

Abstract
Electrical activity in neurons requires a seamless functional coupling between plasmalemmal ion channels and ion transporters. Although ion channels have been studied intensively for several decades, research on ion transporters is in its infancy. In recent years, it has become evident that one family of ion transporters, cation-chloride cotransporters (CCCs), and in particular K+–Cl− cotransporter 2 (KCC2), have seminal roles in shaping GABAergic signalling and neuronal connectivity. Studying the functions of these transporters may lead to major paradigm shifts in our understanding of the mechanisms underlying brain development and plasticity in health and disease.
Most mature neurons in the CNS actively extrude Cl− and thus exhibit a low intracellular Cl− concentration ([Cl−]i). This unique specialization is a necessary, but not sufficient, condition for the generation of hyperpolarizing inhibitory postsynaptic potentials (IPSPs)1 by GABAA receptors (GABAARs) and glycine receptors (GlyRs). The low [Cl−]i comes about owing to an upregulation of the neuron-specific K+–Cl− cotransporter 2 (KCC2) during CNS neuron maturation and is maintained in adult central neurons2–6 (FIG. 1; TABLE 1). This developmental decrease in [Cl−]i does not occur in other cell types6,7. Thus, although neurobiologists generally regard immature neurons, which have a high [Cl−]i (REFS 8–11) and exhibit depolarizing GABAAR responses, as aberrant, it is actually the adult CNS neurons with their low internal levels of Cl− that are exceptional.
Figure 1. Developmental expression profiles, functions and secondary structures of CCCs.

a | Line plots showing the average exon array signal intensity of cation-chloride cotransporter (CCC) transcripts in the human neocortex from the early fetal period to late adulthood. The profiles are qualitatively similar for the hippocampus, amygdala and cerebellar cortex. Of the five CCCs expressed in the human neocortex (K+–Cl− cotransporter 1 (KCC1), KCC2, KCC3, KCC4 and Na+ – K+–2Cl− cotransporter 1 (NKCC1)), only mRNA encoding KCC2 undergoes robust developmental upregulation. NKCC1 shows moderate postnatal upregulation, and KCC3 and NKCC1 are expressed at high levels throughout life while expression of KCC1 and KCC4 is low. The levels of NKCC2 and Na+–Cl− cotransporter (NCC) are below the level (6 on the y axis) for a gene to be considered as expressed according to the criteria described in REF. 295. Only the expression of KCC2 is neuron-specific. Expression data are from the data bank described in REF. 295 and accessible at the Human Brain Transcriptome website. b | NKCC1 and KCC2 control intracellular Cl− concentration ([Cl−]i) in many central neurons. In immature neurons, the Na+ gradient generated by the Na+/K+ ATPase drives cellular uptake of Cl− via NKCC1 and KCC2 has a minor role. This generates a depolarizing Cl− current across GABAA receptors (GABAARs). During neuronal maturation, functional KCC2 attains a high level of expression and the Cl− current becomes hyperpolarizing. Cl− extrusion by KCC2 is fuelled by the Na+/K+ ATPase-dependent K+ gradient. NKCC1 and KCC2, like all CCCs, are electroneutral, in other words, they do not generate any current by themselves. c | Upregulation of KCC2 facilitates the structural and functional development of cortical dendritic spines in an ion-transport-independent manner, probably through effects on the actin–spectrin cytoskeleton. d | Secondary structures of human KCC2b and NKCC1b splice isoforms, highlighting residues critical for function. Dephosphorylation of KCC2 at threonine residues T906 and T1007 and phosphorylation at serine residue S940 are associated with functional activation. Phosphorylation by protein kinase C (PKC) at S940 promotes KCC2 membrane stability. Phosphorylation of NKCC1 by SPAK and OSR1 (oxidative stress responsive kinase 1) at the depicted N-terminal residues leads to functional activation. KCC2 transport function is inhibited by phosphorylation of T906 and T1007 and dephosphorylation at S940. Phosphorylation at T1007 and dephosphorylation at S940 are likely to be mediated by SPAK/OSR1 kinases, and protein phosphatase 1 (PP1), respectively. An arginine-to-histidine mutation at residue 952 (R952H), discovered in humans, results in loss of both transport activity and spinogenesis by KCC2. The approximate (~30 kDa) C-terminal fragment of KCC2 cleaved by calpain is shown in blue. Dephosphorylation of NKCC1 by PP1 at N-terminal threonine residues renders it transport-deficient. Putative glycosylation sites in KCC2 and NKCC1 are highlighted. AMPAR, AMPA receptor. Part d is adapted from 2-D models provided by B.Forbush, Yale University, New Haven, Connecticut, USA.
Table 1.
Effects of manipulation of CCC expression on neuronal chloride homeostasis
| CCC | Model | Age | Effect | Refs |
|---|---|---|---|---|
| KCC2 | Knockdown of gene encoding KCC2 in CA1 pyramidal neurons in rat organotypic HC slices | P11–13 (DIV 2–5) | Loss of hyperpolarizing DFGABA | 2 |
| KCC2-null mouse spinal motor neurons | E18.5 | Excitatory responses to GABA and glycine in knockout but not wild-type neurons | 73 | |
| KCC2b-null mouse spinal motor neurons | P5–8 | EIPSP more positive than in wild-type neurons | 178 | |
| KCC2b-null mouse cultured cortical neurons | DIV 18–21 | Lack of hyperpolarizing responses to GABA | 4 | |
| CA1 pyramidal neurons of compound-heterozygous KCC2 mice with lack of one allele and reduced expression of the second allele (mice retain ~20% of normal KCC2 brain expression) | P30 | EGABA more positive than in wild-type neurons | 177 | |
| KCC2 overexpression in cultured rat HC neurons | DIV 5–7 | Hyperpolarizing shift in EGABA to values observed in mature neurons; decrease in GABA-elicited Ca2+ responses | 148,180 | |
| KCC2 overexpression via in utero electroporation of rat or mouse layer 2/3 pyramidal neurons | P1–7 | Hyperpolarizing shift in EGABA to values observed in mature neurons; enhancement of Cl− extrusion capacity | 31,140, 181 | |
| Knockdown of gene encoding KCC2 in cultured rat HC neurons | DIV 23–24 | Loss of Cl− extrusion capacity | 29,151 | |
| Cell-specific KCC2-null, Cre–lox mouse cerebellar Purkinje neurons | P25–64 | ~60% decrease in hyperpolarizing DFGABA | 5 | |
| Cell-specific KCC2-null, Cre–lox mouse cerebellar granule cells | P30–62 | Depolarizing shift in both Vm and EGABA | 5 | |
| KCC2 overexpression in embryonic zebrafish spinal cord neurons | 26–32 HPF | Shift from depolarizing to hyperpolarizing DFGly | 182 | |
| Knockdown of gene encoding KCC2 in zebrafish retinal ganglion cells | 2.5–6 DPF | Loss of somatodendritic and inter-dendritic EGABA gradients | 179 | |
| KCC3 | Cell-specific KCC3-null, Cre–lox mouse cerebellar Purkinje neurons | P25–64 | No change in EGABA or DFGABA | 5 |
| KCC3-null mouse cerebellar Purkinje neurons | P12–14 | EGABA more positive than in wild-type neurons | 116 | |
| KCC3-null mouse DRG sensory neurons | Adult | Increase in [Cl−]i compared to wild-type neurons | 51 | |
| NKCC1 | Knockdown of gene encoding NKCC1 in newborn DGCs of adult mouse | P49–56 | Loss of depolarizing DFGABA | 296 |
| NKCC1-null mouse CA1 pyramidal neurons | P1 | Decrease in depolarizing DFGABA ; decrease in GABA-elicited Ca2+ responses | 94 | |
| NKCC1-null mouse CA1 pyramidal neurons | P3–4 | Loss of depolarizing DFGABA | 10 | |
| NKCC1-null mouse CA3 pyramidal neurons | P6–7 | Loss of depolarizing DFGABA | 97 | |
| NKCC1-null mouse DGCs | P16–20 | Loss of axosomatic EGABA gradient | 185 |
CCC, cation-chloride cotransporter; [Cl−]i, intracellular Cl− concentration; DFGABA, the driving force of the GABAA receptor (GABAAR)-mediated current; DGC, dentate granule cell; DIV, day in vitro; DPF, days post-fertilization; DRG, dorsal root ganglion; E, embryonic day; EGABA, the reversal potential of GABAAR-mediated responses; EIPSP, reversal potential of inhibitory postsynaptic potentials (IPSPs); HC, hippocampal; HPF, hours post-fertilization; KCC, K+–Cl− cotransporter; NKCC, Na+–K+–2Cl− cotransporter; P, postnatal day; Vm, membrane potential.
As a substrate for plasmalemmal Cl− transporters, Cl− is used to maintain basic cellular parameters, such as cell volume and intracellular pH (pHi). The low [Cl−]i in mature central neurons means that their capacity to respond effectively to volume or pHi disturbances is compromised1,12. There is also a high metabolic cost associated with maintenance of low [Cl−]i and the generation of hyperpolarizing Cl− currents during neuronal signalling, particularly when the neuron faces an energy crisis, such as that associated with recurrent seizures or stroke13–15. Clearly, these important trade-offs must be taken into account when judging whether a given alteration in [Cl−]i regulation has a disease-promoting (maladaptive) or an adaptive role15. For instance, the frequently reported downregulation of KCC2 following neuronal trauma16–23 may be part of a general adaptive cellular response that facilitates neuronal survival by reducing the energetic costs that are needed to preserve low [Cl−]i (REFS 14,15) and by facilitating functional recovery through the removal of GABAergic inhibitory constraints on neuroplasticity24,25.
The solute carrier 12 (SLC12) family of electroneutral cation-chloride cotransporters (CCCs) includes four K+– Cl− cotransporters (KCCs), KCC1 (encoded by SLC12A4), KCC2 (encoded by SLC12A5), KCC3 (encoded by SLC12A6) and KCC4 (encoded by SLC12A7); two Na+–K+–2Cl− cotransporters (NKCCs), NKCC1 (encoded by SLC12A2) and NKCC2 (encoded by SLC12A1); and an Na+–Cl− cotransporter (NCC; encoded by SLC12A3). Two members of the SLC12 family, KCC2 and NKCC1, are known to have cell-autonomous functions in central neurons, including a major role in setting the reversal potential and driving force of the anion currents mediated by GABAARs and GlyRs 6 as well as by non-ligand-gated Cl− channels26,27. Based on their developmental, cellular and subcellular patterns of functional expression, CCCs have turned out to be highly versatile factors in the control of GABAergic and glycinergic signalling. The importance of CCCs in brain function is underscored by the recent discovery of an ion-transport-independent structural role for KCC2 in the morphological and functional maturation of cortical dendritic spines28–32. Thus, KCC2 is in a key position to control and coordinate the development of both GABAergic and glutamatergic transmission. It is not surprising that CCC dysfunctions are likely to be associated with a wide range of neurological and psychiatric disorders15,33–41.
Here, we summarize recent progress in our understanding of the roles of CCCs in neuronal development, plasticity and disease, with a particular focus on the functions of KCC2 and NKCC1 in synaptic signalling. Of the wide range of diseases to which CCCs have been linked, we focus on epilepsy and chronic pain because research on these disorders has shed light on the fundamental roles of CCCs in neuronal signalling.
Expression of CCCs in the CNS
CCCs are glycoproteins that have a core molecular weight of ~110–130 kDa. Each CCC has a similar predicted structure of 12 transmembrane segments and intracellular amino- and carboxy-terminal domains33,42 (FIG. 1d). With the exception of a high-resolution structure that has been obtained for the C-terminal domain of a bacterial CCC protein43, the tertiary structures of CCCs are not known. In terms of quaternary structure, the CCCs are likely to form dimers42. However, the available data are not conclusive regarding the manner in which multimerization affects CCC functions6,42,44. CCCs are expressed in all organ systems and, in addition to being instrumental in neuronal signalling, they are involved in a range of physiological processes, including cell volume regulation (BOX 1), transepithelial ion transport, neuroendocrine signalling and blood pressure regulation6,7,33,45–48.
Box 1. Control of neuronal and glial volume by CCCs.
Following an acute perturbation, mechanisms that restore neuronal and glial volume are needed to cope with cell swelling. Isosmotic swelling results from an activity-dependent increase in the ionic load and is often seen in dendrites as a result of channel-mediated net influx of Na+, Cl− and osmotically obliged water. As the intracellular Na+ concentration ([Na+]i) and [Cl−]i are low under normal conditions, such an influx leads to a large relative increase in the concentrations of these ions, despite the accompanying water influx; and to a significant rise in the equilibrium potential of chloride (ECl). In some pathological states (such as water intoxication or hyponatraemia), neurons and glia are subject to hyposmotic swelling, as water flows from the vasculature into the extracellular space (ECS). In this case, cell swelling results exclusively from net water influx and intracellular ion concentrations will decrease.
The membrane lipid bilayer cannot expand elastically beyond 3%260, and acute swelling is therefore largely attributable to unfolding of the cell plasmalemma261. In neurons, with their highly complex morphology, local differences in hydrostatic pressure effects as well as in Ca2+-sensitive cytoskeletal mechanisms will lead to differential susceptibility to swelling in distinct subcellular compartments.
The ability of neurons to recover volume differs dramatically depending on whether the volume change results from isosmotic or hyposmotic swelling. One important volume regulatory mechanism involves net K+ and Cl− efflux via K+–Cl− cotransporters (KCCs). Volume recovery in mature CNS neurons upon hyposmotic swelling is severely compromised because of their low [Cl−]i, especially if mechanisms that replenish intracellular Cl− without generating a further osmotic load (such as plasmalemmal HCO3−–Cl− exchange) are inefficient. By contrast, recovery from isosmotic swelling by KCCs is facilitated by the increased [Cl−]i.
All KCC isoforms are thought to be activated by hyposmotic swelling, largely based on data obtained using heterologous expression in Xenopus laevis oocytes33,141,262. However, the intracellular volume-sensitive cascades and their coupling to cation-chloride cotransporter (CCC) functions are unlikely to be identical in amphibian oocytes263,264 and neurons. In addition, a distinction should be made between an ion transporter that is volume-sensitive and one that is genuinely volume-regulatory. In the latter case, the net ion flux will be a graded function of the volume perturbation265. We do not yet have sufficient data to judge whether CCCs are genuinely volume-regulatory in mammalian neurons12. This is also true for astrocytes, which are thought to control the volume of the ECS266. The ECS volume has an important modulatory effect on neuronal excitability267. Somewhat surprisingly, recent work has shown that Na+–K+–2Cl− cotransporter 1 (NKCC1) is not a major factor in the recovery of ECS volume following activity-induced cellular swelling268.
Central neurons are probably devoid of water-permeable aquaporins266, and it has been shown that their soma and dendrites do not swell in response to a 20-minute exposure to moderately hypotonic solutions269. By contrast, peripheral nervous system neurons express water-permeable aquaporins and maintain a high [Cl−]i (REF. 270), which implies that they have a higher sensitivity to swelling but also a better capacity for Cl−-dependent volume regulation271. Moreover, water can also move passively through ion channels272, thereby influencing short-term and long-term homeostasis of volume.
There are two functional branches of the CCC phylogenetic tree46: the Na+-dependent and the Na+-independent CCCs. Uptake of Cl− by NKCC1, NKCC2 and NCC is fuelled by the inwardly directed plasmalemmal Na+ gradient, which is maintained by the Na+/K+ ATPase. By contrast, KCC1, KCC2, KCC3 and KCC4 typically mediate net Cl− efflux, which is driven by the respective outwardly directed K+ gradient (BOX 2). NCC and NKCC2 are predominantly expressed in the kidney49,50, whereas all other CCCs exhibit varied spatiotemporal patterns of expression in the mammalian CNS (FIG. 1a) and some, such as NKCC1 and KCC3, are also expressed in the peripheral nervous system (PNS)6,51,52.
Box 2. Energetics of chloride regulation.
K+–Cl− cotransport is at thermodynamic equilibrium when all the energy available from the outward movement of K+ across the membrane is used to extrude Cl− (see the figure)6. This can be expressed in terms of ion concentrations or of equilibrium potentials (E) (see Supplementary information S2 (text)):
| (1) |
where o and i refer to extracellular and intracellular. Equation 1 also defines the equilibrium conditions at which net K+–Cl− transport by K+–Cl− cotransporters (KCCs) will reverse. In a resting neuron with constitutively active KCC2, ECl will be close to EK, and an increase in [K+]o will drive Cl− into the neuron. Conversely, if a conductive influx of Cl− increases intraneuronal [Cl−], thereby promoting K+–Cl− efflux via KCC2, [K+]o will increase101.
The equilibrium condition for Na+–K+–2Cl− cotransporters (NKCCs) is obtained in an analogous manner:
| (2) |
With typical values of EK = −90 mV and ENa = +74 mV164,224, KCCs could reduce [Cl−]i down to 4.4 mM (ECl = −90 mV), and NKCCs could increase [Cl−]i up to >90 mM (ECl of −8 mV). However, such theoretical limits can be achieved only in the absence of opposing Cl− fluxes such as ‘leaks’ across channels. For instance, in the simultaneous presence of KCC-mediated Cl− extrusion and GABAA receptor (GABAAR)-mediated Cl− influx, a dynamic steady state will be established in which changes in Cl− conductance or KCC activity will be reflected in [Cl−]i. This classical pump–leak relationship273 is made use of in experiments addressing the transport capacity of KCC2 and NKCC1 by experimentally inducing an exogenous excess or deficit of Cl−, respectively, in a neuron5,146,185,274,275.
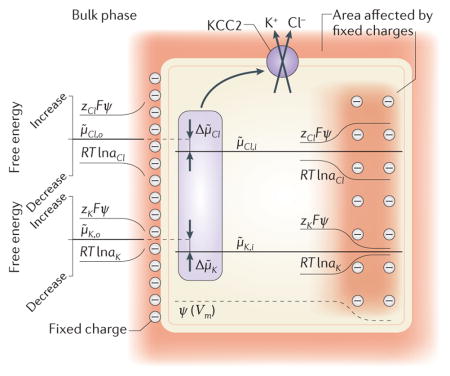
Equations 1 and 2 depend on bulk ion concentrations. However, binding of ions to transporters and channels takes place in the aqueous layer, where local ion concentrations may differ substantially compared with those in the bulk phase because of fixed charges located on the phospholipid or protein surfaces272,276,277. This has been shown to increase transport rates (conductance) in ion channels by increasing the availability of permeant ions at the pore mouth272 but has no effect on thermodynamic equilibria. This is because, at equilibrium, a local electric field has an equal but opposite effect on the local concentration-dependent (more precisely, ion activity-dependent) and electrical potential-dependent components (RTlna and zFψ, respectively) of the free energy of mobile ions. Therefore, there is no change in the electrochemical potential of Cl− or K+ (μ̃Cl,o, μ̃K,o) in the vicinity of, for instance, a negatively charged external membrane surface (see the figure). With regard to KCCs, the local electrostatic effects on [Cl−] and [K+] are inversely proportional, keeping the product, [K+]·[Cl−], constant. Thus, there is no change in the KCC equilibrium as defined by equation 1. An analogous situation arises within the cell if a spatial gradient of immobile charge density generates an electric field (a shift in the local membrane potential (Vm)) along which mobile ions such as Cl− and K+ equilibrate (see the figure). Again, there is no change in the electrochemical potential of mobile ions (μ̃Cl,i, μ̃K,i); and their transmembrane gradients (Δμ̃Cl, Δμ̃K), the product [K+]·[Cl−] and the KCC equilibrium remain unaffected. In other words, the local shifts in Vm and ECl (and EK) are equal, which keeps the transmembrane driving force of Cl− (and K+) constant (see Supplementary Information S2 (text))278,279. Similarly, the equilibrium of NKCCs is insensitive to local electrostatic effects (equation 2). Thus, in contrast to a recent study280, the equilibrium and reversal of Cl− transport by cation-chloride cotransporters are correctly predicted on the basis of bulk ion concentrations281.
KCC2
The main Cl− extruder to promote fast hyperpolarizing postsynaptic inhibition in the brain is KCC2 (TABLE 1). It is abundantly expressed in most mature mammalian central neurons, with very little or no expression in PNS neurons and non-neuronal cell types2,6,53–55. KCC2 is the only KCC isoform that is not expressed in glia53,56,57. The two N-terminally spliced variants, KCC2a and KCC2b, have similar ion-transport properties when expressed in human embryonic kidney (HEK) cells58. The expression of KCC2a remains relatively low throughout life, whereas KCC2b is strongly upregulated during postnatal life in mice and rats3,58,59 and constitutes up to ~90% of total KCC2 protein in the adult murine cortex58,59. Of note, most studies on KCC2 expression have used mRNA probes and antibodies that detect both KCC2a and KCC2b58. Thus, unless stated otherwise, ‘KCC2’ here refers collectively to both splice variants. Certain subpopulations of adult CNS neurons, such as the dopaminergic neurons of the substantia nigra60 and most neurons of the thalamic reticular nucleus61–63, lack KCC2.
The upregulation of KCC2 expression is part of neuronal differentiation54,64–66, and this is consistent with a developmental gradient in the onset of KCC2 expression from caudal-to-rostral regions of the CNS3,54,67. In the spinal cord and brainstem of rodents, perinatal KCC2 expression levels are high and comparable to those observed in older animals3,59,68,69, whereas in more rostral parts such as the cortex, robust upregulation of KCC2 commences by the time of birth2,3,54,67,69 and undergoes a steep increase during the first postnatal month2,10,67,70,71. Interestingly, low levels of Slc12a5 mRNA and protein can already be detected at early embryonic stages54,65,72. Indeed, knockout and overexpression studies point to a critical structural role of KCC2 in the maturation of neurons in the fetal nervous system65,73,74.
In keeping with the timing of other milestones of CNS development75, the developmental upregulation of KCC2 with regard to birth is both brain region- and species-specific2. While rats are born with very low KCC2 expression and depolarizing GABAAR-mediated responses in cortical neurons, in the guinea pig KCC2 is upregulated in utero and cortical neurons show hyperpolarizing GABAAR responses at birth2. In the human neocortex, SLC12A5 mRNA undergoes robust upregulation during the second half of gestation37,41,76 (FIG. 1a), and there are immunohistochemical data indicating that from the 25th postconceptional week onwards, most cortical neurons express KCC2 (REFS 77,78). In contrast with the conclusions of a widely cited study that suggested that KCC2 is predominantly expressed postnatally79, the above data collectively indicate that, unlike in rodents, massive upregulation of KCC2 in the human neocortex begins prenatally.
Neuron-specific expression of KCC2, as studied in mice, is ensured by multiple mechanisms, including two neuron-restrictive silencing elements (NRSEs) associated with Slc12a5 (REFS 80–82) and transcription factors of the early growth response (EGR) family70,83 (FIG. 2). Pathways regulating neuron-restrictive silencing factor (NRSF), which binds to NRSEs, may contribute to the downregulation of KCC2 (see below) during epileptogenesis84 (BOX 3). Brain-derived neurotrophic factor (BDNF) and its receptor tropomyosin-related kinase B (TRKB) are thought to promote the developmental upregulation of mRNA encoding KCC2b83,85 (FIG. 2c) through extracellular signal-regulated kinase 1/2 (ERK1/2)-dependent expression of EGR4 (REF. 83). However, total KCC2 expression decreased by less than ~50% when EGR4 signalling was blocked70, suggesting that KCC2 transcription is under the control of additional mechanisms, including upstream stimulating factor 1 (USF1) and USF2 (REF. 86). There is also evidence for steroid hormone-dependent effects on KCC2 expression, which has important implications for gender differences in the propensity to early-life seizures87,88.
Figure 2. Mechanisms of ionic plasticity and their temporal domains.

Cation-chloride cotransporters (CCCs) control ionic plasticity across various overlapping time scales that range from seconds to weeks, and beyond. a | Short-term ionic plasticity of GABAergic signalling is based on fast, activity-dependent transmembrane movements of Cl− and HCO3−, which alter the driving force and polarity of GABA-induced currents and thereby cause changes in the postsynaptic membrane potential (Vm). On the left, a single presynaptic event leads to a hyperpolarization of Vm. On the right, repetitive activation of GABAergic terminals evokes a biphasic Vm response. In biphasic Vm responses, the depolarizing HCO3− current leads to an increase in GABAA receptor (GABAAR)-mediated uptake of Cl− and an increase in intracellular Cl− concentration ([Cl−]i). The consequent K+–Cl− cotransporter 2 (KCC2)-mediated extrusion of K+ increases the extracellular K+ concentration ([K+]o) and has a depolarizing or even functionally excitatory action on Vm. b | Fast functional regulation of CCCs on a time scale of minutes to hours is mediated by post-translational mechanisms, including (de)phosphorylation of key residues on the intracellular domains of KCC2 and Na+–K+–2Cl− cotransporter 1 (NKCC1) (see also FIG. 1d) and calpain-mediated cleavage of KCC2. Constitutive membrane recycling (dashed arrows) of KCC2 is regulated by the phosphorylation state of the C-terminal serine residue S940. Phosphorylation of S940 by protein kinase C (PKC) limits clathrin-mediated endocytosis of KCC2. By contrast, protein phosphatase 1 (PP1)-dependent dephosphorylation of S940 leads to internalization of KCC2 and a reduction in neuronal Cl− extrusion capacity following intense activation of NMDA receptors (NMDARs) and an increase in [Ca2+]. Under such conditions, KCC2 is also C-terminally cleaved by the Ca2+- and brain-derived neurotrophic factor (BDNF)-activated protease calpain, which results in irreversible inactivation of KCC2 (BOX 3). NKCC1 is kinetically regulated through a Cl−-sensing cascade involving WNK (with no lysine kinase) and the STE20-related kinases SPAK and OSR1 (oxidative stress responsive kinase 1; not shown). WNKs are allosterically modulated by Cl−, with a decrease in [Cl−]i leading to activation of SPAK and OSR1 by WNKs. Consequent SPAK-mediated phosphorylation of key N-terminal threonine residues (for example, T212 and T217; see also FIG. 1d) of NKCC1 results in its activation and Cl− accumulation. By contrast, dephosphorylation of these residues by PP1 inactivates NKCC1. Reciprocal regulation of transport activities of NKCC1 and KCC2 by the WNK SPAK and WNK OSR1 cascade has been demonstrated in heterologous expression systems and may also take place in neurons. At least one SPAK and OSR1 phosphorylation site (T1007) is found in KCC2b, the principal KCC2 splice variant found in neurons. However, it is not clear how phosphorylation of KCC2 by SPAK and OSR1 affects neuronal Cl− extrusion capacity. c | Neuron-specific expression of KCC2 is ensured via multiple transcriptional mechanisms, including the actions of neuron-restrictive elements (NRSEs; also known as RE1), which silence SLC12A5 (encoding KCC2) in non-neuronal cells and neuron-enriched transcription factors (TFs). Neuron-enriched TFs, for example, members of the early growth response (EGR; not shown) family, are sensitive to signalling by neurotrophic factors, such as BDNF and its receptor tropomyosin-related kinase B (TRKB), which exert qualitatively different effects on SLC12A5 transcript expression in immature and mature neurons. The TRKB-mediated cascades may reverse during neuronal trauma, resulting in recapitulation of immature-like Cl− homeostasis in diseased neurons (BOX 3). Long-term consolidation of changes in KCC2 following trauma or during epileptogenesis is likely to be mediated to an extent by the above transcriptional mechanisms.
Box 3. Regulation of KCC2 by TRKB and calpain.
Deficits in K+–Cl− cotransporter 2 (KCC2) expression, which are often associated with decreased efficacy of GABAergic inhibition and the emergence of depolarizing GABAA receptor (GABAAR)-mediated currents, have been documented following experimental seizures17,20,282,283 and in models of cerebral ischaemia22,284, traumatic brain injury16,19,285 and neuropathic pain18,21,23. The downregulation of KCC2 is part of a spectrum of changes associated with cellular de-differentiation following neuronal trauma15,176. In parallel with, or as a result of, changes in its serine 940 phosphorylation status32,141, KCC2 is cleaved at its C-terminal domain by the Ca2+- and brain-derived neurotrophic factor (BDNF)-activated protease calpain, leading to a loss of a ~20–40 kDa fragment23,150,151,286 (FIG. 1d) and reduction of both total and surface-expressed KCC2 protein. The part of the C-terminal domain that is removed contains many of the sites that are critical for the function and regulation of KCC2 (REFS 141,215) (FIG. 1d), and it is thus not surprising that calpain-mediated cleavage of KCC2 compromises neuronal Cl− extrusion capacity150,151. Furthermore, both serine 940 dephosphorylation and calpain activation have been suggested to regulate the lateral mobility of KCC2 within the plasma membrane, with potential consequences for the synaptic localization of AMPA receptors29,32,151. As the C-terminal domain of KCC2 is also important for its ion- transport-independent functions30 (including formation of the mature spine phenotype28,29), calpain cleavage of KCC2 is likely to contribute to the changes in dendritic spines associated with calpain activation (REFS 32,287,288).
In cortical and spinal cord neurons, trauma- or seizure-induced downregulation of KCC2 involves activation of NMDA receptors (NMDARs), tropomyosin-related kinase B (TRKB), calpain and probably protein phosphatase 1 (REFS 17,23,149,150,152). Downregulation of KCC2 is absent in mice carrying a point mutation that uncouples TRKB from phospholipase Cγ1 (PLCγ1)152, and, at least in the hippocampus, loss of KCC2 protein triggered by brain-derived neurotrophic factor (BDNF) also takes place in the presence of NMDAR inhibitors17. This is intriguing, as PLCγ1 activation seems to be one of the most well-defined ways in which TRKB activation promotes epileptogenesis289,290. In models of seizures and trauma, the most prominent loss of KCC2 mRNA and protein takes place in regions with the highest increase in BDNF and TRKB expression17,92.
In immature neurons, experimentally-increased BDNF–TRKB signalling promotes KCC2 expression83,85 (see also REF. 291). This contrast to the situation in the adult brain is likely to be related to the qualitative difference in TRKB phosphorylation and activation of its downstream cascades, such as PLCγ1, in immature and mature cortical neurons292,293. Interestingly, in damaged mature neurons, BDNF may resume its ability to promote KCC2 expression soon after an acute insult21,294. This supports the idea that acute downregulation of KCC2 associated with excitotoxicity and an energy crisis, such as ischaemia and seizures, reflects an adaptive response to promote neuronal survival and potential for rewiring15. Indeed, recent work has shown that reinduction of synaptic plasticity by chronic exposure to the serotonin reuptake inhibitor fluoxetine involves upregulation of BDNF and downregulation of KCC2 (REF. 25).
An important question is whether developmental upregulation of KCC2 is influenced by neuronal activity89. Chronic exposure to glutamatergic, GABAergic or voltage-gated Na+ channel antagonists had no major effect on KCC2 levels in cortical neurons in vitro71,90 (see also REFS 10,91–95). Likewise, upregulation of KCC2 is unaffected in VIAAT (vesicular inhibitory amino acid transporter)-knockout mice, despite the complete absence of GABAergic synaptic transmission96.
NKCC1
NKCC1 is the principal transport mechanism responsible for Cl− uptake and for the depolarizing GABAAR responses of immature neurons10,94,97 (TABLE 1). By contrast, in some other neuronal populations, such as the brainstem superior olivary complex, other — probably HCO3−-dependent — mechanisms of Cl− uptake are important69. For example, depolarizing GABA actions in dendrites of mature pyramidal neurons98,99 are fully attributable to HCO3−-mediated GABAAR currents and associated depolarizing extracellular K+ concentration ([K+]o) transients100,101 (see below).
In line with the nearly ubiquitous expression of NKCC1 (REFS 102,103), Slc12a2 includes a promoter region that is characteristic of a housekeeping gene104. A substantial portion of Slc12a2 mRNA in the adult rodent CNS and PNS is found in glial cells105,106. NKCC1 has been suggested to undergo a global downregulation during development in the rat brain and human brain79,107,108. However, other studies have reported a developmental upregulation of mRNA encoding NKCC1 in the CNS of rodents and humans37,50,67,69,109–112 (FIG. 1a). The gene encoding NKCC1 produces two splice variants, NKCC1a and NKCC1b104,112,113. Expression of NKCC1b, which lacks exon 21, is higher than that of NKCC1a in the adult human brain112,113. Therefore, the apparent downregulation of NKCC1 (REFS 79,107,108) may be explained by the use of probes and antibodies targeting exon 21 and thus detecting NKCC1a but not NKCC1b (see REFS 41,50), of which the latter undergoes more robust developmental upregulation112.
KCC3
KCC3 mRNA and protein are widely expressed throughout development in the rodent CNS114,115. In the mouse CNS, KCC3 is mainly, but not exclusively, expressed in neurons115 and appears to undergo developmental upregulation in the rodent CNS in parallel with KCC2 (REFS 114,115). In the human cortex, mRNA encoding KCC3 is highly expressed throughout life (FIG. 1a). Nevertheless, the physiological roles of KCC3 in neurons remain largely unknown5,6,51,116,117 (TABLE 1). The idea that KCC3 has important roles in the healthy nervous system is strongly supported by the fact that peripheral neuropathy associated with agenesis of the corpus callosum (ACCPN; also known as Andermann syndrome) is caused by genetic impairment of KCC3 (REFS 46,116,118). Interestingly, neuron-specific knockout of Slc12a6 in mice has been demonstrated to recapitulate most of the neuropathic features of ACCPN119. Of the two main splice variants, KCC3a and KCC3b, KCC3a is preferentially expressed in the CNS and PNS56,114,115.
KCC1 and KCC4
In the rodent CNS, mRNA encoding KCC1 is expressed in neurons and non-neuronal cells at relatively low levels2,61. In the embryonic brain, it is exclusively detected in the choroid plexus54. mRNA encoding KCC4 is abundant in the embryonic ventricular zone54 but is present at low levels in the adult rodent CNS, with the exception of the suprachiasmatic nucleus in the rat56. In the adult mouse brain, KCC4 protein is mainly detected in cranial nerves, brainstem and spinal cord120. There is no obvious CNS phenotype in mice lacking KCC1 (REF. 121) or KCC4 (REF. 122). In the human cortex, the expression of both KCC1 and KCC4 is very low (FIG. 1a).
Post-translational regulation of CCCs
The mere presence of CCC protein, even in abundant quantities in the plasma membrane, does not imply that the transporter is active11,68,69,123. Like a closed ion channel, an ion transporter may be completely inactive despite the presence of a strong driving force. Once activated, a transporter’s capacity is determined by the number of operational transporters in the plasma membrane as well as the unitary transport rate (also known as ion-turnover or ion-translocation rate). Membrane trafficking, (de)phosphorylation of key residues and proteolytic cleavage all kinetically regulate the transport capacity of CCCs (FIGS 1d,2b).
The kinetic regulation of NKCC1 has been extensively studied in secretory epithelia, where it has been shown to be under phosphorylation-dependent control by secretagogues7. NKCC1 is sensitive to changes in a cell’s [Cl−]i (REFS 7,124). A fall in [Cl−]i below the physiological ‘set point’ leads to direct phosphorylation of NKCC1 at specific N-terminal residues, its functional activation and thereby restoration of [Cl−]i (REFS 124,125). Conversely, an increase in [Cl−]i promotes Cl− extrusion by KCC2 (REF. 126), indicating that NKCC1 and KCC2 are reciprocally regulated by [Cl−]i. An interesting hypothesis is that WNKs (with no lysine kinases), acting through STE20-type kinases (SPAK and OSR1 (oxidative stress-responsive kinase 1)), may function as part of the Cl−-sensing127 regulatory pathway that alters the phosphorylation states, and consequently the activities, of KCC2 and NKCC1 in a physiologically concerted manner128–130. Upon their activation by upstream kinases, such as WNKs, SPAK and OSR1 bind to and phosphorylate the cytosolic N-terminal tail of NKCC1 and stimulate Cl− uptake, whereas protein phosphatase 1 (PP1)-mediated dephosphorylation of NKCC1 has the opposite effect131–138 (FIG. 1d). C-terminal phosphorylation of KCC2 by SPAK and OSR1 is likely to decrease KCC2-mediated K+–Cl− cotransport129,139,140. The exact signalling pathways remain to be identified in neurons44,141.
In immature cortical neurons, a substantial part of the total cellular KCC2 protein pool resides in cytosolic vesicles or in an inactive state in the plasmalemma11,142,143. Dephosphorylation of threonine residues 906/1007 in the C terminus139 is likely to be involved in the activation of KCC2 and in the consequent development of hyperpolarizing GABAAR signalling 141 (see also REF. 144). In immature hippocampal neurons, neonatal seizures trigger a fast and pronounced enhancement of KCC2-mediated Cl− extrusion, leading to ‘precocious’ hyperpolarizing GABAAR responses 11. An analogous situation has been described in spinal cord injury145. This effect is caused at least in part by an activity-induced increase in the insertion of KCC2 into the plasma membrane and is likely to act as an endogenous safety mechanism in immature neurons to counteract the massive increases in intracellular Cl− loads induced by seizures and trauma. Changes in KCC2 membrane trafficking involve TRKB, protein kinase C (PKC) and 5-HT2A serotonin receptors11,145,146, and phosphorylation of KCC2 by PKC at serine 940 decreases the rate of internalization of KCC2 from the plasma membrane147.
By contrast, in mature neurons, prolonged intense neuronal firing results in sustained increases in [Ca2+]i and leads to downregulation of KCC2 (REF. 148). The underlying mechanisms are likely to include PP1-dependent dephosphorylation of KCC2 at serine 940 and calpain-mediated cleavage149–151 (see also REF. 148) (BOX 3). Plasmalemmal KCC2 undergoes rapid endocytosis and proteolysis in response to seizures, resulting in loss of hyperpolarizing responses to GABA150,152. The qualitatively opposite changes in KCC2 ion-transport functions in response to seizures in mature versus neonatal neurons are probably attributable to the age-dependence of TRKB activation (BOX 3) and of CNS energy consumption75. The basal turnover of total cellular KCC2 protein in mature neurons is slow, lasting several hours, as seen in hippocampal slices and cultures2,150. The rate of turnover of plasmalemmal KCC2 in healthy neurons has not been measured so far (see REF. 150) but, of note, following overexpression of KCC2 in HEK-293 cells, the entire functional cell-surface pool is recycled every 10 minutes147.
Given that optimizing energy metabolism is a major determinant of the evolution and overall ‘design’ of the CNS14,153, it is interesting that the brain-type creatine kinase (CKB) interacts with and activates both KCC2 and KCC3 (REFS 154,155). There is also evidence for co-regulation of and a direct interaction between KCC2 and the Na+/K+ ATPase11,40,156, suggesting that they may form an ion-transport metabolon15.
CCCs regulate inhibitory signalling
A CNS neuron typically receives input from thousands of excitatory and inhibitory synapses. An inhibitory synaptic input decreases the spiking probability of the target neuron, whereas an excitatory synapse has the opposite effect. The increases in the anion conductance that are gated by GABAARs are spatially restricted according to the cell type-specific innervation patterns of distinct interneuron types157. Moreover, the GABAergic conductances — as well as the corresponding inhibitory postsynaptic currents (IPSCs) — are temporally restricted by the receptor channels’ kinetics. There are two ways in which GABAAR activity can inhibit the target neuron. In a short-circuiting process known as shunting inhibition, which can take place at any level of the membrane potential (Vm), the increase in conductance that results from GABAAR activation suppresses the spatial and temporal summation of depolarizing membrane responses to excitatory postsynaptic currents (EPSCs) and functionally excitatory intrinsic currents158,159. Voltage inhibition is caused by hyperpolarizing IPSPs and is enabled by KCC2. The hyperpolarizing IPSP outlasts the original conductance change and the associated shunting inhibition. As it spreads along the neuronal membrane, the IPSP is attenuated in a manner dictated by the membrane space and time constants160,161.
If GABAAR channels were exclusively selective for Cl−, all neurons with effective Cl− extrusion would produce hyperpolarizing IPSPs because the equilibrium potential of Cl− (ECl) would be more negative than the resting membrane potential (Vrest). However, GABAARs and GlyRs mediate currents carried not only by Cl− but also by HCO3−, with a HCO3−/Cl− permeability ratio of 0.2–0.4 (REFS 161,162). The equilibrium potential of HCO3 − (EHCO3) is maintained at a much more positive level than ECl, at around −10 to −20 mV by pH-regulatory mechanisms (Supplementary information S1 (figure)). Therefore, the reversal potentials (EGABA and EGly) of GABAAR currents and GlyR currents are more positive than ECl, and this deviation becomes progressively larger with a decrease in [Cl−]i (REFS 1,161). In neurons with high [Cl−]i, the HCO3− component has little influence on EGABA, and the driving force of the GABAAR-mediated current (DFGABA) is depolarizing. In a neuron with functionally active KCC2 and a consequent low [Cl−]i, ECl is more negative than Vrest and DFGABA can either be hyperpolarizing or depolarizing, depending on the levels of the internal and external Cl− and HCO3− concentrations163,164 (Supplementary information S1 (figure), Supplementary information S2 (text, equation 9)). Certain types of adult neurons, such as neocortical pyramidal neurons and dentate granule cells, equipped with highly active KCC2-mediated Cl− extrusion, exhibit moderately depolarizing HCO3−-dependent IPSPs, even under resting (quiescent) conditions163,164.
Slightly depolarizing postsynaptic GABAAR responses can sometimes exert a stronger inhibitory action than hyperpolarizing ones as a result of the inactivation of voltage-gated conductances. Strongly depolarizing GABAergic synaptic responses can be functionally excitatory, and they are termed ‘GABA PSPs’. Such responses are based on a high [Cl−]i, and they are mainly seen in immature or diseased neurons40,48,165–168.
Neurons also harbour various extrasynaptic GABAARs and GlyRs, which have an exceptionally high agonist affinity. They are found in all subcellular compartments of neurons, and current data suggest that the axon proper (the entire axon with the exception of the axon initial segment (AIS) and presynaptic boutons) contains only such high-affinity GABAARs 169. Extrasynaptic GABAARs and GlyRs mediate tonic inhibition170,171, which is often considered to be of a purely shunting type. However, NKCC1-dependent depolarizing and excitatory tonic GABAAR currents have been identified in somatic recordings in immature neurons172,173 and in axons of adult neurons169.
Development and subcellular targets of inhibition
Endogenous network activity promoted by NKCC1-dependent depolarizing GABAAR signalling is thought to be crucial for the development of neuronal connections before the maturation of sensory inputs6,9,174,175. Depolarizing GABAAR signalling, typically acting in concert with glutamatergic mechanisms and intrinsic excitatory currents172, generates intracellular Ca2+ transients that activate downstream cascades with trophic functions9,174. The developmental upregulation of KCC2 reduces the depolarizing action of GABA, ending its trophic effect; however, this may be resumed during post-traumatic dedifferentiation of neurons15,176. The mechanisms and consequences of depolarizing GABA actions during neuronal maturation have been extensively reviewed6,9,174,175.
The causal role of KCC2 in the ontogenesis of GABAAR-mediated hyperpolarization was first demonstrated by Slc12a5 knockdown in cortical pyramidal neurons2. This finding was corroborated in subsequent studies4,5,29,73,151,177–179 (TABLE 1). Furthermore, early over-expression of KCC2 is sufficient to establish precocious hyperpolarizing GABAAR-mediated signalling 148,180–182 (TABLE 1). Work on adult cerebellar Purkinje neurons demonstrated no change in Cl− extrusion capacity in mature neurons with a targeted loss of KCC3, whereas cell-specific knockout of the gene encoding KCC2 dramatically reduced the ability of Purkinje neurons to extrude Cl− in the absence and presence of experimental Cl− loading5.
Recent in vitro studies demonstrated the existence of spatial differences in the values of EGABA and DFGABA (REFS 183,184) that are attributable to subcellular heterogeneity of CCC localization185,186. In neocortical and hippocampal principal neurons, EGABA and DFGABA shift towards more hyperpolarizing values from the AIS to the soma as a result of high NKCC1 levels and negligible levels of KCC2 in the AIS183,185–187 (but see REF. 188). Although the AIS-targeting axo-axonal interneurons have a depolarizing effect, this does not immediately imply that they are functionally excitatory. A moderately depolarized EGABA value in the AIS will render inhibition purely shunting near the action potential threshold. Accomplishing inhibition with minimal voltage responses may explain the functional significance of NKCC1 expressed in the AIS. Notably, GABAARs at the AIS have been shown to act as gatekeepers that control pyramidal cell firing189.
The above data indicate that NKCC1 and KCC2 are targeted to different compartments during neuronal maturation. Exon 21, which is not present in NKCC1b, contains a dileucine motif that is implicated in the basolateral versus apical targeting of NKCCs in polarized epithelial cells190. In neuronal differentiation, this would tentatively suggest (see REF. 191) that NKCC1a and NKCC1b are targeted to the dendrites and axons, respectively. Moreover, silencing the expression of the cytoskeletal scaffold protein ankyrin G dismantles the AIS and causes axons to acquire properties of dendrites, including the expression of KCC2 and, strikingly, even the formation of ‘axonal spines’ (REF. 187).
Why hyperpolarizing inhibition?
Merely shunting the postsynaptic membrane in the absence of any voltage change will decrease the probability of spiking192. Therefore, an obvious question that emerges is: what is achieved by equipping neurons with a Cl− extruder such as KCC2 that enables hyperpolarizing IPSPs? Most of the data on hyperpolarizing IPSPs have been obtained in experiments on quiescent in vitro preparations and, therefore, two important factors must be considered. First, the continuous network activities of neuronal circuits lead to activity-dependent Cl− loading; and, second, there is no well-defined Vrest under in vivo conditions. Both of these factors will have a powerful impact on DFGABA, as explained below.
By definition, a shunt reduces input resistance and excitation of the plasma membrane by providing a route for outwardly directed current. Thus, DFGABA always drives inhibitory outward currents when EGABA is more negative than the momentary Vm. In a neuron engaged in network activity, inward currents generated by excitatory synapses and voltage-gated channels induce profound membrane depolarizations that are transient in space and time. For GABAAR signalling to be functionally inhibitory at a given moment, the local level of EGABA must therefore be maintained by active extrusion of Cl− at a level that is sufficiently negative to promote outwardly directed GABAAR currents. If the membrane conductance were to be increased by opening GABAARs in the absence of Cl− extrusion, the depolarizing drive of excitatory inputs would lead to an unremitting GABAAR-mediated uptake of Cl− (REF. 14), rendering EGABA progressively more depolarizing and, finally, resulting in an excitatory action of GABA. Thus, extrusion of Cl− by KCC2 is necessary for the maintenance of efficient GABAAR-mediated inhibition during network activity. This also means that the amplitudes of hyperpolarizing IPSPs or hyperpolarizing EGABA values observed in quiescent neurons do not provide valid estimates of their counterparts and of DFGABA under the dynamic conditions in vivo.
The above discussion leads to further important corollaries. The non-uniform plasmalemmal distribution of ion channels and transporters6 implies that the temporal dynamics of DFGABA must be distinct in different subcellular compartments of a neuron, as dictated by the local ionic loads and leaks; by the diameter (or surface-to-volume ratio) of a given compartment; and by the presence or absence of cytosolic carbonic anhydrase activity, which affects the rate of the channel-mediated Cl−–HCO3− shuttle and thereby the local-level EGABA and DFGABA (REFS 6,176,193) (Supplementary information S2 (text)). Various types of interneurons show astonishingly variable, anatomically distinct patterns of innervation of their targets157. Therefore, it is likely that the subcellular localization and efficacy of plasmalemmal KCC2 and other anion-regulatory proteins add to the multitude of mechanisms that are known to underlie the integration of excitatory and inhibitory synaptic signals. Because of technical reasons, intracellular recordings in vivo are typically carried out in cell bodies, and therefore they do not provide correct estimates of EGABA and DFGABA in dendrites during network activities194. Thus, at the moment, tackling the above ideas is largely restricted to using computational models195. Experimental approaches would require reliable dyes for in vivo imaging of neuronal Cl−, combined with simultaneous measurements of the Vm. As pointed out by several researchers, including pioneers on its development, Clomeleon196 — a widely used first-generation optogenetic sensor197 — has a Cl− affinity that is far beyond the physiologically relevant concentration range196. Fortunately, there are promising methodological developments to solve such problems196,198.
CCCs control ionic plasticity
Neurons in the living CNS are never at rest, and their plasmalemmal Cl− and HCO3− gradients are determined moment by moment by the pump–leak relationship between CCCs and GABAARs, as well as other channels and transporters (Supplementary information S2 (text))1,15. These mechanisms underlie the phenomenon of ionic plasticity6,15,176,199,200, a hallmark of GABAAR-mediated transmission that is based on the dynamic nature of DFGABA (REFS 100,201,202).
Repetitive stimulation of GABAergic inputs in hippocampal pyramidal neurons reduces the amplitude of hyperpolarizing IPSPs or IPSCs, and this is often followed by a change in their polarity100,202,203. Dendrites, with their high surface-to-volume ratio, are highly prone to such fast activity-dependent ECl shifts. In hippocampal CA1 neurons that show robust hyperpolarizing IPSPs in vitro, functionally excitatory GABAergic responses can be induced by high-frequency stimulation100. Such stimulation causes intense interneuronal firing, a consequent HCO3−-dependent increase in [Cl−]i and a large depolarizing shift in DFGABA in the target pyramidal neurons, which is sufficient for the activation of NMDA receptors (NMDARs)1,202. Because CO2 readily permeates neuronal membranes, intraneuronal HCO3 − that is lost owing to net efflux through GABAARs is replenished by the activity of cytosolic carbonic anhydrases193,204 (Supplementary information S2 (text)). Under these conditions, the increase in [Cl−]i leads to enhanced K+–Cl− extrusion by KCC2, and to a consequent increase in [K+]o, which further depolarizes both neurons and glia in a non-synaptic manner101 (FIG. 2a).
The mechanisms described above are likely to promote tetanus-induced long-term potentiation203 as well as highly synchronized spontaneous network events, including seizures (see REFS 15,204), and may contribute to neuropathic pain205. Notably, carbonic anhydrase inhibitors, which are well known for their anticonvulsant actions, strongly inhibit activity-dependent ECl shifts and GABAergic excitation in slice preparations100,101 and reduce neuropathic pain206,207.
A large increase in membrane conductance leads to a fall in the membrane time constant, which can change a neuron’s integrative properties from an integrate-and-fire mode to coincidence detection208. An intriguing, novel scenario that emerges here is that the high shunting conductance and the consequent rapid positive shift in ECl that are caused by pronounced activity of GABAergic inputs may counteract each other: a high inhibitory conductance can silence the cell, but this effect is opposed by the functionally pro-excitatory positive shift in ECl. Thus, fast ionic plasticity (FIG. 2a) may turn out to be a key parameter in the dynamic control of coincidence detection.
In addition to fast ionic shifts, EGABA and EGly are subject to transcriptional and post-translational modifications of CCCs (see above)6, thereby extending the temporal domains of ionic plasticity to a lifelong timespan that is relevant to neuronal development, ageing and trauma15 (FIG. 2).
CCCs in dendritic spine formation
Most excitatory synapses are formed on dendritic spines, and most inhibitory inputs are made onto dendritic shafts157,209. The steep developmental increase in KCC2 expression in the rodent cortex3,71,143,210 is associated with intense synaptogenesis30,143,211,212, and a considerable proportion of the KCC2 in cortical neurons is located in, or in the vicinity of, dendritic spines29,142,143,186. In humans, the onset of massive cortical synaptogenesis213,214 and robust upregulation of KCC2 commences at the third trimester of pregnancy76–78. Recent work has demonstrated a role for KCC2 in the formation of dendritic spines that is not dependent on its ability to transport ions28,30,31 (FIG. 1c). Neurons in cortical cultures from Slc12a5−/ − mice exhibited elongated filopodium-like dendritic protrusions and a reduced number of functional excitatory synapses28. Strikingly, transfection of Slc12a5−/ − neurons with either wild-type or an N-terminally deleted transport-deficient KCC2 construct (KCC2-ΔNTD) rescued spines28,31. Furthermore, in vivo overexpression of KCC2, KCC2-ΔNTD or the cytosolic C-terminal domain of KCC2 during the late embryonic period induced a lifelong increase in the number of spines in cortical pyramidal neurons30,31. Although both the intracellular N- and C-terminal domains of KCC2 are necessary for its function as a K+–Cl− cotransporter28,31,150,215, the above data indicate that the structural role of KCC2 in spine formation is mediated by its C-terminal domain. It has been suggested that the actin-associated protein 4.1N (also known as band 4.1-like protein 1) directly interacts with the C terminus of KCC2 (REFS 28,65). Preventing expression of 4.1N or of actin depolymerization in mature neurons increased the lateral diffusion of KCC2 away from excitatory synapses in cultured hippocampal neurons151. Changes in the localization of KCC2 between spines and the dendritic shaft may regulate the efficacy of excitatory synapses by physically constraining AMPA receptors in spine heads29,151 (FIG. 1c).
The molecular cascades that underlie the developmental upregulation of KCC2 in spines remain poorly understood6,32,44,216. Embryonic overexpression of BDNF results in precocious upregulation of KCC2 and in an increased number of GABAergic and non-GABAergic synapses85. The cell adhesion molecule neuroligin 2 (NLGN2)217,218 appears to maintain dendritic spines by promoting KCC2 expression71. Alterations in the regulation of NLGN2 and/ or KCC2 may turn out to be relevant in developmental neuropsychiatric disorders, such as autism and schizophrenia37–39,219. Whether the recently discovered interactions between KCC2 and the kainate-type glutamate receptor (KAR) auxiliary subunit NETO2 (REF. 220), as well as between KCC2 and KARs themselves221, are important for the morphogenetic roles of KCC2 is not known.
Some interneurons have been shown to target spines222. In particular, somatostatin-expressing interneurons exert a GABAAR-mediated inhibitory effect on glutamatergic Ca2+ transients in individual spines223. Although much of the KCC2 in this location may have a structural role, there are no data available on the presence of KCC2-mediated Cl− extrusion in spines. Notably, activity-dependent increases in [Na+]i within spines can reach levels of up to 100 mM224, which must be associated with comparable decreases in intraspine [K+]. This would immediately reverse the direction of net Cl− transport by KCC2 from extrusion to uptake, thereby producing a positive-feedback loop between the amount of excitation and net accumulation of both Cl− and Na+, as well as net influx of water. Thus, it is possible that the spinogenesis-promoting KCC2 protein fraction is not transport-active. This is an important novel hypothesis that has to be tested in future research.
CCCs in seizures and epilepsy
Experimentally induced defects in GABAergic transmission can provoke seizures in vivo and seizure-like activity in vitro. However, as discussed recently15, the standard concept of excitation/inhibition balance (E/I balance) is unable to explain the aetiology and manifestation of epilepsies, which encompass a wide and heterogeneous spectrum of brain malfunctions and adaptive mechanisms225. Although the E/I balance concept has turned out to be useful in, for instance, understanding neuronal firing characteristics during distinct cortical states226, it is obvious that suggesting an E/I imbalance as a cause of epileptogenesis and epilepsy is based on a circular argument (see REFS 15,227): the presence of seizures is taken as an indicator of an altered E/I ratio228.
Because of the key functions of KCC2 and NKCC1 in controlling the efficacy of inhibition, many studies have addressed the roles of these ion transporters in the acute generation of seizures and in epileptogenesis (reviewed in REFS 15,36,40,41,228). The above E/I misconception has extended to the use of the ‘NKCC1-to-KCC2 ratio’ (as measured in quantifications of total mRNA and protein levels in samples of brain tissue) as a parameter of proneness to epilepsy and other neurological disorders. However, among other caveats (see REF. 15), the total amounts of CCC mRNA or protein do not directly translate into functional efficacy.
The multiple and context-dependent roles of CCC functions in epilepsy are evident in light of the enhanced susceptibility to seizures that follows genetic impairment of KCC2 expression in mouse models229,230 and in human patients31,231, resulting in the loss of both its ion-transport-dependent and ion-transport-independent functions28,31. Observations on adult rat dentate granule cells show that, under normal conditions, spiking of these cells is strongly suppressed by KCC2-dependent GABAergic mechanisms20,100,101. Progressive downregulation of KCC2 after pilocarpine-induced status epilepticus decreased the efficacy of inhibition and abolished the function of the dentate gyrus as a hippocampal barrier against seizure activity arising in the entorhinal cortex20. In sharp contrast to the above two examples, a seizure-promoting action by KCC2 is seen in mature neurons under conditions that lead to GABA-mediated excitation paralleled by an increase in [K+]o, that are sufficient to trigger ictal events100,101,204. Notably, almost all interneurons that target perisomatic regions of the hippocampal CA1 area are activated during epileptiform discharges232.
In hippocampal brain slices from adult patients with temporal lobe epilepsy, downregulation of KCC2 and upregulation of NKCC1 leads to depolarizing GABAAR responses in a subpopulation of subicular principal neurons. This has been implicated in the generation of spontaneous interictal-like (but not ictal-like) activity233–235 (see also REF. 167). Inhibition of NKCC1 by bumetanide blocks interictal activity in vitro (for example, see REF. 234), but this does not imply that the drug would block ictal events. Indeed, the evidence for an anticonvulsant effect of bumetanide in neonates and adults is meagre40,41. Novel compounds, perhaps including CNS-permeant prodrugs of bumetanide236 as well as activators of KCC2 (REF. 237), are needed for further research on CCCs as pharmacotherapeutic targets.
After the cloning of KCC2 (REF. 53), no variations in SLC12A5 in human disease were reported for nearly two decades. Recently, a rare SLC12A5 point variant (KCC2-R952H) was identified in an Australian family with early childhood onset of febrile seizures31. This missense variant led to a defect in neuronal Cl− extrusion capacity and cortical dendritic spine formation in rodent neurons31 (FIG. 1c,d). These data suggest that this is a bona fide susceptibility variant for febrile seizures, which might lead to other kinds of seizure disorders later in life. Support for this conclusion was gained from subsequent identification of KCC2-R952H and another point variant, KCC2-R1049H, in a French-Canadian cohort with idiopathic generalized epilepsy231. Although it is possible that an impairment of GABAAR-mediated inhibition underlies the enhanced seizure susceptibility associated with KCC2-R952H, the decrease in the number of functional dendritic spines may lead to desynchronization at the level of neuronal networks, thereby promoting seizures15,238.
CCCs and chronic pain
Epilepsy and neuropathic pain bear striking resemblances. Anticonvulsant agents are among the only effective treatments for neuropathic pain239, and increasing evidence points to BDNF–TRKB- and calpain-mediated regulation of CCC function as common causes of epilepsy and neuropathic pain23,240,241 (BOX 3).
The spinal dorsal horn is richly endowed with GABAergic and glycinergic interneurons, which provide inputs to dorsal horn neurons. The central terminals of primary afferents contain GABAARs but are devoid of GlyRs242. The gate control theory of pain243, which describes modulation of nociceptive processing at the first synapses of the pain pathway, was based, in part, on evidence that presynaptic inhibition of afferent fibres is mediated by interneurons in lamina I/II of the dorsal horn. This GABAAR-mediated action results in depolarization of the presynaptic terminals of primary afferent fibres (primary afferent depolarization (PAD)) and the suppression of nociceptive signalling244. A small depolarization of the Vm, which is typical of PAD, produces presynaptic inhibition by inactivating voltage-gated Na+ channels and consequently reducing transmitter release at the synapse245.
Dorsal root ganglion (DRG) and trigeminal ganglion neurons express NKCC1, but not KCC2, providing an explanation for the high [Cl−]i required for PAD18,106. Thus, CCCs are at the heart of the gate control theory of pain. NKCC1 expression in DRG neurons is also crucial for nociceptive functions that do not involve PAD. Notably, the Ca2+-activated Cl− channel anoctamin 1 depolarizes DRG neurons in response to nociceptive stimuli that evoke Ca2+ release from intracellular stores246. Plasticity of GABAAR responses in the presynaptic terminals of primary afferents may contribute to pathological pain in at least two ways: through a loss of GABAAR-mediated inhibitory PAD caused by decreased expression of GABAARs and through increased [Cl−]i as a result of altered regulation of NKCC1, leading PAD to be converted to frank excitation of afferent fibres (FIG. 3a). In the case of peripheral nerve injury (PNI), a loss of GABAAR-mediated responses has been observed in DRG neurons247 and is paralleled by a reduction in PAD. There is also evidence for increased NKCC1 expression in response to peripheral inflammation245, and inhibition of NKCC1 with intrathecally delivered bumetanide has been shown to reduce injury-induced nociceptive hypersensitivity248–250. However, it has not been technically possible to measure [Cl−]i in afferent terminals, and it is not known whether injury can lead to functional upregulation of NKCC1 at this critical subcellular site for nociception.
Figure 3. Cation-chloride cotransporters in pain.

There are two major theories concerning the role of cation-chloride cotransporters (CCCs) in chronic pain, and more specifically in touch-evoked pain (allodynia). Under normal conditions (part a), the activation of Aβ fibres, the myelinated fibres responsible for light touch sensation, leads to primary afferent depolarization in C fibres and consequent presynaptic shunting inhibition of pain-conducting C fibres by dorsal horn GABAergic interneurons. This requires the expression of Na+–K+–2Cl− cotransporter 1 (NKCC1) in dorsal root ganglion (DRG) C fibre neurons. The inhibition blocks the signalling of the C fibres to lamina I projection neurons. Following injury, signalling cascades lead to phosphorylation and thereby kinetic activation of NKCC1 (FIG. 1d) in C fibre terminals. This may be linked to an increase in [Cl−]i, potentially converting Aβ fibre-mediated inhibition into frank excitation of C fibres and leading to activation of pain signalling via lamina I projection neurons. This provides a neurophysiological explanation for allodynia. Very recent evidence suggests that nociceptive-specific (NS) neurons in the deep laminae of the dorsal horn (lamina V) lose KCC2 expression following peripheral nerve injury (PNI), thereby altering the response of these neurons to GABA and unmasking an Aβ fibre-mediated input to NS neurons, effectively converting them to wide dynamic range (WDR) neurons (part b). As in the scenario outlined in part a, this gives the Aβ fibre pathway access to pain signalling through feedforward activation of projection neurons in lamina I/II. Importantly, conversion of NS neurons to WDR neurons following PNI is reversed by positive modulation of KCC2 and this treatment also reduces touch-evoked pain in behavioural assays. GABAAR, GABAA receptor.
GABAAR- and GlyR-mediated postsynaptic inhibition, which depends on KCC2 in projection neurons, is another feature of the dorsal horn pain gate. Following experimental PNI, a positive shift in EGABA and EGly, which is attributable to loss of KCC2 activity in a subpopulation of lamina I and II neurons, generates depolarizing instead of hyperpolarizing GlyR- and GABAAR-mediated responses18. Likewise, Slc12a5 knockdown results in touch-evoked pain or allodynia18. An important question is whether allodynia is explained by touch-activated afferents gaining access to a nociception-specific pathway or by an amplification of postsynaptic responses in dorsal horn neurons that normally receive noxious and innocuous inputs. Recent evidence strongly suggests that nociception-specific neurons acquire a novel input in the innocuous range after PNI251. This demonstrates how innocuous sensory signals gain access to a pain- dedicated input pathway to the CNS (FIG. 3b). Interestingly, these effects are rapidly reversed by positive modulation of KCC2 (REFS 237,251). Other studies have pointed to changes in KCC2 expression in the spinal dorsal horn as a prominent feature of inflammation245, metabolic nerve injury252 and even opioid-induced hyperalgesia253. Similar changes in KCC2 expression have been demonstrated in spinal cord injury-induced spasticity and pain, suggesting a KCC2-based approach to therapeutics for these clinical conditions21,145,254.
In PNI and opioid-induced hyperalgesia, there is strong evidence that microglial cell-derived BDNF has an essential role in modulating EGABA and EGly through decreased KCC2 expression240,255. As in the post-traumatic cortex, the PNI-induced positive shift in EGly, and presumably EGABA, parallels an influx of Ca2+ through NMDARs, resulting in calpain-mediated cleavage of KCC2 (REF. 23). Whether this involves activation of TRKB receptors by BDNF is unclear. However, BDNF signalling results in a loss of effective GABAergic inhibition in the spinal dorsal horn, resulting in nociceptive hypersensitivity256. Importantly, whereas blockade of spinal GABAARs in naïve animals produces a neuropathic pain-like behavioural phenotype, GABAAR blockade following spinal application of exogenous BDNF reverses this BDNF-induced nociceptive hypersensitivity257. A similar mechanism may be at play in the brainstem, where chronic pain promotes a depolarization of EGABA, through BDNF-mediated downregulation of KCC2, in descending pain facilitation neurons258.
Summary and future directions
Beyond the view that functional CCCs are probably dimers of 12 transmembrane-domain monomers, there is little structural information about these transporters42. Although the high-resolution structure has been obtained for the C terminus of a bacterial CCC protein43, the three-dimensional structure of the large and critically important C terminus of KCC2 has not been determined. There is also a striking lack of information about the intrinsic ion-transport rates of CCCs and their modulation by intracellular signalling cascades. Such data are crucial not only for understanding the fundamental molecular mechanisms and properties of CCC functions but also for the rational design of drugs targeting these transporters.
Accumulating evidence shows that CCCs are key molecules in shaping neuronal signalling and structure throughout an individual’s lifespan. Research themes of particular future interest include CCC functions in short- and long-term plasticity. Explorations of the functions of excitatory synapses have yielded useful parameters such as synaptic weights that can be used in modelling of network events158. Much less analogous data are available for inhibitory signalling, and ionic plasticity based on CCCs adds a novel and exciting aspect to future research in this area. How does EGABA change at subcellular sites in an individual neuron during network events? Do such changes have resonance frequencies of their own, thereby enhancing or suppressing various frequency bands during oscillatory activity? And would CCC-related abnormalities therein provide another link to neurological and psychiatric diseases, ranging from epilepsy to autism and schizophrenia? In addition, given the role of the hypothalamic–pituitary–adrenal axis in seizure disorders259, research on the differential expression patterns of CCCs in neuroendocrine cells will also help to further clarify the molecular aetiologies of these and other types of CNS diseases.
The above is but a brief list of possible directions for future studies on CCCs, intended to provoke creative, high-risk and high-impact approaches in this rapidly expanding field of research.
Supplementary Material
Acknowledgments
The authors thank B. Forbush for kindly providing the KCC2 and NKCC1 two-dimensional models that were adapted for figure 1d, and P. Blaesse, T. Z. Deeb, M. S. Gold, E. Ruusuvuori, P. Seja, R.-L. Uronen and L. Vutskits for comments and suggestions on an early version of this manuscript. The authors original research work is funded by the European Research Council Advanced Grant, Academy of Finland (AoF), AoF (ERA-Net NEURON II CIPRESS), the Sigrid Jusélius Foundation, the Jane and Aatos Erkko Foundation (K.K., M.P. and J.V.), US National Institutes of Health grants NS065926, GM102575 (T.J.P.) and NS36296 (J.A.P.).
Glossary
- Inhibitory postsynaptic potentials (IPSPs)
Synaptic potentials elicited by GABA or glycine that inhibit postsynaptic excitation and the generation of action potentials
- Reversal potential
The membrane potential at which a channel-mediated current reverses its polarity
- Driving force
The electrical potential difference that drives a conductive current. The driving force is calculated as the difference between the membrane potential and either the equilibrium (Nernst) potential of a single ion species or the reversal potential of a channel-mediated current
- Bulk ion concentrations
Ion concentrations in intracellular and extracellular compartments in which the vicinity of the membrane surface has no effect. All intracellular microelectrode measurements of membrane potentials yield data on the voltage between these bulk phases
- Shunting inhibition
Suppression of postsynaptic excitation that results from an increase in neuronal membrane conductance that is caused by activation of GABAA receptors or glycine receptors
- Excitatory postsynaptic currents (EPSCs)
Inward currents elicited by excitatory neurotransmitters (typically glutamate) that depolarize the neuron to enhance spiking probability
- Space and time constants
Reflect the passive electrical properties of neurons. The time constant (τm) is the product of membrane resistance and capacitance (τm = Rm·Cm) and defines the rate of change of a passive membrane potential (Vm) response evoked by a current pulse. The space constant (λ) quantifies the spatial extent of passively spreading signals in an elongated structure, such as a dendrite. Note that inducing an inhibitory conductance produces a decrease in both τm and λ
- Equilibrium potential
The membrane potential at which a single ion species is at equilibrium across the membrane; given by the Nernst equation
- Axon initial segment (AIS)
A structurally and functionally specialized region between the neuronal soma and axon proper that has a low voltage threshold for action potential generation and therefore often acts as the main site of spike initiation
- Tonic inhibition
Inhibition resulting from activation of extrasynaptic high-affinity GABAA receptors
- Integrate-and-fire
A situation in which excitatory inputs impinging on a neuron’s dendritic tree are summed up both spatially (from different locations) and temporally (during a high-frequency sequence of excitatory signals) to trigger spiking
- Coincidence detection
A situation in which spiking occurs in response to near-simultaneous, spatially distinct excitatory signals. If a large increase in the neuron’s conductance (a fall in time and space constants) takes place (for example, because of GABAergic inhibition), an integrate-and-fire mode of operation will change into coincidence detection
- Excitation/inhibition balance (E/I balance)
The relative quantitative contributions of excitatory and inhibitory synaptic signals at the level of a single neuron or a neuronal network
Footnotes
Note added in proof
Our recent comment281 on Glykys et al.280 explains why immobile anions do not account for DFGABA. In their response297 they now suggest that DFGABA is due to Donnan-equilibration of Cl− plus an energy-requiring Vm shift. If so, Cl− leaks would dissipate DFGABA, thereby necessitating never-ending Vm shifts. As described here, DFGABA and its spatial gradients are maintained by CCCs.
Competing interests statement
The authors declare no competing interests.
References
- 1.Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 2.Rivera C, et al. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. This was the first study to demonstrate the causal role of KCC2 upregulation in the development of hyperpolarizing inhibition. [DOI] [PubMed] [Google Scholar]
- 3.Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hübner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- 4.Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol. 2005;93:1557–1568. doi: 10.1152/jn.00616.2004. [DOI] [PubMed] [Google Scholar]
- 5.Seja P, et al. Raising cytosolic Cl− in cerebellar granule cells affects their excitability and vestibulo-ocular learning. EMBO J. 2012;31:1217–1230. doi: 10.1038/emboj.2011.488. This elegant study used cell-specific genetic targeting of KCC2 and KCC3 to clarify their contribution to Cl− regulation and spine morphology in the mature cerebellar cortex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 7.Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- 8.Achilles K, et al. Kinetic properties of Cl uptake mediated by Na+-dependent K+-2Cl cotransport in immature rat neocortical neurons. J Neurosci. 2007;27:8616–8627. doi: 10.1523/JNEUROSCI.5041-06.2007. The authors show how to rigorously quantify NKCC1-mediated Cl− uptake using an electro-physiological approach. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- 10.Sipilä ST, et al. Compensatory enhancement of intrinsic spiking upon NKCC1 disruption in neonatal hippocampus. J Neurosci. 2009;29:6982–6988. doi: 10.1523/JNEUROSCI.0443-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khirug S, et al. A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J Neurosci. 2010;30:12028–12035. doi: 10.1523/JNEUROSCI.3154-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Payne JA. In: Physiology and Pathology of Chloride Transporters and Channels in the Nervous System. Alvarez-Leefmans FJ, Delpire E, editors. Academic Press; 2009. pp. 333–356. [Google Scholar]
- 13.Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiol Rev. 1985;65:101–148. doi: 10.1152/physrev.1985.65.1.101. [DOI] [PubMed] [Google Scholar]
- 14.Buzsaki G, Kaila K, Raichle M. Inhibition and brain work. Neuron. 2007;56:771–783. doi: 10.1016/j.neuron.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaila K, Ruusuvuori E, Seja P, Voipio J, Puskarjov M. GABA actions and ionic plasticity in epilepsy. Curr Opin Neurobiol. 2014;26:34–41. doi: 10.1016/j.conb.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Nabekura J, et al. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivera C, et al. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coull JA, et al. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. The first paper to clearly delineate a role for loss of KCC2 function in the dorsal horn in the generation of neuropathic pain. [DOI] [PubMed] [Google Scholar]
- 19.Jin X, Huguenard JR, Prince DA. Impaired Cl− extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. This study demonstrates that measurements of EGABA obtained in the absence of experimental Cl− loading are not necessarily sufficient to prove compromised Cl− extrusion following neuronal damage. [DOI] [PubMed] [Google Scholar]
- 20.Pathak HR, et al. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. A seminal study on the erosion of inhibition and loss of the dentate ‘seizure barrier’ following seizure-induced downregulation of KCC2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boulenguez P, et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nature Med. 2010;16:302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- 22.Jaenisch N, Witte OW, Frahm C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke. 2010;41:e151–e159. doi: 10.1161/STROKEAHA.109.570424. [DOI] [PubMed] [Google Scholar]
- 23.Zhou HY, et al. N-methyl-D-aspartate receptor- and calpain-mediated proteolytic cleavage of K+-Cl− cotransporter-2 impairs spinal chloride homeostasis in neuropathic pain. J Biol Chem. 2012;287:33853–33864. doi: 10.1074/jbc.M112.395830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468:305–309. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karpova NN, et al. Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science. 2011;334:1731–1734. doi: 10.1126/science.1214592. This study opens up a novel field of research for studies on CCC functions in so-called induced plasticity brought about by antidepressant drugs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rinke I, Artmann J, Stein V. ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J Neurosci. 2010;30:4776–4786. doi: 10.1523/JNEUROSCI.6299-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ratte S, Prescott SA. ClC-2 channels regulate neuronal excitability, not intracellular chloride levels. J Neurosci. 2011;31:15838–15843. doi: 10.1523/JNEUROSCI.2748-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, et al. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. This study was the first to show an ion-transport-independent role of KCC2 in cortical dendritic spines. [DOI] [PubMed] [Google Scholar]
- 29.Gauvain G, et al. The neuronal K-Cl cotransporter KCC2 influences postsynaptic AMPA receptor content and lateral diffusion in dendritic spines. Proc Natl Acad Sci USA. 2011;108:15474–15479. doi: 10.1073/pnas.1107893108. This study demonstrated that KCC2 regulates the efficacy of glutamatergic synapses by constraining AMPA receptors in spines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiumelli H, et al. An ion transport-independent role for the cation-chloride cotransporter KCC2 in dendritic spinogenesis in vivo. Cereb Cortex. 2013;23:378–388. doi: 10.1093/cercor/bhs027. [DOI] [PubMed] [Google Scholar]
- 31.Puskarjov M, et al. A variant of KCC2 from patients with febrile seizures impairs neuronal Cl− extrusion and dendritic spine formation. EMBO Rep. 2014;15:723–729. doi: 10.1002/embr.201438749. This work was the first to identify and functionally characterize a human SLC12A5 disease mutation, which is a candidate susceptibility gene for febrile seizures. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blaesse P, Schmidt T. K-Cl cotransporter KCC2 — a moonlighting protein in excitatory and inhibitory synapse development and function. Pflugers Arch. 2014 doi: 10.1007/s00424-014-1547-6. http://dx.doi.org/10.1007/s00424-014-1547-6. [DOI] [PubMed]
- 33.Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005;85:423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 34.Price TJ, Cervero F, De Koninck Y. Role of cation-chloride-cotransporters (CCC) in pain and hyperalgesia. Curr Top Med Chem. 2005;5:547–555. doi: 10.2174/1568026054367629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Koninck Y. Altered chloride homeostasis in neurological disorders: a new target. Curr Opin Pharmacol. 2007;7:93–99. doi: 10.1016/j.coph.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 36.Kahle KT, et al. Roles of the cation-chloride cotransporters in neurological disease. Nature Clin Pract Neurol. 2008;4:490–503. doi: 10.1038/ncpneuro0883. [DOI] [PubMed] [Google Scholar]
- 37.Hyde TM, et al. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci. 2011;31:11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JY, et al. Interplay between DISC1 and GABA signaling regulates neurogenesis in mice and risk for schizophrenia. Cell. 2012;148:1051–1064. doi: 10.1016/j.cell.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tao R, et al. Transcript-specific associations of SLC12A5 (KCC2) in human prefrontal cortex with development, schizophrenia, and affective disorders. J Neurosci. 2012;32:5216–5222. doi: 10.1523/JNEUROSCI.4626-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Löscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. 2013;69:62–74. doi: 10.1016/j.neuropharm.2012.05.045. [DOI] [PubMed] [Google Scholar]
- 41.Puskarjov M, Kahle KT, Ruusuvuori E, Kaila K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia. 2014;55:806–818. doi: 10.1111/epi.12620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Payne JA. Molecular operation of the cation chloride cotransporters: ion binding and inhibitor interaction. Curr Top Membr. 2012;70:215–237. doi: 10.1016/B978-0-12-394316-3.00006-5. [DOI] [PubMed] [Google Scholar]
- 43.Warmuth S, Zimmermann I, Dutzler R. X-ray structure of the C-terminal domain of a prokaryotic cation-chloride cotransporter. Structure. 2009;17:538–546. doi: 10.1016/j.str.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 44.Medina I, et al. Current view on the functional regulation of the neuronal K-Cl cotransporter KCC2. Front Cell Neurosci. 2014;8:27. doi: 10.3389/fncel.2014.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 46.Gagnon KB, Delpire E. Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically-engineered mouse knockouts. Am J Physiol Cell Physiol. 2013;304:C693–C714. doi: 10.1152/ajpcell.00350.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inoue W, Bains JS. Beyond inhibition: GABA synapses tune the neuroendocrine stress axis. Bioessays. 2014;36:561–569. doi: 10.1002/bies.201300178. [DOI] [PubMed] [Google Scholar]
- 48.Kim YB, et al. GABAergic excitation of vasopressin neurons: possible mechanism underlying sodium-dependent hypertension. Circ Res. 2013;113:1296–1307. doi: 10.1161/CIRCRESAHA.113.301814. This pioneering work shows that GABAergic inhibition is converted into NKCC1-dependent excitation of vasopressin-releasing hypothalamic neurons in a rat model of hypertension. This also implies that CCCs are promising drug targets for the treatment of hypertension. [DOI] [PubMed] [Google Scholar]
- 49.Gamba G, et al. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J Biol Chem. 1994;269:17713–17722. [PubMed] [Google Scholar]
- 50.Clayton GH, Owens GC, Wolff JS, Smith RL. Ontogeny of cation-Cl− cotransporter expression in rat neocortex. Dev Brain Res. 1998;109:281–292. doi: 10.1016/s0165-3806(98)00078-9. [DOI] [PubMed] [Google Scholar]
- 51.Lucas O, Hilaire C, Delpire E, Scamps F. KCC3-dependent chloride extrusion in adult sensory neurons. Mol Cell Neurosci. 2012;50:211–220. doi: 10.1016/j.mcn.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 52.Mao S, et al. Molecular and functional expression of cation-chloride cotransporters in dorsal root ganglion neurons during postnatal maturation. J Neurophysiol. 2012;108:834–852. doi: 10.1152/jn.00970.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem. 1996;271:16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- 54.Li H, Tornberg J, Kaila K, Airaksinen MS, Rivera C. Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur J Neurosci. 2002;16:2358–2370. doi: 10.1046/j.1460-9568.2002.02419.x. [DOI] [PubMed] [Google Scholar]
- 55.Gagnon KB, Di FM. A molecular analysis of the Na+-independent cation chloride cotransporters. Cell Physiol Biochem. 2013;32:14–31. doi: 10.1159/000356621. [DOI] [PubMed] [Google Scholar]
- 56.Le Rouzic P, et al. KCC3 and KCC4 expression in rat adult forebrain. Brain Res. 2006;1110:39–45. doi: 10.1016/j.brainres.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 57.Gagnon KB, Adragna NC, Fyffe RE, Lauf PK. Characterization of glial cell K-Cl cotransport. Cell Physiol Biochem. 2007;20:121–130. doi: 10.1159/000104160. [DOI] [PubMed] [Google Scholar]
- 58.Uvarov P, et al. A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J Biol Chem. 2007;282:30570–30576. doi: 10.1074/jbc.M705095200. KCC2b was identified as the main KCC2 splice variant, which is responsible for hyperpolarizing GABA actions. [DOI] [PubMed] [Google Scholar]
- 59.Uvarov P, et al. Coexpression and heteromerization of two neuronal K-Cl cotransporter isoforms in neonatal brain. J Biol Chem. 2009;284:13696–13704. doi: 10.1074/jbc.M807366200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gulacsi A, et al. Cell type-specific differences in chloride-regulatory mechanisms and GABAA receptor-mediated inhibition in rat substantia nigra. J Neurosci. 2003;23:8237–8246. doi: 10.1523/JNEUROSCI.23-23-08237.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kanaka C, et al. The differential expression patterns of messenger RNAs encoding K-Cl cotransporters (KCC1,2) and Na-K-2Cl cotransporter (NKCC1) in the rat nervous system. Neuroscience. 2001;104:933–946. doi: 10.1016/s0306-4522(01)00149-x. [DOI] [PubMed] [Google Scholar]
- 62.Bartho P, Payne JA, Freund TF, Acsady L. Differential distribution of the KCl cotransporter KCC2 in thalamic relay and reticular nuclei. Eur J Neurosci. 2004;20:965–975. doi: 10.1111/j.1460-9568.2004.03562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun YG, et al. GABAergic synaptic transmission triggers action potentials in thalamic reticular nucleus neurons. J Neurosci. 2012;32:7782–7790. doi: 10.1523/JNEUROSCI.0839-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song LY, et al. Molecular, functional, and genomic characterization of human KCC2, the neuronal K-Cl cotransporter. Mol Brain Res. 2002;103:91–105. doi: 10.1016/s0169-328x(02)00190-0. [DOI] [PubMed] [Google Scholar]
- 65.Horn Z, Ringstedt T, Blaesse P, Kaila K, Herlenius E. Premature expression of KCC2 in embryonic mice perturbs neural development by an ion transport-independent mechanism. Eur J Neurosci. 2010;31:2142–2155. doi: 10.1111/j.1460-9568.2010.07258.x. [DOI] [PubMed] [Google Scholar]
- 66.Talos DM, et al. Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann Neurol. 2012;71:539–551. doi: 10.1002/ana.22696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang C, et al. Developmental changes in KCC1, KCC2, and NKCC1 mRNA expressions in the rat brain. Brain Res Dev Brain Res. 2002;139:59–66. doi: 10.1016/s0165-3806(02)00536-9. [DOI] [PubMed] [Google Scholar]
- 68.Blaesse P, et al. Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. J Neurosci. 2006;26:10407–10419. doi: 10.1523/JNEUROSCI.3257-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Balakrishnan V, et al. Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J Neurosci. 2003;23:4134–4145. doi: 10.1523/JNEUROSCI.23-10-04134.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Uvarov P, Ludwig A, Markkanen M, Rivera C, Airaksinen MS. Upregulation of the neuron-specific K+/Cl− cotransporter expression by transcription factor early growth response 4. J Neurosci. 2006;26:13463–13473. doi: 10.1523/JNEUROSCI.4731-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun C, Zhang L, Chen G. An unexpected role of neuroligin-2 in regulating KCC2 and GABA functional switch. Mol Brain. 2013;6:23. doi: 10.1186/1756-6606-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bayatti N, et al. A molecular neuroanatomical study of the developing human neocortex from 8 to 17 postconceptional weeks revealing the early differentiation of the subplate and subventricular zone. Cereb Cortex. 2008;18:1536–1548. doi: 10.1093/cercor/bhm184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hübner CA, et al. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. The first paper on a full Slc12a5-knockout mouse. The authors concluded that the perinatally lethal phenotype is attributable to lack of functional inhibition and consequent lack of central motor programmes, especially those sustaining breathing. [DOI] [PubMed] [Google Scholar]
- 74.Khalilov I, et al. Enhanced synaptic activity and epileptiform events in the embryonic kcc2 deficient hippocampus. Front Cell Neurosci. 2011;5:23. doi: 10.3389/fncel.2011.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Erecinska M, Cherian S, Silver IA. Energy metabolism in mammalian brain during development. Prog Neurobiol. 2004;73:397–445. doi: 10.1016/j.pneurobio.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 76.Vanhatalo S, et al. Slow endogenous activity transients and developmental expression of K+-Cl− cotransporter 2 in the immature human cortex. Eur J Neurosci. 2005;22:2799–2804. doi: 10.1111/j.1460-9568.2005.04459.x. [DOI] [PubMed] [Google Scholar]
- 77.Robinson S, Mikolaenko I, Thompson I, Cohen ML, Goyal M. Loss of cation-chloride cotransporter expression in preterm infants with white matter lesions: implications for the pathogenesis of epilepsy. J Neuropathol Exp Neurol. 2010;69:565–572. doi: 10.1097/NEN.0b013e3181dd25bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sedmak G, Jovanov-Milosevic N, Puskarjov M, Kaila K, Judas M. Expression patterns of K-Cl cotransporter KCC2 in the human fetal and adult brain. FENS Abstr. 2014;2398 [Google Scholar]
- 79.Dzhala VI, et al. NKCC1 transporter facilitates seizures in the developing brain. Nature Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- 80.Karadsheh MF, Delpire E. Neuronal restrictive silencing element is found in the KCC2 gene: molecular basis for KCC2-specific expression in neurons. J Neurophysiol. 2001;85:995–997. doi: 10.1152/jn.2001.85.2.995. [DOI] [PubMed] [Google Scholar]
- 81.Uvarov P, Pruunsild P, Timmusk T, Airaksinen MS. Neuronal K+/Cl− co-transporter (KCC2) transgenes lacking neurone restrictive silencer element recapitulate CNS neurone-specific expression and developmental up-regulation of endogenous KCC2 gene. J Neurochem. 2005;95:1144–1155. doi: 10.1111/j.1471-4159.2005.03434.x. [DOI] [PubMed] [Google Scholar]
- 82.Yeo M, Berglund K, Augustine G, Liedtke W. Novel repression of Kcc2 transcription by REST-RE-1 controls developmental switch in neuronal chloride. J Neurosci. 2009;29:14652–14662. doi: 10.1523/JNEUROSCI.2934-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ludwig A, et al. Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J Neurosci. 2011;31:644–649. doi: 10.1523/JNEUROSCI.2006-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goldberg EM, Coulter DA. Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nature Rev Neurosci. 2013;14:337–349. doi: 10.1038/nrn3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aguado F, et al. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/ Cl− co-transporter KCC2. Development. 2003;130:1267–1280. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- 86.Markkanen M, Uvarov P, Airaksinen MS. Role of upstream stimulating factors in the transcriptional regulation of the neuron-specific K-Cl cotransporter KCC2. Brain Res. 2008;1236:8–15. doi: 10.1016/j.brainres.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 87.Galanopoulou AS. Sexually dimorphic expression of KCC2 and GABA function. Epilepsy Res. 2008;80:99–113. doi: 10.1016/j.eplepsyres.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kight KE, McCarthy MM. Using sex differences in the developing brain to identify nodes of influence for seizure susceptibility and epileptogenesis. Neurobiol Dis. 2014 doi: 10.1016/j.nbd.2014.05.027. http://dx.doi.org/10.1016/j.nbd.2014.05.027. [DOI] [PMC free article] [PubMed]
- 89.Ganguly K, Schinder AF, Wong ST, Poo MM. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell. 2001;105:521–532. doi: 10.1016/s0092-8674(01)00341-5. [DOI] [PubMed] [Google Scholar]
- 90.Ludwig A, Li H, Saarma M, Kaila K, Rivera C. Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur J Neurosci. 2003;18:3199–3206. doi: 10.1111/j.1460-9568.2003.03069.x. [DOI] [PubMed] [Google Scholar]
- 91.Titz S, et al. Hyperpolarizing inhibition develops without trophic support by GABA in cultured rat midbrain neurons. J Physiol. 2003;550:719–730. doi: 10.1113/jphysiol.2003.041863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kanold PO, Shatz CJ. Subplate neurons regulate maturation of cortical inhibition and outcome of ocular dominance plasticity. Neuron. 2006;51:627–638. doi: 10.1016/j.neuron.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 93.Liu Z, Neff RA, Berg DK. Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science. 2006;314:1610–1613. doi: 10.1126/science.1134246. [DOI] [PubMed] [Google Scholar]
- 94.Pfeffer CK, et al. NKCC1-dependent GABAergic excitation drives synaptic network maturation during early hippocampal development. J Neurosci. 2009;29:3419–3430. doi: 10.1523/JNEUROSCI.1377-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lacoh CM, Bodogan T, Kaila K, Fiumelli H, Vutskits L. General anaesthetics do not impair developmental expression of the KCC2 potassium-chloride cotransporter in neonatal rats during the brain growth spurt. Br J Anaesth. 2013;110 (Suppl 1):i10–i18. doi: 10.1093/bja/aet063. [DOI] [PubMed] [Google Scholar]
- 96.Wojcik SM, et al. A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron. 2006;50:575–587. doi: 10.1016/j.neuron.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 97.Nardou R, et al. Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain. 2011;134:987–1002. doi: 10.1093/brain/awr041. [DOI] [PubMed] [Google Scholar]
- 98.Alger BE, Nicoll RA. GABA-mediated biphasic inhibitory responses in hippocampus. Nature. 1979;281:315–317. doi: 10.1038/281315a0. [DOI] [PubMed] [Google Scholar]
- 99.Alger BE, Nicoll RA. Feed-forward dendritic inhibition in rat hippocampal pyramidal cells studied in vitro. J Physiol. 1982;328:105–123. doi: 10.1113/jphysiol.1982.sp014255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high- frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Viitanen T, Ruusuvuori E, Kaila K, Voipio J. The K+-Cl cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol. 2010;588:1527–1540. doi: 10.1113/jphysiol.2009.181826. This study showed that, paradoxically, KCC2 can promote seizures by contributing to pro-ictal extracellular K+ transients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR. Molecular cloning and chromosome localization of a putative basolateral Na+-K+-2Cl− cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem. 1994;269:25677–25683. [PubMed] [Google Scholar]
- 103.Payne JA, et al. Primary structure, functional expression, and chromosomal localization of the bumetanide-sensitive Na-K-Cl cotransporter in human colon. J Biol Chem. 1995;270:17977–17985. doi: 10.1074/jbc.270.30.17977. [DOI] [PubMed] [Google Scholar]
- 104.Randall J, Thorne T, Delpire E. Partial cloning and characterization of Slc12a2: the gene encoding the secretory Na+-K+-2Cl- cotransporter. Am J Physiol. 1997;273:C1267–C1277. doi: 10.1152/ajpcell.1997.273.4.C1267. [DOI] [PubMed] [Google Scholar]
- 105.Hübner CA, Lorke DE, Hermans-Borgmeyer I. Expression of the Na-K-2Cl-cotransporter NKCC1 during mouse development. Mech Dev. 2001;102:267–269. doi: 10.1016/s0925-4773(01)00309-4. [DOI] [PubMed] [Google Scholar]
- 106.Price TJ, Hargreaves KM, Cervero F. Protein expression and mRNA cellular distribution of the NKCC1 cotransporter in the dorsal root and trigeminal ganglia of the rat. Brain Res. 2006;1112:146–158. doi: 10.1016/j.brainres.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA’s excitatory role in immature brain. J Neurobiol. 1997;33:781–795. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 108.Aronica E, et al. Differential expression patterns of chloride transporters, Na+-K+-2Cl—cotransporter and K+-Cl−-cotransporter, in epilepsy-associated malformations of cortical development. Neuroscience. 2007;145:185–196. doi: 10.1016/j.neuroscience.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 109.Yan Y, Dempsey RJ, Sun D. Expression of Na+-K+-Cl− cotransporter in rat brain during development and its localization in mature astrocytes. Brain Res. 2001;911:43–55. doi: 10.1016/s0006-8993(01)02649-x. [DOI] [PubMed] [Google Scholar]
- 110.Marty S, Wehrle R, Alvarez-Leefmans FJ, Gasnier B, Sotelo C. Postnatal maturation of Na+, K+, 2Cl- cotransporter expression and inhibitory synaptogenesis in the rat hippocampus: an immunocytochemical analysis. Eur J Neurosci. 2002;15:233–245. doi: 10.1046/j.0953-816x.2001.01854.x. [DOI] [PubMed] [Google Scholar]
- 111.Mikawa S, et al. Developmental changes in KCC1, KCC2 and NKCC1 mRNAs in the rat cerebellum. Dev Brain Res. 2002;136:93–100. doi: 10.1016/s0165-3806(02)00345-0. [DOI] [PubMed] [Google Scholar]
- 112.Morita Y, et al. Characteristics of the cation cotransporter NKCC1 in human brain: alternate transcripts, expression in development, and potential relationships to brain function and schizophrenia. J Neurosci. 2014;34:4929–4940. doi: 10.1523/JNEUROSCI.1423-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vibat CRT, Holland MJ, Kang JJ, Putney LK, O’Donnell ME. Quantitation of Na+-K+-2Cl− cotransport splice variants in human tissues using kinetic polymerase chain reaction. Anal Biochem. 2001;298:218–230. doi: 10.1006/abio.2001.5398. [DOI] [PubMed] [Google Scholar]
- 114.Pearson MM, Lu J, Mount DB, Delpire E. Localization of the K-Cl-cotransporter, KCC3, in the central and peripheral nervous systems: expression in the choroid plexus, large neurons and white matter tracts. Neuroscience. 2001;103:481–491. doi: 10.1016/s0306-4522(00)00567-4. [DOI] [PubMed] [Google Scholar]
- 115.Shekarabi M, et al. Cellular expression of the K+-Cl− cotransporter KCC3 in the central nervous system of mouse. Brain Res. 2011;1374:15–26. doi: 10.1016/j.brainres.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 116.Boettger T, et al. Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J. 2003;22:5422–5434. doi: 10.1093/emboj/cdg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Byun N, Delpire E. Axonal and periaxonal swelling precede peripheral neurodegeneration in KCC3 knockout mice. Neurobiol Dis. 2007;28:39–51. doi: 10.1016/j.nbd.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Howard HC, et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nature Genet. 2002;32:384–392. doi: 10.1038/ng1002. [DOI] [PubMed] [Google Scholar]
- 119.Shekarabi M, et al. Loss of neuronal potassium/ chloride cotransporter 3 (KCC3) is responsible for the degenerative phenotype in a conditional mouse model of hereditary motor and sensory neuropathy associated with agenesis of the corpus callosum. J Neurosci. 2012;32:3865–3876. doi: 10.1523/JNEUROSCI.3679-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Karadsheh MF, Byun N, Mount DB, Delpire E. Localization of the KCC4 potassium chloride cotransporter in the nervous system. Neuroscience. 2004;123:381–391. doi: 10.1016/j.neuroscience.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 121.Rust MB, et al. Disruption of erythroid K-Cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J Clin Invest. 2007;117:1708–1717. doi: 10.1172/JCI30630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boettger T, et al. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature. 2002;416:874–878. doi: 10.1038/416874a. [DOI] [PubMed] [Google Scholar]
- 123.Zhang LL, Fina ME, Vardi N. Regulation of KCC2 and NKCC during development: membrane insertion and differences between cell types. J Comp Neurol. 2006;499:132–143. doi: 10.1002/cne.21100. [DOI] [PubMed] [Google Scholar]
- 124.Lytle C, Forbush B. Regulatory phosphorylation of the secretory Na-K-Cl cotransporter: modulation by cytoplasmic Cl. Am J Physiol. 1996;270:C437–C448. doi: 10.1152/ajpcell.1996.270.2.C437. [DOI] [PubMed] [Google Scholar]
- 125.Haas M, McBrayer D, Lytle C. Cl-dependent phosphorylation of the Na-K-Cl cotransport protein of dog tracheal epithelial cells. J Biol Chem. 1995;270:28955–28961. doi: 10.1074/jbc.270.48.28955. [DOI] [PubMed] [Google Scholar]
- 126.Williams JR, Payne JA. Cation transport by the neuronal K+-Cl− cotransporter KCC2: thermodynamics and kinetics of alternate transport modes. Am J Physiol. 2004;287:C919–C931. doi: 10.1152/ajpcell.00005.2004. [DOI] [PubMed] [Google Scholar]
- 127.Piala AT, et al. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal. 2014;7:ra41. doi: 10.1126/scisignal.2005050. This work suggests that WNK1 acts as a sensor of the intracellular Cl− level through direct binding of a Cl− ion to its active site, inhibiting autophosphorylation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Delpire E, Gagnon KBE. SPAK and OSR1: STE20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. Biochem J. 2008;409:321–331. doi: 10.1042/BJ20071324. [DOI] [PubMed] [Google Scholar]
- 129.de los Heros P, et al. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl− co-transporters. Biochem J. 2014;458:559–573. doi: 10.1042/BJ20131478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Alessi DR, et al. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal. 2014;7:re3. doi: 10.1126/scisignal.2005365. [DOI] [PubMed] [Google Scholar]
- 131.Darman RB, Flemmer A, Forbush B. Modulation of ion transport by direct targeting of protein phosphatase type 1 to the Na-K-Cl cotransporter. J Biol Chem. 2001;276:34359–34362. doi: 10.1074/jbc.C100368200. [DOI] [PubMed] [Google Scholar]
- 132.Flemmer AW, Gimenez I, Dowd BFX, Darman RB, Forbush B. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem. 2002;277:37551–37558. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- 133.Darman RB, Forbush B. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem. 2002;277:37542–37550. doi: 10.1074/jbc.M206293200. [DOI] [PubMed] [Google Scholar]
- 134.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1) J Biol Chem. 2002;277:50812–50819. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 135.Dowd BFX, Forbush B. PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1) J Biol Chem. 2003;278:27347–27353. doi: 10.1074/jbc.M301899200. [DOI] [PubMed] [Google Scholar]
- 136.Filippi BM, et al. MO25 is a master regulator of SPAK/OSR1 and MST3/MST4/YSK1 protein kinases. EMBO J. 2011;30:1730–1741. doi: 10.1038/emboj.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gagnon KB, Delpire E. Multiple pathways for protein phosphatase 1 (PP1) regulation of Na-K-2Cl cotransporter (NKCC1) function: the N-terminal tail of the Na-K-2Cl cotransporter serves as a regulatory scaffold for Ste20-related proline/alanine-rich kinase (SPAK) AND PP1. J Biol Chem. 2010;285:14115–14121. doi: 10.1074/jbc.M110.112672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Markadieu N, Delpire E. Physiology and pathophysiology of SLC12A1/2 transporters. Pflugers Arch. 2014;466:91–105. doi: 10.1007/s00424-013-1370-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rinehart J, et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 2009;138:525–536. doi: 10.1016/j.cell.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Inoue K, et al. Taurine inhibits K+-Cl− cotransporter KCC2 to regulate embryonic Cl− homeostasis via with-no-lysine (WNK) protein kinase signaling pathway. J Biol Chem. 2012;287:20839–20850. doi: 10.1074/jbc.M111.319418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kahle KT, et al. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013;36:726–737. doi: 10.1016/j.tins.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gulyas AI, Sik A, Payne JA, Kaila K, Freund TF. The KCl cotransporter, KCC2, is highly expressed in the vicinity of excitatory synapses in the rat hippocampus. Eur J Neurosci. 2001;13:2205–2217. doi: 10.1046/j.0953-816x.2001.01600.x. This the first study to report the surprising finding that KCC2 levels are particularly high in and around spines. This boosted subsequent work on a possible structural role for KCC2 in spine formation. [DOI] [PubMed] [Google Scholar]
- 143.Kovacs K, Basu K, Rouiller I, Sik A. Regional differences in the expression of K+–Cl− 2 cotransporter in the developing rat cortex. Brain Struct Funct. 2013;219:527–538. doi: 10.1007/s00429-013-0515-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Weber M, Hartmann AM, Beyer T, Ripperger A, Nothwang HG. A novel regulatory locus of phosphorylation in the C terminus of the potassium chloride cotransporter KCC2 that interferes with N-ethylmaleimide or staurosporine-mediated activation. J Biol Chem. 2014;289:18668–18679. doi: 10.1074/jbc.M114.567834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bos R, et al. Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc Natl Acad Sci USA. 2013;110:348–353. doi: 10.1073/pnas.1213680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Khirug S, et al. Distinct properties of functional KCC2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur J Neurosci. 2005;21:899–904. doi: 10.1111/j.1460-9568.2005.03886.x. [DOI] [PubMed] [Google Scholar]
- 147.Lee HHC, et al. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J Biol Chem. 2007;282:29777–29784. doi: 10.1074/jbc.M705053200. [DOI] [PubMed] [Google Scholar]
- 148.Fiumelli H, Cancedda L, Poo MM. Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron. 2005;48:773–786. doi: 10.1016/j.neuron.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 149.Lee HH, Deeb TZ, Walker JA, Davies PA, Moss SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nature Neurosci. 2011;14:736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Puskarjov M, Ahmad F, Kaila K, Blaesse P. Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J Neurosci. 2012;32:11356–11364. doi: 10.1523/JNEUROSCI.6265-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chamma I, et al. Activity-dependent regulation of the K/Cl transporter KCC2 membrane diffusion, clustering, and function in hippocampal neurons. J Neurosci. 2013;33:15488–15503. doi: 10.1523/JNEUROSCI.5889-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rivera C, et al. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Laughlin SB, Sejnowski TJ. Communication in neuronal networks. Science. 2003;301:1870–1874. doi: 10.1126/science.1089662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Inoue K, Yamada J, Ueno S, Fukuda A. Brain-type creatine kinase activates neuron-specific K+-Cl− co-transporter KCC2. J Neurochem. 2006;96:598–608. doi: 10.1111/j.1471-4159.2005.03560.x. [DOI] [PubMed] [Google Scholar]
- 155.Salin-Cantegrel A, et al. HMSN/ACC truncation mutations disrupt brain-type creatine kinase-dependant activation of K+/Cl− cotransporter 3. Hum Mol Genet. 2008;17:2703–2711. doi: 10.1093/hmg/ddn172. [DOI] [PubMed] [Google Scholar]
- 156.Ikeda K, et al. Malfunction of respiratory-related neuronal activity in Na+, K+-ATPase α2 subunit-deficient mice is attributable to abnormal Cl− homeostasis in brainstem neurons. J Neurosci. 2004;24:10693–10701. doi: 10.1523/JNEUROSCI.2909-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sjöström PJ, Rancz EA, Roth A, Häusser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- 159.Gidon A, Segev I. Principles governing the operation of synaptic inhibition in dendrites. Neuron. 2012;75:330–341. doi: 10.1016/j.neuron.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 160.Jack JJB, Noble D, Tsien RW. Electric Current Flow in Excitable Cells. Oxford Univ. Press; 1975. [Google Scholar]
- 161.Farrant M, Kaila K. The cellular, molecular and ionic basis of GABAA receptor signalling. Prog Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- 162.Fatima-Shad K, Barry PH. Anion permeation in GABA- and glycine-gated channels of mammalian cultured hippocampal neurons. Proc R Soc Lond B. 1993;253:69–75. doi: 10.1098/rspb.1993.0083. [DOI] [PubMed] [Google Scholar]
- 163.Misgeld U, Deisz RA, Dodt HU, Lux HD. The role of chloride transport in postsynaptic inhibition of hippocampal neurons. Science. 1986;232:1413–1415. doi: 10.1126/science.2424084. [DOI] [PubMed] [Google Scholar]
- 164.Kaila K, Voipio J, Paalasmaa P, Pasternack M, Deisz RA. The role of bicarbonate in GABAA receptor-mediated IPSPs of rat neocortical neurones. J Physiol. 1993;464:273–289. doi: 10.1113/jphysiol.1993.sp019634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen G, Trombley PQ, van den Pol AN. Excitatory actions of GABA in developing rat hypothalamic neurones. J Physiol. 1996;494:451–464. doi: 10.1113/jphysiol.1996.sp021505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.van den Pol AN, Obrietan K, Chen G. Excitatory actions of GABA after neuronal trauma. J Neurosci. 1996;16:4283–4292. doi: 10.1523/JNEUROSCI.16-13-04283.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Köhling R, et al. Spontaneous sharp waves in human neocortical slices excised from epileptic patients. Brain. 1998;121:1073–1087. doi: 10.1093/brain/121.6.1073. [DOI] [PubMed] [Google Scholar]
- 168.Kilb W, et al. Glycine receptors mediate excitation of subplate neurons in neonatal rat cerebral cortex. J Neurophysiol. 2008;100:698–707. doi: 10.1152/jn.00657.2007. [DOI] [PubMed] [Google Scholar]
- 169.Christie JM, Jahr CE. Selective expression of ligand-gated ion channels in L5 pyramidal cell axons. J Neurosci. 2009;29:11441–11450. doi: 10.1523/JNEUROSCI.2387-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nature Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- 171.Brickley SG, Mody I. Extrasynaptic GABAA receptors: their function in the CNS and implications for disease. Neuron. 2012;73:23–34. doi: 10.1016/j.neuron.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sipilä ST, Huttu K, Soltesz I, Voipio J, Kaila K. Depolarizing GABA acts on intrinsically bursting pyramidal neurons to drive giant depolarizing potentials in the immature hippocampus. J Neurosci. 2005;25:5280–5289. doi: 10.1523/JNEUROSCI.0378-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Demarque M, et al. Paracrine intercellular communication by a Ca2+- and SNARE-independent release of GABA and glutamate prior to synapse formation. Neuron. 2002;36:1051–1061. doi: 10.1016/s0896-6273(02)01053-x. [DOI] [PubMed] [Google Scholar]
- 174.Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nature Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- 175.Luhmann HJ, Kirischuk S, Sinning A, Kilb W. Early GABAergic circuitry in the cerebral cortex. Curr Opin Neurobiol. 2014;26:72–78. doi: 10.1016/j.conb.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 176.Rivera C, Voipio J, Kaila K. Two developmental switches in GABAergic signalling: the K+-Cl− cotransporter KCC2 and carbonic anhydrase CAVII. J Physiol. 2005;564:953. doi: 10.1113/jphysiol.2004.077495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Riekki R, et al. Altered synaptic dynamics and hippocampal excitability but normal long-term plasticity in mice lacking hyperpolarizing GABAA receptor-mediated inhibition in CA1 pyramidal neurons. J Neurophysiol. 2008;99:3075–3089. doi: 10.1152/jn.00606.2007. [DOI] [PubMed] [Google Scholar]
- 178.Stil A, et al. Contribution of the potassium-chloride co-transporter KCC2 to the modulation of lumbar spinal networks in mice. Eur J Neurosci. 2011;33:1212–1222. doi: 10.1111/j.1460-9568.2010.07592.x. [DOI] [PubMed] [Google Scholar]
- 179.Zhang RW, Zhang SY, Du JL. KCC2-dependent subcellular ECl difference of ON-OFF retinal ganglion cells in larval zebrafish. Front Neural Circuits. 2013;7:103. doi: 10.3389/fncir.2013.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lee H, Chen CXQ, Liu YJ, Aizenman E, Kandler K. KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of GABA responses. Eur J Neurosci. 2005;21:2593–2599. doi: 10.1111/j.1460-9568.2005.04084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cancedda L, Fiumelli H, Chen K, Poo MM. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Reynolds A, et al. Neurogenic role of the depolarizing chloride gradient revealed by global overexpression of KCC2 from the onset of development. J Neurosci. 2008;28:1588–1597. doi: 10.1523/JNEUROSCI.3791-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Szabadics J, et al. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. The authors show that EGABA is spatially non-uniform in cortical pyramidal neurons. Whether the depolarizing GABAergic input at the AIS is functionally excitatory is being debated. [DOI] [PubMed] [Google Scholar]
- 184.Waseem T, Mukhtarov M, Buldakova S, Medina I, Bregestovski P. Genetically encoded Cl-sensor as a tool for monitoring of Cl-dependent processes in small neuronal compartments. J Neurosci Methods. 2010;193:14–23. doi: 10.1016/j.jneumeth.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 185.Khirug S, et al. GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J Neurosci. 2008;28:4635–4639. doi: 10.1523/JNEUROSCI.0908-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Baldi R, Varga C, Tamas G. Differential distribution of KCC2 along the axo-somato-dendritic axis of hippocampal principal cells. Eur J Neurosci. 2010;32:1319–1325. doi: 10.1111/j.1460-9568.2010.07361.x. [DOI] [PubMed] [Google Scholar]
- 187.Hedstrom KL, Ogawa Y, Rasband MN. Ankyrin G is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183:635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Glickfeld LL, Roberts JD, Somogyi P, Scanziani M. Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nature Neurosci. 2009;12:21–23. doi: 10.1038/nn.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Viney TJ, et al. Network state-dependent inhibition of identified hippocampal CA3 axo-axonic cells in vivo. Nature Neurosci. 2013;16:1802–1811. doi: 10.1038/nn.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Carmosino M, Gimenez I, Caplan M, Forbush B. Exon loss accounts for differential sorting of Na-K-Cl cotransporters in polarized epithelial cells. Mol Biol Cell. 2008;19:4341–4351. doi: 10.1091/mbc.E08-05-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Dotti CG, Simons K. Polarized sorting of viral glycoproteins to the axon and dendrites of hippocampal neurons in culture. Cell. 1990;62:63–72. doi: 10.1016/0092-8674(90)90240-f. [DOI] [PubMed] [Google Scholar]
- 192.Fatt P, Katz B. The effect of inhibitory nerve impulses on a crustacean muscle fibre. J Physiol. 1953;121:374–389. doi: 10.1113/jphysiol.1953.sp004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ruusuvuori E, Kaila K. In: Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications. Frost S, McKenna R, editors. Springer; 2014. pp. 271–290. [Google Scholar]
- 194.Chadderton P, Schaefer AT, Williams SR, Margrie TW. Sensory-evoked synaptic integration in cerebellar and cerebral cortical neurons. Nature Rev Neurosci. 2014;15:71–83. doi: 10.1038/nrn3648. [DOI] [PubMed] [Google Scholar]
- 195.Doyon N, et al. Efficacy of synaptic inhibition depends on multiple, dynamically interacting mechanisms implicated in chloride homeostasis. PLoS Comput Biol. 2011;7:e1002149. doi: 10.1371/journal.pcbi.1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Grimley JS, et al. Visualization of synaptic inhibition with an optogenetic sensor developed by cell-free protein engineering automation. J Neurosci. 2013;33:16297–16309. doi: 10.1523/JNEUROSCI.4616-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kuner T, Augustine GJ. A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron. 2000;27:447–459. doi: 10.1016/s0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- 198.Raimondo JV, et al. A genetically-encoded chloride and pH sensor for dissociating ion dynamics in the nervous system. Front Cell Neurosci. 2013;7:202. doi: 10.3389/fncel.2013.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Raimondo JV, Markram H, Akerman CJ. Short-term ionic plasticity at GABAergic synapses. Front Synaptic Neurosci. 2012;4:5. doi: 10.3389/fnsyn.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wright R, Raimondo JV, Akerman CJ. Spatial and temporal dynamics in the ionic driving force for GABAA receptors. Neural Plast. 2011;2011:728395. doi: 10.1155/2011/728395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Thompson SM, Gähwiler BH. Activity-dependent disinhibition. I Repetitive stimulation reduces IPSP driving force and conductance in the hippocampus in vitro. J Neurophysiol. 1989;61:501–511. doi: 10.1152/jn.1989.61.3.501. [DOI] [PubMed] [Google Scholar]
- 202.Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- 203.Taira T, Lamsa K, Kaila K. Posttetanic excitation mediated by GABAA receptors in rat CA1 pyramidal neurons. J Neurophysiol. 1997;77:2213–2218. doi: 10.1152/jn.1997.77.4.2213. [DOI] [PubMed] [Google Scholar]
- 204.Ruusuvuori E, et al. Neuronal carbonic anhydrase VII provides GABAergic excitatory drive to exacerbate febrile seizures. EMBO J. 2013;32:2275–2286. doi: 10.1038/emboj.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ruscheweyh R, Sandkuhler J. Epileptiform activity in rat spinal dorsal horn in vitro has common features with neuropathic pain. Pain. 2003;105:327–338. doi: 10.1016/s0304-3959(03)00248-3. This work demonstrated that the dorsal horn can generate epileptiform activity in a GABAAR-dependent manner. [DOI] [PubMed] [Google Scholar]
- 206.Asiedu M, Ossipov MH, Kaila K, Price TJ. Acetazolamide and midazolam act synergistically to inhibit neuropathic pain. Pain. 2010;148:302–308. doi: 10.1016/j.pain.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Asiedu MN, Mejia GL, Hubner CA, Kaila K, Price TJ. Inhibition of carbonic anhydrase augments GABA receptor-mediated analgesia via a spinal mechanism of action. J Pain. 2014;15:395–406. doi: 10.1016/j.jpain.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- 209.Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nature Rev Neurosci. 2001;2:880–888. doi: 10.1038/35104061. [DOI] [PubMed] [Google Scholar]
- 210.Takayama C, Inoue Y. Developmental localization of potassium chloride co-transporter 2 (KCC2), GABA and vesicular GABA transporter (VGAT) in the postnatal mouse somatosensory cortex. Neurosci Res. 2010;67:137–148. doi: 10.1016/j.neures.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 211.De Felipe J, Marco P, Fairen A, Jones EG. Inhibitory synaptogenesis in mouse somatosensory cortex. Cereb Cortex. 1997;7:619–634. doi: 10.1093/cercor/7.7.619. [DOI] [PubMed] [Google Scholar]
- 212.Juraska JM. The development of pyramidal neurons after eye opening in the visual cortex of hooded rats: a quantitative study. J Comp Neurol. 1982;212:208–213. doi: 10.1002/cne.902120210. [DOI] [PubMed] [Google Scholar]
- 213.Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997;387:167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 214.Petanjek Z, et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci USA. 2011;108:13281–13286. doi: 10.1073/pnas.1105108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Mercado A, Broumand V, Zandi-Nejad K, Enck AH, Mount DB. A C-terminal domain in KCC2 confers constitutive K+-Cl− cotransport. J Biol Chem. 2006;281:1016–1026. doi: 10.1074/jbc.M509972200. [DOI] [PubMed] [Google Scholar]
- 216.Chamma I, Chevy Q, Poncer JC, Levi S. Role of the neuronal K-Cl co-transporter KCC2 in inhibitory and excitatory neurotransmission. Front Cell Neurosci. 2012;6:5. doi: 10.3389/fncel.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Varoqueaux F, Jamain S, Brose N. Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol. 2004;83:449–456. doi: 10.1078/0171-9335-00410. [DOI] [PubMed] [Google Scholar]
- 218.Tyagarajan SK, Fritschy JM. Gephyrin: a master regulator of neuronal function? Nature Rev Neurosci. 2014;15:141–156. doi: 10.1038/nrn3670. [DOI] [PubMed] [Google Scholar]
- 219.Sun C, et al. Identification and functional characterization of rare mutations of the neuroligin-2 gene (NLGN2) associated with schizophrenia. Hum Mol Genet. 2011;20:3042–3051. doi: 10.1093/hmg/ddr208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ivakine EA, et al. Neto2 is a KCC2 interacting protein required for neuronal Cl− regulation in hippocampal neurons. Proc Natl Acad Sci USA. 2013;110:3561–3566. doi: 10.1073/pnas.1212907110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mahadevan V, et al. Kainate receptors coexist in a functional complex with KCC2 and regulate chloride homeostasis in hippocampal neurons. Cell Rep. 2014;7:1762–1770. doi: 10.1016/j.celrep.2014.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kubota Y, Hatada S, Kondo S, Karube F, Kawaguchi Y. Neocortical inhibitory terminals innervate dendritic spines targeted by thalamocortical afferents. J Neurosci. 2007;27:1139–1150. doi: 10.1523/JNEUROSCI.3846-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Chiu CQ, et al. Compartmentalization of GABAergic inhibition by dendritic spines. Science. 2013;340:759–762. doi: 10.1126/science.1234274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Rose CR, Konnerth A. NMDA receptor-mediated Na+ signals in spines and dendrites. J Neurosci. 2001;21:4207–4214. doi: 10.1523/JNEUROSCI.21-12-04207.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Jensen FE. Epilepsy as a spectrum disorder: implications from novel clinical and basic neuroscience. Epilepsia. 2011;52 (Suppl 1):1–6. doi: 10.1111/j.1528-1167.2010.02904.x. [DOI] [PubMed] [Google Scholar]
- 226.Haider B, McCormick DA. Rapid neocortical dynamics: cellular and network mechanisms. Neuron. 2009;62:171–189. doi: 10.1016/j.neuron.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Uusisaari M, Smirnov S, Voipio J, Kaila K. Spontaneous epileptiform activity mediated by GABAA receptors and gap junctions in the rat hippocampal slice following long-term exposure to GABAB antagonists. Neuropharmacology. 2002;43:563–572. doi: 10.1016/s0028-3908(02)00156-9. [DOI] [PubMed] [Google Scholar]
- 228.Pavlov I, Kaila K, Kullmann DM, Miles R. Cortical inhibition, pH and cell excitability in epilepsy: what are optimal targets for antiepileptic interventions? J Physiol. 2013;591:765–774. doi: 10.1113/jphysiol.2012.237958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Woo NS, et al. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- 230.Tornberg J, Voikar V, Savilahti H, Rauvala H, Airaksinen MS. Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur J Neurosci. 2005;21:1327–1337. doi: 10.1111/j.1460-9568.2005.03959.x. [DOI] [PubMed] [Google Scholar]
- 231.Kahle KT, et al. Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 2014;15:766–774. doi: 10.15252/embr.201438840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Marchionni I, Maccaferri G. Quantitative dynamics and spatial profile of perisomatic GABAergic input during epileptiform synchronization in the CA1 hippocampus. J Physiol. 2009;587:5691–5708. doi: 10.1113/jphysiol.2009.179945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- 234.Huberfeld G, et al. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. This work showed that a depolarized EGABA and consequent interictal activity in subicular tissue from patients with temporal lobe epilepsy are based on a loss of KCC2 in a subpopulation of principal neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Miles R, Blaesse P, Huberfeld G, Wittner L, Kaila K. In: Jasper’s Basic Mechanisms of the Epilepsies. 4. Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Oxford Univ. Press; 2012. pp. 581–590. [Google Scholar]
- 236.Töllner K, et al. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann Neurol. 2014;75:550–562. doi: 10.1002/ana.24124. [DOI] [PubMed] [Google Scholar]
- 237.Gagnon M, et al. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nature Med. 2013;19:1524–1528. doi: 10.1038/nm.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Jiruska P, et al. Synchronization and desynchronization in epilepsy: controversies and hypotheses. J Physiol. 2013;591:787–797. doi: 10.1113/jphysiol.2012.239590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Bialer M. Why are antiepileptic drugs used for nonepileptic conditions? Epilepsia. 2012;53 (Suppl 7):26–33. doi: 10.1111/j.1528-1167.2012.03712.x. [DOI] [PubMed] [Google Scholar]
- 240.Ferrini F, De Koninck Y. Microglia control neuronal network excitability via BDNF signalling. Neural Plast. 2013;2013:429815. doi: 10.1155/2013/429815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Doyon N, Ferrini F, Gagnon M, De KY. Treating pathological pain: is KCC2 the key to the gate? Expert Rev Neurother. 2013;13:469–471. doi: 10.1586/ern.13.40. [DOI] [PubMed] [Google Scholar]
- 242.Lorenzo LE, et al. Gephyrin clusters are absent from small diameter primary afferent terminals despite the presence of GABAA receptors. J Neurosci. 2014;34:8300–8317. doi: 10.1523/JNEUROSCI.0159-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150:971–979. doi: 10.1126/science.150.3699.971. This pioneering theoretical paper set the stage for much of the current work done on the role of CCCs in pain. [DOI] [PubMed] [Google Scholar]
- 244.Willis WD. Jr Dorsal root potentials and dorsal root reflexes: a double-edged sword. Exp Brain Res. 1999;124:395–421. doi: 10.1007/s002210050637. [DOI] [PubMed] [Google Scholar]
- 245.Price TJ, Cervero F, Gold MS, Hammond DL, Prescott SA. Chloride regulation in the pain pathway. Brain Res Rev. 2009;60:149–170. doi: 10.1016/j.brainresrev.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Cho H, et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nature Neurosci. 2012;15:1015–1021. doi: 10.1038/nn.3111. [DOI] [PubMed] [Google Scholar]
- 247.Kingery WS, Fields RD, Kocsis JD. Diminished dorsal root GABA sensitivity following chronic peripheral nerve injury. Exp Neurol. 1988;100:478–490. doi: 10.1016/0014-4886(88)90033-7. [DOI] [PubMed] [Google Scholar]
- 248.Granados-Soto V, Arguelles CF, Alvarez-Leefmans FJ. Peripheral and central antinociceptive action of Na+-K+-2Cl- cotransporter blockers on formalin-induced nociception in rats. Pain. 2005;114:231–238. doi: 10.1016/j.pain.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 249.Pitcher MH, Price TJ, Entrena JM, Cervero F. Spinal NKCC1 blockade inhibits TRPV1-dependent referred allodynia. Mol Pain. 2007;3:17. doi: 10.1186/1744-8069-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Pitcher MH, Cervero F. Role of the NKCC1 co-transporter in sensitization of spinal nociceptive neurons. Pain. 2010;151:756–762. doi: 10.1016/j.pain.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 251.Lavertu G, Cote SL, De KY. Enhancing K-Cl co-transport restores normal spinothalamic sensory coding in a neuropathic pain model. Brain. 2014;137:724–738. doi: 10.1093/brain/awt334. [DOI] [PubMed] [Google Scholar]
- 252.Jolivalt CG, Lee CA, Ramos KM, Calcutt NA. Allodynia and hyperalgesia in diabetic rats are mediated by GABA and depletion of spinal potassium-chloride co-transporters. Pain. 2008;140:48–57. doi: 10.1016/j.pain.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ferrini F, et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl− homeostasis. Nature Neurosci. 2013;16:183–192. doi: 10.1038/nn.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Jean-Xavier C, Pflieger JF, Liabeuf S, Vinay L. Inhibitory postsynaptic potentials in lumbar motoneurons remain depolarizing after neonatal spinal cord transection in the rat. J Neurophysiol. 2006;96:2274–2281. doi: 10.1152/jn.00328.2006. [DOI] [PubMed] [Google Scholar]
- 255.Keller AF, Beggs S, Salter MW, De Koninck Y. Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Mol Pain. 2007;3:27. doi: 10.1186/1744-8069-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Coull JAM, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 257.Lee-Kubli CA, Calcutt NA. Altered rate-dependent depression of the spinal H-reflex as an indicator of spinal disinhibition in models of neuropathic pain. Pain. 2014;155:250–260. doi: 10.1016/j.pain.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zhang Z, Wang X, Wang W, Lu YG, Pan ZZ. Brain-derived neurotrophic factor-mediated downregulation of brainstem K+-Cl− cotransporter and cell-type-specific GABA impairment for activation of descending pain facilitation. Mol Pharmacol. 2013;84:511–520. doi: 10.1124/mol.113.086496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Maguire J, Salpekar JA. Stress, seizures, and hypothalamic–pituitary–adrenal axis targets for the treatment of epilepsy. Epilepsy Behav. 2013;26:352–362. doi: 10.1016/j.yebeh.2012.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Nichol JA, Hutter OF. Tensile strength and dilatational elasticity of giant sarcolemmal vesicles shed from rabbit muscle. J Physiol. 1996;493:187–198. doi: 10.1113/jphysiol.1996.sp021374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Gauthier NC, Masters TA, Sheetz MP. Mechanical feedback between membrane tension and dynamics. Trends Cell Biol. 2012;22:527–535. doi: 10.1016/j.tcb.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 262.de los Heros P, et al. WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc Natl Acad Sci USA. 2006;103:1976–1981. doi: 10.1073/pnas.0510947103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Payne JA. Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am J Physiol. 1997;42:C1516–C1525. doi: 10.1152/ajpcell.1997.273.5.C1516. [DOI] [PubMed] [Google Scholar]
- 264.Strange K, Singer TD, Morrison R, Delpire E. Dependence of KCC2 K-Cl cotransporter activity on a conserved carboxy terminus tyrosine residue. Am J Physiol. 2000;279:C860–C867. doi: 10.1152/ajpcell.2000.279.3.C860. [DOI] [PubMed] [Google Scholar]
- 265.Cala PM. In: Chloride Channels and Carriers in Nerve, Muscle, and Glial Cells. Alvarez-Leefmans FJ, Russell JM, editors. Plenum; 1990. pp. 67–83. [Google Scholar]
- 266.Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nature Rev Neurosci. 2003;4:991–1001. doi: 10.1038/nrn1252. [DOI] [PubMed] [Google Scholar]
- 267.Jefferys JG. Nonsynaptic modulation of neuronal activity in the brain: electric currents and extracellular ions. Physiol Rev. 1995;75:689–723. doi: 10.1152/physrev.1995.75.4.689. [DOI] [PubMed] [Google Scholar]
- 268.Larsen BR, et al. Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia. 2014;62:608–622. doi: 10.1002/glia.22629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Andrew RD, Labron MW, Boehnke SE, Carnduff L, Kirov SA. Physiological evidence that pyramidal neurons lack functional water channels. Cereb Cortex. 2007;17:787–802. doi: 10.1093/cercor/bhk032. [DOI] [PubMed] [Google Scholar]
- 270.Alvarez-Leefmans FJ, Gamino SM, Giraldez F, Nogueron I. Intracellular chloride regulation in amphibian dorsal root ganglion neurones studied with ion-selective microelectrodes. J Physiol. 1988;406:225–246. doi: 10.1113/jphysiol.1988.sp017378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Shields SD, Mazario J, Skinner K, Basbaum AI. Anatomical and functional analysis of aquaporin 1, a water channel in primary afferent neurons. Pain. 2007;131:8–20. doi: 10.1016/j.pain.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 272.Hille B. Ionic Channels of Excitable Membranes. 3. Sinauer Associates; 2001. [Google Scholar]
- 273.Tosteson DC, Hoffman JF. Regulation of cell volume by active cation transport in high and low potassium sheep red cells. J Gen Physiol. 1960;44:169–194. doi: 10.1085/jgp.44.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Jarolimek W, Lewen A, Misgeld U. A furosemide-sensitive K+-Cl− cotransporter counteracts intracellular Cl− accumulation and depletion in cultured rat midbrain neurons. J Neurosci. 1999;19:4695–4704. doi: 10.1523/JNEUROSCI.19-12-04695.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Cordero-Erausquin M, Coull JA, Boudreau D, Rolland M, De Koninck Y. Differential maturation of GABA action and anion reversal potential in spinal lamina I neurons: impact of chloride extrusion capacity. J Neurosci. 2005;25:9613–9623. doi: 10.1523/JNEUROSCI.1488-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.McLaughlin SG, Szabo G, Eisenman G. Divalent ions and the surface potential of charged phospholipid membranes. J Gen Physiol. 1971;58:667–687. doi: 10.1085/jgp.58.6.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 278.Kaila K, Pasternack M, Saarikoski J, Voipio J. Influence of GABA-gated bicarbonate conductance on potential, current and intracellular chloride in crayfish muscle fibres. J Physiol. 1989;416:161–181. doi: 10.1113/jphysiol.1989.sp017755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Armstrong CM. The Na/K pump, Cl ion, and osmotic stabilization of cells. Proc Natl Acad Sci USA. 2003;100:6257–6262. doi: 10.1073/pnas.0931278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Glykys J, et al. Local impermeant anions establish the neuronal chloride concentration. Science. 2014;343:670–675. doi: 10.1126/science.1245423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Voipio J, et al. Comment on “Local impermeant anions establish the neuronal chloride concentration”. Science. 2014;345:1130. doi: 10.1126/science.1252978. [DOI] [PubMed] [Google Scholar]
- 282.Lee HH, Jurd R, Moss SJ. Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol Cell Neurosci. 2010;45:173–179. doi: 10.1016/j.mcn.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Barmashenko G, Hefft S, Aertsen A, Kirschstein T, Kohling R. Positive shifts of the GABAA receptor reversal potential due to altered chloride homeostasis is widespread after status epilepticus. Epilepsia. 2011;52:1570–1578. doi: 10.1111/j.1528-1167.2011.03247.x. [DOI] [PubMed] [Google Scholar]
- 284.Papp E, Rivera C, Kaila K, Freund TF. Relationship between neuronal vulnerability and potassium-chloride cotransporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience. 2008;154:677–689. doi: 10.1016/j.neuroscience.2008.03.072. [DOI] [PubMed] [Google Scholar]
- 285.Bonislawski DP, Schwarzbach EP, Cohen AS. Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol Dis. 2007;25:163–169. doi: 10.1016/j.nbd.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Jantzie LL, et al. Erythropoietin attenuates loss of potassium chloride co-transporters following prenatal brain injury. Mol Cell Neurosci. 2014;61:152–162. doi: 10.1016/j.mcn.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Amini M, et al. Conditional disruption of calpain in the CNS alters dendrite morphology, impairs LTP, and promotes neuronal survival following injury. J Neurosci. 2013;33:5773–5784. doi: 10.1523/JNEUROSCI.4247-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Andres AL, et al. NMDA receptor activation and calpain contribute to disruption of dendritic spines by the stress neuropeptide CRH. J Neurosci. 2013;33:16945–16960. doi: 10.1523/JNEUROSCI.1445-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.He XP, Pan E, Sciarretta C, Minichiello L, McNamara JO. Disruption of TrkB-mediated phospholipase Cγ signaling inhibits limbic epileptogenesis. J Neurosci. 2010;30:6188–6196. doi: 10.1523/JNEUROSCI.5821-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.He XP, Wen R, McNamara JO. Impairment of kindling development in phospholipase Cγ1 heterozygous mice. Epilepsia. 2014;55:456–463. doi: 10.1111/epi.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Carmona MA, et al. Age-dependent spontaneous hyperexcitability and impairment of GABAergic function in the hippocampus of mice lacking trkB. Cereb Cortex. 2006;16:47–63. doi: 10.1093/cercor/bhi083. [DOI] [PubMed] [Google Scholar]
- 292.Knusel B, Rabin SJ, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Di Lieto A, et al. The responsiveness of TrkB to BDNF and antidepressant drugs is differentially regulated during mouse development. PLoS ONE. 2012;7:e32869. doi: 10.1371/journal.pone.0032869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Shulga A, et al. Posttraumatic GABAA-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. J Neurosci. 2008;28:6996–7005. doi: 10.1523/JNEUROSCI.5268-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Kang HJ, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Ge S, et al. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. 2006;439:589–593. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Glykys J, et al. Response to Comments on “Local impermeant anions establish the neuronal chloride concentration”. Science. 2014;345:1130. doi: 10.1126/science.1253146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.