

Background: Alcadein α (Alcα) forms a ternary complex with APP and X11L.
Results: Transport into the nerve terminus and metabolism of APP were facilitated in Alcα CTF transgenic mice, along with an increase in Aβ.
Conclusion: Alcα ICD, a product of γ-secretase cleavage of Alcα CTF, enhanced APP trafficking from the ternary complex into a late secretory pathway.
Significance: Novel function of Alcadein α results from regulated intramembrane proteolysis.
Keywords: Alzheimer Disease, Amyloid Precursor Protein (APP), Amyloid-beta (AB), Gamma-secretase, Proteolysis
Abstract
The neural type I membrane protein Alcadein α (Alcα), is primarily cleaved by amyloid β-protein precursor (APP) α-secretase to generate a membrane-associated carboxyl-terminal fragment (Alcα CTF), which is further cleaved by γ-secretase to secrete p3-Alcα peptides and generate an intracellular cytoplasmic domain fragment (Alcα ICD) in the late secretory pathway. By association with the neural adaptor protein X11L (X11-like), Alcα and APP form a ternary complex that suppresses the cleavage of both Alcα and APP by regulating the transport of these membrane proteins into the late secretory pathway where secretases are active. However, it has not been revealed how Alcα and APP are directed from the ternary complex formed largely in the Golgi into the late secretory pathway to reach a nerve terminus. Using a novel transgenic mouse line expressing excess amounts of human Alcα CTF (hAlcα CTF) in neurons, we found that expression of hAlcα CTF induced excess production of hAlcα ICD, which facilitated APP transport into the nerve terminus and enhanced APP metabolism, including Aβ generation. In vitro cell studies also demonstrated that excess expression of Alcα ICD released both APP and Alcα from the ternary complex. These results indicate that regulated intramembrane proteolysis of Alcα by γ-secretase regulates APP trafficking and the production of Aβ in vivo.
Introduction
Alcadein (Alc)2 is a brain abundant type I membrane protein family comprised of Alcα, Alcβ, and Alcγ (1), which are also identified as the Ca2+-binding protein calsyntenin (2, 3). They share two cadherin repeats, a concanavalin A-like lectin/glucanase superfamily domain in their amino-terminal extracellular region, an acidic domain, a kinesin light chain-binding WD motif and an X11L (X11-like)-binding NP sequence in their carboxyl-terminal cytoplasmic region (1, 4). Originally, we isolated Alcα as an X11L-interacting molecule (1, 5). X11L is a neuron-specific cytoplasmic adaptor protein and was also identified as a binding partner of amyloid β-protein precursor (APP) (6). Both Alcα and APP interact with the phosphotyrosine interaction/phosphotyrosine-binding domain of X11L and form a ternary complex comprised of Alcα, X11L, and APP (1, 5).
In neurons, APP695, a neuron-specific isoform, undergoes N-glycosylation in the endoplasmic reticulum, producing immature APP (7). The immature APP is transported to the Golgi and further modified by O-glycosylation to form mature APP (mAPP). The mAPP enters into the late secretory pathway and localizes to the plasma membrane, whereas some mAPP also enter endosomal recycling pathways. During the late secretory pathway, APP is subject to consecutive cleavages (8). Alcα/calsyntenin-1 is also subject to intracellular trafficking and metabolism and participates in neural functions, similar to APP (4, 9–11). Calsyntenins have also been reported to mediate exit of APP from the TGN (12).
It is well known that APP undergoes primary proteolytic cleavage at juxtamembrane α- or β-sites by α- or β-secretase and that membrane-associated APP C-terminal fragments (APP CTFs) are further cleaved at γ/ϵ-sites by γ-secretase (8). When APP is cleaved by a combination of α- and γ-secretases, a metabolically labile p3 peptide is generated, whereas neurotoxic amyloid β (Aβ) peptide is generated when APP is cleaved by β- and γ-secretases. The Aβ peptide is known as a causative molecule of Alzheimer disease (13, 14). Alcα is also subject to proteolytic cleavage by α-secretase and remains a membrane-associated C-terminal fragment (Alcα CTF), which is further cleaved by γ-secretase to secrete p3-Alcα and generate an intracellular domain fragment (Alcα ICD) (10, 15).
X11L associates with both APP and Alcα in the Golgi and also in the late secretory pathway (16, 17). In the Golgi, X11L is thought to regulate the formation of APP and Alcα cargo vesicles (17). Formation of the ternary complex composed of APP, X11L, and Alcα also regulates the entry of APP into lipid rafts where β-secretase is active (16). Additionally, X11L is thought to regulate γ-cleavage of APP CTFs directly (18).
Aβ production is suppressed when APP is expressed with X11L, and we reported that suppressed Aβ production by X11L was further enhanced when full-length Alcα was also coexpressed (1, 5, 6) due to ternary complex formation. In vitro, binding of APP to X11L is stabilized when Alcα is coexpressed, and this enhanced interaction of APP with X11L mediated by Alcα is thought to further stabilize APP metabolism as well as regulate intracellular APP trafficking because the cleavage of APP by secretases occurs in the late secretory pathway. In vivo, X11L/X11β transgenic mice expressing human APP suppressed amyloidogenic processing of APP (19). Furthermore, X11L gene knock-out (X11L-KO) mice showed enhanced generation of endogenous Aβ in the brain (20), and human APP transgenic mice lacking the X11L gene exhibited enhanced amyloid plaque formation in the brain (21). However, the role of Alcα metabolites in APP metabolism and Aβ generation remain unclear in vivo. To determine the function of Alcα CTF and its intracellular metabolic fragment Alcα ICD, we generated a transgenic mouse line expressing human Alcα-CTF under the control of the PDGF-β promoter and examined its effect on the metabolism of APP in vivo.
EXPERIMENTAL PROCEDURES
Generation of hAlcα CTF Transgenic Mouse Lines
cDNA encoding human Alcα CTF (amino acids 817 to 971 of the hAlcα1 isoform) (1, 10) was connected to the signal sequence (amino acids 1 to 28 of hAlcα1). The construct was inserted into a vector with a 5′-PDGF-β promoter and a 3′-SV40 poly(A) tail to produce the TgPDhAlcαCTF plasmid. For DNA microinjection, linearized DNA was prepared by digestion with restriction enzymes, as illustrated (see Fig. 1A). Mice were purchased from CLEA-Japan (Tokyo, Japan), and all of the animal studies were conducted in compliance with the guidelines of the Animal Studies Committee of Hokkaido University. Linearized DNA (SalI/NotI fragment) was micro-injected into fertilized eggs produced by mating between BDF1 mice (F1 hybrid of C57BL/6 and DBA/2 mice) according to standard procedures (22). In brief, BDF1 females (6–8 weeks of age) that had been super-ovulated by injection of pregnant mare serum gonadotropin (serotropin, Asuka Pharmaceutical Co.) and human chorionic gonadotropin (Asuka Pharmaceutical Co.) were mated with males of the same strain. Pronuclear stage embryos were collected from pregnant females, and DNA fragments were injected into the male pronuclei of the zygotes. The embryos were then cultured in potassium simplex optimized medium at 37 °C in an atmosphere of 5% CO2 and 95% humidity. The surviving embryos were transplanted into the oviducts of pseudo-pregnant females (Multi Cross Hybrid (Institute of Cancer Research), 8–12 weeks of age). Transgenic founders were identified by PCR and Southern blot analysis of genomic DNA extracted from tail biopsies. Genotyping PCR was performed using a set of sense (Alcα CTF, 5′-cct gac cat cac cgt caa cc-3′) and antisense (SV40, 5′-cac ctc ccc ctg aac ctg aa-3′) primers with ExTaq DNA polymerase (Takara-bio). For Southern blot analysis, genomic DNA was digested with EcoRI, separated by agarose gel electrophoresis, and transferred onto a Nylon membrane. The membrane was incubated with a probe prepared from the DNA sequence of an injected construct, and signals were detected by CDP-Star (GE Healthcare). Founder mice were backcrossed with C57BL/6 mice, and offspring were backcrossed over 10 times. Non-transgenic littermates derived from the same crosses were used as controls.
FIGURE 1.

Generation and characterization of hAlcα CTF transgenic mouse lines. A, construct for generation of hAlcα CTF transgenic mouse lines. The cDNA sequence encoding amino acids 817–971 of human Alcα1 (971 amino acids) was ligated with cDNA encoding the signal sequence (amino acids 1 to 28) and cloned into a vector with the 5′-PDGF-β promoter sequence and 3′-SV40 poly(A) sequence. DNA fragments prepared by digestion with SalI and NotI were used for injection into fertilized eggs. B, identification of human p3-Alcα secreted from HEK293 cells expressing hAlcα CTF. Amino acid sequence of human p3-Alcα35 and a representative MS spectrum of immunoprecipitate recovered from medium of HEK293 cells expressing human Alcα CTF with anti-p3-Alcα antibody UT135 are shown. Numbers in parentheses indicate p3-Alcα species (35 indicates p3-Alcα35, a major p3-Alcα species). Intens, Intensity; a.u., arbitrary unit. C, establishment of hAlcα CTF Tg mouse founders. A summary of the individual numbers at the respective experimental stages is shown. D, genomic Southern blot analysis of six transgenic lines. Genomic DNA of six founders was analyzed by Southern blotting with DNA fragments prepared by SalI/NotI digestion (A) as a probe. The transgenic lines used are numbered, along with non-transgenic mouse (Non-Tg) DNA and 10 copies of injected DNA. E, expression levels of hAlcα CTF in three Tg mice lines. Protein expression of hAlcα CTF, a transgenic product, was quantified by immunoblotting. A brain region, including the cerebral cortex and hippocampus of Tg18, Tg47, and Tg54 mice along with their non-Tg littermates (N), was lysed and analyzed by immunoblotting with anti-Alcα no. 958 (top) and anti-flotillin-1 (bottom) antibodies. Band densities of Alcα CTF were quantified and standardized with respect to the band density of flotillin-1. Expression levels in Tg mice (filled bars) are shown as a ratio relative to the endogenous Alcα CTF level of non-Tg littermates (open bars), which was set at 1.0. Data represent mean ± S.E. (n = 3). Specificity of the anti-Alcα no. 958 antibody is shown in Fig. 2A.
MALDI-TOF/MS Analysis of p3-Alcα in the Medium of Cells Expressing Human Alcα CTF
Tg construct was recloned into pcDNA3.1 to generate pcDNA3.1-hAlcαCTF. The p3-Alcα peptides secreted into the medium of HEK293 cells transiently transfected with pcDNA3.1-hAlcαCTF was recovered by immunoprecipitation with anti-p3-Alcα UT135 antibody and protein G-Sepharose beads (10). The beads were washed and sample was eluted with trifluoroacetic acid/acetonitrile/water (1:20:20) saturated with sinapinic acid. The dissolved sample was subject to MALDI-TOF/MS analysis with a UltraflexII TOF/TOF (Bruker Daltonics, Bremen, Germany) (10).
Immunohistochemistry
Mouse brain sections (25-μm thick) were prepared as described (20), and the sections were stored at −30 °C until use. Frozen sections were washed with PBS for 20 min and then incubated in PBS containing 1% (v/v) Triton X-100 for 20 min to permeabilize the membranes. Tissue sections were then incubated with PBS containing 0.3% (v/v) H2O2 for 10 min to inactivate endogenous peroxidase activity and washed three times with PBS. The sections were blocked with PBS containing 5% (v/v) normal goat serum for 1 h at room temperature and then incubated with no. 958 antibody (serum diluted to 1:8,000) for ∼8 h at 4 °C. After three washes with PBS, sections were incubated with an anti-rabbit IgG peroxidase-linked species-specific whole antibody (GE Healthcare, dilution 1:500) for 30 min at room temperature. The signal was visualized with a VECTASTAIN kit (Vector Laboratories) following the manufacturer's protocol.
Subcellular Fractionation
The cytosolic fraction of mouse brain tissue was prepared as described (23) and subjected to immunoprecipitation with no. 958 antibody. The synaptosome fraction was prepared as described (24) with some modifications. In brief, the cerebral cortex and hippocampus regions of 12-month-old Tg54 and non-Tg mice were homogenized in buffer A (10 mm HEPES (pH 7.4) containing 0.32 m sucrose, 5 μg/ml chymostatin, and 5 μg/ml leupeptin), and this fraction was used as total lysate. The lysate was then centrifuged at 1,000 × g for 10 min, and a clarified supernatant (S1 fraction) was recovered. The S1 fraction was further centrifuged at 13,800 × g for 20 min, and the supernatant (S2 fraction) and pellet (P2 fraction) were used for assays. The P2 fraction was suspended again in buffer A and overlaid on a discontinuous sucrose gradient prepared with 1.2 m, 1 m, and 0.85 m sucrose solution and centrifuged at 82,500 × g for 2 h with an SW41 rotor (Beckman Coulter). After centrifugation, the layer between 1.0 m and 1.2 m sucrose was collected and resuspended in 6 mm Tris-HCl (pH 8.0) buffer containing 0.5% (v/v) Triton X-100 to prepare the synaptosome fraction.
Antibodies, Immunoprecipitation, and Immunoblot Analysis
A rabbit polyclonal anti-Alcα carboxyl-terminal domain antibody, no. 958, was raised against a synthesized peptide, 948GEQGDPQNATRQQQL962, of human Alcα1. IgG was affinity purified with antigen-coupled beads and used for analyses. This antibody specifically recognizes Alcα of human and mouse almost equivalently but does not show cross-reactivity with Alcβ and Alcγ of human (see Fig. 2A) or mouse (data not shown).
FIGURE 2.

Expression of hAlcα CTF in the brains of Tg54 mice. A, specificity of anti-Alcα carboxyl-terminal domain antibody no. 958. Lysates of HEK293 cells expressing human FLAG-Alcα (lane 2), human FLAG-Alcβ (lane 3), and human FLAG-Alcγ (lane 4) along with plasmid alone (lane 1) were analyzed by immunoblotting with anti-FLAG M2 (left) and anti-Alcα no. 958 (right) antibodies. The arrow indicates FLAG-tagged Alcα, Alcβ, and Alcγ, and the arrowhead indicates Alcα CTF. Numbers indicate protein standards (kDa). The asterisk indicates a nonspecific product. B, expression of hAlcα CTF in the brain regions of Tg54 mice. Expression of hAlcα CTF in brain regions of Tg54 mice was examined by immunoblotting with no. 958 antibody. Brain tissue from 3-month-old Tg54 (Tg) and non-Tg (N) littermates were dissected into the indicated brain regions, and lysates obtained from these sections were analyzed by immunoblotting with no. 958 (top) and anti-β-actin (bottom) antibodies. OB, olfactory bulb; CC, cerebral cortex; Hipp, hippocampus; Th/Hy, thalamus/hypothalamus; St, striatum; Mid, midbrain; Me, medulla; Ce, cerebellum; Sc, spinal cord. C, localization of hAlcα CTF in the brains of Tg54 mice. Localization of hAlcα CTF in brain regions was examined by immunostaining with no. 958 antibody. Sections of the cerebral cortex (a and d), hippocampus (b and e), and olfactory bulb (c and f) were prepared from 3-month-old Tg54 (d–f) and non-Tg (a–c) littermates and immunostained. Py, pyramidal cells; Gr, granule cells; DG, dentate gyrus; MCL, mitral cell layer; GL, granule cell layer. D, quantification of p3-Alcα in the brains of Tg54 mice. The total amount of p3-Alcα in the brains of 6-month-old Tg54 (Tg, filled bar) and non-Tg (N, open bar) littermates were quantified by sELISA. Quantified values are given as the mean ± S.E. (n = 4). ***, p < 0.005, Student's t test. #, below detectable levels. E, detection of hAlcα ICD in the cytosolic fraction of mice brains. Brain lysates prepared from 2-month-old Tg54 (Tg) and non-Tg (N) mice brains were subjected to immunoprecipitation with no. 958 antibody, and the precipitates were detected by immunoblotting with the same antibody. Arrows indicate hAlcα ICD fragments. Numbers on the left side of the panel indicate the molecular mass (kDa). Asterisks indicate IgG heavy and light chains. F, schematic structure of p3-Alcα and hAlcα ICD. Alcα CTF is first cleaved at the ϵ-site by γ-secretase to release Alcα ICD into the cytoplasm. Next, γ-secretase cleavages reach to the γ-site to secrete p3-Alcα into the extracellular milieu (15).
Immunoprecipitation was performed as described (1) using a previously described plasmid for FLAG-X11L (6). For the APP-GFP construct, human APP695 cDNA was cloned into pEGFP-N3 (Clontech) between the HindIII/NotI sites. For the hAlcα ICD construct, cDNA encoding the cytoplasmic region of hAlcα from 868 to 971 amino acids was cloned into pcDNA3.1. Amino acid position 868 of Alcα1 is the major N terminus of Alcα ICD, which is generated by ϵ-cleavage of Alcα CTF by γ-secretase (15). Brain and cell extracts used in immunoblot analyses were prepared by homogenizing samples in eight volumes of RIPA buffer containing 5 μg/ml chymostatin and 5 μg/ml leupeptin. The lysates were centrifuged, and supernatants were used for immunoprecipitation analysis. The procedures used for immunoblotting and the identification of APP CTFs were described previously (25).
Antibodies used in immunoblot analysis were as follows: rabbit polyclonal anti-Alcα (no. 958) and anti-APP (no. 8717, Sigma), mouse monoclonal anti-FLAG (M2, Sigma), anti-flotillin-1 (BD Biosciences), anti-synaptophysin (SY38, DAKO), anti-α-tubulin (sc-32293, Santa Cruz Biotechnology), anti-GM130 (BD Biosciences), anti-GFP (no. 598, MBL), and anti-β-actin (ab8226, Abcam). For quantification of immunoreactive proteins, a VersaDoc system (Bio-Rad) or LAS-4000 mini (Fujifilm) was used. The intensities of the protein bands of Alcα CTF, APP full-length, and APP CTFs were normalized to the values of flotillin-1.
sELISA for Quantification of Mouse Aβ and p3-Alcα
To quantify Aβ in mice brains, the cerebral cortex and hippocampus were excised from 12- to 15-month-old mice, and TBS-soluble and -insoluble fractions were prepared as described (16). Mouse Aβ1–40 and Aβ1–42 in the TBS-soluble and -insoluble fractions were examined with an sELISA kit (Aβ1–40, no. 27720; Aβ1–42, no. 27721; IBL, Takasaki, Japan). Quantification of p3-Alcα was described previously (26).
Immunofluorescence Microscopy Analysis and Quantification of Fluorescence Intensity
The procedures in immunofluorescence experiments shown in Fig. 5 were followed as described previously (27). The images were analyzed by a fluorescence microscope (Keyence, BZ-700X). Fluorescence quantification was performed using the imaging software ImageJ (http://imagej.nih.gov/ij/, National Institutes of Health). The images were acquired with a resolution of 640 × 480 pixels using a 100× objective lens, which covered the whole cell region. The region of interest that contained the cell body-neurite junction area was selected at a resolution of 132 × 48 pixels, in which the junction was set to a middle region manually using a transparent image obtained by transmission light. The fluorescence intensities in the proximal region of neurite (“neurite,” right half of a selected image) and the hillock of cell body (“cell body,” left half of a selected image) were then measured. The fluorescence intensities where no cell region was observed in the right and left half images were also measured as background. The background intensity was subtracted from the respective intensity of cell body and neurite, and the values were indicated as a ratio to average intensity of neurite plus cell body.
FIGURE 5.

Alcα ICD facilitates the trafficking of APP into neurites. A, the proximal region of neurites for analysis of APP trafficking. Neuro2a cells were observed under a transmission light and the hillock of cell body (asterisk, cell body) and the proximal region of neurite (triangle, neurite) were analyzed for EGFP-APP fluorescence as described under “Experimental Procedures.” B, representative images of the fluorescence of EGFP-APP. The fluorescence of EGFP-APP was observed in living cells expressing HA-X11L and Alcα CTF in the presence (top) or absence (bottom) of Alcα ICD expression. Analyses were performed in the presence or absence of DAPT treatment (10 μm for 4 h). C, quantifications of fluorescence intensities of EGFP-APP at the proximal region of neurites. The intensities of EGFP-APP in B were quantified. The left panel indicates the intensity of cell body, and the right panel indicates the intensity of neurite. The fluorescence intensity of cell body or neurite is shown as a ratio to average intensity of cell body plus neurite. Black (control) and gray (DAPT-treated) lines are shown. The values are indicated with mean ± S.E. (n = 9–14). *, p < 0.05, Student's t test. D, levels of protein expression in cells. Lysates of Neuro2a cells with or without DAPT treatment were analyzed by immunoblotting with anti-GFP no. 598 (MBL), anti-HA 12CA5 (Roche Applied Science), and anti-Alcα 958 (for Alcα CTF and Alcα ICD) antibodies. Detected proteins are indicated with arrowheads. Transfection of plasmids is indicated with a plus sign, whereas a minus indicates the use of empty plasmid alone. Numbers indicate the protein molecular mass standards.
RESULTS
Generation of Human Alcα Carboxyl-terminal Fragment Transgenic Mice
A cDNA sequence encoding the carboxyl-terminal region of human Alcα1 together with the signal peptide sequence was expressed under the regulation of the mouse PDGF-β promoter (Fig. 1A). The signal sequence was cleaved correctly by signal peptidase when expressed in HEK293 cells, producing the human Alcα carboxyl-terminal fragment (hAlcα CTF) with the correct amino-terminal sequence. hAlcα CTF then underwent intramembrane cleavage by γ-secretase to generate p3-Alcα with the same amino acid sequence observed in vivo (10). MALDI-TOF/MS analysis of p3-Alcα secreated from HEK293 cells showed that the p3-Alcα35 (amino acid sequence indicated in B) was generated as a major p3-Alcα form along with several minor species (Fig. 1B). The result indicates that exogenously expressed hAlcα CTF was processed in a manner similar to that of endogenously generated Alcα CTF.
We generated six transgenic mouse founder lines using this construct (Fig. 1C). Genomic Southern blot analysis of the six lines shows a few to several hundred copies of exogenous gene incorporated into the genome (Fig. 1D). The protein expression level of hAlcα CTF in the brain region, including the cerebral cortex and hippocampus was analyzed in the three lines with the highest trans-gene dose by immunoblotting with an anti-Alcα antibody (Fig. 1E). Of these founders, line no. 54, which showed a 4-fold increase in hAlcα CTF expression compared with endogenous levels of the protein, was chosen for use in further analyses. Tg54 was subjected to genetic back-cross with C57BL/6 over 10 generations.
Expression of hAlcα CTF in Tg54 Mouse Brain
Alcadein family proteins Alcα, Alcβ, and Alcγ share a similar structure in the cytoplasmic region. Therefore, we developed an antibody raised against the cytoplasmic sequence of Alcα. This antibody, designated no. 958, recognizes specifically Alcα and its CTF but not Alcβ or Alcγ. An anti-FLAG antibody was used to confirm the levels of expression in cells expressing these FLAG-tagged constructs (Fig. 2A).
Using this antibody, we analyzed the expression of hAlcα CTF in 3-month-old Tg54 (Tg) and non-Tg (N) mice (Fig. 2, B and C). Under the regulation of the PDGF-β promoter, increased expression of hAlcα CTF was observed in the cerebral cortex, hippocampus, and olfactory bulb in Tg compared with non-Tg mice (Fig. 2B). Immunohistochemical analysis of endogenous Alcα and hAlcα CTF expression agreed well with the results of immunoblot analysis. An immuno-reactive signal was observed throughout the cerebral cortex (Fig. 2C, panels a and d), in pyramidal cells in CA1 to CA3 along with granule cells in the dentate gyrus of the hippocampus (Fig. 2C, panels b and e), and in layers of mitral and granule cells of the olfactory bulb (Fig. 2C, panels c and f). These signals were enhanced in Tg54 mouse brains (Fig. 2C, panels d–f). Immunoreactivity of tissue staining is specific because this antibody does not react to the tissue staining of Alcα-KO mouse brain (data not shown). Taken together, we concluded that the Tg line expressing hAlcα CTF, Tg54, was successfully established. Tg54 mice exhibit normal growth and fertility (data not shown).
We next measured the amounts of p3-Alcα, which is generated by γ-secretase cleavage of Alcα CTF (illustrated in Fig. 2F). In brains of Tg54 mice, the total amount of p3-Alcα greatly increased, whereas in non-Tg mice, it was below the level of detection (Tg, 0.33 ± 0.03 pmol/g tissue; n = 3, Fig. 2D). Corresponding to the increased amount of p3-Alcα in brain tissue, a significant amount of hAlcα ICD, which was detected as a doublet, was also detected in the cytosolic fraction of Tg54 mouse brain tissue, whereas only very low levels were detected in non-Tg mice (Fig. 2E). These findings indicate that exogenously expressed hAlcα CTF is physiologically cleaved by γ-secretase in the brain to generate p3-Alcα along with Alcα ICD (10, 15).
Facilitation of Intracellular Trafficking and Metabolism of Endogenous Mouse APP in Tg54 Mice
To reveal the effect of Alcα CTF on the metabolism of APP in vivo, the amounts of endogenous Aβ1–40 and Aβ1–42 in brain lysate isolated from the cerebral cortex and hippocampus were quantified in 12- to 15-month-old Tg54 and non-Tg mice. Unexpectedly, the amounts of both Aβ species were significantly greater in Tg54 than non-Tg mice (TBS insoluble Aβ1–40, non-Tg = 0.61 ± 0.04 pmol/g tissue; Tg54 = 0.72 ± 0.02 pmol/g tissue (p = 0.0017); TBS insoluble Aβ1–42, non-Tg = 0.18 ± 0.00 pmol/g tissue, Tg54 = 0.22 ± 0.01 pmol/g tissue (p = 0.0105)) (Fig. 3A). The amount of Aβ yielded in the TBS-soluble fraction was too small to detect a significant change. These results suggest that the metabolism of APP is facilitated in the brains of Tg54 mice.
FIGURE 3.
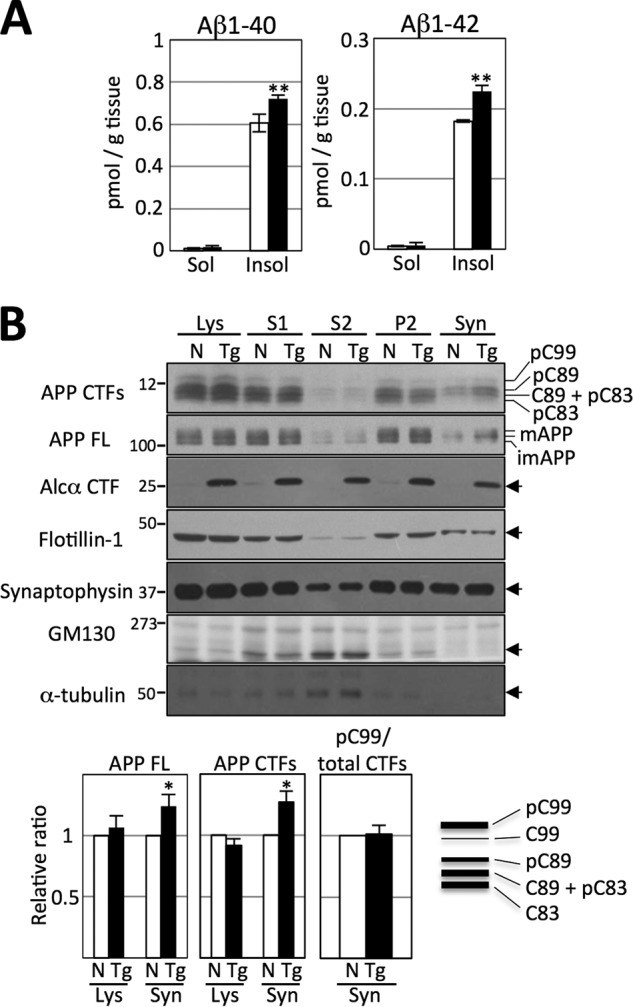
Changes in APP metabolism in the brains of Tg54 mice. A, quantification of Aβ1–40 and Aβ1–42 in brain tissue of Tg54 and non-Tg mice. Brain regions, including the cerebral cortex and hippocampus of 12- to 15-month-old Tg54 (filled bars) and non-Tg (open bars) mice, were dissected, and endogenous Aβ1–40 (left) and Aβ1–42 (right) in the TBS-soluble and -insoluble fractions were quantified by sELISA. Results are given as the means ± S.E. Asterisks indicate statistical significance as determined by Student's t test (n = 12, **, p < 0.01). B, subcellular localization of APP and APP CTFs in Tg54 and non-Tg brains. Brain regions, including the cerebral cortex and hippocampus of 12-month-old Tg54 (Tg) and non-Tg (N) littermates, were fractionated and analyzed by immunoblotting with antibodies against the indicated proteins: anti-APP (no. 8717, Sigma) for APP CTFs and APP FL, anti-Alcα (no. 958) for Alcα CTF, anti-flotillin-1 (BD Biosciences), anti-synaptophysin (SY38, DAKO), anti-GM130 (BD Biosciences), and anti-α-tubulin (sc-32293, Santa Cruz Biotechnology). Full-length APP (APP FL), mature (mAPP with N- and O-glycosylation) and immature (imAPP with N-glycosylation) APP695 are indicated. C99 and C89 indicate CTFβ, and C83 indicates CTFα. The pC99, pC89, and pC83 are CTFs phosphorylated at Thr-668. (A schematic blot of detected CTFs is indicated in the lower right panel.) Lys, total lysate; S1, post-nuclear supernatant; S2 and P2, supernatant and pellet of S1 fraction that was subjected to further centrifugation; Syn, synaptosome fraction. Numbers on the left represent protein molecular mass standards. Arrows on the right indicate the protein detected with antibodies. The bar graphs show the results of quantification of protein bands in the total lysate (Lys) and synaptosome fractions (Syn). Quantified values of APP FL (far left graph) and APP CTFs (middle graph) in the lysate and synaptosome fractions are indicated. The pC99 was also quantified, and the ratio of pC99/total CTFs (right graph) is shown. Values were normalized to the values of flotillin-1. Values obtained from non-Tg mice were set to 1.0 and are given as the mean ± S.E. Asterisks indicate statistical significance as determined by Student's t test (n = 5; *, p < 0.05).
Cleavage of APP in neurons occurs during or after its axonal transport; in other words, in the late secretory pathway. Therefore, the brains of Tg54 and non-Tg mice were fractionated to isolate the synaptosome fraction, which includes membrane vesicles of the late secretory pathway. As shown in Fig. 3B, the amounts of full-length APP, especially mAPP in synaptosomes, was significantly greater in Tg54 mice (synaptosome = 1.24 ± 0.19 (n = 5, p = 0.0458) when the value in non-Tg mice was set at 1.0). However, total APP, which includes mAPP and immature APP, was not changed significantly in the total lysate of either Tg or non-Tg mice (lysate = 1.06 ± 0.19, n = 5, p = 0.4904). APP CTFs were also significantly more abundant in the synaptosome fraction of Tg54 mice compared with non-Tg mice (1.27 ± 0.09 (n = 7, p = 0.0261) when the value in non-Tg mice was set at 1.0), whereas the amounts were equivalent in total lysate of Tg54 and non-Tg mice (0.92 ± 0.05, n = 7, p = 0.1843). These results suggest that increased amounts of APP undergo enhanced trafficking into the nerve terminus where it is cleaved by primary secretases. Increased CTFs in the late secretory pathway are then further cleaved by γ-secretase in Tg54 mice, thus facilitating the production of Aβ in the brain. This is not the result of enhanced amyloidogenic processing of APP. Aβ1–40/1–42 is derived from C99 among three CTF species; C99 (CTFβ), C89 (CTFβ), and C83 (CTFα). These CTFs are phosphorylated at Thr-668 in brain, and the phosphorylated C99 (pC99) is a major C99 rather than non-phosphorylated form in mouse brain (7). Therefore, an increase of pC99 level can indicate the enhanced amyloidogenic cleavage of APP. However, in Fig. 3B, the ratio of pC99 to total CTF was equivalent in the synaptosome fraction of Tg54 and non-Tg mice brains, although total amounts of APP CTFs increased significantly in the synaptosome fraction of Tg54. Therefore, we understand that the increased Aβ production in Tg54 is due to the facilitated trafficking of APP into the late secretory pathway rather than the enhanced amyloidogenic cleavage of APP. Identification of mature and immature APP and APP CTF species in the brain was described in detail previously (7).
We then considered the possibility that hAlcα ICD derived from hAlcα CTF could play a role in processing the increased amounts of APP entering the late secretory pathway, because the hAlcα ICD was free from membrane association and highly abundant in Tg54 (Fig. 2F). APP and Alcα are associated in the Golgi apparatus via interaction with X11 family proteins. Interaction of APP with X11L, a neuron-specific protein, is enhanced by association of Alcα with X11L. APP in this ternary complex is thought to suppress further trafficking into the late secretory pathway, thus stabilizing APP for cleavage by secretases. In Tg54 mice, excess expression of hAlcα CTF is likely to relieve this suppression. hAlcα CTF is then quickly cleaved by γ-secretase to generate hAlcα ICD along with p3-Alcα (Fig. 2, D–E). Thus, hAlcα ICD could perform a function different from full-length Alcα in stabilizing APP metabolism in the presence of X11L.
To assess this possibility, hAlcα ICD was coexpressed in Neuro2a cells, together with GFP-tagged hAPP (hAPP-GFP), full-length hAlcα (hAlcα FL), and FLAG-tagged X11L (FLAG-X11L), which form a ternary complex (Fig. 4). In the absence of hAlcα ICD, co-immunoprecipitation with an anti-FLAG antibody recovered FLAG-X11L together with hAPP-GFP and hAlcα FL (Fig. 4A, lane 5). However, increased expression of hAlcα ICD decreased the recovery of both hAPP-GFP and hAlcα FL significantly (Fig. 4A, lanes 6–8, and 4B), although hAPP-GFP, hAlcα FL, and FLAG-X11L are expressed at equal levels (Fig. 4A, lanes 1–4). These results indicate that Alcα ICD performs a novel function that releases both APP and Alcα from X11L and is different from the function of Alcα FL. Thus, it is possible to conclude that the increased levels of APP and its metabolic fragments APP CTFs and Aβ in the synaptosome fraction of the brains of Tg54 mice is due to enhanced release and anterograde trafficking of APP to the nerve terminus from the cell body. In summary, Alcα ICD regulates the trafficking of APP and Alcα itself. This may be a novel function involved in regulated intramembrane proteolysis by γ-secretase (28, 29).
FIGURE 4.
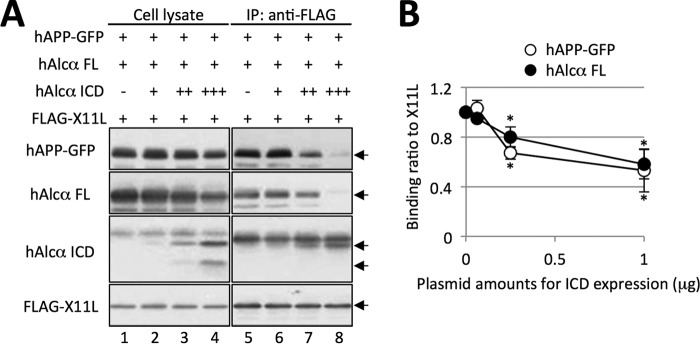
Facilitation of APP and Alcα release from the APP/X11L/Alcα ternary complex by Alcα ICD. A, co-immunoprecipitation of ternary complex components in the presence of hAlcα ICD. Neuro2a cells expressing human APP-GFP (hAPP-GFP), full-length Alcα (Alcα FL), and FLAG-X11L with or without hAlcα ICD were analyzed for formation of a ternary complex composed of APP, X11L, and Alcα. Proteins in cell lysates (lanes 1–4) were subjected to immunoprecipitation with anti-FLAG antibody and co-immunoprecipitated proteins (IP) were analyzed by immunoblotting with antibodies (lanes 5–8); anti-GFP (no. 598, MBL), anti-Alcα (UT83) (see Ref. 1) and anti-FLAG (M2, Sigma). B, quantification of APP and Alcα recovered by co-immunoprecipitation assay. Protein band densities of hAPP-GFP (open circle) and hAlcα FL (filled circle) co-immunoprecipitated with FLAG-X11L were quantified and are shown relative to the value obtained in the absence of hAlcα ICD expression (0, set to 1.0). The horizontal axis represents the amount of pcDNA3-hAlcα ICD (μg) co-transfected with pcDNA3-hAPP-GFP (1 μg), pcDNA3-hAlcα (1 μg), and pcDNA3-FLAG-X11L (0.5 μg). Results are given as the means ± S.E. Asterisks indicate statistical significance as determined by Student's t test (n = 3; *, p < 0.05).
Alcα ICD Regulates the Trafficking of APP into Neurites
To analyze the function of Alcα ICD on the trafficking of APP into neurites, we used Neuro2a cells that exhibit the extensions of neurites under a normal culture condition. In neuronal cells expressing EGFP-APP, APP appears the proximal region of extending neurites because APP is subject to an anterograde transport into the late secretory pathway (4). Therefore, we analyzed quantitatively the fluorescence signals of EGFP-APP in the proximal region of neurites (Fig. 5).
When Alcα CTF was expressed in Neuro2a cells expressing EGFP-APP and HA-X11L, fluorescence signal of APP appears in the proximal neurite, whereas the signal reduced significantly in the presence of 10 mm DAPT (γ-secretase inhibitor: (3,5-difluorophenylacetyl)-Ala-Phg-OBut) (Fig. 5, B and C, upper panels). Cells treated with DAPT showed the increased Alcα CTF levels (Fig. 5D), suggesting the production of Alcα ICD by γ-secretase cleavage of Alcα CTF was inhibited.
When Alcα ICD was further expressed in the cells along with Alcα CTF expression, EGFP-APP fluorescence in the proximal region was not decreased by DAPT treatment (Fig. 5, B and C, lower panels). These observations indicate that Alcα ICD, but not Alcα CTF, facilitates the trafficking of APP into the late secretory pathway.
DISCUSSION
Because it was documented that the generation of Aβ from APP and the formation of neurotoxic Aβ oligomers in neurons are closely linked to the etiology of Alzheimer disease, many efforts have been made to identify the modulator of APP metabolism, including Aβ generation and clearance (14). With regard to APP trafficking and metabolism, we and others have reported previously that X11 family proteins (X11s) are regulators of APP metabolism (6, 16, 20, 30–33). Excess expression of X11s suppresses APP processing, including Aβ generation and APP trafficking, via binding of X11L to APP. Furthermore, X11s function in the regulation of γ-cleavage of APP CTF directly or indirectly (18) or of β-cleavage of APP indirectly (16). Importantly, X11s-KO mice showed a significant increase in endogenous Aβ generation in the brain and increased formation of amyloid plaques was observed in the brains of hAPP-Tg/X11s-defective mice, indicating that X11s play an important role in APP regulation at various stages of APP metabolism and trafficking in vivo (16, 20, 21, 34, 35).
This effect is further enhanced in the presence of Alcα by the formation of a metabolically stable ternary APP·X11L·Alcα complex. This suggests that along with X11L, Alcα may be a key molecule for APP metabolism and trafficking in vivo. Because our previous in vitro study indicated that not only Alcα, but also Alcα CTF, could form a ternary complex together with X11L and APP in cultured cells (1), we hypothesized that membrane-associated full-length Alcα and Alcα CTF would suppress the production of Aβ in the presence of X11L.
However, we observed that the brains of Tg54 mice expressing excess amounts of hAlcα CTF showed a significant increase in Aβ due to enhanced transport of APP into the late secretory pathway. Moreover, our analysis showed remarkable production of p3-Alcα and Alcα ICD, which are the products of Alcα CTF following cleavage by γ-secretase. This observation indicates that in vivo, introduced Alcα CTF is quickly cleaved by γ-secretase to release Alcα ICD, which is free from membrane association. Indeed, in vitro experiments showed that Alcα ICD performs a novel function in releasing APP and Alcα from X11L, different from the function of membrane-associated Alcα and Alcα CTF.
Strikingly, both full-length APP and Alcα are released from X11L by excess expression of Alcα ICD in cultured cells. These studies in vivo and in vitro strongly indicate that Alcα ICD has different binding properties than full-length Alcα and may be a key molecule for the regulation of APP trafficking and metabolism. As shown in Figs. 2E and 4, we detected Alcα ICD as a doublet. Interestingly, only the slower migrating band interacted with X11L in co-immunoprecipitation assays (Fig. 4). Although we determined that molecular weight of the faster migrating protein band was the expected size of hAlcα ICD (data not shown), we have yet to identify the modification of slower-migrating protein band and determine why it interacts differently with X11L.
Several previous reports indicated that stalling of APP transport increases the production of Aβ because of the increased probability of association with secretases (36–38). The source of Aβ is still controversial. A recent report suggested that majority of intracellular APP is transported into lysosome from Golgi to generate Aβ (39). In contrast, it has been thought that cell surface APP is a significant source of Aβ (40, 41). Our observation that the increased Aβ in Tg mice brain is due to the increased synaptosomal APP level may be consistent with a hypothesis that cell surface APP contributes to the increased production of Aβ. We revealed here that increased trafficking of APP into the late secretory pathway facilitated APP metabolism, including Aβ generation, which was regulated by Alcα ICD derived from Alcα CTF by γ-secretase cleavage. Although the precise molecular mechanism is still under investigation, it is clear that dysfunctional APP intracellular trafficking is closely related to the aberrant production of Aβ. Moreover, the role of Alcα ICD in the regulation of membrane protein trafficking is a novel function of regulated intramembrane proteolysis.
This work was supported in part by Grants-in-aid for Scientific Research 26293010 (to T. S.) and 24790062 (to S. H.) from the Ministry of Education, Culture, Sports, Science, and Technology in Japan and by Bilateral Joint Research Projects (to S. H.) and an Asian Core Program (to T. S.) of the Japan Society for the Promotion of Science.
- Alc
- Alcadein
- APP
- amyloid β-protein precursor
- CTF
- carboxyl-terminal fragment
- ICD
- intracellular domain fragment
- X11L
- X11-like
- hAlc
- human Alc
- Aβ
- amyloid β-protein
- mAPP
- mature APP(s)
- Tg
- transgenic
- DAPT
- (3,5-difluorophenylacetyl)-Ala-Phg-OBut
- hAlca FL
- full-length hAlca
- sELISA
- sandwich ELISA.
REFERENCES
- 1. Araki Y., Tomita S., Yamaguchi H., Miyagi N., Sumioka A., Kirino Y., Suzuki T. (2003) Novel cadherin-related membrane proteins, Alcadeins, enhance the X11-like protein mediated stabilization of amyloid β-protein precursor metabolism. J. Biol. Chem. 278, 49448–49458 [DOI] [PubMed] [Google Scholar]
- 2. Vogt L., Schrimpf S. P., Meskenaite V., Frischknecht R., Kinter J., Leone D. P., Ziegler U., Sonderegger P. (2001) Calsyntenin-1, a proteolytically processed postsynaptic membrane protein with a cytoplasmic calcium-binding domain. Mol. Cell Neurosci. 17, 151–166 [DOI] [PubMed] [Google Scholar]
- 3. Hintsch G., Zurlinden A., Meskenaite V., Steuble M., Fink-Widmer K., Kinter J., Sonderegger P. (2002) The calsyntenins – a family of postsynaptic membrane proteins with distinct neuronal expression patterns. Mol. Cell. Neurosci. 21, 393–409 [DOI] [PubMed] [Google Scholar]
- 4. Araki Y., Kawano T., Taru H., Saito Y., Wada S., Miyamoto K., Kobayashi H., Ishikawa H. O., Ohsugi Y, Yamamoto T., Matsuno K., Kinjo M., Suzuki T. (2007) The novel cargo receptor Alcadein induces vesicle association of kinesin-1 motor components and activates axonal transport. EMBO J. 26, 1475–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Araki Y., Miyagi N., Kato N., Yoshida T., Wada S., Nishimura M., Komano H., Yamamoto T., De Strooper B., Yamamoto K., Suzuki T. (2004) Coordinated metabolism of Alcadein and amyloid β-protein precursor regulates FE65-dependent gene transactivation. J. Biol. Chem. 279, 24343–24354 [DOI] [PubMed] [Google Scholar]
- 6. Tomita S., Ozaki T., Taru H., Oguchi S., Takeda S., Yagi Y., Sakiyama S., Kirino Y., Suzuki T. (1999) Interaction of a neuron-specific protein containing PDZ domains with Alzheimer's amyloid precursor protein. J. Biol. Chem. 274, 2243–2254 [DOI] [PubMed] [Google Scholar]
- 7. Suzuki T., Nakaya T. (2008) Regulation of amyloid β-protein precursor by phosphorylation and protein interactions. J. Biol. Chem. 283, 29633–29637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thinakaran G., Koo E. (2008) Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Konecna A., Frischknecht R., Kinter J., Ludwig A., Steuble M., Meskenaite V., Indermühle M., Engel M., Cen C., Mateos J. M., Streit P., Sonderegger P. (2006) Calsyntenin-1 docks vesicular cargo to kinesin-1. Mol. Biol. Cell 17, 3651–3663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hata S., Fujishige S., Araki Y., Kato N., Araseki M., Nishimura M., Hartmann D., Saftig P., Fahrenholz F., Taniguchi M., Urakami K., Akatsu H., Martins R. N., Yamamoto K., Maeda M., Yamamoto T., Nakaya T., Gandy S., Suzuki T. (2009) Alcadein cleavages by APP α- and γ-secretases generate small peptides p3-Alcs indicating Alzheimer disease-related γ-secretase dysfunction. J. Biol. Chem. 284, 36024–36033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hata S., Fujishige S., Araki Y., Taniguchi M., Urakami K., Peskind E., Akatsu H., Araseki M., Yamamoto K., Martins R. N., Maeda M., Nishimura M., Levey A., Chung K. A., Montine T., Leverenz J., Fagan A., Goate A., Bateman R., Holtzman D. M., Yamamoto T., Nakaya T., Gandy S., Suzuki T. (2011) Alternative γ-secretase processing of γ-secretase substrates in common forms of mild cognitive impairment and Alzheimer disease: Evidence for γ-secretase dysfunction. Ann. Neurol. 69, 1026–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ludwig A., Blume J., Diep T. M., Yuan J., Mateos J. M., Leuthäuser K., Steuble M., Streit P., Sonderegger P. (2009) Calsyntenins mediate TGN exit of APP in a kinesin-1-dependent manner. Traffic 10, 572–589 [DOI] [PubMed] [Google Scholar]
- 13. Glabe C. G. (2008) Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benilova I., Karran E., De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 15. Piao Y., Kimura A., Urano S., Saito Y., Taru H., Yamamoto T., Hata S., Suzuki T. (2013) Mechanism of intramembrane cleavage of Alcadeins by γ-secretase. PLoS One 8, e62431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saito Y., Sano Y., Vassar R., Gandy S., Nakaya T., Yamamoto T., Suzuki T. (2008) X11 proteins regulate the translocation of amyloid β-protein precursor (APP) into detergent-resistant membrane and suppress the amyloidogenic cleavage of APP by β-site-cleaving enzyme in brain. J. Biol. Chem. 283, 35763–35771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saito Y., Akiyama M., Araki Y., Sumioka A., Shiono M., Taru H., Nakaya T., Yamamoto T., Suzuki T. (2011) Intracellular trafficking of the amyloid β-protein precursor (APP) regulated by novel function of X11-like. PLoS One 6, e22108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lau K. F., McLoughlin D. M., Standen C., Miller C. C. (2000) X11α and X11β interact with presenilin-1 via their PDZ domains. Mol. Cell. Neurosci. 16, 557–565 [DOI] [PubMed] [Google Scholar]
- 19. Lee J. H., Lau K. F., Perkinton M. S., Standen C. L., Rogelj B., Falinska A., McLoughlin D. M., Miller C. C. (2004) The neuronal adaptor protein X11β reduces amyloid β-protein levels and amyloid plaque formation in the brains of transgebic mice. J. Biol. Chem. 279, 49099–49104 [DOI] [PubMed] [Google Scholar]
- 20. Sano Y., Nakaya T., Pedrini S., Takeda S., Iijima-Ando K., Iijima K., Mathews P. M., Itohara S., Gandy S., Suzuki T. (2006) Physiological mouse brain Aβ levels are not related to the phosphorylation state of threonine 668 of Alzheimer APP. PLoS One 1, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kondo M., Shiono M., Itoh G., Takei N., Matsushima T., Maeda M., Taru H., Hata S., Yamamoto T., Saito Y., Suzuki T. (2010) Increased amyloidogenic processing of transgenic human APP in X11-like deficient mouse brain. Mol. Neurodegener. 5, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hogan B., Bedding R., Constantini F., Lacy E. (1986) Manipulating the Mouse Embryo: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, New York [Google Scholar]
- 23. Nakaya T., Suzuki T. (2006) Role of APP phosphorylation in Fe65-dependent gene transactivation mediated by AICD. Genes Cells 11, 633–645 [DOI] [PubMed] [Google Scholar]
- 24. Carlin R. K., Grab D. J., Cohen R. S., Siekevitz P. (1980) Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J. Cell Biol. 86, 831–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matsushima T., Saito Y., Elliott J. I., Iijima-Ando K., Nishimura M., Kimura N., Hata S., Yamamoto T., Nakaya T., Suzuki T. (2012) Membrane-microdomain localization of amyloid β-precursor protein (APP) C-terminal fragments is regulated by phosphorylation of the cytoplasmic Thr668 residue. J. Biol. Chem. 287, 19715–19724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Konno T., Hata S., Hamada Y., Horikoshi-Sakuraba Y., Nakaya T., Saito Y., Yamamoto T., Yamamoto T., Maeda M., Ikeuchi T., Gandy S., Akatsu H., Suzuki T., and Japanese Alzheimer's Disease Neuroimaging Initiative (2011) Coordinate increase of γ-secretase reaction products in the plasma of some female Japanese sporadic Alzheimer's disease patients: quantitative analysis with a new ELISA system. Mol. Neurodegener. 6, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakaya T., Kawai T., Suzuki T. (2008) Regulation of FE65 nuclear translocation and function by amyloid β-protein precursor in osmotically stressed cells. J. Biol. Chem. 283, 19119–19131 [DOI] [PubMed] [Google Scholar]
- 28. Brown M. S., Ye J., Rawson R. B., Goldstein J. L. (2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 100, 391–398 [DOI] [PubMed] [Google Scholar]
- 29. Morohashi Y., Tomita T. (2013) Protein trafficking and maturation regulate intramembrane proteolysis. Biochim. Biophys. Acta 1828, 2855–2861 [DOI] [PubMed] [Google Scholar]
- 30. McLoughlin D. M., Irving N. G., Brownlees J., Brion J. P., Leroy K., Miller C. C. (1999) Mint2/X11-like colocalizes with the Alzheimer's disease amyloid precursor protein and is associated with neuritic plaques in Alzheimer's disease. Eur. J. Neurosci. 11, 1988–1994 [DOI] [PubMed] [Google Scholar]
- 31. Xie Z., Romano D. M., Tanzi R. E. (2005) RNA interference-mediated silencing of X11α and X11β attenuates amyloid β-protein levels via differential effects on β-amyloid precursor protein processing. J. Biol. Chem. 280, 15413–15421 [DOI] [PubMed] [Google Scholar]
- 32. Ho A., Liu X., Südhof T. C. (2008) Deletion of Mint proteins decreases amyloid production in transgenic mouse models of Alzheimer's disease. J. Neurosci. 28, 14392–14400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chaufty J., Sullivan S. E., Ho A. (2012) Intracellular amyloid precursor protein sorting and amyloid-β secretion are regulated by Src-mediated phosphorylation of Mint2. J. Neurosci. 32, 9613–9625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saluja I., Paulson H., Gupta A., Turner R. S. (2009) X11α haploinsufficiency enhances Aβ amyloid deposition in Alzheimer's disease transgenic mice. Neurobiol. Dis. 36, 162–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mitchell J. C., Ariff B. B., Yates D. M., Lau K. F., Perkinton M. S., Rogelj B., Stephenson J. D., Miller C. C., McLoughlin D. M. (2009) X11β rescues memory and long-term potentiation deficits in Alzheimer's disease APPswe Tg2576 mice. Hum. Mol. Genet. 18, 4492–4500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kamal A., Stokin G. B., Yang Z., Xia C. H., Goldstein L. S. (2000) Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 28, 449–459 [DOI] [PubMed] [Google Scholar]
- 37. Stokin G. B., Lillo C., Falzone T. L., Brusch R. G., Rockenstein E., Mount S. L., Raman R., Davies P., Masliah E., Williams D. S., Goldstein L. S. (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307, 1282–1288 [DOI] [PubMed] [Google Scholar]
- 38. Almenar-Queralt A., Falzone T. L., Shen Z., Lillo C., Killian R. L., Arreola A. S., Niederst E. D., Ng K. S., Kim S. N., Briggs S. P., Williams D. S., Goldstein L. S. (2014) UV irradiation accelerates amyloid precursor protein (APP) processing and disrupts APP axonal transport. J. Neurosci. 34, 3320–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tam J. H., Seah C., Pasternak S. H. (2014) The amyloid precursor protein is rapidly transported from the Golgi apparatus to the lysosome and where it is processed into beta-amyloid. Mol. Brain 7, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koo E. H., Squazzo S. L. (1994) Evidence that prodiction and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 269, 17386–17389 [PubMed] [Google Scholar]
- 41. Grbovic O. M., Mathews P. M., Jiang Y., Schmidt S.D., Dinakar R., Summers-Terio N. B., Ceresa B. P., Nixon R. A., Cataldo A. M. (2003) Rab5-stimulated up-regulation of the endocytic pathway increases intracellular β-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Aβ production. J. Biol. Chem. 278, 31261–31268 [DOI] [PubMed] [Google Scholar]