

Background: TNFR1 signaling has been intensively studied, but it remains unclear how TNFR2 transduces TNF-α signal under inflammatory conditions.
Results: TNFR2 associates with IL-17RD, resulting in receptor aggregation and TRAF2 recruitment, leading to promotion of NF-κB signaling in renal tubular epithelial cells.
Conclusion: TNFR2 cooperates with IL-17RD to activate NF-κB.
Significance: The TNFR2·IL-17RD heteromer might be implicated in nephritis.
Keywords: NF-κ B (NF-KB), Receptor Regulation, Signal Transduction, TNF Receptor-associated Factor (TRAF), Tumor Necrosis Factor (TNF), IL-17RD/Sef, TNFR2
Abstract
TNF receptor 2 (TNFR2) exerts diverse roles in the pathogenesis of inflammatory and autoimmune diseases. Here, we report that TNFR2 but not TNFR1 forms a heteromer with interleukin-17 receptor D (IL-17RD), also named Sef, to activate NF-κB signaling. TNFR2 associates with IL-17RD, leading to mutual receptor aggregation and TRAF2 recruitment, which further activate the downstream cascade of NF-κB signaling. Depletion of IL-17RD impaired TNFR2-mediated activation of NF-κB signaling. Importantly, IL-17RD was markedly increased in renal tubular epithelial cells in nephritis rats, and a strong interaction of TNFR2 and IL-17RD was observed in the renal epithelia. The IL-17RD·TNFR2 complex in activation of NF-κB may explain the role of TNFR2 in inflammatory diseases including nephritis.
Introduction
TNF-α exerts its biological effects via two functionally distinct receptors, TNFR13 (also known as p55 or TNFRSF1A) and TNFR2 (also known as p75 or TNFRSF1B). Although TNFR1, through boosting NF-κB activation, was found to response to broad inflammation stimulation in various diseases (1), TNFR2 was considered to function preferably on several special kidney diseases (2–4). Recently TNFR2 was reported to be activated by TNF-α and to further activate NF-κB in certain types of cells (5–7). It appears that TNFR2 induces activation of NF-κB signaling differentially under a specific inflammatory condition with an assistance of some co-factors.
Renal tubular epithelial cells (RTECs) are damaged by inflammation stimulation and also produce cytokines and chimokines to exacerbate local inflammation in tubulointerstitial renal diseases (8). A growing body of evidence indicated that RTECs expressed a high level of TNFR2 but not TNFR1 (2, 9–11), suggesting an important role of TNFR2 in renal diseases (2, 3, 12). However, it remains unknown how TNFR2 functions in RTECs in renal diseases. Here, we show that TNFR2 interacts with IL-17RD, also named Sef (similar expression to FGF), an orphan receptor in IL-17R family, resulting in enhanced NF-κB signaling in RTECs.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
HK-2, HEK293T, 786-O, and COS-7 cells were kept in our laboratories. Media and serum were purchased from Invitrogen. An antibody against IL-17RD (3F12) was generated in our laboratory. Antibodies against TNF-α, TNFR2, TRAF2, Myc, and GFP were from Santa Cruz. Antibodies against β-actin and FLAG were from Sigma. Antibodies conjoined with Fluorescent (goat anti-rabbit IgG and goat anti-mouse IgG) were from Jackson ImmunoResearch Laboratories. TNF-α was from Cell Signaling.
Plasmid Constructs
Myc-IL-17RD, Myc-IL-17RA, Myc-IL-17RD-ECD, Myc-IL-17RD-ΔECD, and GFP-IL-17RD plasmids (where IL-17RD was of human origin) were described previously (13, 14). Myc-TNFR1, Myc-TNFR2, FLAG-TNFR2, FLAG-TRAF2, pGL3/NF-κB-luc, and pRL-TK luciferase reporter were all kept in our laboratory. TNFR2mut (19), which fails to interact with TRAF2, was constructed by our laboratory. EYFP-TNFR1, EYFP-TNFR2, ECFP-IL-17RD, and ECFP-IL-17RA were constructed using pECFP-N1 and pEYFP-N1. pcDNA3.1-TNFR2-ECD-FLAG and pcDNA3.1-TNFR2-ICD-FLAG (ICD is intracellular domain) were also constructed for interaction mapping assays. TNF80(sc)-FLAG-TNC and TNF(mut60)-FLAG-TNC were all kindly provided by Dr. Harald Wajant. Ad-hIL-17RDi-1 and Ad-hIL-17RDi-2, adenovirus vectors carrying shRNAs against human IL-17RD by targeting sequences 5′-GCATGTGATTGCTGACGCC-3′ and 5′-GTCGGAGGGAAGACAGTGC-3′, respectively, and an adenovirus vector (Ad-GFPi) carrying an shRNA against green fluorescent protein (GFP) targeting TGACCACCCTGACCTACGGCGTGCAGTGC were generated in our laboratory.
Cell Culture and Transfection
HK-2, HEK293T, 786-O, and COS-7 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum (FBS) and antibiotics. All the cells were kept at 37 °C in a 5% CO2-containing atmosphere. Transient transfection experiments were carried out using Genejuice regent (VigoFect) (Vigofect Inc. Beijing, China) according to the manufacturer's protocol.
Luciferase Assay
HEK293T and HK-2 cells were transiently transfected with the indicated plasmids using Vigofect, according to the manufacturer's instructions. Briefly, 0.1 μg of reporter plasmid pGL3/NF-κB-luc together with 5 ng of the internal control plasmid pRL-TK and the indicated amounts of plasmids were co-transfected into the cells. The empty plasmid was used to balance the total amount of DNA for equal transfection efficiency. All the experiments were performed in triplicate. Luciferase activity was assayed after 24 h of transfection using a Dual-Luciferase reporter assay system. The luciferase activity was normalized by firefly against Renilla luciferase activity and presented as the mean ± S.D. The relative luciferase activity was shown.
Immunoprecipitation, Western Blotting, and Cell Fractionation
Cells or renal tissue of mice were lysed with lysis buffer I (50 mm Tris-HCl, 150 mm NaCl, 50 mm EDTA, 0.5% Nonidet P-40, pH 7.5) containing a protease inhibitors mixture. The lysates were cleared by centrifugation and incubated with the appropriate antibodies for 4 h and then with protein G-agarose beads for 2 h at 4 °C with rotary agitation. Immunoprecipitants and 5% of lysates were separated by 10% SDS-PAGE, and Western blot analyses were performed for the indicated proteins. Cell fractionation was performed by lysis buffer I containing a protease inhibitor mixture without DTT followed by spinning down at 10,000 × g for 15 min and lysis buffer II (50 mm Tris-HCl, 150 mm NaCl, 0.25% SDS, 0.1% Nonidet P-40, pH 7.5) containing a protease inhibitor mixture without DTT. The supernatant was taken as soluble protein, and the pellets were subsequently solubilized in 100 μl of lysis buffer II as insoluble fraction. Western blot for the aggregation of receptors were under non-reducing condition.
Immunofluorescence
COS-7 and HK-2 cells were seeded on coverslips and transfected with or without the indicated plasmids. 24 h after transfection cells were washed with PBS and fixed with 4% paraformaldehyde for 20 min. After blocking with 10% FBS for 1 h, sections and cells were incubated with the appropriate primary antibodies at 37 °C for 1 h followed by incubation with second antibodies conjugated with Fluorescent for 1 h and counterstained with DAPI for 10 min. Images were obtained with a confocal laser scanning microscope (OLYMPUS BX61).
Fluorescence Resonance Energy Transfer (FRET) Microscopy
FRET measurements were performed in COS-7 cells. After transfected with the appropriate expression constructs for 24 h, FRET were evaluated in living cells using the acceptor photobleaching method in a confocal laser scanning microscope (A1Rsi; Nikon, Inc.). For the acceptor photobleaching method, the donor signal in defined regions of interest were bleached with a 515-nm light at 100% power for 50 iterations to ensure >80% bleaching efficiency. The intensity in each region of interest at 488-nm excitation before and after the bleach was measured. Similar calculations were made in non-photobleached cells in the same culture. The averaged intensity for each protein pair was measured from at least 25 cells in three different experiments. FRET analysis was based on all pixels in the selected regions of interest. FRET efficiency was calculated from the summary of the fluorescence intensities from individual pixels by normalizing the difference of the donor post- and pre-bleach intensity by the post-bleach intensity according FRET all algorithms implemented in the custom-developed FRETcalc plugin.
Immunohistochemistry and Scoring Method
Immunohistochemical detection of IL-17RD, TNFR1, and TNFR2 was performed on kidney sections from IgA nephropathy rats, normal wild-type rats, 5/6 nephrectomy rats, and control rats induced by sham operation. 12 rats were used in each group. All studies on animal tissues were approved by the Institutional Review Board of Tsinghua University. Paraffin sections (3-mm thick) were treated with 0.3% hydrogen peroxidase/methanol and incubated with antibodies against IL-17RD (1:2000), TNFR1 (1:250), or TNFR2 (1:50) followed by incubation with secondary antibodies (biotin-labeled; Santa Cruz) and third antibodies (peroxidase-labeled; Santa Cruz). Samples were developed using 3,3′-diaminobenzidine as substrates (Santa Cruz). Ten fields were selected randomly using a light microscope (200× magnification) for scoring the positive rate of IL-17RD expression in RTECs. Cells with IL-17RD showing positive staining were counted based on the total cell number in the field. The stained areas were rated as follows: 0, no staining or positive staining cells <10%; 1, positive staining cells between 10 and 35%; 2, positive staining cells between 35 and 70%; 3, positive staining cells >70%.
Reverse Transcriptase PCR (RT-PCR)
Total cellular RNA was prepared from HK-2 cells using TRIzol reagent (Invitrogen). Reverse transcription was done using a Quantscript RT kit (TIANGEN Biotech, Beijing, China). RT-PCR was performed using a RealMasterMix (SYBR Green) kit (TIANGEN Biotech, Beijing, China) and carried out on a Bio-Rad iCycler. The primers for human IL-17RD gene were AGAACACGGGCCTGTGACCTGT and AGTCCTCCGCCTGAAGGGGC, and primers for GAPDH gene were AGACCACAGTCCATGCCATC and TTGCCCACAGCTTGGCAG. All the experiments were performed in triplicate.
Protein Expression and Purification
The extracellular domain of human IL-17RD (hIL-17RD; residues 16–299), human TNFR1 (residues 22–211), and human TNFR2 (residues 23–255) with a modified N-terminal gp67 secretion signal sequence and C-terminal His6 tag were inserted into pFastBacI vector. The constructs were transformed into bacterial DH10Bac component cells, and extracted bacmid was then transfected into Sf9 cells with Cellfectin II Reagent (Invitrogen). The low titer viruses were harvested and then amplified. High titer virus was used to infect 1 liter of Sf9 cells at a density of 2 × 106 cells/ml. The supernatant of the cell culture containing secreted hIL-17RD, TNFR1, or TNFR2 was harvested 48–72 h after infection and concentrated and buffer-exchanged to HBS (10 mm HEPES, pH 7.2, 150 mm NaCl). Protein was captured by nickel resin (GE Healthcare), washed with HBS containing 20 mm imidazole, and eluted with HBS containing 500 mm imidazole. Protein was then purified by gel filtration chromatography using Superdex 200 column (GE Healthcare) in HBS buffer. Fractions containing hIL-17RD, TNFR1, or TNFR2 were collected and pooled together for further assays.
Affinity Measurement
Interaction of hIL-17RD-ECD, TNFR1-ECD, or TNFR2-ECD with TNF-α were analyzed by surface plasmon resonance using Biacore 3000 (GE Healthcare) at 298 K. TNF-α was immobilized on sensor chip CM5 using the standard amine-coupling method (GE Healthcare) to ∼120 response units. To collect data for kinetic and affinity analysis, IL-17RD-ECD was diluted serially between 1 and 32 μm in binding buffer (HBS plus 0.005% Tween 20), and TNFR1-ECD (0.1 μm) or TNFR2-ECD (0.1 μm) was injected over the chip at a flow rate of 30 μl/min. The complex was allowed to associate for 60 s and dissociate for 60 s. Regeneration was accomplished by passing binding buffer over the chip surface until dissociation completed. Data were analyzed with Biacore 3000 evaluation software by fitting to a 1:1 Langmuir binding fitting model.
Enzyme-linked Immunosorbent Assay (ELISA)
To determinate the affinity of IL-17RD-ECD to TNFR2 in the presence of TNF-α, amine-binding 96-well microtiter plates were coated with different concentrations of TNFR2-ECD in PBS at 4 °C overnight. Nonspecific binding was blocked by incubation with 3% (w/v) BSA at 37 °C for 1 h. IL-17RD-ECD with or without TNF-α was added to each well, and the plates were incubated for 2 h at room temperature. The plates were then incubated with 3F12 monoclonal antibody, and bound 3F12 was detected after a 2-h incubation using a 1:10,000 dilution of HRP-conjugated anti-mouse antibody.
Statistics
The data presented in this study were obtained from at least three independent experiments for each experimental condition. Data are expressed as the means ± S.D., and their statistical significance was analyzed by one-way analysis of variance analysis. Multiple comparisons were performed by Duncan's test.
RESULTS
IL-17RD Is Up-regulated in the Epithelia of Renal Tubules in Nephritis Rats
A previous report showed that IL-17RD mRNA was abundant in ductal epithelial cells in the collecting tubules of the kidney (15). To examine IL-17RD protein levels in the kidney, a monoclonal antibody against the extracellular domain of hIL-17RD (Fig. 1A) was raised, which specifically recognized both the endogenous IL-17RD (rat, mouse, and human IL-17RD) (Fig. 1, B and C) and ectopically expressed IL-17RD (Fig. 1, B–D), but not IL-17RA, another receptor of the IL-17 family (Fig. 1D). An immunohistochemistry assay showed that IL-17RD was highly expressed in RTECs of kidneys from IgA nephropathy rats, which suffered from immune nephritis (16), and from 5/6 nephrectomy rats, which showed renal failure because of renal injury (17), in comparison with that of control rats (Fig. 1E). A quantitative analysis indicated that the expression of IL-17RD was significantly increased in RTECs after the rats suffered from two types of nephritis (Fig. 1F). These results suggested that IL-17RD is up-regulated in RTECs under inflammatory conditions.
FIGURE 1.
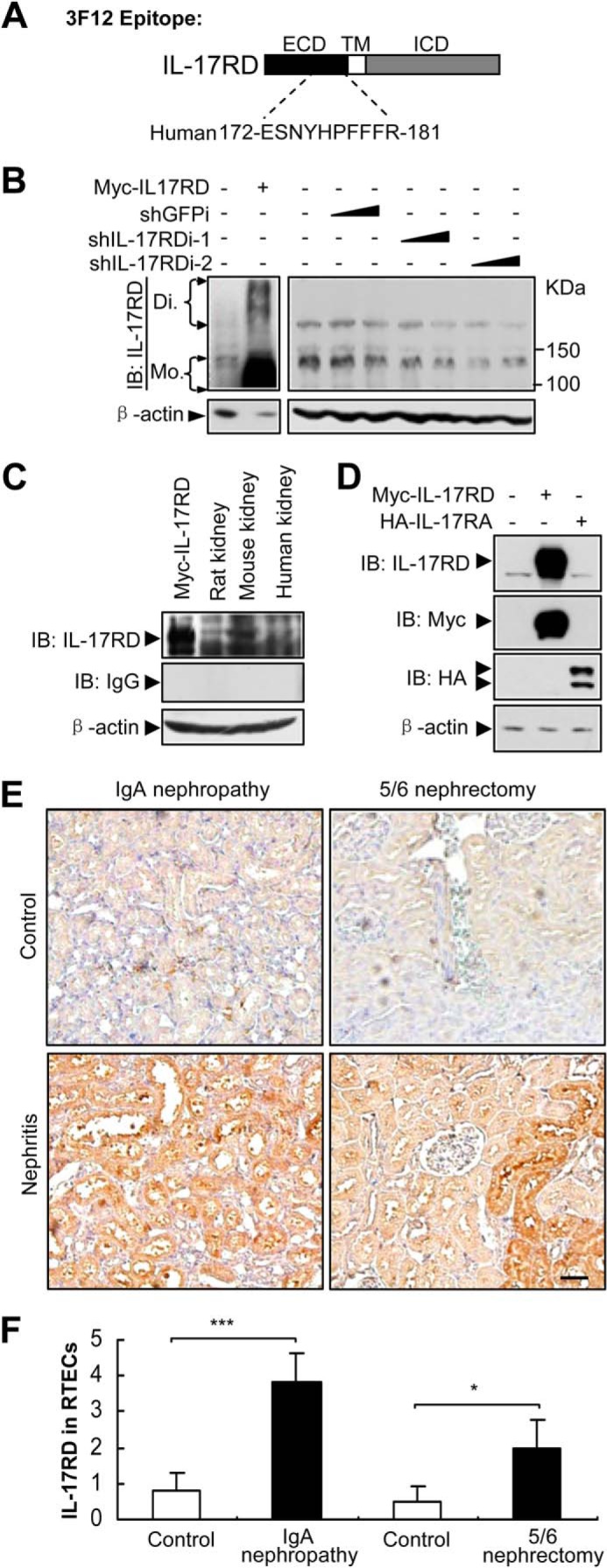
IL-17RD is up-regulated in RTECs. A, schematic of an epitope of a monoclonal antibody against IL-17RD (3F12). ICD, intracellular domain; TM, transmembrane. B–D, Western blot (IB) assays for validating the anti-IL-17RD antibody. HKE293T cells transfected with the indicated plasmids or shRNAs against IL-17RD (named shIL-17RDi-1 and shIL-17RDi-2) and tissue lysates from rat, mouse, and human were collected, and IL-17RD was examined by Western blot using the anti-IL-17RD antibody (3F12). Di, dimer; Mo, monomer. E, IL-17RD was up-regulated in RTECs of nephritis rats. Immunohistochemical staining for IL-17RD in sections from kidneys of IgA nephropathy and a control and 5/6 nephrectomized rats and sham controls are shown (bars, 200 μm). F, quantification of the IL-17RD expression in RTECs. Scores of IL-17RD positive cells in RTECs were calculated, and averaged scores were used to present the IL-17RD levels. p < 0.05 (*) and p < 0.001 (***).
IL-17RD Is Required for NF-κB Signaling in RTECs
Because NF-κB is activated in RTECs during nephritis, we questioned whether up-regulated IL-17RD contributes to the NF-κB activation. A luciferase reporter assay showed that overexpression of hIL-17RD promoted the NF-κB transcriptional activity in the presence or absence of TNF-α in HK-2 cells (a human cell line of RTEC feature), similar to the role of TNFR2 (Fig. 2A). Consistently, depletion of endogenous hIL-17RD by two adenovirus-driven shRNAs (Fig. 2B) in HK-2 cells almost completely suppressed the TNF-α-induced NF-κB transcriptional activity (Fig. 2C). Furthermore, depletion of hIL-17RD reduced the level of p-IκB-α, as observed 5 min after TNF-α treatment (Fig. 2D), and nuclear localized p65, as observed by the ratio of nuclear/cytoplasmic p65 (Fig. 2, E and F). These results suggested that hIL-17RD is critical for the TNF-α-induced activation of NF-κB in HK-2 cells. However, IL-17RD was reported to function as an inhibitor for NF-κB during the induction by IL-1β through interacting with NF-κB (p65) and blocking its nuclear localization (18). Indeed, we observed that overexpression of IL-17RD inhibited (Fig. 2G), but depletion of IL-17RD facilitated (Fig. 2H) the TNF-α-induced NF-κB activation in HEK293T cells, a human kidney embryo cell line where TNFR2 expression was absent (Fig. 2I). Interestingly, we observed that IL-17RD(Y330F), a membrane-bound mutant (13), abrogated its inhibitory role on NF-κB activation (Fig. 2G), consistent with a previous report (see Figs. 4C and 5A in Fuchs et al. (18)). Indeed, we also found that IL-17RD interacted with p65 in HEK293T cells, whereas overexpression of TNFR2 suppressed the association of IL-17RD with p65 (Fig. 2J), suggesting that TNFR2 disrupts the interaction of IL-17RD and p65. In addition, we found that IL-17RD failed to associate with p65 in HK-2 cells (Fig. 2K), where TNFR2 was abundant (Fig. 2I). We reasoned that the role of IL-17RD on the NF-κB activity depends on TNFR2 level and the membrane-bound status. Furthermore, the protein level of TNFR2 was obviously enhanced in RTECs of kidneys from IgA nephropathy rats, whereas the TNFR1 protein level decreased in comparison with that of control rats (Fig. 2L). Together, these results indicated that consistent up-regulation of IL-17RD and TNFR2 in RTECs may play a vital role in NF-κB activation during nephritis.
FIGURE 2.

IL-17RD is critical for NF-κB activation in RTECs. A, overexpression of IL-17RD enhances the NF-κB transcriptional activity in HK-2 cells. HK-2 cells transfected with NF-κB luciferase reporter, pRL-TK, and Myc-IL-17RD or Myc-TNFR2 were stimulated with TNF-α for 6 h. Results presented are the average from a triplicate experiment. **, p < 0.01. B and C, depletion of IL-17RD suppresses NF-κB activation in HK-2 cells. After depletion of IL-17RD using adenovirus-driven shRNA against IL-17RD (named Ad-hIL-17RDi-1 and Ad-hIL-17RDi-2), the IL-17RD mRNA was examined by an RT-PCR. Adenovirus-driven shRNA against GFP (named Ad-GFPi) was used as a control. GAPDH was used as a control. The ratio represents the relative density of IL-17RD over GAPDH (B). HK-2 cells were transfected with NF-κB luciferase reporter, pRL-TK, and infected with indicated adenovirus. *, p < 0.05; ***, p < 0.001 (C). D, depletion of hIL-17RD suppressed the phosphorylation levels of IκB-α in HK-2 cells. Bottom, densitometry of the bands, showing the p-IκB-α level, normalized to β-actin, compared with Ad-GFPi control. *, p < 0.05. Data are representative of at least three experiments. E and F, depletion of hIL-17RD inhibits the TNF-α-induced p65 nuclear translocalization. HK-2 cells affected with Ad-hIL-17RDi-1 and Ad-GFPi were stimulated with TNF-α for 5 min. The cytoplasmic (Cyt) and nuclear (Nuc) fractions of HK-2 cells were extracted and blotted with an anti-p65 antibody (E). Quantification of the ratio of nuclear/cytoplasmic p65, normalized by tubulin and c-Jun respectively, are shown (F). G, IL-17RDY330F abrogates the IL-17RD inhibition of TNF-α-induced NF-κB in HEK293T cells. Cells were treated as in A. H, depletion of IL-17RD promotes TNF-α-induced NF-κB transcriptional activity in HEK293T cells. I, a Western blot assay for the protein levels of IL-17RD and TNFR2 or TNFR1 in HEK293T and HK-2 cells. J, TNFR2 suppresses the association of IL-17RD and p65 in HEK293T cells. The protein complex was immunoprecipitated (IP) by an anti-FLAG antibody and resolved by an anti-Myc or anti-FLAG antibody. IB, immunoblot. K, IL-17RD fails to interact with NF-κB (p65) in HK-2 cells. An endogenous IP experiment was performed for HK-2 cells. L, immunohistochemical staining for TNFR2 and TNFR1 in sections from kidneys of IgA nephropathy and its control (bars, 500 μm).
FIGURE 4.

TNF-α facilitates the association of IL-17RD and TNFR2. A–C, IL-17RD exists in vivo as a complex with endogenous TNFR2. Endogenous IL-17RD interacts with TNFR2 in HK-2, 786-O cells, and renal tissues of mouse (A). HK-2 cells treated with or without TNF-α were used for IP experiments (B). IB, immunoblot. The relative density of TNFR2 precipitated from three independent IP experiments was quantified and presented as average ± S.D. **, p < 0.01 (C). D, an ELISA assay for the binding ability of IL-17RD-ECD with TNFR2-ECD (500 ng/well) in the absence or presence of equal amounts of TNF-α. *, p < 0.05. E and F, IL-17RD co-localizes with TNFR2 in RTECs. HK-2 cells stimulated without (control) or with TNF-α (E) and the sections from kidneys of IgA nephropathy and 5/6 nephrectomy rats and the controls (F) were immunostained with anti-TNFR2 (or anti-TNFR1) and anti-IL-17RD antibodies. Arrows indicate the co-localization of IL-17RD and TNFR2. Scale bars: E, 10 μm; F, 20 μm. G and H, surface plasmon resonance analyses for the binding affinities of TNF-α to IL-17RD, TNFR1, and TNFR2. Proteins of TNFR1-ECD (0.1 μm) or TNFR2-ECD (0.1 μm) and different concentrations of IL-17RD-ECD (1, 2, 4, 8, 16, 32 μm) were injected using FastStep injection, and dissociation of analyte-ligand complexes was monitored. KD for each interaction is indicated. RU, response units. I, co-localization of IL-17RD with TNFR2 and TNF80(sc). COS-7 cells were co-transfected with indicated plasmids. Cells were analyzed by double immunostaining with anti-Myc/FLAG antibodies. Scale bar: upper panel, 25 μm; lower panel, 10 μm.
FIGURE 5.

IL-17RD coordinates with TNFR2 to activate NF-κB via facilitating TNFR2 aggregation. A–E, IL-17RD and TNFR2 mutually promote their aggregation (A–C). Aggregated proteins were indicated by oligomers from Western blots (IB). ECD of IL-17RD was sufficient to promote aggregation of TNFR2 (D). Depletion of IL-17RD suppresses TNFR2 aggregation in HK-2 cells (E). Different aggregated forms of IL-17RD and TNFR2 were examined by Western blots using the indicated antibodies from soluble and insoluble fractions of HEK293T cells co-expressed with Myc-IL-17RD and Myc-TNFR2 (A–C), with FLAG-TNFR2 and full-length or its mutants of Myc-IL-17RD (FL, ECD, and ΔECD) (D), or HK-2 cells where IL-17RD was depleted by an shRNA (IL-17RDi-1) (E). F–I, luciferase assays. The experiments have been performed as in Fig. 2. IL-17RD cooperates with TNFR2 (F) but not TNFR1 (G) to activate NF-κB signaling. ECD of IL-17RD was sufficient to enhance TNFR2-mediated NF-κB activity (H). Depletion of IL-17RD suppresses TNFR2-mediated NF-κB transcriptional activity in HK-2 cells (I). The luciferase activity was analyzed. n.s., not significant. *, p < 0.05; **, p < 0.01.
IL-17RD Specifically Associates with TNFR2 in Vitro
Given the similar expression pattern of IL-17RD and TNFR2 on RTECs during nephritis, we examined whether IL-17RD associates with TNFR2. Immunoprecipitation (IP) results demonstrated that IL-17RD interacted with TNFR2 but not TNFR1 (Fig. 3, A and B), suggesting that IL-17RD specifically associates with TNFR2. However, IL-17RA, a homolog of IL-17RD, failed to associated with either TNFR1 or TNFR2 (Fig. 3B), consistently suggesting the specific interaction of IL-17RD with TNFR2. Interestingly, we also found that the interaction of IL-17RD with TNFR2 decreased in the presence of IL-17RA (Fig. 3C, line 3).
FIGURE 3.

IL-17RD associates with TNFR2 in vitro. A, IL-17RD interacts with TNFR2 in HEK293T cells. The protein complex was immunoprecipitated (IP) by an anti-GFP antibody and resolved by an anti-Myc or anti-GFP antibody. IB, immunoblot. B, IL-17RA fails to interact with TNFR1 and TNFR2. An IP experiment was performed for HEK293T cells. C, IL-17RA interferes with the association of TNFR2 and IL-17RD. D and E, extracellular domains of IL-17RD and TNFR2 are responsible for the IL-17RD·TNFR2 interaction. IPs were performed using an anti-Myc (D) or anti-FLAG (E) antibody, respectively. Immunoblotting was performed using the indicated antibodies. ICD, intracellular domain; TM, transmembrane. F, TNFR2-ECD interacts with endogenous IL-17RD in HK-2 cells. The protein of His-TNFR2-ECD was added to the cell lysates from HK-2 cells, and the complex was precipitated with His column and resolved by an anti-IL-17RD antibody. G, an ELISA assay for binding of IL-17RD-ECD to TNFR2-ECD or TNFR1-ECD. Microtiter plates were coated with various amounts (0–500 ng) of TNFR2-ECD or TNFR1-ECD. After blocking, 500 ng of IL-17RD-ECD were added to each well, and the bound protein from the liquid phase was detected by an antibody against IL-17RD (3F12) followed by a secondary antibody conjugated with horseradish peroxidase. H and I, FRET assays in COS-7 cells. IL-17RD was engineered into ECFP, and TNFR2 was engineered into EYFP. The plasmids were co-transfected in COS-7 cells. IL-17RD and TNFR2 FRET signals were taken before photobleaching (a–c) and after photobleaching (d–f). Scale bars, 10 μm (H). The emission spectra of ECFP-IL-17RD and EYFP-TNFR2 were shown (I). J, a statistic analysis of the averaged FRET efficiency (%). n indicates the numbers of cells observed. Data are presented as the average ± S.D. *, p < 0.05. EYFP, enhanced yellow fluorescent protein; ECFP, enhanced cyan fluorescent protein.
To examine which domain is responsible for the IL-17RD·TNFR2 interaction, another set of IP experiments was performed. The results showed that the extracellular domain (ECD) of IL-17RD (Myc-IL-17RD-ECD) interacted with FLAG-TNFR2 (Fig. 3D) and ECD of TNFR2 (FLAG-TNFR2-ECD or His-TNFR2-ECD protein) interacted with Myc-IL-17RD (Fig. 3E) or endogenous IL-17RD (Fig. 3F), indicating that IL-17RD interacts with TNFR2 via their respective extracellular domains. Intriguingly, the transmembrane and intracellular domain of IL-17RD failed to associate with full-length TNFR2 (Fig. 3D, last lane), and the full-length IL-17RD did not interact with the intracellular domain of TNFR2 (Fig. 3E, last lane), suggesting that the intracellular domains of the receptors are insufficient for their interaction. To further test the direct interaction of IL-17RD-ECD and TNFR2-ECD, we purified the extracellular domains of hIL-17RD, human TNFR2, and human TNFR1 (named IL-17RD-ECD, TNFR2-ECD, and TNFR1-ECD) and examined their binding abilities in vitro. ELISA assays using an anti-IL-17RD antibody showed that the bound IL-17RD-ECD proteins were correlated to the amounts of mounted TNFR2-ECD significantly higher than that of TNFR1-ECD (Fig. 3G). These results suggested that IL-17RD-ECD binds preferably to TNFR2-ECD. Collectively, these results indicated that the interaction of IL-17RD and TNFR2 occurs at their ECDs.
To observe the interaction of IL-17RD and TNFR2 in living cells, FRET experiments were performed. An acceptor photobleaching FRET microscopy showed that after photobleaching EYFP-TNFR2 (as the acceptor), the donor (ECFP-IL-17RD) emission was increased (Fig. 3H, compare e with b, and I), accompanied by diminished emission from EYFP-TNFR2 (Fig. 3H, compare d with a, and 3I), suggesting that IL-17RD associates with TNFR2 in situ. Further statistical analyses indicated that the averaged relative FRET efficiency between ECFP-IL-17RD and EYFP-TNFR2 reached to 40%, whereas the FRET efficiencies between ECFP-IL-17RD and EYFP-TNFR1, ECFP-IL-17RA and EYFP-TNFR2, and ECFP-IL-17RA and EYFP-TNFR1 were maintained at very low levels (<10%) (Fig. 3J). Taken together, these results suggested that IL-17RD and TNFR2 interact directly and specifically in situ.
TNF-α Facilitates the Association of IL-17RD and TNFR2
To address the interaction of endogenous IL-17RD and TNFR2, we performed IP experiments in cells where both IL-17RD and TNFR2 were co-expressed. The result showed that IL-17RD formed a complex with TNFR2 in HK-2, 786-O cells (a human epithelial cell line of kidney carcinoma) or renal tissue of mice (Fig. 4A), suggesting that the interaction of IL-17RD with TNFR2 occurs under physiological conditions. Intriguingly, we observed that the IL-17RD·TNFR2 complex was increased upon TNF-α stimulation (Fig. 4B). A quantitative analysis indicated that TNFR2 was highly presented in the complex of IL-17RD when TNF-α was added for different times in HK-2 cells (Fig. 4C). An ELISA experiment further confirmed that the association of IL-17RD-ECD and TNFR2-ECD was enhanced in the presence of TNF-α (Fig. 4D). Consistently, an immunostaining experiment showed that endogenous IL-17RD and TNFR2 were co-localized on the cell membrane of HK-2 (Fig. 4E, Control), and the co-localization was enhanced and shifted into the intracellular perinuclear compartment after TNF-α treatment (Fig. 4E, TNF-α). These results indicated that IL-17RD and TNFR2 are associated with each other in response to TNF-α in RTECs. To further examine whether IL-17RD co-localizes with TNFR2 under pathological conditions, immunostaining assays for the sections from IgA nephropathy and 5/6 nephrectomy rats were performed. The results showed that IL-17RD mainly co-localized with TNFR2 in the epithelial cells of the proximal and distal convoluted tubule, and their co-localization was obviously increased in the nephritis rats (Fig. 4F, top panel). Furthermore, we found that IL-17RD failed to co-localize with TNFR1 in RTECs in the sections from IgA nephropathy and their controls (Fig. 4F, bottom panel). These results implied that the specific complex of IL-17RD and TNFR2 in RTECs may function under physiological (Control) or pathological (kidney inflammatory diseases) conditions.
As the interaction of IL-17RD and TNFR2 occurs via their ECDs and is enhanced by TNF-α, we questioned whether TNF-α directly binds to orphan receptor IL-17RD. A surface plasmon resonance assay showed that IL-17RD-ECD remained a weak interaction with TNF-α (1.06 × 10−6 m) (Fig. 4H), whereas TNFR2 associated with TNF-α as strongly as did TNFR1 (Fig. 4G). To define the specificity of TNF-α in the complex of IL-17RD and TNFR2, we used TNF80(sc), a TNF-α mutant specifically binding to TNFR2 (19), and TNF(mut60), a TNF-α mutant specifically binding to TNFR1 (19), to compare the complex formation by an immunostaining experiment. The results showed that IL-17RD co-localized with TNF80(sc) and TNFR2 (Fig. 4I, top panel) but not with TNF(mut60), which colocalized with TNFR1 (Fig. 4I, bottom panel). These results suggested that IL-17RD prefers to interact with TNF-α-associated TNFR2. We speculated that IL-17RD might be a specific co-receptor for TNF-α in recognizing TNFR2.
IL-17RD Coordinates with TNFR2 to Mediate NF-κB Activation via Facilitating Receptor Aggregation
TNFRs aggregate after TNF-α stimulation to activate their signaling (20). To address whether IL-17RD influences the aggregation of TNFR2 after their interaction, we examined the levels of different oligomers of TNFR2. The results showed that the level of monomer TNFR2 in the soluble fraction of the cells was decreased dramatically (Fig. 5A, left panel), but the oligomer TNFR2 in the insoluble fraction of the cells was significantly increased (Fig. 5A, right panel) when IL-17RD was co-expressed. Another Western blot analysis using antibodies against TNFR2 (Fig. 5B) or IL-17RD (Fig. 5C) demonstrated that both IL-17RD and TNFR2 forms more oligomers when they were co-expressed. These results suggest that interaction of IL-17RD and TNFR2 resulted in their aggregation. Moreover, we found that ECD but not the transmembrane and intracellular domain of IL-17RD facilitated TNFR2 aggregation (Fig. 5D). Furthermore, we observed that depletion of endogenous IL-17RD impaired the aggregation of endogenous TNFR2 in the insoluble fraction of HK-2 cells (Fig. 5E). All these results suggested that IL-17RD and TNFR2 mutually promotes receptor aggregation.
We next sought to determine whether IL-17RD regulates TNFR2-induced transcription. Luciferase assays using a NF-κB reporter in HEK293T cells demonstrated that co-expression of IL-17RD dose-dependently enhanced the TNFR2 (Fig. 5F)- but not TNFR1 (Fig. 5G)-induced NF-κB activity, and ECD of IL-17RD was sufficient to enhance the TNFR2-mediated NF-κB activity (Fig. 5H), consistent with the aforementioned observations that ECD of IL-17RD formed a stabilized complex with TNFR2 but not TNFR1 (Figs. 3 and 4). On the other hand, we observed that depletion of IL-17RD, using two siRNAs, impaired the activity of TNFR2 in activating NF-κB (Fig. 5I), suggesting that IL-17RD is required for TNFR2 signaling in HK-2 cells. All these results indicated that IL-17RD is involved in TNFR2-mediated activation of NF-κB.
IL-17RD and TNFR2 Mutually Promote the TRAF2 Recruitment
TNFR2 has been shown to recruit TRAF2 in the initiation of NF-κB signaling (21). To address whether the IL-17RD·TNFR2 complex transmits NF-κB signal through TRAF2, we examined the luciferase activity of the NF-κB reporter. The results showed that the effect of IL-17RD on TNFR2 in NF-κB activation was almost completely abolished by ΔTRAF2, a dominant-negative mutant of TRAF2 (Fig. 6, A and B), indicating that TRAF2 mediates IL-17RD·TNFR2 complex-activated NF-κB. To illustrate the role of TNFR2 in the complex of IL-17RD·TNFR2 on the transduction of NF-κB signal though TRAF2, we generated a TNFR2 mutant (TNFR2mut), which lost the interaction with TRAF2 (19). The results showed that TNFR2mut failed to bind to TRAF2 (Fig. 6C) and could not cooperate with IL-17RD to enhance the transcriptional activity of NF-κB in the control group (Fig. 6D, Vector). Interestingly, whereas TNFR2 further enhanced the transcriptional activity under overexpression of TRAF2, TNFR2mut still failed to enhance the activity (Fig. 6D, compare the eighth column to the sixth column). We also observed that over-expression of both TRAF2 and IL-17RD showed greater effect on the transcription activity compared with that from individually over-expressed TRAF2 or IL-17RD (Fig. 6D, compare the sixth column to the second and fifth columns). These results indicate that TRAF2 is required for TNFR2 and IL-17RD in the IL-17RD·TNFR2 complex-mediated NF-κB activation.
FIGURE 6.

IL-17RD coordinates with TNFR2 to recruit TRAF2. A and B, TRAF2 is required for the IL-17RD·TNFR2 complex-mediated NF-κB transcriptional activity. HEK293T cells co-transfected with indicated plasmids were stimulated without (A) or with TNF-α (B). The luciferase activity was analyzed. **p < 0.01; ***p < 0.001. C, TNFR2mut fails to interact with TRAF2. An IP experiment was performed for HEK293T cells. IB, immunoblot. D, TNFR2mut cannot cooperate with IL-17RD to activate the NF-κB transcriptional activity. HEK293T cells co-expressed with the indicated plasmids. The luciferase activity was analyzed. *, p < 0.05; **, p < 0.01. n.s., not significant. E, co-localization of the IL-17RD·TNFR2·TRAF2 ternary complex. COS-7 cells transfected with indicated plasmids were stimulated without (Control) or with TNF-α and analyzed by double immunostaining with anti-Myc/FLAG antibodies. Scale bar, 20 μm. F and G, IL-17RD promotes TRAF2 recruitment to TNFR2 in co-IP assays. The relative density of precipitated TRAF2/β-actin was used to present the TNFR2·TRAF2 complex (F). Quantitative results are presented as the mean ± S.D. from three independent repeats of G. *, p < 0.05; **, p < 0.01. H, IL-17RD interacts with TRAF2 in HK-2 cells. HK-2 cells treated with or without TNF-α were used to immunoprecipitate complexes after lysis. I–K, TNFR2 facilitates TRAF2 binding to IL-17RD in co-IP assays. The relative density of precipitated IL-17RD/β-actin (J) and TNFR2/β-actin (K), respectively, presented the IL-17RD/TRAF2 and TNFR2·TRAF2 complexes. Quantitative results are presented as the mean ± S.D. from three independent repeats of I. *, p < 0.05; **, p < 0.01.
The complex of IL-17RD·TNFR2 with TRAF2 is further demonstrated by an immunostaining experiment showing co-localization of IL-17RD, TNFR2, and TRAF2 in the absence or presence TNF-α (Fig. 6E). IP experiments revealed that IL-17RD promoted the complex formation of TNFR2 and TRAF2 under a normal culture condition (Fig. 6, F and G, and Fig. 6, I and K). The endogenous TRAF2 and IL-17RD complex was enhanced by TNF-α stimulation in HK-2 cells (Fig. 6H). Furthermore, TNFR2 promoted the complex formation of IL-17RD and TRAF2 (Fig. 6, I and J). Taken together, all the results suggested that the association of IL-17RD with TNFR2 mutually promotes TRAF2 recruitment for the initiation of downstream signaling.
DISCUSSION
NF-κB activation in RTECs plays an important role in tubulointerstitial inflammation, a hallmark of most renal diseases. However, it remains to be elucidated to identify specific factors for the NF-κB activation in RTECs. In this study we reported that the complex of TNFR2 and IL-17RD activates the NF-κB activation. We proposed that IL-17RD preferentially cooperates with TNFR2, but not TNFR1, to enhance NF-κB activation in RTECs through promotion of receptor aggregation and TRAF2 recruitment. We attributed the role of IL-17RD on the activation of NF-κB to the interaction of TNFR2, which presented in a specific pathological condition, for instance in the nephritis where TNFR2 and IL-17RD co-expressed abundantly. We propose that the elevated IL-17RD and TNFR2 in the RTECs will lead to an increase of inflammatory factors, which then results in the damages of kidney. Given the important role of TNFR2 in various renal diseases (2, 3, 12), this finding provided an explanation of the specific role of TNFR2 on NF-κB activation under the condition of inflammatory including nephritis.
A previous report demonstrated that TNF-α induced the heterocomplex formation between TNFR2 and TNFR1 (22). The ligand-induced heterocomplex formation is transient and occurs at a physiologically relevant concentration of TNF-α (22). In contrast to this, our findings suggest that the heterocomplex of IL-17RD and TNFR2 persists in the RTECs (Fig. 4) and is enhanced by TNF-α but can occur without TNF-α treatment (Fig. 5). Our observations explained why NF-κB could be activated by up-regulated TNFR2 in a TNF-α-independent manner under certain inflammation conditions (23–25). Based on our strong evidence and other's observations (23–25), we proposed that the presence of IL-17RD might play a critical role in the TNFR2-mediated activation of NF-κB without TNF-α treatment.
It had been suggested that TNF-α can be transferred from TNFR2 to TNFR1 after its association with TNFR2, which was regarded as a “ligand passing” mechanism (26). In our study we observed that TNF-α facilitated the complex formation of TNFR2 and IL-17RD (Fig. 4). We did not know whether the complex of TNF-α, IL-17RD, and TNFR2 could be interrupted by TNFR1. We also did not understand whether the TNF-α-promoted association of TNFR2 with IL-17RD will affect the binding of TNF-α with TNFR1. Further studies are needed to address the impact of TNFR1 on the specific interaction of IL-17RD and TNFR2. Nevertheless, we have shown that TNF-α enhanced the association of IL-17RD with TNFR2. Because no ligand was identified for the IL-17RD, we speculated that IL-17RD might be a specific co-receptor for TNFR2.
Our study revealed a mechanism by which IL-17RD enhances the NF-κB signaling through TNFR2 in RTECs. However, IL-17RD, by an interaction with p50/p65, a cellular complex in the activation of NF-κB cascade, was reported to block the nuclear translocation of p50/p65 (18). We speculate that IL-17RD may mainly function to assist its partners. For instance, Il-17RD forms a complex with TNFR2 to facilitate the NF-κB activation and with p65 to attenuate the signaling. We provided a case that IL-17RD, an orphan receptor, functions as either an inhibitor or activator for NF-κB activation during different inflammations. Furthermore, we and others have revealed that, in addition to associating with FGFR (27–29) and EGFR (30), IL-17RD formed a heterodimer with IL-17RA (14), and eventually inhibited FGF-induced Erk1/2 activation but also activated EGF and IL-17A signaling. Therefore, we envision that IL-17RD might be an adaptor receptor that generates different effects on the downstream event depending on its partnership and pools of membrane-bound receptors.
In conclusion, we have shown that the association of IL-17RD and TNFR2 mediates NF-κB activation through the adaptor protein TRAF2. Our study addressed a specific role of IL-17RD in promoting TNFR2 activity in nephritis. IL-17RD·TNFR2 heteromerization provides a new mechanism for the pathogenic properties of TNF-α in inflammatory diseases including nephritis.
Acknowledgment
We are grateful to Dr. Chen Dong for helpful discussion and comments.
This work was supported by grants from the 973 Project (2011CB910502), the National Natural Science Foundation of China (30871286, 31071225, 31030040, and 91229203), the Tsinghua Science Foundation (20121080018), the Natural Science Foundation of Beijing (511003), the Major Program (2013ZX08011-006), and the 863 project (2012AA021703) in China.
- TNFR1
- tumor necrosis factor receptor 1
- TNFR2
- tumor necrosis factor receptor 2
- IL-17RD
- interleukin-17 receptor D
- hIL-17RD
- human IL-17RD
- TRAF2
- TNF receptor-associated factor 2
- ECD
- extracellular domain
- RTEC
- renal tubular epithelial cell
- IP
- immunoprecipitation.
REFERENCES
- 1. Wajant H., Pfizenmaier K., Scheurich P. (2003) Tumor necrosis factor signaling. Cell Death Differ 10, 45–65 [DOI] [PubMed] [Google Scholar]
- 2. Vielhauer V., Stavrakis G., Mayadas T. N. (2005) Renal cell-expressed TNF receptor 2, not receptor 1, is essential for the development of glomerulonephritis. J. Clin. Invest. 115, 1199–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ramesh G., Reeves W. B. (2003) TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am. J. Physiol. Renal Physiol. 285, F610–F618 [DOI] [PubMed] [Google Scholar]
- 4. Horiuchi T., Kiyohara C., Tsukamoto H., Sawabe T., Furugo I., Yoshizawa S., Ueda A., Tada Y., Nakamura T., Kimoto Y., Mitoma H., Harashima S., Yoshizawa S., Shimoda T., Okamura S., Nagasawa K., Harada M. (2007) A functional M196R polymorphism of tumour necrosis factor receptor type 2 is associated with systemic lupus erythematosus: a case-control study and a meta-analysis. Ann. Rheum. Dis. 66, 320–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bruggeman L. A., Drawz P. E., Kahoud N., Lin K., Barisoni L., Nelson P. J. (2011) TNFR2 interposes the proliferative and NF-κB-mediated inflammatory response by podocytes to TNF-α. Lab. Invest. 91, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ait-Ali D., Turquier V., Tanguy Y., Thouënnon E., Ghzili H., Mounien L., Derambure C., Jégou S., Salier J. P., Vaudry H., Eiden L. E., Anouar Y. (2008) Tumor necrosis factor (TNF)-α persistently activates nuclear factor-κB signaling through the type 2 TNF receptor in chromaffin cells: implications for long-term regulation of neuropeptide gene expression in inflammation. Endocrinology 149, 2840–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vallabhapurapu S., Karin M. (2009) Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733 [DOI] [PubMed] [Google Scholar]
- 8. de Haij S., Bakker A. C., van der Geest R. N., Haegeman G., Vanden Berghe W., Aarbiou J., Daha M. R., van Kooten C. (2005) NF-κB-mediated IL-6 production by renal epithelial cells is regulated by c-jun NH2-terminal kinase. J. Am. Soc. Nephrol. 16, 1603–1611 [DOI] [PubMed] [Google Scholar]
- 9. Al-Lamki R. S., Wang J., Skepper J. N., Thiru S., Pober J. S., Bradley J. R. (2001) Expression of tumor necrosis factor receptors in normal kidney and rejecting renal transplants. Lab. Invest. 81, 1503–1515 [DOI] [PubMed] [Google Scholar]
- 10. Aten J., Roos A., Claessen N., Schilder-Tol E. J., Ten Berge I. J., Weening J. J. (2000) Strong and selective glomerular localization of CD134 ligand and TNF receptor-1 in proliferative lupus nephritis. J. Am. Soc. Nephrol. 11, 1426–1438 [DOI] [PubMed] [Google Scholar]
- 11. Al-Lamki R. S., Wang J., Vandenabeele P., Bradley J. A., Thiru S., Luo D., Min W., Pober J. S., Bradley J. R. (2005) TNFR1- and TNFR2-mediated signaling pathways in human kidney are cell type-specific and differentially contribute to renal injury. FASEB J. 19, 1637–1645 [DOI] [PubMed] [Google Scholar]
- 12. Ernandez T., Mayadas T. N. (2009) Immunoregulatory role of TNFα in inflammatory kidney diseases. Kidney Int. 76, 262–276 [DOI] [PubMed] [Google Scholar]
- 13. Ren Y., Li Z., Rong Z., Cheng L., Li Y., Wang Z., Chang Z. (2007) Tyrosine 330 in hSef is critical for the localization and the inhibitory effect on FGF signaling. Biochem. Biophys. Res. Commun. 354, 741–746 [DOI] [PubMed] [Google Scholar]
- 14. Rong Z., Wang A., Li Z., Ren Y., Cheng L., Li Y., Wang Y., Ren F., Zhang X., Hu J., Chang Z. (2009) IL-17RD (Sef or IL-17RLM) interacts with IL-17 receptor and mediates IL-17 signaling. Cell Res. 19, 208–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang R. B., Ng C. K., Wasserman S. M., Kömüves L. G., Gerritsen M. E., Topper J. N. (2003) A novel interleukin-17 receptor-like protein identified in human umbilical vein endothelial cells antagonizes basic fibroblast growth factor-induced signaling. J. Biol. Chem. 278, 33232–33238 [DOI] [PubMed] [Google Scholar]
- 16. Zhang S., Xin H., Li Y., Zhang D., Shi J., Yang J., Chen X. (2013) Skimmin, a coumarin from Hydrangea paniculata, slows down the progression of membranous glomerulonephritis by anti-inflammatory effects and inhibiting immune complex deposition. Evid. Based Complement Alternat. Med. 2013, 819296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ozcan A., Ware K., Calomeni E., Nadasdy T., Forbes R., Satoskar A. A., Nadasdy G., Rovin B. H., Hebert L. A., Brodsky S. V. (2012) 5/6 nephrectomy as a validated rat model mimicking human warfarin-related nephropathy. Am. J. Nephrol. 35, 356–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fuchs Y., Brunwasser M., Haif S., Haddad J., Shneyer B., Goldshmidt-Tran O., Korsensky L., Abed M., Zisman-Rozen S., Koren L., Carmi Y., Apte R., Yang R. B., Orian A., Bejar J., Ron D. (2012) Sef is an inhibitor of proinflammatory cytokine signaling, acting by cytoplasmic sequestration of NF-κB. Dev. Cell 23, 611–623 [DOI] [PubMed] [Google Scholar]
- 19. Rodríguez M., Cabal-Hierro L., Carcedo M. T., Iglesias J. M., Artime N., Darnay B. G., Lazo P. S. (2011) NF-κB signal triggering and termination by tumor necrosis factor receptor 2. J. Biol. Chem. 286, 22814–22824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chan F. K., Chun H. J., Zheng L., Siegel R. M., Bui K. L., Lenardo M. J. (2000) A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 288, 2351–2354 [DOI] [PubMed] [Google Scholar]
- 21. Rothe M., Sarma V., Dixit V. M., Goeddel D. V. (1995) TRAF2-mediated activation of NF-κB by TNF receptor 2 and CD40. Science 269, 1424–1427 [DOI] [PubMed] [Google Scholar]
- 22. Pinckard J. K., Sheehan K. C., Schreiber R. D. (1997) Ligand-induced formation of p55 and p75 tumor necrosis factor receptor heterocomplexes on intact cells. J. Biol. Chem. 272, 10784–10789 [DOI] [PubMed] [Google Scholar]
- 23. Haridas V., Darnay B. G., Natarajan K., Heller R., Aggarwal B. B. (1998) Overexpression of the p80 TNF receptor leads to TNF-dependent apoptosis, nuclear factor-κ B activation, and c-Jun kinase activation. J. Immunol. 160, 3152–3162 [PubMed] [Google Scholar]
- 24. Douni E., Kollias G. (1998) A critical role of the p75 tumor necrosis factor receptor (p75TNF-R) in organ inflammation independent of TNF, lymphotoxin α, or the p55TNF-R. J. Exp. Med. 188, 1343–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Akassoglou K., Douni E., Bauer J., Lassmann H., Kollias G., Probert L. (2003) Exclusive tumor necrosis factor (TNF) signaling by the p75TNF receptor triggers inflammatory ischemia in the CNS of transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 100, 709–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tartaglia L. A., Pennica D., Goeddel D. V. (1993) Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J. Biol. Chem. 268, 18542–18548 [PubMed] [Google Scholar]
- 27. Fürthauer M., Lin W., Ang S. L., Thisse B., Thisse C. (2002) Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat. Cell Biol. 4, 170–174 [DOI] [PubMed] [Google Scholar]
- 28. Tsang M., Friesel R., Kudoh T., Dawid I. B. (2002) Identification of Sef, a novel modulator of FGF signalling. Nat. Cell Biol. 4, 165–169 [DOI] [PubMed] [Google Scholar]
- 29. Xiong S., Zhao Q., Rong Z., Huang G., Huang Y., Chen P., Zhang S., Liu L., Chang Z. (2003) hSef inhibits PC-12 cell differentiation by interfering with Ras-mitogen-activated protein kinase MAPK signaling. J. Biol. Chem. 278, 50273–50282 [DOI] [PubMed] [Google Scholar]
- 30. Ren Y., Cheng L., Rong Z., Li Z., Li Y., Zhang X., Xiong S., Hu J., Fu X. Y., Chang Z. (2008) hSef potentiates EGF-mediated MAPK signaling through affecting EGFR trafficking and degradation. Cell. Signal. 20, 518–533 [DOI] [PMC free article] [PubMed] [Google Scholar]