

Background: Parkin mitochondrial recruitment upon CCCP treatment requires active glucose metabolism.
Results: ATP is a key regulator of PINK1-mediated mitophagy by controlling PINK1 translation levels.
Conclusion: PINK1 levels decrease in response to low ATP, resulting in inactivation of Parkin-mediate mitophagy.
Significance: Short half-life of PINK1 renders it sensitive to metabolic changes and ATP level. The finding offers insight into bioenergetics of the PINK1-Parkin pathway.
Keywords: ATP; Glucose Metabolism; Mitophagy; Parkin; Parkinson Disease (Autosomal Recessive, Early Onset) 7 (PARK7); PTEN-induced Putative Kinase 1 (PINK1)
Abstract
Mutations in several genes, including PINK1 and Parkin, are known to cause autosomal recessive cases of Parkinson disease in humans. These genes operate in the same pathway and play a crucial role in mitochondrial dynamics and maintenance. PINK1 is required to recruit Parkin to mitochondria and initiate mitophagy upon mitochondrial depolarization. In this study, we show that PINK1-dependent Parkin mitochondrial recruitment in response to global mitochondrial damage by carbonyl cyanide m-chlorophenylhydrazine (CCCP) requires active glucose metabolism. Parkin accumulation on mitochondria and subsequent Parkin-dependent mitophagy is abrogated in glucose-free medium or in the presence of 2-deoxy-d-glucose upon CCCP treatment. The defects in Parkin recruitment correlate with intracellular ATP levels and can be attributed to suppression of PINK1 up-regulation in response to mitochondria depolarization. Low levels of ATP appear to prevent PINK1 translation instead of affecting PINK1 mRNA expression or reducing its stability. Consistent with a requirement of ATP for elevated PINK1 levels and Parkin mitochondrial recruitment, local or individual mitochondrial damage via photoirradiation does not affect Parkin recruitment to damaged mitochondria as long as a pool of functional mitochondria is present in the photoirradiated cells even in glucose-free or 2-deoxy-d-glucose-treated conditions. Thus, our data identify ATP as a key regulator for Parkin mitochondrial translocation and sustaining elevated PINK1 levels during mitophagy. PINK1 functions as an AND gate and a metabolic sensor coupling biogenetics of cells and stress signals to mitochondria dynamics.
Introduction
As the center of cellular energy, maintenance of salubrious mitochondrion is essential for cell function and sustainability (1). A mechanism for sequestering and eliminating malfunctioning mitochondria is vital in the cell, which otherwise may produce harmful reactive oxygen species. PINK1 and Parkin have been found to work in the same autophagic pathway in removing damaged mitochondria from the cell (2–7). As a testament to their significance in cellular health, mutations in PINK1 (8, 9) and Parkin (10, 11) have been implicated in autosomal recessive Parkinsonism. The current paradigm suggests that if the mitochondrial membrane potential is intact, the serine-threonine kinase PINK1 is translocated to the inner mitochondrial membrane to undergo proteolytic cleavage by PARL (presenilin-associated rhomboid-like protein) (12, 13) followed by proteasomal degradation (14). If the mitochondrial membrane potential is dissipated, PINK1 accrues on the outer mitochondrial membrane as its 63-kDa full-length isoform to recruit cytosolic Parkin (2, 4, 5, 15), an E3 ubiquitin ligase, which ubiquitylates numerous OMM proteins leading to autophagosome engulfment of the ubiquitin-tagged depolarized mitochondria and subsequent lysosomal degradation, i.e. mitophagy (2, 7, 16–22).
Although several studies have verified this observation in neuronally derived (4, 23) and non-neuronal immortalized cell lines (2, 4, 5), the PINK1/Parkin mitophagy pathway has been proven to be less robust in neurons (24). To date, research in this field predominantly relied on ionophores and inhibitor drugs to compromise the mitochondrial integrity (3, 25–27). Consequently, the differing bioenergetics resulting from this large scale mitochondrial damage may contribute to this observed variance, in which immortalized cell lines exhibit the Warberg effect to rely on glycolysis for a significant portion of their ATP production (28–31), whereas neurons rely primarily on oxidative phosphorylation for ATP production (32). Although the involvement of ATP was suggested to influence the mitophagy pathway in previous studies (24, 33), definitive evidence demonstrating the necessity for ATP in the PINK1/Parkin pathway, as well as the mechanism that explains this phenomenon, has yet to be explored.
Here we demonstrate the requirement of ATP to be present for carbonyl cyanide m-chlorophenylhydrazine (CCCP)-induced3 Parkin mitochondrial recruitment while uncovering the mechanistic link between PINK1 up-regulation with ATP levels. Although the level of 63-kDa full-length PINK1 was previously shown to increase when cells are incubated with a working concentration of CCCP (4, 12), we show that this increase can only be expressed when a threshold concentration of glucose is met. Otherwise, the PINK1 levels remain low, consequently restricting mitophagic responses. The dynamic range in which PINK1 levels exhibit its response to glucose strongly correlates ATP levels under the same glucose concentrations. This observation was verified using various concentrations of 2-deoxy-d-glucose to control cellular ATP levels. By demonstrating that PINK1 senses both mitochondrial damage and ATP to influence the activation or deactivation of the mitophagic response, our findings offer the novel characteristic of PINK1 as an AND gate (34) that acts as a molecular switch whose up-regulation upon mitochondrial depolarization ultimately determines the triggering of mitochondrial damage response.
Although the role of the PINK1/Parkin pathway in eliminating depolarized mitochondria via mitophagy has been well documented (2, 16–19), there are still many unanswered questions about its molecular mechanisms. For example, the signaling mechanisms upstream of PINK1/Parkin that trigger mitophagy remain poorly characterized (35). There is significant variability in Parkin recruitment in neurons in part because of culture conditions or antioxidant levels in the cultured medium (35). To elucidate the pathway as a function of culture conditions, we began our studies with the most basic of cellular functions: glucose metabolism. Because CCCP treatment effectively collapses the electron transport chain (3) and forces the cell to rely exclusively on fermentation for energy production, we hypothesized that the PINK1/Parkin pathway may integrate, or be influenced by, glycolytic metabolism to induce mitophagy upon irreversible mitochondrial damage.
EXPERIMENTAL PROCEDURES
Western Blot Analysis
To analyze protein levels, HeLa and mouse embryonic fibroblast (MEF) cells were grown on 10-cm plates to be harvested and lysed using a standard RIPA buffer mixed with cOmplete Protease inhibitor mixture (Roche). Total cellular extracts in the presence of a protein standard (Bio-Rad) were resolved by 12% SDS-PAGE and transferred to a 0.22-μm nitrocellulose membrane and incubated with specific antibodies overnight at 4 °C. The antibodies used in this investigation were: mouse anti-Parkin (1:5000; clone PRK8, Sigma-Aldrich), rabbit anti-PINK1 (1:500; BC100-494, Novus Biologicals), rabbit monoclonal antibody (D8G3) (6946S, Cell Signaling Technology), mouse anti-EZRIN (1:5000, Sigma-Aldrich), and mouse anti-GAPDH (1:20,000, Santa Cruz Biotechnology).
Plasmids, Cell Culture, and Transfection
Gateway recombination technology (Invitrogen) was used to generate constructs. PINK1 was cloned into CSII-EF-DEST-IRES-Hygromycin lentiviral vectors (gift from Dr. Hiroyuki Miyoshi). Parkin was cloned into pREX-Venus-DEST-IRES-Blasticidin retroviral vector. The mitochondria marker, pMSCV-CMV-puro-IMS-RFP, was a gift from Dr. Sabrina Spencer. The HeLa and HEK293T cell lines were obtained from the American Type Culture Collection. MEF cells that are PINK1 or Parkin null or derivatives that stably express human PINK1 or Parkin were previously described (36). All cell lines were maintained in DMEM containing 4.5 g/liter glucose, 1 mm l-glutamine, 1 mm sodium pyruvate, 100 units/ml penicillin/streptomycin, and 10% fetal bovine serum at 37 °C in 5% CO2. All the stable cell lines were generated by lentivirus and retrovirus transduction and selected with 100 μg/ml hygromycin (Alexis Biochemicals), 5 μg/ml blasticidin (Invitrogen), or 2 μg/ml puromycin (Sigma).
Live Cell Image Acquisition and Quantification
Cells were grown on Costar 96-well plates, and ImageXpress XL (Molecular Devices) was used to screen the plates to collect data. Parkin localization in mitochondria was assessed with MetaXpress application module transfluor Cell Scoring Application Module (Molecular Devices) with more than 10,000 cells normalized via Hoechst 33258 stained nucleus. Standard deviations were determined from at least three sets of data. Confocal images were obtained on Nikon A1R Confocal and total internal reflection fluorescence using 100×/1.45 objectives at 37 °C in 5% CO2. For the photodamage experiments, the region of interest (ROI) encompassing ∼5–10 mitochondrion on three different cells were bleached using a 488-nm (20-mW) laser line for MEF cells and a 405-nm (20-mW) laser line for HeLa cells at 100% power for 4 s of stimulation each. Subsequent image acquisition followed every 1 min for 2–5 h (125–305 cycles over the length of the experiment).
RNA Extraction and Quantitative PCR Assays
RNA was isolated with TRIzol reagent (Invitrogen) following standard phenol extraction protocol. Quantitative PCR amplification was performed in a final volume of 15 μl, containing 1 μl of cDNA, 5 μm of each respective primer, and 7.5 μl of Fast SybrGreen Master Mix (Applied Biosystems). The primers used for RT-PCR are: human PINK1 forward (5′-GGACGCTGTTCCTCGTTA-3′), human PINK1 reverse (5′-ATCTGCGATCACCAGCCA-3′), human GAPDH forward (5′-GAAGGTGAAGGTCGGAGT-3′), and human GAPDH reverse (5′-GAAGATGGTGATGGGATTTC-3′). The amplifications were performed in optical grade 96-well plates on a StepOnePlus real time PCR system with an initial step at 95 °C for 20 s, followed by 40 cycles of 95 °C for 10 s and 60 °C for 20 s. All samples were probed in triplicate. The CT was automatically determined by the instrument.
ATP Bioluminescent Assay
Cells were grown on Costar 96-well plates. After treatment and 2 h of incubation, the medium was removed and replaced with boiling 100 mm Tris, 4 mm EDTA, pH 7.75. When the samples reached room temperature, Luciferase reagent from ATP Bioluminescence assay kit CLS II (catalog no. 11699695001; Roche) was added at a 1:1 ratio. Luminescence was read by a microplate reader (Synergy H1).
Statistical Analysis
All Parkin localization in mitochondria are presented as means ± S.D. by visually scoring >1000 cells per stable cell line from at least three independent experiments in each group at each time point. ATP Bioluminescent Assay is presented as the means ± S.E. from two independent experiments. PINK1 quantification and immunoblots were performed by at least three independent biological replicate experiments. Sigmaplot 11.0 was used to fit the dose-response curve using four-parameter logistic function.
RESULTS
Glucose Is Required for CCCP-induced Mitophagic Response
To investigate the need for glucose metabolism in mitophagy, human epithelial cancer cells (HeLa) stably expressing fluorescent protein tagged wild type Parkin were treated with 20 μm CCCP in FBS-free DMEM containing 4.5 g/liter of glucose or no glucose for 8 h. Under physiological conditions, Parkin is predominantly localized in the cytosol, whereas cells eliciting mitophagic responses display an increased localization of Parkin to the mitochondria. Under our experimental conditions, the response was monitored via fluorescent microscopy. Increased Parkin mitochondrial localization in medium containing 4.5 g/liter glucose was observed within 2 h, whereas no mitochondrial localization was observed in medium containing no glucose and ultimately led to a significant loss in structural integrity within 4 h (Fig. 1A). These results indicate that, in conjunction with mitochondrial damage, glucose is a necessary variable for the PINK1/Parkin-dependent mitophagy pathway.
FIGURE 1.

CCCP-induced Parkin mitochondrial translocation requires glucose. A, the absence of either CCCP or glucose abrogates Parkin mitochondrial translocation. HeLa cells expressing Venus-Parkin-WT and RFP-smac-mts were left treated or were treated with 20 μm CCCP and incubated for 2 h with or without 4.5 g/liter glucose. Parkin mitochondrial translocation was visualized by fluorescent microscopy. B, HeLa cells were treated with CCCP and incubated for 2 h in a glucose concentration gradient between 0 and 1.0 mg/ml. C and D, quantitation of Venus-Parkin accumulation on mitochondria reveals a conserved glucose concentration range between 0.05 and 0.2 mg/ml for both HeLa (C) and MEF (D) cells.
To further characterize the response to glucose in the PINK1/Parkin mitophagy pathway, HeLa cells stably expressing Venus-Parkin and MEF cells stably expressing Venus-Parkin and overexpressing human PINK1 and were incubated in different concentrations of glucose, ranging from 0 mg/ml to 1.0 mg/ml, while being treated with 20 μm CCCP and incubated for 2 h. Full Parkin mitochondrial localization was observed in both HeLa (Fig. 1B) and MEF cells (data not shown) above a comparable threshold glucose concentration range of 0.05 to 0.2 mg/ml, where EC50 = 0.098 mg/ml for HeLa cells (Fig. 1C) and EC50 = 0.111 mg/ml for MEF cells (Fig. 1D). However, the kinetics between the two cell lines was different, exhibiting full Parkin mitochondrial localization after ∼1.5 h in HeLa cells and ∼20 min in MEF cells. The differing kinetics can likely be attributed to the differing expression levels of PINK1 between the cell lines, where the ectopically expressing PINK1 levels of MEF cells are higher than the endogenous PINK1 levels of HeLa cells, leading to a kinetically favorable triggering of Parkin mitochondrial localization. Furthermore, our data showed no correlation in Parkin mitochondrial localization between cell density and glucose concentration (data not shown), indicating that the uptake of glucose is dependent on concentration and not on the availability of glucose molecules per cell. These results indicate that exceeding a conserved glucose concentration threshold elicits the mitophagic response between different cell lines upon mitochondrial damage, and the sensitivity of the PINK1/Parkin mitophagy pathway to glucose is characteristic of the pathway itself and not a phenomenon exclusive to a particular cell line.
Elevated PINK1 Levels upon CCCP Treatment Are Glucose-dependent
Upon mitochondrial depolarization, the inner mitochondrial membrane transport of PINK1 is restricted, and thus the ensuing PARL cleavage and proteasomal degradation is also restricted and leads to stabilization of the 63-kDa PINK1 at mitochondrial outer membrane (2, 4, 5, 15). To identify the glucose-sensitive response mechanism in the PINK1/Parkin mitophagy pathway, we assessed the protein levels and/or response of ectopically expressing PINK1 and Venus-Parkin in MEF cells by immunoblotting upon 2 h of incubation in 20 μm CCCP in a glucose gradient ranging from 0 to 1.5 mg/ml. Our results indicate an increase in PINK1 expression levels between glucose concentrations of 0.1 and 1.0 mg/ml with EC50 = 0.211 mg/ml (Fig. 2A). Parkin expression levels remained constant, whereas mobility shifts were observed (red arrowhead), which is likely due to elevated PINK1 activity. To further determine the specificity of glucose starvation on protein levels, we also measured the expression levels of GRP75/Mortalin, a member of the heat shock protein 70 (HSP70) class of proteins predominantly localized to mitochondria that is involved in cellular stress responses (37). Like Parkin and GAPDH, GRP75 expression levels are independent of glucose amount in the medium, suggesting that the decline in PINK1 levels is relatively specific. To verify the sensitivity toward glucose concentration for endogenous PINK1, HeLa cells were incubated in the same conditions and immunoblotted. The results were concurrent with MEF cells where an increase in PINK1 expression levels was observed within a similar range of glucose concentrations of 0.1 and 1.0 mg/ml with EC50 = 0.351 mg/ml (Fig. 2B). These results indicate that stabilization of PINK1 in response to CCCP treatment requires glucose. Persistent low levels of PINK1 in the absence of glucose may be responsible for the defects seen in Parkin recruitment and activation of mitophagy pathway.
FIGURE 2.

PINK1 expression levels alter in response to glucose. A and B, PINK1 levels under CCCP treatment display sigmoidal rise at higher glucose concentration within a cell type-conserved range of 0.1–1.0 mg/ml. PINK1−/− Parkin−/− MEF cells stably expressing Venus-Parkin-WT and hPINK1-WT (A) and HeLa cells stably expressing Venus-Parkin-WT (B) were treated with 20 μm CCCP, incubated in a glucose concentration gradient between 0 and 1.5 mg/ml, and collected after 2 h. PINK1, Parkin, Grp75, Ezrin, and GAPDH were monitored by immunoblotting with their respective antibodies. PINK1 quantitation was based on relative chemiluminescence intensity. C, mutation of native gatekeeper residue Met-318 to Ala in hPINK1 renders kinase activity susceptible to inhibition via ATP analog 1-NA-PP1. PINK1−/− Parkin−/− MEF cells stably expressing Venus-Parkin-WT and either hPINK1-WT or hPINK1-M318A were treated with 20 μm CCCP in the presence or absence of 2 μm 1-NA-PP1. D, the expression level of PINK1 in response to glucose is independent of its kinase activity. MEF cells stably expressing Venus-Parkin-WT and hPINK1-M318A were treated with 20 μm CCCP and 2 μm 1-NA-PP1, incubated in a glucose concentration gradient between 0 and 1.5 mg/ml, and collected after 2 h. PINK1, Parkin, and Ezrin were monitored by immunoblotting with their respective antibodies. E, hPINK1 mRNA are unaffected by glucose concentration. HeLa cells were left untreated or were treated with 20 μm CCCP, incubated in a glucose concentration gradient between 0 and 1.0 mg/ml for 2 h, and collected for RNA extraction and subsequent quantitative PCR for hPINK1 and GAPDH. CT and primer efficiency were measured and quantified via the Pfaffl method. F, the stability of full-length PINK1 is not compromised upon glucose depletion in the presence of CCCP. MEF cells were treated with 20 μm CCCP and incubated for 1 h in full medium, followed by treatment with 5 μm CHX in full medium or glucose-free medium. The cells were collected at 30-min intervals for 1.5 h, and PINK1, Grp75, and Ezrin degradation levels were monitored via immunoblot. PINK1 was monitored by using rabbit anti-PINK1 antibody (Novus). G, full-length PINK1 degradation is independent of glucose in the absence of CCCP. MEF cells were treated with 5 μm CHX in full medium or glucose-free medium. The cells were collected at 30-min intervals for 1.5 h, and PINK1 and Ezrin levels were monitored via immunoblot. PINK1 was monitored by using anti-PINK1 mAb D8G3 antibody, which is more sensitive in detecting the native PINK1 levels without CCCP treatment.
We have recently shown that the Met-318 residue of hPINK1 is the ATP gatekeeper residue, and mutation to Ala renders the kinase activity susceptible to ATP analogs (38). Specifically, in the presence of an ATP analog such as 1-NA-PP1, the kinase activity of hPINK1-M318A is effectively shut down, restricting Parkin mitochondrial translocation, whereas wild type PINK1 is insensitive (Fig. 2C). To test whether or not the kinase activity is important in maintaining PINK1 levels in response to glucose, MEF cells stably expressing hPINK1-M318A and Venus-Parkin were incubated in 20 μm CCCP for 2 h in the presence of 2 μm 1-NA-PP1 and collected for immunoblotting. Although PINK1 kinase activity was inhibited, which is confirmed by the lack of mobility shift in Parkin, PINK1 exhibited a change in up-regulation in response to differing glucose concentrations (Fig. 2D), which appears to be analogous to the response of PINK1-WT when kinase activity was intact (Fig. 2A). Therefore, our results demonstrate that the ability of PINK1 to sense glucose concentration and alter expression levels is independent of its kinase activity.
To assess whether PINK1 expression regulation occurred at the level of transcription or protein synthesis, quantitative PCR was conducted for endogenous PINK1 in HeLa cells left untreated or treated with 20 μm CCCP in different glucose concentrations for 2 h using GAPDH as controls. Our results indicate that mRNA expression levels for PINK1 remain largely unchanged in the differing glucose concentrations whether or not CCCP was present (Fig. 2E). Therefore, lower PINK1 levels in the absence of glucose is unlikely due to suppression of its mRNA expression at the transcriptional level.
Low levels of PINK1 in the absence of glucose could be a result of excessive degradation of the 63-kDa PINK1. To test this hypothesis, we measured stability of PINK1 with or without glucose in the presence of 5 μm cycloheximide (CHX) upon CCCP treatment. As shown in Fig. 2F, the half-life of PINK1 is ∼30 min in the glucose medium. Remarkably in glucose-free medium, the full-length (63-kDa) PINK1 is very stable. This result suggests that the full-length PINK1, upon OMM accumulation, is not targeted for excessive degradation when glucose is depleted, and to the contrary, the levels are sustained. As a control, we also measured the stability of PINK1 in glucose and glucose-free medium without CCCP treatment. The result shown in Fig. 2G suggests that there is no significant difference in PINK1 stability in the presence or absence of glucose. These results demonstrate that the decrease in full-length PINK1 levels is not due to decreased stability as a function of glucose concentration. Taken together, our data suggest that the decrease in PINK1 levels seen in glucose withdrawal is most likely due to translational suppression.
Mitochondrial Depolarization-induced Parkin Mitochondrial Recruitment and Elevated PINK1 Levels Correlate with Intracellular ATP Levels
Previous studies have shown that the rapid loss of ATP after mitochondrial depolarization could be one of the reasons behind poor Parkin-mitochondrial translocation in neurons or HeLa cells forced into dependence on mitochondrial respiration (24). Because HeLa cells and immortalized cell lines generally utilize glycolytic metabolism for energy production, it is our expectation that glucose withdrawal coupled with mitochondrial depolarization would severely suppress intracellular ATP levels. To test this hypothesis, HeLa and MEF cells expressing PINK1 and Parkin were incubated in a glucose gradient ranging from 0 to 4.5 mg/ml in the presence or absence of 20 μm CCCP for 2 h. The ATP levels were assayed via luciferase luminescence. Our data indicate that ATP levels in HeLa and MEF cells are not significantly altered within the glucose gradient in the 2 h time frame when no CCCP is present. However, when CCCP was present, a drop in ATP levels in HeLa and MEF cells was observed at low glucose concentrations, whereas ATP levels appear to dramatically increase and remain at CCCP-untreated levels under high glucose concentrations (39). The glucose concentration range where the ATP level shift occurs is consistent in both HeLa and MEFs at 0.1 and 1.0 mg/ml, where EC50 = 0.543 mg/ml for HeLa (Fig. 3A) and EC50 = 0.233 mg/ml for MEFs (Fig. 3B). Interestingly, the glucose concentration range where ATP levels shift occurs is highly comparable with where the PINK1 expression levels shift, suggesting a positive quantitative correlation.
FIGURE 3.
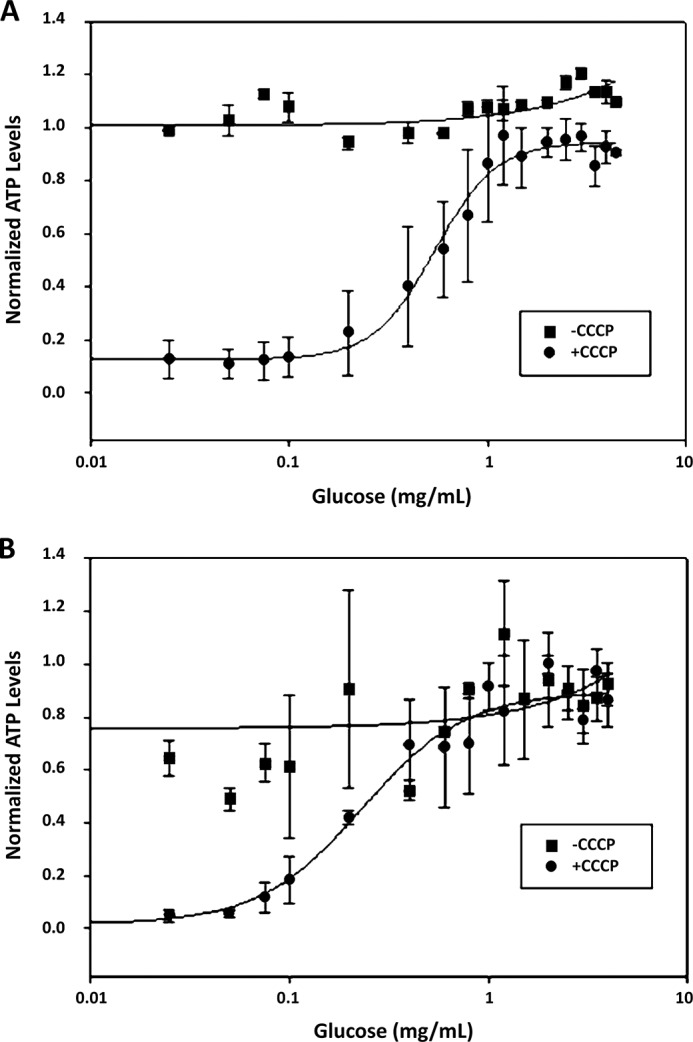
ATP level response to glucose concentration and CCCP treatment. A and B, ATP levels under CCCP treatment display parallel sigmoidal response to PINK1 in glucose concentrations range between 0.1 and 1.0 mg/ml. HeLa cells (A) and MEF cells (B) were incubated in a glucose gradient ranging from 0 to 4.5 mg/ml in the presence or absence of 20 μm CCCP for 2 h. ATP levels were quantified via luciferase assay and normalized to absolute cell count.
Glucose Metabolism, Not Glucose per se, Is Required for Elevated PINK1 Levels
Mechanistically, the requirement of glucose for PINK1 induction by CCCP could be attribute to glucose per se as a signaling molecule or breakdown of glucose for energy production. To differentiate these possibilities, we treated HeLa cells with 2-deoxy-d-glucose (2-DG), an inhibitor that is known to block glucose metabolism. HeLa cells stably expressing Venus-Parkin-WT were treated with different concentrations of 2-DG ranging from 0 to 50 mm in DMEM containing 4.5 g/liter of glucose. After the 2 h of incubation, the cells were treated with 20 μm CCCP and incubated for another 2 h, followed by observation via fluorescent microscopy. Full Parkin mitochondrial localization was observed in low 2-DG concentrations, whereas higher 2-DG concentrations fully inhibited Parkin mitochondrial localization (Fig. 4A) between the range of 3 to 10 mm with IC50 = 6.56 mm (Fig. 4B). These results not only validate that an active glycolytic pathway is necessary to trigger Parkin mitochondrial localization but also rules out the possibility that extracellular sensing of glucose levels is a key component in PINK1/Parkin mitophagy pathway. Therefore, glucose alone is not a necessary variable, but an active glycolytic pathway is necessary for activation of the PINK1/Parkin mitophagy pathway.
FIGURE 4.

CCCP-induced Parkin mitochondrial translocation is abrogated in the presence of 2-deoxy-d-glucose. A, treatment of 2-DG abrogates CCCP-induced Parkin mitochondrial translocation. HeLa cells expressing Venus-Parkin-WT and RFP-smac-mts were incubated in a 2-DG dose response between 0 and 50 mm for 2 h, followed by treatment with 20 μm CCCP and incubated for another 2 h. Parkin mitochondrial translocation was visualized by fluorescent microscopy. B, quantitation of Venus-Parkin accumulation on mitochondria reveals a 2-DG inhibitory response range between 3 and 10 mm. C, ATP levels in response to inhibition of glycolysis and oxidative phosphorylation decline within a range between 1 and 5 mm. HeLa cells were incubated in a 2-DG dose response ranging between 0 and 40 mm for 2 h, followed by the presence or absence of 20 μm CCCP or 10 μm oligomycin (Oligo) for another 2 h. ATP levels were quantified via luciferase assay and normalized to absolute cell count. D, PINK1 expression levels under 2-DG and CCCP treatment display parallel inhibitory response to ATP in 2-DG dose range between 1 and 5 mm. HeLa cells were incubated in a 2-DG dose response between 0 and 20 mm for 2 h, followed by 20 μm CCCP treatment and were incubated for another 2 h and collected. PINK1, Parkin, Grp75, and GAPDH were monitored by immunoblotting with their respective antibodies. PINK1 quantitation was based on relative chemiluminescence intensity.
To confirm that the level of intracellular ATP is altered by 2-DG and mitochondria damagers, HeLa cells expressing PINK1 and Parkin were incubated in a 2-DG concentration gradient ranging from 0 to 40 mm for 2 h, followed by treatment or nontreatment with 20 μm CCCP or 10 μm oligomycin to be incubated for another 2 h. The ATP levels were assayed via luciferase luminescence. Our data indicate that ATP levels in HeLa cells drop significantly upon CCCP and oligomycin treatment within a 2-DG concentration range between 1 to 5 mm. Our data demonstrate that IC50 = 3.09 mm for cells treated with CCCP and IC50 = 1.81 mm for cells treated with oligomycin. Cells untreated with CCCP or oligomycin also exhibited a drop in ATP levels within a 2-DG concentration range between 0 and 20 mm with IC50 = 7.75 mm but remained higher than cells treated with CCCP while maintaining a steady level of ATP at ∼40% in comparison with ATP levels with low/no 2-DG treatment (Fig. 4C).
To further validate the mechanistic response of 2-DG in the PINK1/Parkin mitophagy pathway, we assessed the protein levels and/or response of endogenous PINK1 and ectopic Venus-Parkin of HeLa cells by immunoblotting the cells incubated with 2-DG concentration gradient ranging from 0 to 20 mm for 2 h, followed by 20 μm CCCP treatment and incubation for another 2 h. Concurrent with our previous glucose gradient immunoblot data, our results substantiate that the mechanistic perpetrator is PINK1, indicated by a decrease in expression levels between 2-DG concentrations of 1 to 5 mm with IC50 = 1.72 mm (Fig. 4D). Parkin expression levels remained unchanged, whereas mobility shifts were observed (red arrowhead) wherever PINK1 levels were high. When comparing the dose response that PINK1 and ATP exhibit in the 2-DG concentration range with CCCP treatment, the conserved range of 1 to 5 mm where PINK1 expression levels and ATP levels drop, as well as the highly comparable IC50 values, further confirm a positive correlation between PINK1 and ATP. These results validate PINK1 to be responsible for sensing glycolytic activity to alter its expression level to influence its kinase activity, ultimately leading to the activation or inactivation of the mitophagy pathway.
Parkin Recruitment to Individually Damaged Mitochondrion Can Occur Independent of Glucose
We show that despite incubation in a glucose-free medium, as long as functional mitochondria within a cell are present, oxidative ATP production is active (Fig. 3, A and B). If the levels of ATP are critical, we would expect that mitophagy and Parkin recruitment could still occur if there is sufficient healthy mitochondria that undergo oxidative phosphorylation. Although CCCP is known to induce large scale mitochondrial depolarization, photoirradiation of individual mitochondria with 400- to 500-nm lights causes oxygen-dependent inactivation of flavoproteins and succinate dehydrogenase that is mediated by production of reactive oxygen species (40, 41). Photoirradiation with high doses of 488-nm light induces irreversible mitochondrial depolarization and mitophagy. Consistent with reactive oxygen species-mediated mitophagy, cells stably expressing mitochondrial targeted photosensitizer KillerRed, which effectively produces reactive oxygen species in response to low levels of 559-nm light, undergo Parkin-mediated mitophagy with appearance of Parkin aggregation near the site of photoirradiation (40, 42). We verified the photoirradiation technique in HeLa (supplemental Movie S1) and MEF cells (supplemental Movie S3) expressing Venus-Parkin in medium containing 4.5 mg/ml of glucose to observe robust Parkin localization and aggregate formation on, or within a close proximity to, the photoirradiated mitochondria via confocal microscopy (Figs. 5A and 6A). Whereas both 488-nm light and 405-nm light are equally effective in triggering Parkin aggregation in MEF cells, 405-nm light appears to be more effective in HeLa cells. Next, we photoirradiated individual mitochondria in HeLa and MEF cell lines in medium containing no glucose. Consistent with our hypothesis, MEF cells (supplemental Movie S4) exhibited Parkin localization toward the photoirradiated mitochondria (Fig. 6B). However, HeLa cells did not exhibit mitochondrial localization of Parkin and underwent apoptosis after ∼3 h, seen by RFP-smac release (data not shown). To constrain apoptotic responses, HeLa cells were supplemented with 10 μm caspase inhibitor Ac-DEVD-CHO in a glucose-free medium, which we then proceeded to photoirradiate individual mitochondria. Under these conditions, HeLa cells (supplemental Movie S2) displayed Parkin localization on the photoirradiated mitochondria (Fig. 5B). Taken together, our results demonstrate that mitochondrial localization of Parkin is not specific to glucose when ATP production remains active.
FIGURE 5.

HeLa cells display ATP-dependent Parkin mitochondrial translocation. A and B, individual mitochondrial damage can induce Parkin mitochondrial translocation in ATP-dependent/glucose-independent manner in HeLa cells. HeLa cells expressing Venus-Parkin-WT and RFP-smac-mts were subjected to individual mitochondrial photoirradiation with 405-nm light (indicated by dotted white outlines) and incubated in control medium (A) or glucose-free medium (B). Parkin accumulation (indicated by arrows) is visualized in the zoomed in view of the dotted square regions within the left panels. Parkin aggregation fold difference in fluorescence intensity versus background was quantified by acquiring ratio between bright versus dim ROI.
FIGURE 6.

MEF cells display ATP-dependent Parkin mitochondrial translocation. A and B, individual mitochondrial damage can induce Parkin mitochondrial translocation in ATP-dependent/glucose-independent manner in MEF cells. PINK1−/− Parkin−/− MEF cells expressing hPINK1-WT and Venus-Parkin-WT were stained with mitochondrial dye TMRE and subjected to individual mitochondrial photoirradiation with 405-nm light (indicated by dotted white outlines) and incubated in control medium (A) or glucose-free medium (B). Parkin accumulation (indicated by arrows) is visualized in the zoomed in view of the dotted square regions within the left panels. Parkin aggregation fold difference in fluorescence intensity versus background was quantified by acquiring ratio between bright versus dim ROI.
Suppression of ATP Production Is Sufficient to Abrogate Parkin Recruitment to Individually Damaged Mitochondrion
To further corroborate our hypothesis that glucose is dispensable for Parkin recruitment to individually damaged mitochondrion, HeLa cells expressing Venus-Parkin was incubated with 30 mm 2-DG for 2 h in medium containing 4.5 mg/ml of glucose (supplemental Movie S5). In this scenario, 2-DG shuts down the glycolytic pathway without disturbing mitochondrial integrity, which maintains a constant, albeit lower, production of ATP via oxidative phosphorylation (Fig. 4C) (39, 43, 44). Although 30 mm 2-DG was more than sufficient in restricting Parkin mitochondrial localization by large scale mitochondrial damage through CCCP treatment, selective mitochondrial damage via photoirradiation resulted in robust Parkin localization and aggregate formation on, or within a close proximity to, the photoirradiated mitochondria after ∼90 min (Fig. 7A). This result is in excellent agreement with our hypothesis that glucose or glycolytic pathway is dispensable for locally damaged mitochondrial mitophagy responses and suggests that Parkin mitochondrial localization is possible if an active pool of mitochondria in the cell can generate ATP.
FIGURE 7.

ATP-dependent PINK1 expression mediate Parkin mitochondrial translocation. A–C, inhibition of cellular bioenergetics at different stages verifies dependence on ATP to instigate Parkin mitochondrial translocation. HeLa cells expressing Venus-Parkin-WT and RFP-smac-mts were incubated in either 30 mm 2-DG (A), 10 μm oligomycin (B), or both (C) for 2 h, followed by individual mitochondrial photoirradiation with 405-nm light (indicated by dotted white outlines) and visualized for Parkin accumulation (indicated by arrows) in the zoomed in view of the dotted square regions within the left panels. Parkin aggregation fold difference in fluorescence intensity versus background was quantified by acquiring ratio between bright versus dim ROI.
To determine whether shutdown of oxidative phosphorylation-dependent ATP production can abrogate Parkin recruitment to individually damaged mitochondrion, we subjected cells to oligomycin, which is known to inhibit ATP synthase. HeLa cells expressing Venus-Parkin was incubated with 10 μm oligomycin for 2 h in medium containing 4.5 mg/ml of glucose (supplemental Movie S6). At this concentration, oligomycin is known to shut down mitochondrial ATP productions without impacting the glycolytic pathway, allowing the cells to rely on anaerobic glycolysis to produce ATP (Fig. 4C) (39). Under the same photoirradiation conditions (in terms of ROI area and laser strength) as seen with 2-DG, robust Parkin localization and aggregate formation was observed on, or within a close proximity to, the photoirradiated mitochondria after ∼90 min (Fig. 7B). This result is consistent with the notion that when ATP levels are maintained in the cells, Parkin mitochondrial localization appears to be functional.
Although 2-DG and oligomycin individually did not lead to significant perturbation of Parkin mitochondrial recruitment by photoirradiation, we further tested the effects of combination of both treatments, which is expected to shut down ATP production in cells (Fig. 4C), on photoirradiation-induced Parkin recruitment (supplemental Movie S7). HeLa cells expressing Venus-Parkin were incubated with both 30 mm 2-DG and 10 μm oligomycin for 2 h in medium containing 4.5 mg/ml of glucose prior to photoirradiation. Neither Parkin localization nor aggregation was detected after observation up to 6 h (Fig. 7C). Taken together, our data demonstrate and validate that Parkin mitochondrial localization is dependent on the presence of anaerobic and/or aerobic ATP production, and this is linked to diminished PINK1 levels.
DISCUSSION
Here we show that induction of Parkin translocation to mitochondria and subsequent Parkin-dependent mitophagy upon mitochondrial depolarization require glucose metabolism in HeLa and MEF cells. The defects of Parkin recruitment correlates with declining ATP levels by combined glucose withdrawal and CCCP treatment, suggesting that CCCP-induced Parkin mitochondrial translocation is ATP-sensitive. Furthermore, we demonstrate that defects in Parkin recruitment is linked to PINK1 induction because low levels of ATP suppress elevation of PINK1 in response to global depolarization of mitochondria by CCCP treatment. Suppression of PINK1 induction is most likely due to translation inhibition by low levels of ATP because neither mRNA levels or stability of PINK1 can account for decreased PINK1 levels. Consistent with the requirement of ATP for PINK1 induction and Parkin recruitment, Parkin mitochondrial translocation and mitophagy induced by local damage of individual mitochondrion via photoirradiation is insensitive to glucose withdrawal or inhibition of glycolytic metabolism as long as a pool of healthy mitochondria in the same cells can supply adequate ATP. Perturbation of oxidative phosphorylation alone has little effect on these processes. However, when both oxidative phosphorylation and glycolytic metabolism are perturbed, Parkin translocation and mitophagy are effectively abrogated. Our results suggest that stress-induced PINK1 elevation requires ATP. PINK1 functions as an AND gate by integrating bioenergetics of cells and stress signals to specify mitochondrial damage response decisions.
The importance of bioenergetics for depolarization-induced Parkin mitochondrial recruitment has been observed in previous studies (24, 45). HeLa cells that normally prefer glycolytic metabolism exhibit robust Parkin translocation upon CCCP treatment. However, when they are forced into dependence on mitochondrial respiration, minimal or no Parkin recruitment can be observed upon mitochondrial depolarization. It was suggested that the reason little to no Parkin translocation and mitophagy is seen in primary neurons upon CCCP treatment is associated with their reliance on oxidative phosphorylation for energy production (24). Cookson and co-workers (45) recently identified hexokinase (HK) as a novel modifier of depolarization-induced Parkin recruitment because knockdown of both HK1 and HK2 led to a stronger block in Parkin relocalization and conversely expression of HK2 in primary neurons promoted YFP-Parkin recruitment to depolarized mitochondria. It appears that Parkin translocation in response to mitochondrial depolarization occurs most robustly in cells that can operate both glycolytic and oxidative phosphorylation. Our studies are in agreement with this notion. Furthermore, we show that the restraint in Parkin recruitment is linked to ATP levels rather than glycolytic pathway per se. Robust glycolytic pathway can compensate inhibition of oxidative phosphorylation to maintain cellular ATP levels that are permissive for Parkin recruitment. Without sufficient intracellular ATP, stress-induced PINK1 elevation is abrogated, which may be a key reason why Parkin accumulation on mitochondria is restricted. Our results provide at least one explanation for variable Parkin recruitment as a function of bioenergetics.
Minimal PINK1 induction by CCCP in the absence of glucose showed strong correlation with cellular ATP levels. Our data suggest that the decrease in PINK1 levels at different glucose concentrations under large scale mitochondrial damage is likely due to translational inhibition. We show that PINK1 mRNA levels exhibit little change as a function of glucose concentration whether or not CCCP is present. The short half-life of PINK1 makes the PINK1-Parkin pathway particularly susceptible to changes in translation rate. A corollary to this observation is that translational or transcriptional inhibitors can effectively shut down PINK1-dependent mitochondrial damage repair pathway. We directly tested this prediction by investigating the effect of translation inhibitor CHX on PINK1-dependent Parkin translocation. As expected, CHX incubation prior to CCCP treatment suppresses Parkin recruitment to mitochondria (Fig. 8, A and B). In combination with our collective data suggesting translational repression of PINK1, this direct approach using CHX further supports a mechanism that suggests low ATP results in translational repression of PINK1, thereby abrogating Parkin translocation (Fig. 8, A–C). Our studies uncovered the intracellular ATP levels as a key regulator for maintaining elevated PINK1 in the mitochondrial outer membrane. However, we cannot exclude the possibility that the rates of PINK1 transport, import, or degradation are also affected by a decline in intracellular ATP levels. Although these molecular events may contribute to the defects in activation of Parkin-dependent mitophagy, regulation of PINK1 levels appears to be the rate-limiting step in ATP sensitivity (Fig. 8C).
FIGURE 8.

A model for ATP-dependent translation inhibition of PINK1. A and B, treatment with CHX blocks CCCP-induced Parkin mitochondrial translocation. HeLa cells expressing Venus-Parkin-WT and RFP-smac-mts were left untreated or were treated with 10 μg/ml CHX for 1.5 h and incubated with 20 μm CCCP. Parkin mitochondrial localization was visualized by fluorescent microscopy (A) and quantified with respect to increasing CHX concentration (B). C, proposed model for ATP-dependent translation inhibition of PINK1 as a regulatory mechanism that suppresses Parkin mitochondrial translocation.
Whether or not ATP has an immediate input in PINK1 expression levels remains unclear. The possibility remains that a protein(s) upstream of PINK1 is responsible for the direct sensing of ATP levels to regulate PINK1 expression levels. The PI3K/Akt/mTOR pathway has been implicated in sensing the cellular AMP/ATP ratio to regulate protein synthesis (46–48). Specifically, under low cellular ATP conditions, mTOR is known to unfetter its inhibition of 4EBP1, thereby allowing 4EBP1 to inhibit eIF4E and restrict cap-dependent mRNA translation (49, 50). This occurrence may particularly impact any protein with a high turnover rate, such as PINK1, that undergoes constant degradation and maintain low steady-state levels in physiological conditions (4, 5). Inhibition of Akt by LY294002 and FPA-124 has been shown to decrease Parkin recruitment after CCCP treatment (45). It would be interesting to test whether Akt targets PINK1 translation to regulate Parkin recruitment.
The PINK1-Parkin pathway can mediate repair, mitophagy, and apoptotic responses depending on the nature of the stimuli (36). The results from this study also suggest that the cell fate decision in response to mitochondrial damaging agent such as CCCP also depends on cellular ATP levels, which are controlled by the configuration of glycolytic and oxidative phosphorylation pathways. In the absence of glucose, HeLa and MEF cells cannot undergo Parkin-dependent mitophagy in response to CCCP treatment. Our findings demonstrate a characteristic of PINK1 that simultaneously senses two separate stimuli: mitochondria depolarization and ATP levels, to regulate the PINK1/Parkin-dependent mitophagy pathway. The absence of either stimuli results in the deactivation of the pathway and ultimately results in different mitochondrial damage response outcomes. Such a characteristic of PINK1 is representative of an AND gate and provides a novel insight into the poorly understood control mechanism upstream of the PINK1/Parkin pathway.
Supplementary Material
Acknowledgments
We thank Kevin Dean for technical help with photoirradiation experiment and Douglas Chapnick and Eric Bunker for help with ImageXpress and data analysis. We also thank members of Xuedong Liu's laboratory for discussion.
This work was supported, in whole or in part, by National Institutes of Health Grant CA107098 (to X. L.) and S10 RR026680 (to ImageXpress MicroXL). This work was also supported by the Butch Award of University of Colorado and a grant from the Cancer League of Colorado (to X. L.).

This article contains supplemental Movies S1–S7.
- CCCP
- carbonyl cyanide m-chlorophenylhydrazine
- 2-DG
- 2-deoxy-d-glucose
- MEF
- mouse embryonic fibroblast
- ROI
- region of interest
- CHX
- cycloheximide
- HK
- hexokinase.
REFERENCES
- 1. Nunnari J., Suomalainen A. (2012) Mitochondria: in sickness and in health. Cell 148, 1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Geisler S., Holmström K. M., Skujat D., Fiesel F. C., Rothfuss O. C., Kahle P. J., Springer W. (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131 [DOI] [PubMed] [Google Scholar]
- 3. Narendra D., Tanaka A., Suen D.-F., Youle R. J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Narendra D. P., Jin S. M., Tanaka A., Suen D.-F., Gautier C. A., Shen J., Cookson M. R., Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsuda N., Sato S., Shiba K., Okatsu K., Saisho K., Gautier C. A., Sou Y. S., Saiki S., Kawajiri S., Sato F., Kimura M., Komatsu M., Hattori N., Tanaka K. (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Youle R. J., Narendra D. P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McLelland G. L., Soubannier V., Chen C. X., McBride H. M., Fon E. A. (2014) Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rogaeva E., Johnson J., Lang A. E., Gulick C., Gwinn-Hardy K., Kawarai T., Sato C., Morgan A., Werner J., Nussbaum R., Petit A., Okun M. S., McInerney A., Mandel R., Groen J. L., Fernandez H. H., Postuma R., Foote K. D., Salehi-Rad S., Liang Y., Reimsnider S., Tandon A., Hardy J., St George-Hyslop P., Singleton A. B. (2004) Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch. Neurol. 61, 1898–1904 [DOI] [PubMed] [Google Scholar]
- 9. Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., Albanese A., Nussbaum R., González-Maldonado R., Deller T., Salvi S., Cortelli P., Gilks W. P., Latchman D. S., Harvey R. J., Dallapiccola B., Auburger G., Wood N. W. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160 [DOI] [PubMed] [Google Scholar]
- 10. Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 [DOI] [PubMed] [Google Scholar]
- 11. Shimura H., Hattori N., Kubo S., Mizuno Y., Asakawa S., Minoshima S., Shimizu N., Iwai K., Chiba T., Tanaka K., Suzuki T. (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305 [DOI] [PubMed] [Google Scholar]
- 12. Jin S. M., Lazarou M., Wang C., Kane L. A., Narendra D. P., Youle R. J. (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meissner C., Lorenz H., Weihofen A., Selkoe D. J., Lemberg M. K. (2011) The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 117, 856–867 [DOI] [PubMed] [Google Scholar]
- 14. Muqit M. M., Abou-Sleiman P. M., Saurin A. T., Harvey K., Gandhi S., Deas E., Eaton S., Payne Smith M. D., Venner K., Matilla A., Healy D. G., Gilks W. P., Lees A. J., Holton J., Revesz T., Parker P. J., Harvey R. J., Wood N. W., Latchman D. S. (2006) Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J. Neurochem. 98, 156–169 [DOI] [PubMed] [Google Scholar]
- 15. Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R. L., Kim J., May J., Tocilescu M. A., Liu W., Ko H. S., Magrané J., Moore D. J., Dawson V. L., Grailhe R., Dawson T. M., Li C., Tieu K., Przedborski S. (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chan N. C., Salazar A. M., Pham A. H., Sweredoski M. J., Kolawa N. J., Graham R. L., Hess S., Chan D. C. (2011) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Narendra D., Kane L. A., Hauser D. N., Fearnley I. M., Youle R. J. (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tanaka A., Cleland M. M., Xu S., Narendra D. P., Suen D.-F., Karbowski M., Youle R. J. (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshii S. R., Kishi C., Ishihara N., Mizushima N. (2011) Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ashrafi G., Schlehe J. S., LaVoie M. J., Schwarz T. L. (2014) Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 206, 655–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bingol B., Tea J. S., Phu L., Reichelt M., Bakalarski C. E., Song Q., Foreman O., Kirkpatrick D. S., Sheng M. (2014) The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375 [DOI] [PubMed] [Google Scholar]
- 22. Sheng Z. H. (2014) Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 204, 1087–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gegg M. E., Cooper J. M., Chau K. Y., Rojo M., Schapira A. H., Taanman J. W. (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Van Laar V. S., Arnold B., Cassady S. J., Chu C. T., Burton E. A., Berman S. B. (2011) Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum. Mol. Genet. 20, 927–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin W., Kang U. J. (2008) Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 106, 464–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kondapalli C., Kazlauskaite A., Zhang N., Woodroof H. I., Campbell D. G., Gourlay R., Burchell L., Walden H., Macartney T. J., Deak M., Knebel A., Alessi D. R., Muqit M. M. (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2, 120080–120080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rakovic A., Shurkewitsch K., Seibler P., Grünewald A., Zanon A., Hagenah J., Krainc D., Klein C. (2013) Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem. 288, 2223–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vander Heiden M. G., Cantley L. C., Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gogvadze V., Zhivotovsky B., Orrenius S. (2010) The Warburg effect and mitochondrial stability in cancer cells. Mol. Aspects Med. 31, 60–74 [DOI] [PubMed] [Google Scholar]
- 30. Rossignol R., Gilkerson R., Aggeler R., Yamagata K., Remington S. J., Capaldi R. A. (2004) Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 64, 985–993 [DOI] [PubMed] [Google Scholar]
- 31. Reitzer L. J., Wice B. M., Kennell D. (1979) Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 254, 2669–2676 [PubMed] [Google Scholar]
- 32. Bolaños J. P., Almeida A., Moncada S. (2010) Glycolysis: a bioenergetic or a survival pathway? Trends Biochem. Sci. 35, 145–149 [DOI] [PubMed] [Google Scholar]
- 33. Kanki T., Klionsky D. J. (2008) Mitophagy in yeast occurs through a selective mechanism. J. Biol. Chem. 283, 32386–32393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anderson J. C., Voigt C. A., Arkin A. P. (2007) Environmental signal integration by a modular AND gate. Mol. Syst. Biol. 3, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Trempe J. F., Fon E. A. (2013) Structure and function of Parkin, PINK1, and DJ-1, the three musketeers of neuroprotection. Front. Neurol. 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang C., Lee S., Peng Y., Bunker E., Giaime E., Shen J., Zhou Z., Liu X. (2014) PINK1 triggers autocatalytic activation of Parkin to specify cell fate decisions. Curr. Biol. 24, 1854–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee A. S. (2014) Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 14, 263–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang C., Lee S., Peng Y., Bunker E., Shen C., Giaime E., Shen J., Shen J., Zhou Z., Liu X. (2014) A chemical genetic approach to probe the function of PINK1 in mitochondrial dynamics. Cell Res., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu M., Neilson A., Swift A. L., Moran R., Tamagnine J., Parslow D., Armistead S., Lemire K., Orrell J., Teich J., Chomicz S., Ferrick D. A. (2007) Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 292, C125–C136 [DOI] [PubMed] [Google Scholar]
- 40. Kim I., Lemasters J. J. (2011) Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid. Redox. Signal. 14, 1919–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aggarwal B. B., Quintanilha A. T., Cammack R., Packer L. (1978) Damage to mitochondrial electron transport and energy coupling by visible light. Biochim. Biophys. Acta 502, 367–382 [DOI] [PubMed] [Google Scholar]
- 42. Yang J. Y., Yang W. Y. (2011) Spatiotemporally controlled initiation of Parkin-mediated mitophagy within single cells. Autophagy 7, 1230–1238 [DOI] [PubMed] [Google Scholar]
- 43. Ihrlund L. S., Hernlund E., Khan O., Shoshan M. C. (2008) 3-Bromopyruvate as inhibitor of tumour cell energy metabolism and chemopotentiator of platinum drugs. Mol. Oncol. 2, 94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peng X., Guo X., Borkan S. C., Bharti A., Kuramochi Y., Calderwood S., Sawyer D. B. (2005) Heat shock protein 90 stabilization of ErbB2 expression is disrupted by ATP depletion in myocytes. J. Biol. Chem. 280, 13148–13152 [DOI] [PubMed] [Google Scholar]
- 45. McCoy M. K., Kaganovich A., Rudenko I. N., Ding J., Cookson M. R. (2014) Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum. Mol. Genet. 23, 145–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang X., Proud C. G. (2006) The mTOR pathway in the control of protein synthesis. Physiology 21, 362–369 [DOI] [PubMed] [Google Scholar]
- 47. Hahn-Windgassen A., Nogueira V., Chen C. C., Skeen J. E., Sonenberg N., Hay N. (2005) Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J. Biol. Chem. 280, 32081–32089 [DOI] [PubMed] [Google Scholar]
- 48. Vadlakonda L., Dash A., Pasupuleti M., Anil Kumar K., Reddanna P. (2013) The paradox of Akt-mTOR interactions. Front. Oncol. 3, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gingras A. C., Kennedy S. G., O'Leary M. A., Sonenberg N., Hay N. (1998) 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 12, 502–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carrera A. C. (2004) TOR signaling in mammals. J. Cell Sci. 117, 4615–4616 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.