suggested that small liposomes with higher surface curvature had more water-accessible hydrophobic regions and were effective for producing amyloid nucleation. Small liposomes with larger water-accessible hydrophobic regions interact with Aβ monomers weakly and concentrate them adequately to initiate nucleation. Although the binding of Aβ-(1–40) to larger liposomes could also occur, it may be non-productive for amyloid nucleation because the local concentration was not sufficiently high to break supersaturation. Alternatively, although the local concentration was high, bound Aβ-(1–40) molecules were tightly trapped on the surface of membranes and, thus, were unable to diffuse and transform to seed competent conformations. The ITC measurements showed that the affinity of Aβ to the 100-nm liposome was slightly stronger than that to the 30-nm liposome, consistent with the tighter binding and trapping of the Aβ-(1–40) on the larger liposomes. These two types of interactions may propagate beyond liposome surfaces, leading to amyloid fibrillation or amorphous aggregation in bulk solvent.
FIGURE 8.

Schematic models of amyloid nucleation and amorphous aggregation on the surface of liposome membranes. A and B, schematic mechanism of the Aβ-(1–40) fibrillation in the presence of liposomes. In the presence of smaller liposomes, Aβ-(1–40) interacts with the surface of membrane weakly, and this weak interaction promotes the nucleation. Aβ-(1–40) is easy to remove due to this interaction and the elongation reaction is similar to without lipids (A). In the presence of larger liposomes, Aβ-(1–40) interacts with the surface of membrane more strongly than that of smaller liposomes. This stronger interaction makes it difficult to induce nucleation and leads some Aβ peptides to amorphous aggregates. Other Aβ peptides exist in medium, and they form amyloid fibrils or amorphous aggregates (B). C–E, schematic phase diagram of conformational states of Aβ-(1–40) in the absence (C) or presence of small (D) or large vesicles (E). The locations of the protein solutions as studied here are indicated by dots.
The competitive mechanism between productive and non-productive binding (Fig. 8A) was analogous to the competition between amyloid fibrillation and amorphous aggregation in the absence of liposomes, as represented by a phase diagram of amyloid fibrillation dependent on salt and protein concentrations. Amyloid fibrils have been shown to form in the supersaturated solutions of responsible proteins via a nucleation and growth mechanism (41, 54–56). Supersaturated regions can be classified into two types depending on the stability of supersaturation (Fig. 8C) (41). Supersaturation was remarkably stable in the metastable region, at which the solute concentration was slightly above the solubility, and seeding was essential to break supersaturation and make fibrils. Spontaneous fibrillation could occur after a certain lag time in the labile region, in which the driving force of precipitation increased. Various kinds of agitations such as shaking (57), stirring (58), or ultrasonic irradiation (44, 59, 60) have been shown to effectively force spontaneous fibrillation under these supersaturated regions. One possible mechanism responsible for agitation-induced fibrillation is the concentration of amyloidogenic proteins at the hydrophobic air-water interface produced by agitation (41, 61). Ultrasonication is known to produce cavitation bubbles with hydrophobic surfaces, and stirring or shaking will also increase the hydrophobic air-water interface. The hydrophobic air-water interface adsorbs proteins, concentrates them, and produces various aggregates including seed competent aggregates. Once amyloid fibrillation starts on the seeds, it even propagates to bulk regions without a hydrophobic surface.
Water-accessible hydrophobic surfaces produced by membrane curvature may play a similar role in membrane-assisted amyloid fibrillation to the air-water hydrophobic surface of cavitation bubbles induced by ultrasonic irradiation. Aβ monomers may be concentrated effectively by absorption to the hydrophobic surface due to membrane curvature. Thus, the membrane-assisted acceleration of fibrillation might be similar to the agitation-induced acceleration of fibrillation in the labile region (Fig. 8).
In the phase diagram excessively strong precipitation forces produced glassy amorphous aggregates in which molecules were trapped in non-amyloidogenic conformations. One possible mechanism underlying aggregation is that promiscuous interactions between molecules produce various conformational states. In a similar way, various types of interactions on the membrane may trap the monomers, preventing rearrangements to seed-competent conformations. The evenly dispersed charged head groups of phospholipids may contribute to preventing the bound Aβ monomers from associating to the seed-competent conformation. Thus, we considered that membrane-assisted amorphous aggregation corresponded to amorphous aggregation under strong forces of precipitation (Fig. 8).
We previously reported the amyloid fibrillation of Aβ-(1–40) in real time at a single fibrillar level at pH 7.0 with TIRFM combined with the use of ThT (62). We used the surfaces of various chemically modified substrates. Extensive fibrillation was generally observed on surfaces with negative charges. In contrast, fibril growth was largely suppressed on positively charged or hydrophobic surfaces. A previous study reported that Aβ fibrillation was facilitated by negatively charged acidic phospholipids rather than neutral phospholipids (63). Because the pI value of Aβ is ∼5, the net charge was slightly negative under neutral pH conditions. These results indicated that strong electrostatic attraction tightly trapped the Aβ-(1–40) monomers on the surface so that subsequent lateral movement and nucleation were inhibited. The inhibitory role of a strongly hydrophobic surface can be also explained in a similar manner assuming that the hydrophobic surface interacts with the hydrophobic residues of Aβ-(1–40). These results support the view that there are two types of membrane binding, one leading to amyloid fibrillation and the other to amorphous aggregation.
Finally, although lipid rafts have been considered to play an important role in Aβ fibrillation (13, 16–19), the underlying mechanism remains unclear (21, 22). It is possible that the lipid raft carries the site of productive binding, as proposed here more than the other lipid membranes. Our proposal that there are two types of membrane binding for amyloidogenic proteins, one leading to amyloid nucleation and the other to non-productive binding and amorphous trapping, may be common for various amyloidogenic proteins. The specific removal of amyloidogenic proteins may be possible by preparing liposomes with only non-productive binding.
Acknowledgment
M. S. T. is indebted to Dr. T. Terakawa for helpful discussions and continuous encouragement.
This work was supported by the Ministry of Education, Science, Sports, and Culture, Japan, a grant-in-aid for scientific research (to Y. G.), and a grant-in-aid for the Japan Society for the Promotion of Science Fellows (to M. S. T.). We acknowledge financial support from Takeda Science Foundation (to H. Y.).
- Aβ
- amyloid β
- Aβ-(1–40)
- amyloid β peptide composed of 1–40 residues
- AFM
- atomic force microscopy
- ANS
- 8-anilino-1-naphthalenesulfonate
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- DOPS
- 1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- TEM
- transmission electron microscopy
- ThT
- thioflavin T
- TIRFM
- total internal reflection fluorescence microscopy
- ITC
- isothermal titration calorimetry.
REFERENCES
- 1. Dobson C. M. (2003) Protein folding and misfolding. Nature 426, 884–890 [DOI] [PubMed] [Google Scholar]
- 2. Halverson K., Fraser P. E., Kirschner D. A., Lansbury P. T., Jr. (1990) Molecular determinants of amyloid deposition in Alzheimer's disease: conformational studies of synthetic β-protein fragments. Biochemistry 29, 2639–2644 [DOI] [PubMed] [Google Scholar]
- 3. Cohen S. I., Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., Knowles T. P. (2013) Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lomakin A., Chung D. S., Benedek G. B., Kirschner D. A., Teplow D. B. (1996) On the nucleation and growth of amyloid β-protein fibrils: detection of nuclei and quantitation of rate constants. Proc. Natl. Acad. Sci. U.S.A. 93, 1125–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. LaFerla F. M., Green K. N., Oddo S. (2007) Intracellular amyloid-β in Alzheimer's disease. Nat. Rev. Neurosci. 8, 499–509 [DOI] [PubMed] [Google Scholar]
- 6. Bemporad F., Chiti F. (2012) Protein misfolded oligomers: experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem. Biol. 19, 315–327 [DOI] [PubMed] [Google Scholar]
- 7. Vivekanandan S., Brender J. R., Lee S. Y., Ramamoorthy A. (2011) A partially folded structure of amyloid-β(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun. 411, 312–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Selkoe D. J. (2003) Folding proteins in fatal ways. Nature 426, 900–904 [DOI] [PubMed] [Google Scholar]
- 9. Hu X., Crick S. L., Bu G., Frieden C., Pappu R. V., Lee J. M. (2009) Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-β peptide. Proc. Natl. Acad. Sci. U.S.A. 106, 20324–20329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haass C., Schlossmacher M. G., Hung A. Y., Vigo-Pelfrey C., Mellon A., Ostaszewski B. L., Lieberburg I., Koo E. H., Schenk D., Teplow D. B. (1992) Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 359, 322–325 [DOI] [PubMed] [Google Scholar]
- 11. Seubert P., Oltersdorf T., Lee M. G., Barbour R., Blomquist C., Davis D. L., Bryant K., Fritz L. C., Galasko D., Thal L. J. (1993) Secretion of β-amyloid precursor protein cleaved at the amino terminus of the β-amyloid peptide. Nature 361, 260–263 [DOI] [PubMed] [Google Scholar]
- 12. Yagi H., Hasegawa K., Yoshimura Y., Goto Y. (2013) Acceleration of the depolymerization of amyloid β fibrils by ultrasonication. Biochim. Biophys. Acta 1834, 2480–2485 [DOI] [PubMed] [Google Scholar]
- 13. Yanagisawa K., Odaka A., Suzuki N., Ihara Y. (1995) GM1 ganglioside-bound amyloid β-protein (Aβ): a possible form of preamyloid in Alzheimer's disease. Nat Med. 1, 1062–1066 [DOI] [PubMed] [Google Scholar]
- 14. Aisenbrey C., Borowik T., Byström R., Bokvist M., Lindström F., Misiak H., Sani M. A., Gröbner G. (2008) How is protein aggregation in amyloidogenic diseases modulated by biological membranes? Eur. Biophys. J. 37, 247–255 [DOI] [PubMed] [Google Scholar]
- 15. Bokvist M., Gröbner G. (2007) Misfolding of amyloidogenic proteins at membrane surfaces: the impact of macromolecular crowding. J. Am. Chem. Soc. 129, 14848–14849 [DOI] [PubMed] [Google Scholar]
- 16. Gorbenko G. P., Kinnunen P. K. (2006) The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids 141, 72–82 [DOI] [PubMed] [Google Scholar]
- 17. Rushworth J. V., Hooper N. M. (2010) Lipid rafts: linking Alzheimer's amyloid-β production, aggregation, and toxicity at neuronal membranes. Int. J. Alzheimers Dis. 2011, 603052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kakio A., Nishimoto S. I., Yanagisawa K., Kozutsumi Y., Matsuzaki K. (2001) Cholesterol-dependent formation of GM1 ganglioside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J. Biol. Chem. 276, 24985–24990 [DOI] [PubMed] [Google Scholar]
- 19. Matsuzaki K., Horikiri C. (1999) Interactions of amyloid β-peptide (1–40) with ganglioside-containing membranes. Biochemistry 38, 4137–4142 [DOI] [PubMed] [Google Scholar]
- 20. Sciacca M. F., Kotler S. A., Brender J. R., Chen J., Lee D. K., Ramamoorthy A. (2012) Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys. J. 103, 702–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamada T., Morita M., Kishimoto Y., Komatsu Y., Vestergaard M. d., Takagi M. (2010) Biomimetic microdroplet membrane interface: detection of the lateral localization of amyloid β peptides. J. Phys. Chem. Lett. 1, 170–173 [Google Scholar]
- 22. Morita M., Hamada T., Tendo Y., Hata T., Vestergaard M. d. C., Takagi M. (2012) Selective localization of Alzheimer's amyloid β in membrane lateral compartments. Soft Matter 8, 2816–2819 [Google Scholar]
- 23. Kotler S. A., Walsh P., Brender J. R., Ramamoorthy A. (2014) Differences between amyloid-β aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer's disease. Chem. Soc. Rev. 43, 6692–6700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reynolds N. P., Soragni A., Rabe M., Verdes D., Liverani E., Handschin S., Riek R., Seeger S. (2011) Mechanism of membrane interaction and disruption by α-synuclein. J. Am. Chem. Soc. 133, 19366–19375 [DOI] [PubMed] [Google Scholar]
- 25. Pranke I. M., Morello V., Bigay J., Gibson K., Verbavatz J. M., Antonny B., Jackson C. L. (2011) α-Synuclein and ALPS motifs are membrane curvature sensors whose contrasting chemistry mediates selective vesicle binding. J. Cell Biol. 194, 89–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sasahara K., Morigaki K., Okazaki T., Hamada D. (2012) Binding of islet amyloid polypeptide to supported lipid bilayers and amyloid aggregation at the membranes. Biochemistry 51, 6908–6919 [DOI] [PubMed] [Google Scholar]
- 27. Brender J. R., Salamekh S., Ramamoorthy A. (2012) Membrane disruption and early events in the aggregation of the diabetes related peptide IAPP from a molecular perspective. Acc. Chem. Res. 45, 454–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pfefferkorn C. M., Heinrich F., Sodt A. J., Maltsev A. S., Pastor R. W., Lee J. C. (2012) Depth of α-synuclein in a bilayer determined by fluorescence, neutron reflectometry, and computation. Biophys. J. 102, 613–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Knight J. D., Miranker A. D. (2004) Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 341, 1175–1187 [DOI] [PubMed] [Google Scholar]
- 30. Mizuno N., Varkey J., Kegulian N. C., Hegde B. G., Cheng N., Langen R., Steven A. C. (2012) Remodeling of lipid vesicles into cylindrical micelles by α-synuclein in an extended α-helical conformation. J. Biol. Chem. 287, 29301–29311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jiang Z., de Messieres M., Lee J. C. (2013) Membrane remodeling by α-synuclein and effects on amyloid formation. J. Am. Chem. Soc. 135, 15970–15973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smith P. E., Brender J. R., Ramamoorthy A. (2009) Induction of negative curvature as a mechanism of cell toxicity by amyloidogenic peptides: the case of oslet amyloid polypeptide. J. Am. Chem. Soc. 131, 4470–4478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun Y., Lee C. C., Chen T. H., Huang H. W. (2010) Kinetic process of β-amyloid formation via membrane binding. Biophys. J. 99, 544–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ambroggio E. E., Kim D. H., Separovic F., Barrow C. J., Barnham K. J., Bagatolli L. A., Fidelio G. D. (2005) Surface behavior and lipid interaction of Alzheimer β-amyloid peptide 1–42: a membrane-disrupting peptide. Biophys. J. 88, 2706–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pannuzzo M., Milardi D., Raudino A., Karttunen M., La Rosa C. (2013) Analytical model and multiscale simulations of Aβ peptide aggregation in lipid membranes: towards a unifying description of conformational transitions, oligomerization and membrane damage. Phys. Chem. Chem. Phys. 15, 8940–8951 [DOI] [PubMed] [Google Scholar]
- 36. Qu L., Akbergenova Y., Hu Y., Schikorski T. (2009) Synapse-to-synapse variation in mean synaptic vesicle size and its relationship with synaptic morphology and function. J. Comp. Neurol. 514, 343–352 [DOI] [PubMed] [Google Scholar]
- 37. Chauhan A., Ray I., Chauhan V. P. (2000) Interaction of amyloid β-protein with anionic phospholipids: possible involvement of Lys-28 and C-terminus aliphatic amino acids. Neurochem. Res. 25, 423–429 [DOI] [PubMed] [Google Scholar]
- 38. Yagi H., Ban T., Morigaki K., Naiki H., Goto Y. (2007) Visualization and classification of amyloid β supramolecular assemblies. Biochemistry 46, 15009–15017 [DOI] [PubMed] [Google Scholar]
- 39. Tomas S., Milanesi L. (2010) Mutual modulation between membrane-embedded receptor clustering and ligand binding in lipid membranes. Nat. Chem. 2, 1077–1083 [DOI] [PubMed] [Google Scholar]
- 40. Kremer J. M., Esker M. W., Pathmamanoharan C., Wiersema P. H. (1977) Vesicles variable diameter prepared by a modified injection. Biochemistry 16, 3932–3935 [DOI] [PubMed] [Google Scholar]
- 41. Yoshimura Y., Lin Y., Yagi H., Lee Y. H., Kitayama H., Sakurai K., So M., Ogi H., Naiki H., Goto Y. (2012) Distinguishing crystal-like amyloid fibrils and glass-like amorphous aggregates from their kinetics of formation. Proc. Natl. Acad. Sci. U.S.A. 109, 14446–14451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ban T., Hamada D., Hasegawa K., Naiki H., Goto Y. (2003) Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence. J. Biol. Chem. 278, 16462–16465 [DOI] [PubMed] [Google Scholar]
- 43. Ban T., Goto Y. (2006) Direct observation of amyloid growth monitored by total internal reflection fluorescence microscopy. Methods Enzymol. 413, 91–102 [DOI] [PubMed] [Google Scholar]
- 44. Chatani E., Lee Y. H., Yagi H., Yoshimura Y., Naiki H., Goto Y. (2009) Ultrasonication-dependent production and breakdown lead to minimum-sized amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 106, 11119–11124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paravastu A. K., Leapman R. D., Yau W. M., Tycko R. (2008) Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Naiki H., Higuchi K., Hosokawa M., Takeda T. (1989) Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavine T. Anal. Biochem. 177, 244–249 [DOI] [PubMed] [Google Scholar]
- 47. Ban T., Hoshino M., Takahashi S., Hamada D., Hasegawa K., Naiki H., Goto Y. (2004) Direct observation of Aβ amyloid fibril growth and inhibition. J. Mol. Biol. 344, 757–767 [DOI] [PubMed] [Google Scholar]
- 48. Jarrett J. T., Berger E. P., Lansbury P. T., Jr. (1993) The carboxy terminus of the β amyloid protein Is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697 [DOI] [PubMed] [Google Scholar]
- 49. Yagi-Utsumi M., Matsuo K., Yanagisawa K., Gekko K., Kato K. (2010) Spectroscopic characterization of intermolecular interaction of amyloid β promoted on GM1 micelles. Int. J. Alzheimers Dis. 2011, 925073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shimanouchi T., Sasaki M., Hiroiwa A., Yoshimoto N., Miyagawa K., Umakoshi H., Kuboi R. (2011) Relationship between the mobility of phosphocholine headgroups of liposomes and the hydrophobicity at the membrane interface: a characterization with spectrophotometric measurements. Colloids Surf. B. Biointerfaces 88, 221–230 [DOI] [PubMed] [Google Scholar]
- 51. Goto Y., Fink A. L. (1989) Conformational states of β-lactamase: molten-globule states at acidic and alkaline pH with high salt. Biochemistry 28, 945–952 [DOI] [PubMed] [Google Scholar]
- 52. Fusco G., De Simone A., Gopinath T., Vostrikov V., Vendruscolo M., Dobson C. M., Veglia G. (2014) Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 5, 3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lorenzen N., Nielsen S. B., Yoshimura Y., Vad B. S., Andersen C. B., Betzer C., Kaspersen J. D., Christiansen G., Pedersen J. S., Jensen P. H., Mulder F. A., Otzen D. E. (2014) How epigallocatechin gallate can inhibit α-synuclein oligomer toxicity in vitro. J. Biol. Chem. 289, 21299–21310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jarrett J. T., Lansbury P. T., Jr. (1993) Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73, 1055–1058 [DOI] [PubMed] [Google Scholar]
- 55. Harper J. D., Lansbury P. T., Jr. (1997) Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66, 385–407 [DOI] [PubMed] [Google Scholar]
- 56. Wetzel R. (2006) Kinetics and thermodynamics of amyloid fibril assembly. Acc. Chem. Res. 39, 671–679 [DOI] [PubMed] [Google Scholar]
- 57. Platt G. W., Routledge K. E., Homans S. W., Radford S. E. (2008) Fibril growth kinetics reveal a region of β2-microglobulin important for nucleation and elongation of aggregation. J. Mol. Biol. 378, 251–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Giehm L., Otzen D. E. (2010) Strategies to increase the reproducibility of protein fibrillization in plate reader assays. Anal. Biochem. 400, 270–281 [DOI] [PubMed] [Google Scholar]
- 59. Ohhashi Y., Kihara M., Naiki H., Goto Y. (2005) Ultrasonication-induced amyloid fibril formation of β2-microglobulin. J. Biol. Chem. 280, 32843–32848 [DOI] [PubMed] [Google Scholar]
- 60. So M., Yagi H., Sakurai K., Ogi H., Naiki H., Goto Y. (2011) Ultrasonication-dependent acceleration of amyloid fibril formation. J. Mol. Biol. 412, 568–577 [DOI] [PubMed] [Google Scholar]
- 61. Yoshimura Y., So M., Yagi H., Goto Y. (2013) Ultrasonication: an efficient agitation for accelerating the supersaturation-limited amyloid fibrillation of proteins. Jpn. J. Appl. Phys. 52, 07HA01-01–07HA01-08 [Google Scholar]
- 62. Ban T., Morigaki K., Yagi H., Kawasaki T., Kobayashi A., Yuba S., Naiki H., Goto Y. (2006) Real-time and single fibril observation of the formation of amyloid β spherulitic structures. J. Biol. Chem. 281, 33677–33683 [DOI] [PubMed] [Google Scholar]
- 63. Gellermann G. P., Appel T. R., Tannert A., Radestock A., Hortschansky P., Schroeckh V., Leisner C., Lütkepohl T., Shtrasburg S., Röcken C., Pras M., Linke R. P., Diekmann S., Fändrich M. (2005) Raft lipids as common components of human extracellular amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 6297–6302 [DOI] [PMC free article] [PubMed] [Google Scholar]