

Abstract
Polyvalent interactions, where multiple ligands and receptors interact simultaneously, are ubiquitous in nature. Synthetic polyvalent molecules, therefore, have the ability to affect biological processes ranging from protein–ligand binding to cellular signaling. In this review, we discuss recent advances in polyvalent scaffold design and applications. First, we will describe recent developments in the engineering of polyvalent scaffolds based on biomolecules and novel materials. Then, we will illustrate how polyvalent molecules are finding applications as toxin and pathogen inhibitors, targeting molecules, immune response modulators, and cellular effectors.
Introduction
Myriad interactions in nature use polyvalent binding, where multiple ligands on one entity bind to multiple receptors on another: antibody–antigen interactions, virus−cell binding, cell–cell signaling, and others.1 In many cases, the polyvalent presentation of a ligand results in avidities that are orders of magnitude stronger than those for the corresponding monovalent interaction.1,2 Additionally, polyvalency can imbue novel properties to a ligand, such as the ability to cluster cell surface receptors.3,4 Thus, engineered polyvalent molecules can inhibit, enhance, and/or mimic natural processes.5−8 This review will primarily focus on papers written within the last five years that describe the development of novel polyvalent scaffolds and their applications in cellular targeting, toxin and pathogen inhibition, immune modulation, and controlling cell signaling.
1. Polyvalent Scaffolds
Recent strategies to develop polyvalent constructs have focused on improving the controlled display of ligands.9,10 Properties such as ligand density (spacing), valency, and orientation are being probed. Natural biological molecules serve as inspiration for well-defined and even responsive polyvalent display.1 This section reviews recent advances using (1) polypeptides, (2) oligonucleotides, (3) nanoparticles, and (4) stimulus-responsive materials as polyvalent scaffolds for ligand display.
1.1. Polypeptide-Based Scaffolds
Polypeptides are essentially polymers that can be precisely controlled with genetic coding. Protein engineering thus provides a powerful approach to design polyvalent molecules.11−18 Lee et al. recently used recursive directional ligation to engineer polypeptides to contain 20, 40, 60, or 80 integrin-binding RGD motifs spaced by SGSGSGSG linkers.15 After linking these polypeptides to a surface, the authors found that all of the polyvalent constructs provided increased cell-adhesion over the RGD monomer. The precise sequences allowed the group to assess specifically how the valency of the binding motif influenced the strength of cell adhesion under shear stress. The adhesion of the cells increased monotonically with increasing valency of the immobilized polypeptides.
We recently designed and synthesized a polyvalent inhibitor of anthrax toxin where multiple instances of an inhibitory toxin-binding peptide were separated by flexible peptide linkers (Figure 1).16 By independently controlling the valency and linker length, we elucidated key structure–activity relationships of the inhibitor. At the optimal conditions, the designed polyvalent inhibitors were over 4 orders of magnitude more potent than the corresponding monovalent ligands.
Figure 1.
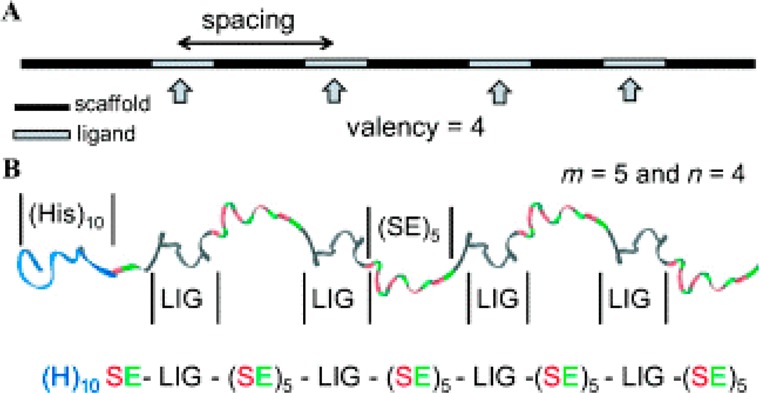
Designing monodisperse inhibitors of anthrax toxin. (A) Schematic representation of key factors affecting activity: spacing and valency. (B) Ribbon diagram for polypeptide inhibitor (H)10-SE[LIG-(SE)5]4. Reproduced with permission from John Wiley and Sons, Inc.16 Copyright 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Hollenbeck et al. also used the idea of a repeating peptide sequence to confer precise valency as well as spacing to polyvalent constructs.17 The ubiquitous and highly stable ankyrin repeat (AR) protein scaffold was used to control the spacing between ligands. The β-turns of each AR are amenable to mutations, and reactive thiols were inserted into selected ARs to give constructs with different spacing. By attaching mannose to two AR backbones with different spacing, the authors were able to probe how the spacing between mannose ligands affected the rate of concanavalin A aggregation. These rigid proteins could provide a framework for studying how the spacing and valency of ligands affects a variety of processes.
The natural assembly of peptides and proteins can be used to create polyvalent scaffolds as well. Brucella abortus Lumazine Synthase (BLS) naturally forms a decamer. Craig et al. fused complementary leucine zipper peptides to BLS monomers and ligand proteins.18 After purification, simply mixing the BLS and ligand protein caused self-assembly into decameric molecules. The globular polyvalent molecules were able to increase the immunogenicity of the murine Staufen protein. Furthermore, the authors verified that this leucine zipper approach worked with ligand proteins of different functions and molecular weights.
Peptidomimetic molecules have also been used to design polyvalent constructs. Levine et al. used a stepwise chemical synthesis technique to create peptoid-based polyvalent constructs.19 Using an N-substituted glycine oligomer scaffold, they produced a library of linear and cyclic scaffolds displaying azide moieties with specified spacing and valency. The highly efficient copper catalyzed alkyne–azide cycloaddition reaction (CuAAC) was then used to attach ethisterone ligands to the azides. The valency, spacing, and cyclization of the polyvalent peptoids all played key roles in modulating the activity of the androgen receptor complex.
1.2. Oligonucleotide-Based Scaffolds
As with polypeptide-based scaffolds, polyvalent molecules based on oligonucleotides can potentially control ligand spacing, valency, and three-dimensional conformation. Zhao et al. demonstrated that manipulating the flexibility and spacing of an RNA linker could reduce the dissociation constant of a polyvalent aptamer by 2 orders of magnitude.20 Heat shock factor 1-binding aptamers were connected by RNA linkers differing in their length and rotational flexibility. Similar to other studies, they found that the affinity of the polyvalent aptamers for Hsf1 increased when both the spacing and orientation of the aptamers matched those for the target. Along the same lines, Ahmad et al. recently showed that the nucleic acid linker between aptamers can be optimized using selection rather than design.21
Zhang et al. created a polyvalent scaffold using polyadenine sequences to prevent nonspecific and undesired base pairing within the backbone.22 They used rolling circle amplification to produce an ssDNA backbone that was complementary to the template plasmid. In this study, biotinylated uracils were evenly spaced throughout the polyadenine backbone, and anti-CD20 antibodies were then conjugated via a neutravidin linkage. The polyvalent antibodies showed enhanced binding to CD20 over the monomeric form. Moreover, the polyvalent constructs clustered CD20 and induced cell apoptosis more rapidly and effectively than the monomers.
Binding additional molecules to a nucleic acid backbone can expand the chemical functionality of nucleic acid–based scaffolds. Motivated by this thought, Englund et al. used l-lysine substituted peptide nucleic acids (PNAs) that hybridized with an ssDNA backbone.23 This strategy allowed for two levels of control: within the PNA structure itself, and in the ssDNA scaffold that the PNAs hybridize with (Figure 2). By independently altering the PNA and ssDNA structure, ligand density and valency could be varied over a large range. The l-lysine substituted PNA enabled the attachment of a wide variety of ligands via the reactive amine handle. Englund et al. used a library of ssDNA:PNA hybrids to examine how density and valency affect the binding and activity of the integrin-binding RGD peptide. In both in vitro cell binding assays and in vivo cancer metastasis studies, the polyvalent constructs improved the effective dose of monovalent RGD peptides by 2 orders of magnitude.
Figure 2.
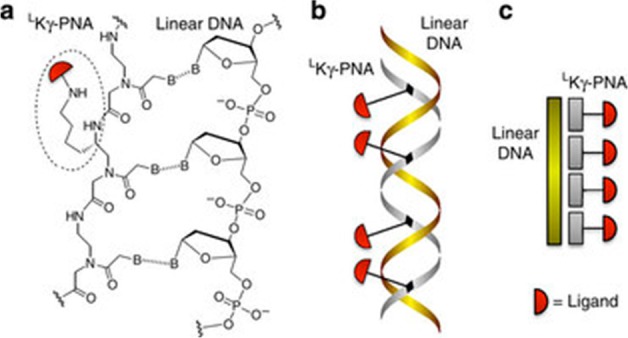
Representations of polyvalent LKγ-PNAs. (a) Chemical, (b) ribbon, and (c) cartoon diagrams of LKγ-PNA bound to DNA. Reprinted by permission from Macmillan Publishers Ltd.: Nature Communications,23 copyright 2012. http://www.nature.com/ncomms/index.html.
1.3. Nanoparticle Scaffolds
Nanoparticles have been used more extensively than any other polyvalent scaffold for purposes ranging from targeted drug delivery to MRI contrast agents.24−27 Often, the surface of a nanoparticle can be decorated with many copies of a ligand, thus enabling polyvalent interactions. Here we highlight some recent accomplishments using nanoparticle scaffolds for polyvalent display.
Hovlid et al. recently used cowpea mosaic virus nanoparticles as scaffolds to display the integrin-binding RGD motif.28 The peptide was introduced by genetic alteration of the coat protein as well as through NHS acylation followed by CuAAC. The genetically and chemically altered particles displayed the RGD peptide, on average, 60 and 80 times, respectively. After conjugating fluorescent tags to the particles, both were effective at labeling cancer cells expressing αν integrins.
Jeon et al. used a protein cage based on human ferritin to polyvalently display an interleukin-4 receptor binding peptide, AP1.29 The human ferritin light chain naturally self-assembles into 12 nm particles. To display AP1, the peptide was fused into an exterior loop of the ferritin chain. The produced nanoparticles successfully bound to IL-4R with a much higher affinity than the peptide ligand itself. The AP1 displaying particles also specifically bound to cells expressing IL-4R. Finally, in an in vivo murine asthma model, the particles were able to reduce airway constriction and the number of inflammatory cells produced.
Gujrati et al. used nanoparticles generated from Escherichia coli.30 Outer membrane vesicles (OMVs) are readily discharged from Gram-negative E. coli and contain cell-surface proteins. OMVs containing antibody-mimetic affibodies specific to HER2 were produced by overexpressing the affibody fused to the toxin protein ClyA, which sends the protein to the outer membrane. The discharged OMVs showed greater binding to HER2 expressing cells than the affibody alone. The nanoparticles were further modified to contain siRNA. The particles successfully targeted tumors in vivo, and the siRNA helped reduce tumor growth even after tumors had been established.
1.4. Stimulus-Responsive Scaffolds
A new class of polyvalent scaffolds includes stimulus-responsive materials. These smart materials can alter their properties in response to stimuli like temperature, light, pH, molecular cues, and magnetic fields.31 Of particular interest to this Review are scaffolds that can modulate their polyvalent presentation of a ligand based on changes in stimuli.
Yu et al. presented the thermal reversal of polyvalent binding based on temperature (Figure 3).32 Using oligo(ethylene oxide) methacrylate and 2-(2-methoxyethoxy) methacrylate, they developed linear copolymers that had lower critical solution temperatures (LCSTs) near 33 °C. Choline phosphate (CP) groups were added via CuAAC after azide modification. At temperatures above the LCST the polymer contracted due to hydrophobic collapse and the CP residues were no longer accessible to bind biomembranes. The polymers could bind to cells below the LCST, and the cells were released upon heating the system above the LCST. Moreover, the process was reversible, enabling cells to be recaptured upon cooling.
Figure 3.
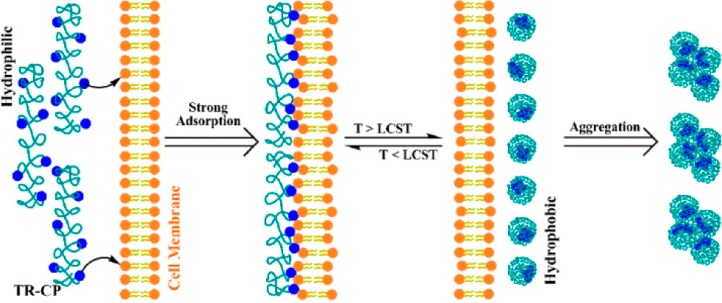
Illustration of temperature-dependent polyvalent binding driven by phase transition. Below the LCST, the choline phosphate groups tightly bind the cell membrane. The binding can be reversed by increasing the temperature above the LCST. Reprinted with permission from ref (32). Copyright 2013 American Chemical Society.
Thermoresponsive materials need not be synthetic polymers. Hassouneh et al. showed that single domain proteins could be assembled into polyvalent micelles by fusing them to thermally responsive elastin-like peptides (ELPs).33 The block copolymer-like structure of hydrophobic and hydrophilic domains within the ELPs drove micelle formation above the critical micelle temperature (CMT); this CMT could be tailored by changing the ELP sequence. Moreover, the transition from monomers to micelles was reversible. When a fibronectin type III domain was fused to the ELPs, the resulting micelles showed enhanced targeting to transfected human leukemia cells that overexpress ανβ3 relative to the monomeric fusion proteins. Future studies could develop polymer-, peptide-, and oligonucleotide-based scaffolds that exhibit or rescind polyvalent effects when triggered by light, chemical cues, and other stimuli.
2. Polyvalency in Targeting
Often the dissociation constant of a ligand can be improved, that is, decreased, from the micromolar to the nanomolar range by using polyvalency.16 This attribute makes polyvalency especially apt for selective targeting. Superselectivity is the phenomenon where the adsorption of a polymer/polyvalent molecule on a surface increases faster than linearly with the density of surface binding sites.34 Such superselectivity could, for instance, enable enhanced discrimination between cells expressing different levels of a target receptor. Dubacheva et al. designed a “guest-host” model to help understand how polyvalency can impart superselective targeting to a system.35 Cyclodextrin molecules served as the hosts for ferrocene ligands (guests) (see Figure 4). The cyclodextrin was conjugated to a hyaluronic acid polymer backbone. Ferrocene monolayers were then attached to a solid support at different surface densities, and the cyclodextrin conjugates were allowed to adsorb onto the surface. Dubacheva et al.35 found that even when the average distances between the individual cyclodextrin and ferrocene molecules were mismatched, there was a strong, positive nonlinear relationship between polymer binding and ligand surface density that has also been seen elsewhere.36 They proposed that both enthalpic and entropic effects played a role, in keeping with previous theoretical models.37,38 Another key aspect contributing toward superselectivity was that the long and flexible polymer chains were capable of interpenetration: they could bend and fold to bind to ligands even when the spacing was mismatched. This flexibility of polyvalent polymeric scaffolds offered enhanced selectivity compared to other scaffolds such as polyvalent nanoparticles.34,35
Figure 4.
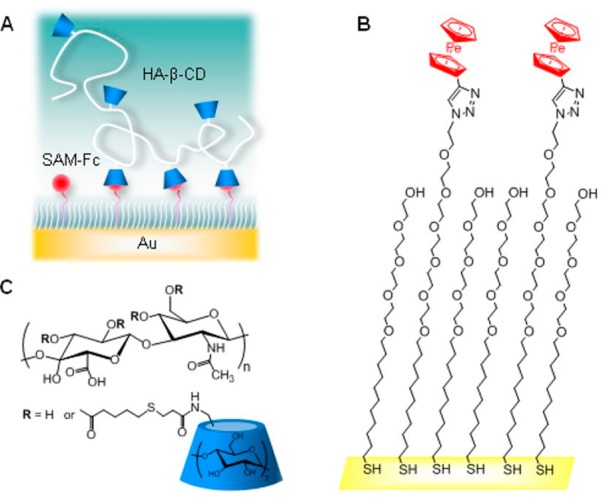
Representation of the “guest-host” model system. (A) Hyaluronic acid-β-cyclodextrin (HA-β-CD), the hosts, polyvalently bind to self-assembled monolayers of ferrocene (SAM-Fc), the guests. Chemical structures of (B) SAM-Fc and (C) HA-β-CD. Reprinted with permission from ref (35). Copyright 2014 American Chemical Society.
A plethora of research on targeting various markers in cancer using polyvalency is producing some promising results. One interesting development has been the use of amphiphilic aptamer–polymer conjugates that are capable of self-assembly in an aqueous environment. Yu et al. designed aptamer-functionalized hyperbranched copolymer conjugates that self-assembled into nanosized micelles with a core–shell structure and a narrow size distribution.39 When displayed on the shell, targeting ligands such as folic acid, antibodies, and aptamers could be used to guide these systems specifically to tumors. The authors used an aptamer-based system that selectively targets the breast cancer cell line MCF-7 and exhibits low cytotoxicity. By conjugating a fluorescein molecule to the core-forming polymer, these particles selectively labeled MCF-7 cells in vitro.
Yang et al. developed a novel approach to graft cancer-cell-targeting DNA aptamers directly into a polyacrylamide backbone.40 By fusing the aptamers to acrydite, mixing with nonactivated or fluorescent acrylamide monomers and polymerizing, polyvalent aptamer conjugates were produced. The sgc8c aptamer that selectively targets a human T-cell lymphoblast cell-line (CEM) showed increased binding in the polyvalent form. Furthermore, the polyvalent aptamer induced internalization of the entire macromolecule. The internalized polyvalent molecules were cytotoxic, whereas the aptamers alone were not. Thus, the authors demonstrated that the polyvalent display not only enhanced the targeting ability of the aptamer, but also imparted a novel function, cytotoxicity.
Zhang et al. have incorporated a leukemia cell-binding aptamer into a Poly-Aptamer-Drug (PAD) system, which is composed of multiple aptamer units synthesized by rolling circle amplification and physically intercalated chemotherapy agents.41 Doxorubicin was used as the drug, which was coupled with a leukemia cell-binding aptamer. This polyvalent system targeted cancer cells with a near 40-fold improved binding affinity as compared to the monomeric aptamer. Furthermore, the PAD internalization induced leukemia cell death. The PAD design was highly tunable because doxorubicin could be replaced by other substitutes in the drug loading domain, and the spacing and valency of the aptamer could be varied.
In another recent article,42 Thomas et al. demonstrated improvements to a system to target cancer cells that they had discussed elsewhere.43−47 Their new system, consisting of a generation 5 dendrimer conjugated to multiple copies of methotrexate and folic acid, had an improved selectivity for the folate receptor, a higher chemotherapeutic index, and a ∼4300 fold increase in affinity over methotrexate alone. These modified conjugates successfully inhibited dihydrofolate reductase and induced apoptosis of cancer cells in vitro.
Brabez et al. created bis-ornithine-based conjugates of melanotropin.48 These trivalent constructs improved the affinity of melanotropin to a model melanoma cell by 350-fold. Josan et al. synthesized heterobivalent ligands to target certain receptor combinations commonly found on cancer cells with high specificity and avidity.49 They optimized heterobivalent ligands that contained analogues of melanocortin- and cholecystokinin-binding peptide ligands joined by linkers of appropriate length (20–50 Å). These ligands exhibited a 24-fold improvement in binding affinity to cells expressing both receptors compared to that for cells expressing either one of the two cognate receptors.
Wang et al. successfully exploited the overexpression of transferrin receptor (TfR) in cancer cells by synthesizing PRINT nanoparticles functionalized with TfR ligands for targeted drug delivery.50 These TfR-based conjugates showed selective uptake by the human tumor cell lines, as compared to negative controls. Elevated caspase 3/7 activity confirmed the activation of apoptosis pathways. The targeting efficiency was a function of particle concentration, ligand density, dosing time, and cell surface receptor expression level.
Liposomes have been used extensively as vehicles to carry drugs and imaging agents to a particular site.51 Mann et al. developed long-circulating liposomes functionalized with a thioaptamer targeting E-selectin (ESTA).52 They evaluated the targeting efficiency and pharmacokinetic parameters, followed by in vitro and in vivo tests. The tests confirmed the efficient uptake of ESTA-conjugated liposomes, as they were shown to accumulate in the tumor vasculature of breast tumor xenografts.
Viral nanoparticles (VNPs) have also been used to target cancer cells. Steinmetz et al. recently exploited the ability of the cowpea mosaic virus (CPMV) to bind to vimentin—a potential tumor marker—to direct these polyvalent nanoparticles to tumors in vivo.53 Pokorski et al. also recently used a VNPs to target cancer cells.54 They produced hybrid Qβ VNPs that expressed endothelial growth factor (EGF). The polyvalent display of EGF targeted the particles to A431 cells that, like many cancer cells, overexpress the EGF receptor.
The endothelial lining of blood vessels is also a viable target for many known diseases. Polymeric filomicelles are filamentous or worm-like micelles made by self-assembly from polymers of suitable block ratios, and they offer the advantage over spherical particles of being long and flexible; thus contributing to a prolonged circulation time and supporting their use for targeted drug delivery to the endothelial cells.55,56 Shuvaev et al. synthesized polymeric filomicelles decorated with antibodies that target certain motifs on the endothelial lining.57 By characterizing these conjugates in vitro and in vivo, the authors demonstrated that the filomicelles not only retained their structural integrity and flexibility, but were also able to adhere to endothelial cells with high specificity.
Vancomycin is a third generation antibiotic that was first discovered in the early 1950s.58 Vancomycin inhibits cell wall synthesis in susceptible bacteria by binding to the d-Ala-d-Ala peptidoglycan subunits and preventing further cross-linking steps.59 The most common obstacle to the long-term success of an antibiotic is the development of resistance. Vancomycin-resistant enterococci (VRE), for example, have modified d-Ala-d-Lac residues on their surface that have a lower affinity for vancomycin than the d-Ala-d-Ala residues on vancomycin-susceptible species.60 To address this shortcoming, Choi et al. designed G5 poly(amidoamine) (PAMAM) dendrimers functionalized with vancomycin at the C-terminus.60 Tests in vancomycin-resistant bacterial cell wall models indicated a 4 to 5 orders of magnitude enhancement in avidity for these conjugates over free vancomycin to d-Ala-d-Lac residues. The higher avidity enables vancomycin to bind VRE cell walls and overcomes the main resistance factor in VRE. This study also demonstrated that the dendrimer conjugates targeting bacteria could be used to coat iron oxide nanoparticles. The authors then exploited this approach along with the speed and convenience of magnetic isolation technology to isolate, enumerate, and sequester bacteria.
3. Polyvalency in Inhibition
Polyvalency is an established effective strategy to design potent inhibitors of toxins and pathogens.1,2,8,61,62 By strictly controlling the valency of the ligands and the spacing between them, polyvalency has enabled the synthesis of inhibitors that are orders of magnitude more active than the corresponding monovalent ligands. Some of the recent advances in this area are described below.
Jacobson et al. synthesized potent Shiga toxin inhibitors.63 They found that a Pk trisaccharide containing a terminal 2-acetamido-2-deoxy-α-d-galactopyranosyl residue instead of the terminal α-d-galactopyranosyl residue displayed preferential binding to the more dangerous Stx2 Shiga toxin (compared to the Stx1 counterpart). The elimination of toxin activity was through supramolecular complex formation between the trisaccharide inhibitor, Stx2 and Human serum amyloid P.
Tran et al. designed cholera toxin inhibitors using polyvalency.64 GM1, a complex glycolipid, is the primary receptor for the cholera toxin. Galactose is the key anchoring residue on GM1 for cholera toxin, but it binds weakly to the toxin, even when displayed polyvalently. To probe the second binding site on GM1, the authors created a library of heterobifunctional ligands composed of an invariable galactose residue and a variable fragment. These heterobifunctional ligands were then made polyvalent by conjugating them to polyacrylamide and dextran backbones. Two of the screened ligands showed a 10-fold reduction in IC50 value over galactose alone. Dextran-based conjugates of the selected heterobifunctional ligands also demonstrated enhanced inhibition of cholera toxin, with IC50 values in the nanomolar range, whereas dextran-galactose conjugates showed no detectable activity.
Maheshwari et al. demonstrated that the manipulation of electrostatic backbone charge, control of the backbone extension, ligand spacing, and saccharide linker interactions were crucial while designing glycopolypeptide-based inhibitors against cholera toxin.65 Peptides with the sequence AXPXG (where X is a positive, negative, or neutral amino acid) were functionalized with propargyl glycine groups and glycosylated using CuAAC. They demonstrated that glycopeptides with a negatively charged backbone showed enhanced inhibition as compared to the other conjugates. Moreover, saccharide linker conformation and the location of charged residues along the polypeptide backbone greatly impacted the potency of the inhibitors.
Saccharide-based polyvalent inhibitors have also been designed against Ebola virus. Ribeiro-Viana et al. used the concept of “nested layers of polyvalency” and constructed glycodendrinanoparticles bearing as many as 1620 glycans that mimic the size and highly glycosylated surfaces of viruses.66 These polyvalent conjugates bound to DC-SIGN receptors on T-lymphocytes and competitively inhibited their infection by the Ebola virus, even at picomolar concentrations (Figure 5).
Figure 5.

Schematic representation of nested polyvalency. (a) Glycodendrons are iteratively synthesized and attached via a tag to multiple protein monomers. (b) Competitive inhibition of a virus (shown in red). Reprinted with permission from Macmillan Publishers Limited: Nature Communications,66 copyright 2012. http://www.nature.com/ncomms/index.html.
A pseudosaccharide is a sugar in which the oxygen in the pyranoid ring has been replaced by a methylene group.67 This modification is advantageous due to the enhanced stability against glycosidase-induced hydrolysis. Luczkowiak et al. developed pseudosaccharide-functionalized dendrimers with an affinity for the DC-SIGN receptor.68 These conjugates inhibited DC-SIGN-dependent, Ebola viral infection of a Jurkat cell line. The researchers also demonstrated that the polyvalent glycomimetic polyester dendrimer systems were nanomolar inhibitors of Ebola viral infections.
Noroviruses bind to their host cells through histo-blood group antigens (HBGAs), and compounds that interfere with this interaction have therapeutic or diagnostic potential.69 Rademacher et al. created polyvalent entry inhibitors against norovirus after identifying small molecules that bind to or adjacent to known HBGA binding sites on the norovirus surface.69 Inhibition assays showed avidity gains of thousand and million fold for two polyvalent inhibitors synthesized relative to a monovalent ligand that had dissociation constants in the millimolar range.
There have been several previous reports of the use of polyvalency to design inhibitors of influenza viruses.1,70−72 Papp et al. studied the functionalization of polyglycerol-based nanoparticles with sialic acid.73 They used biocompatible, hyper-branched polyglycerols and polyglycerol-based nanogels, where size manipulation between the ranges 2–4 nm and 40–100 nm was possible so as to match the receptor multiplicity and size of a virus. The researchers specifically studied the effects of the size of the nanoparticles and the ligand density on the efficiency of inhibition. They found that as the particle size increased, the inhibition was more efficient, with sizes matching that of a virus particle (50–100 nm) being most effective on a per-ligand basis. Increasing the ligand density also increased inhibition up to a “saturation” point, beyond which the inhibition was no longer seen to improve significantly.
The concept of polyvalency has also been used to design inhibitors of HIV infection.8,74−77 Danial et al. synthesized a series of synthetic polymer conjugates presenting peptide sequences derived from the complementarity determining region H3 of the anti-HIV-1 antibody IgG1 b12.78 These conjugates blocked the CD4-binding sites on gp120, thus preventing the entry of HIV-1 into the host cell. Danial et al. found that midsized polymer conjugates showed maximum viral inhibition, while the shorter and longer sized counterparts were not as effective. They hypothesized that the shorter conjugates showed poor inhibition because their length was insufficient to span the distance between the receptors, while the longer conjugates failed to show maximum inhibition due to the high entropic penalty that resulted from their binding to gp120. Even though these conjugates had a higher IC50 value than the IgG1 b12 antibody, some of the advantages of this system included low production and purification costs, high thermal and chemical stability in storage conditions, long half-life in biological tissues, low immunogenicity, and protection from proteolytic degradation.
The external envelope glycoprotein of HIV can also aid in the attachment of the virus to certain glycosphingolipids (GSLs) on the surface of T-cells and peripheral blood mononuclear cells (PBMCs).79 Rosa Borges et al. demonstrated that a dendrimer core to which GSLs such as globotriose and 3′-sialyllactose were attached successfully inhibited the entry of HIV-1 into host cells, with IC50 values ranging from 0.1 to 7.4 μg/mL.80 These multivalent carbohydrates mimicked the clustered carbohydrates on the host cell surface and thereby inhibited the gp120-mediated membrane fusion of the HIV envelope protein with the target cell membrane.
4. Polyvalency and the Immune System
Many if not most of the interactions of the immune system with antigens and pathogens involve polyvalency. Thus, it is not surprising that polyvalent systems have shown promise in immunosuppression applications, such as for inhibiting allergic reactions and autoimmune diseases. In addition, well-defined synthetic polyvalent systems have also been useful for elucidating the mechanisms that determine the biological responses to antigens.
4.1. Immunosuppression
One of the fields in which polyvalent systems are playing an important role is in the study of allergic responses. Allergies are known by the scientific classification “type I hypersensitivity,” and are the result of the adaptive immune system detecting multiple sites on normally harmless substances.81 These polyvalent allergens are bound by multiple IgE antibodies, which results in clustering or cross-linking of the high-affinity IgE receptor, FcεRI.82,83 This clustering in turn initiates a phosphorylation signaling cascade that leads to Ca2+ mobilization and mast cell degranulation. Degranulation causes the release of mediators such as histamine and β-hexosaminidase, which provoke the powerful inflammatory responses that are characteristic of allergic reactions.82,83 Polyvalent constructs are being used to shed light on the key factors that affect the recognition of allergens by IgE and the associated cross-linking of the FcεRI receptors that causes degranulation. Moreover, polyvalent inhibitors are being designed for blocking IgE binding and the subsequent allergic attack.
Several researchers have made use of synthetic antigens based on the hapten 2,4-dinitrophenyl (DNP) for studying mast cell degranulation mechanisms as well as methods for inhibition.82,84−91 Paar et al.84 and Baird et al.85 respectively examined the effects of rigid or flexible linkers for presenting DNP bivalently, which typically led to inhibition of degranulation due to an inability to cross-link more than two IgE-bound FcεRI. More recently, Sil et al.82 used double-stranded DNA to create trivalent degranulation effectors with various arm lengths (Figure 6). These synthetic antigens were employed in a variety of experiments to determine the spatial constraints on the degranulation signaling cascade. Based on the observation that levels of FcεRI phosphorylation decreased progressively after treatment with effectors having increasing arm length, the findings supported a model of FcεRI transphosphorylation in which the presence of multiple FcεRI domains in close proximity amplifies the signal. Furthermore, Ca2+ mobilization and degranulation experiments showed that the effector with the longest arms was less potent than the other three effectors. The authors concluded that there may be a threshold level of transphosphorylation required for further signal propagation.
Figure 6.
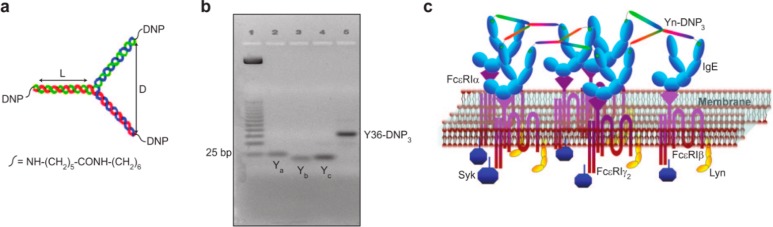
DNA-scaffolded degranulation effectors. (a) Depiction of a rigid trivalent DNA scaffold. (b) Agarose gel characterization of the size of the trivalent, Y-shaped inhibitors. (c) A representation of the proposed interaction between the trivalent synthetic antigens and anti-DNP IgE, leading to FcεRI clustering and the initiation of the degranulation signaling cascade. Reprinted with permission from ref (82). Copyright 2007 American Chemical Society.
In a different system, Huang et al. used gold nanoparticles as a well-defined vehicle for presenting DNP to anti-DNP-IgE-primed mast cells.86 The investigators varied two parameters: the size of the gold nanoparticles, and the density of DNP on the nanoparticle surface. The experimental results showed that for gold nanoparticles with a saturating amount of DNP-thiol conjugated to the nanoparticle surface, there was a positive correlation between the extent of degranulation and nanoparticle size for particles between about 7 and 20 nm in diameter. For particles of between 20 and 50 nm in diameter, the ability to induce degranulation plateaued. The authors argued that only the nanoparticles of 20 nm diameter and larger were able to effectively cross-link the FcεRI receptors.
Next, the researchers varied the density of DNP on the nanoparticle by attaching another small molecule thiol, 3,3′-dithioldipropionic acid, to fill up the available nanoparticle surface between DNP. They found that for 20 nm diameter nanoparticles, the degranulation response decreased with decreasing amounts of DNP on the surface.86 Furthermore, they found that there existed a low DNP surface coverage (10 DNP per nanoparticle) for which the nanoparticles were able to inhibit the normal degranulation response stimulated by a polyvalent antigen: bovine serum albumin conjugated with multiple DNP haptens (BSA-DNP). The authors concluded that the 20 nm diameter nanoparticles with 10 DNP haptens on the surface were able to bind efficiently to the IgE in a manner that was not only unable to cross-link the FcεRI receptors, but was also able to block access of the BSA-DNP antigen to the IgE receptors.
Handlogten et al.87 designed synthetic antigens of a homotetravalent design that was too small to simultaneously bind both arms of a single IgE (i.e., bivalently), but that could simultaneously bind up to four separate IgEs monovalently. The researchers were able to use the homotetravalent scaffold to display a variety of haptens having a range of monovalent affinities, and they found that for this series of synthetic homotetravalent antigens (HmTAs), there was a minimum hapten-antibody affinity below which mast cell degranulation could not be triggered (Kd higher than 105 nM). In addition, the series of HmTAs were used to study how variations of the percentage of hapten-specific IgE versus “orthogonal” IgE on the mast cell surface affected degranulation. For all of the synthetic antigens tested, the maximum amount of degranulation occurred when 25% of the IgE population on the mast cell surface was hapten-specific. The authors note that previous studies have shown an inverse correlation between the size of cross-linked IgE receptor aggregates and the strength of the degranulation response; they hypothesized that the 25% hapten-specific IgE surface coverage allowed for the formation of smaller aggregates capable of initiating the most potent degranulation signal.87
Handlogten et al.88 then furthered these studies by modifying the scaffold to display two copies each of two different haptens, resulting in synthetic heterotetravalent antigens (HtTAs) that were expected to be a more accurate representation of the heterogeneous nature of the antigenic epitopes on naturally occurring allergens. To that end, the haptens were chosen so that there was a difference in hapten-IgE affinity of 2.5-fold (HtTA-1) or 10-fold (HtTA-2).88 The HtTAs were exposed to mast cells which were primed with varying percentages of three different IgEs, including one orthogonal IgE and two hapten-specific IgEs. This experimental system corroborated the previous results with HmTAs, whereby mast cells having significantly less than complete coverage of hapten-specific IgE exhibited more-robust degranulation.88 In addition, homobivalent antigens synthesized using the same haptens and scaffold as the HtTAs were shown to be unable to achieve the level of cross-linking required for degranulation, even for the high-affinity hapten-IgE pair.88 However, the addition of low-affinity haptens to the high-affinity homobivalent antigen enabled the resulting HtTAs to induce degranulation.88 Collectively, these results highlight valency, affinity, and cooperativity as being important components of the allergen-IgE interaction. Furthermore, the authors demonstrated the dependence of degranulation on the availability of both types of IgE, a result that countered previous assumptions that only the high-affinity IgEs were significant.88
The authors then demonstrated the application of this finding for the inhibition of the allergic response by selectively inhibiting the low-affinity hapten-IgE interaction (Figure 7).89 To that end, the researchers employed a heterobivalent inhibitor design that exploited the presence of a conserved, nucleotide-binding site on the Fab of IgEs.89 The inhibitor was synthesized by coupling a nucleotide analogue to the known low-affinity hapten via a flexible linker. The binding affinity of the hapten on the inhibitor was thus enhanced relative to the hapten on the HtTA due to the polyvalent interaction, and this was sufficient to block the cross-linking of IgE by the HtTAs.89 Thus, these studies have not only elucidated mechanistic information about mast cell degranulation, but have also culminated in a proof-of-concept study for the inhibition of the allergic response.
Figure 7.

Inhibition of allergic response using polyvalency. (a) Structure of the melon allergen Cuc m 2. (b) Scheme showing the binding of polyclonal IgE to allergen in a typical allergic reaction. (c) Scheme showing the binding of a combination of low-affinity and high-affinity hapten-specific IgEs to the synthetic antigen HtTA, resulting in mast cell degranulation. (d) Close-up view of an IgG to highlight the locations of the conserved nucleotide- and antigen-binding sites. (e) Scheme showing the inhibition of mast cell degranulation by HtTA due to coadministration of a heterobivalent inhibitor. Reprinted by permission from Macmillan Publishers Ltd.: Nature Chemical Biology,89 copyright 2013. http://www.nature.com/nchembio/index.html.
Similarly, polyvalent conjugates have been designed by Courtney et al. to study the activation and inhibition of B cell activation.90 B cells are a class of lymphocytes that help the immune system distinguish between self-and foreign organisms, primarily through the activation state of a surface protein called the B cell receptor (BCR). While previous work had shown that polymers displaying a larger valency of DNP were more potent activators of B cells than lower valency polymers,91 Courtney et al. were interested in further probing the determinants of BCR activation. To that end, they synthesized linear polymer polyvalent displays of DNP and/or sialylated trisaccharides that would interact specifically with the BCR and/or the inhibitory receptor CD22, respectively.90 CD22 was previously known to be a receptor for glycoconjugates featuring α2,6-linked terminal sialic acid residues, which are also prevalent on the surface of B cells. Interactions between these glycans and CD22 that occur within the same cell membrane are termed “cis” interactions, while those that occur between CD22 and glycans on another cell or antigen are termed “trans” interactions. Previous studies had been unable to demonstrate any noticeable trans interactions or any effects thereof, possibly due to a masking effect of the polyvalent cis interactions.90 Despite this, it was known that CD22 colocalizes with BCR and inhibits the BCR activation signal. However, the precise molecular mechanisms by which this inhibition is achieved is unknown. By treating B cells with polymers polyvalently displaying DNP and/or sialylated trisaccharide, Courtney and colleagues were able to show that the CD22 ligands were able to inhibit B cell activation, but only when they were co-conjugated on the same polymer backbone as DNP.90 That is, polyvalent DNP was able to bind and cluster BCR, which causes cell activation. In contrast, when the CD22 ligands were co-conjugated, CD22 was colocalized due to trans binding interactions, thus preventing the phenomenon of cis binding from sequestering CD22 away from the clustered BCR. The net effect of this trans-induced CD22 and BCR colocalization was inhibition of B cell activation, as tracked by calcium flux, tyrosine phosphorylation, and the presence of other characteristic proteins in the activation pathway.90 Therefore, the idea that CD22 ligands help B cells with self-recognition was reinforced, and this knowledge could be useful for improving tolerance of macromolecular therapeutics or treating autoimmune disorders.
4.2. Bioengineered Vaccines
Viruses and bacteria typically interact with their hosts via polyvalent ligands and receptors, so the immune system may be especially sensitive toward polyvalent displays of biomolecules. Indeed, appropriately designed polyvalent biomolecules may exhibit increased immunogenicity relative to their monovalent or dimeric counterparts.
Wang et al. developed a strategy to generate vaccines that take advantage of the adjuvant effect that polyvalent presentation creates.92 They generated fusion proteins composed of two or three proteins that typically form homodimers, as shown in Figure 8. Depending on whether the fusion proteins were composed of two or three homodimer-forming proteins, the fusions self-assembled into polyvalent complexes with linear or network morphologies, respectively. In a proof-of-concept study, the researchers used dimeric glutathione S-transferase and two different protruding domains of norovirus as the homodimer-forming protein components of the fusion protein “basic unit” from Figure 8. In a follow-up publication, Wang et al.93 made use of the protruding domain of hepatitis E virus. In all cases, the resulting fusions self-assembled into large polyvalent complexes with a linear configuration when the protein fusions were composed of two dimeric proteins (Figure 8A,C), and a network configuration when the protein fusions were composed of three dimeric proteins (Figure 8B,D). After purification, the researchers compared the immunogenicity of the polyvalent complexes to equal amounts of nonfused homodimers. They found that immunization with either type of polyvalent complex could produce significantly higher antibody titers against both types of norovirus protruding domains compared to a mixture of the homodimers of the two domains. Furthermore, the group was able to use the polyvalent complexes as a platform for polyvalent presentation of other monomeric viral antigens, which they demonstrated by creating additional fusions with either the influenza virus peptide epitope M2e or the rotavirus VP8* antigen (Figure 8C,D). These additional antigens were placed at loop 2 of the norovirus protruding domain, an exposed surface loop. Again, the polyvalent complexes displaying the monomeric antigens were significantly more immunogenic than the corresponding free antigens, as measured by assaying the neutralizing activity and the protective immunity of mice against rotavirus and influenza virus. The authors argue that this demonstrates the feasibility of using their technique as a vaccine platform that can simultaneously provide immunization against multiple viruses or viral subtypes.93
Figure 8.
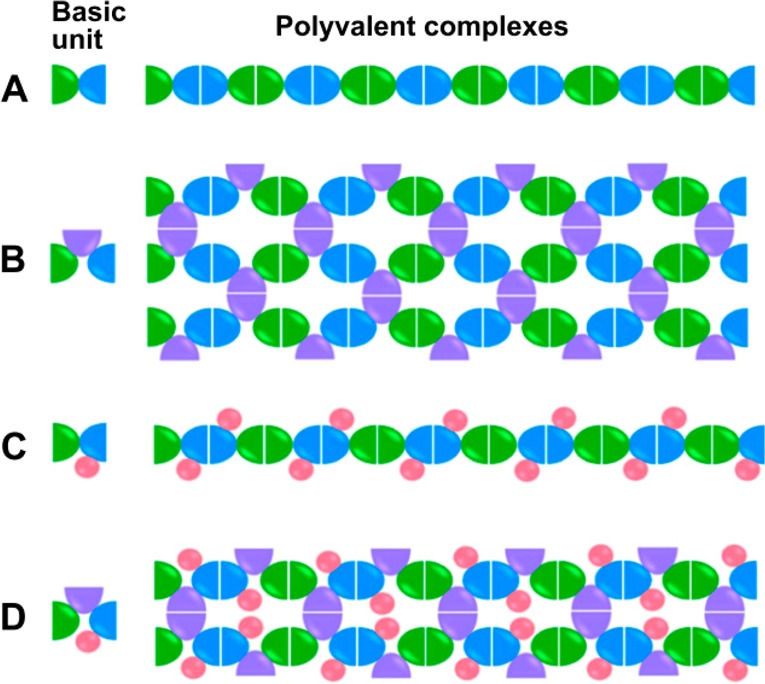
Depiction of polyvalent complex formation. For each type, A–D, the basic unit (one protein fusion) is shown at the left, with each color indicating a part of the fusion that consists of a homodimer-forming protein (blue, green, and purple) or nonhomodimer-forming protein for antigen display (pink). Depending on the number of homodimer-forming proteins included in the basic fusion protein unit, the resulting self-assembled polyvalent complexes were of a linear (A,C) or network (B,D) morphology, as seen in micrographs in the original publication. Reprinted from Wang et al.,92 Copyright 2013 with permission from Elsevier.
Another type of bioengineered protein vaccine that may benefit from a polyvalent display was described by Kanekiyo et al.94 Influenza hemagglutinin protein (HA) fusions with ferritin were used to create nanoparticles composed of 24 identical fusion proteins (Figure 9). The hemagglutinin was oriented such that the hemagglutinin self-assembled into eight trimeric viral spikes. The vaccine outperformed the standard inactivated vaccine at producing hemagglutination inhibition antibody titers in both mice and ferrets, and also produced broadly neutralizing antibodies targeting the conserved stem and receptor binding sites.94 It is possible that the larger interspike angles and oligovalent display allowed for greater exposure of the hemagglutinin trimer epitopes to the adaptive immune system than for a typical inactivated virion.
Figure 9.

Influenza vaccine based on ferritin particles. (a) The residue Asp 5 on ferritin molecules was used to conjugate HA molecules because it is oriented outward and is spaced correctly in the assembled particle to assemble the HA trimer. (b) Electron micrographs of ferritin nanoparticles before and after conjugation with HA. Reprinted by permission from Macmillan Publishers Ltd.: Nature,94 copyright 2013. http://www.nature.com/nature/index.html.
5. Polvalency in Cell Signaling
The simultaneous binding of multiple ligands to multiple cellular receptors can lead to receptor clustering.1,3,4 Many signaling cascades within the cell begin in such a manner.95−97 Polyvalent molecules provide an excellent framework to induce clustering of receptors by increasing the local concentration of ligands as well as by sterically constraining bound receptors closer together.98 This section will review recent advances using polyvalency to affect cellular signaling cascades.
Conway et al. showed that these polyvalent interactions can be used to control stem cell fate both in vitro and in vivo (see Figure 10).99 Eph-Ephrin signaling has been shown to regulate neural stem cell differentiation. Conway et al. covalently attached the ectodomain of ephrin-B2 to monodisperse hyaluronic acid polymers. The polyvalent constructs both clustered receptors and induced neural stem cell differentiation. Furthermore, as the average spacing between the ligands was decreased, receptor clustering was enhanced. The effector molecules were also tested in vivo. After injection into the rodent brain, the polyvalent conjugates increased the neurogenesis over the controls by 60%.
Figure 10.
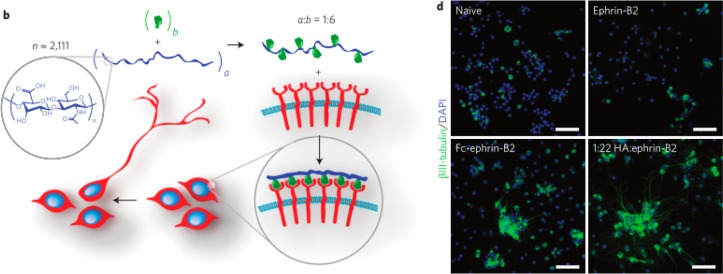
Polyvalent ephrin-B2 conjugates control stem cell fate. (b) Depiction of polyvalent molecules leading to cell differentiation. (d) Images of neural stem cells differentiated in the presence of monovalent, antibody clustered, and polyvalent ephrin-B2. Cells were immunostained for the neuronal marker βIII-tubulin (green) and total nuclei (blue). Adapted by permission from Macmillan Publishers Ltd.: Nature Nanotechnology,99 copyright 2013. http://www.nature.com/nnano/index.html.
Sonic hedgehog (Shh) is another signaling protein responsible for differentiating human pluripotent stem cells (hPSCs) into neurons. Vazin et al. probed the effect that polyvalency has on the strength of Shh signaling.100 Again, the authors covalently attached the protein to hyaluronic acid polymers at a range of stoichiometries. The valencies of the conjugates were determined by size exclusion chromatography coupled to multiangle light scattering. In in vitro assays, polyvalent constructs with higher valencies triggered more cells to differentiate into dopaminergic and GABAergic neurons than did their monomeric counter parts.
Polyvalent ligands based on viral particles have shown the ability to affect cell fate as well. Lee et al. displayed the integrin-binding RGD motif on the tobacco mosaic virus (TMV).101 Surfaces for cell adhesion and growth were either coated with the RGD-presenting TMVs or fibronectin. Bone marrow derived mesenchymal stem cells (BMSCs) were then attached to the surfaces and grown in a selective media with growth factors. After only 2 days, differentiation occurred as analyzed by the presence of key transcription factors as well as cell morphology. This rapid differentiation only occurred when the cells were coated onto the VLPs, while not on fibronectin covered surfaces. The highly patterned and densely packed display of the RGD motif accelerated the differentiation process.
Webber et al. showed the utility of peptide amphiphiles to effect angiogenesis after ischemic tissue damage.102 A peptide that mimics VEGF, a key angiogenic factor, was fused with a C16 tail that promoted self-assembly into cylindrical nanostructures. The peptide amphiphile (PA) was first shown to increase the phosphorylation of VEGF receptor. In vivo studies showed enhanced angiogenesis of embryonic tissue when treated with the PA over the peptide alone. The PA was then tested against full monomeric VEGF protein to repair murine hind limb tissue damage. While both treatments increased motor abilities, the PA treatment increased the capillary density significantly more than the VEGF protein treatment.
Conclusions and Perspectives
Polyvalency has penetrated many areas of biotechnology. Recent efforts in polyvalent scaffold design have allowed researchers to more effectively match the scaffold to the application. Each type of scaffold presents distinct advantages and challenges. With the wide ranging applications of polyvalency in nature, no one scaffold type will work in every situation. On the other hand, there may be applications where multiple types of scaffolds work equally well. Therapeutic use of polyvalent molecules will also require careful evaluation and optimization of key characteristics including immunogenicity, routes of delivery, and pharmacokinetics.
The characterization and optimization of polyvalent interactions will benefit from advances in experimental techniques that provide greater spatial and temporal resolution as well as important structural information. Some of the experimental techniques that have been used to probe polyvalent interactions have been highlighted in previous reviews.1,2,5,6 In the future, super-resolution imaging techniques such as photoactivated localization microscopy (PALM)103 and stochastic optical reconstruction microscopy (STORM)104 will provide a deeper understanding of polyvalent complexes, for instance by helping analyze the interaction of polyvalent molecules with cell-surface receptors.99 The specific binding sites on polyvalent ligands and their receptors can be elucidated using techniques such as saturation transfer difference-NMR and crystallographic methods.105−107 Single-molecule force spectroscopy108 using optical tweezers and atomic force microscopy may provide a more quantitative insight into the strength of polyvalent interactions.109,110 Surface based binding tests like surface plasmon resonance111 and quartz crystal microbalance112 are likewise useful for quantifying affinities and avidities.69,113 Advances in microcontact printing114 are also enabling more in-depth studies of structure–function relationships.36 As the toolbox of analytical techniques continues to grow, many of these can likely be used to provide a deeper understanding of polyvalent interactions.
Polyvalent molecules have already been used extensively to enhance the effectiveness of an inhibitor or effector in a wide variety of biological contexts, often by increasing the avidity of a ligand by several orders of magnitude. For instance, polyvalency has been instrumental in the design and synthesis of inhibitors to various toxins and viruses, and in modulating immune responses and cell-signaling cascades. In the years ahead, the combination of this concept with advances in techniques to discover monomeric ligands will likely result in polyvalent systems that display higher avidity effects, more efficient inhibition, and novel therapeutic and diagnostic avenues currently unavailable. In addition, scaffolds that feature specific numbers of ligands separated by well-defined dimensions will be extremely useful for elucidating the mechanisms that drive biological phenomena such as signaling events. Greater versatility is also being added to polyvalent systems, including the development of stimulus-responsive molecules that can allow spatiotemporal control of polyvalent interactions. It is clear that the polyvalent display of ligands will be a key platform for future diagnostics and therapeutics.
Acknowledgments
We acknowledge support from NIH R01 Grants EB015482 and NS083856.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Mammen M.; Choi S.-K.; Whitesides G. M. Angew. Chem., Int. Ed. 1998, 37, 2754–2794. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy V. M.; Estroff L. A.; Whitesides G. M. In Fragment-Based Approaches in Drug Discovery; Jahnke W.; Erlanson D. A., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA, 2006; pp 11–53. [Google Scholar]
- Kiessling L. L.; Gestwicki J. E.; Strong L. E. Angew. Chem., Int. Ed. 2006, 45, 2348–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiessling L. L.; Gestwicki J. E.; Strong L. E. Curr. Opin. Chem. Biol. 2000, 4, 696–703. [DOI] [PubMed] [Google Scholar]
- Compain P.; Bodlenner A. ChemBioChem 2014, 15, 1239–1251. [DOI] [PubMed] [Google Scholar]
- Fasting C.; Schalley C. a; Weber M.; Seitz O.; Hecht S.; Koksch B.; Dernedde J.; Graf C.; Knapp E.-W.; Haag R. Angew. Chem., Int. Ed. Engl. 2012, 51, 10472–10498. [DOI] [PubMed] [Google Scholar]
- Vorup-Jensen T. Adv. Drug Delivery Rev. 2012, 64, 1759–1781. [DOI] [PubMed] [Google Scholar]
- Danial M.; Klok H.-A. Macromol. Biosci. 2014, 10.1002/mabi.201400298. [DOI] [PubMed] [Google Scholar]
- Levine P. M.; Carberry T. P.; Holub J. M.; Kirshenbaum K. MedChemComm 2013, 4, 493. [Google Scholar]
- Garner A. L.; Park J.; Zakhari J. S.; Lowery C. A.; Struss A. K.; Sawada D.; Kaufmann G. F.; Janda K. D. J. Am. Chem. Soc. 2011, 133, 15934–15937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Gitlin I.; Krishnamurthy V. M.; Vazquez J. A.; Costello C. E.; Whitesides G. M. J. Am. Chem. Soc. 2003, 125, 12392–12393. [DOI] [PubMed] [Google Scholar]
- Davis N. E.; Karfeld-Sulzer L. S.; Ding S.; Barron A. E. Biomacromolecules 2009, 10, 1125–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz H. K.; Plückthun A. Curr. Opin. Biotechnol. 2005, 16, 459–469. [DOI] [PubMed] [Google Scholar]
- Boersma Y. L.; Plückthun A. Curr. Opin. Biotechnol. 2011, 22, 849–857. [DOI] [PubMed] [Google Scholar]
- Lee B. W.; Schubert R.; Cheung Y. K.; Zannier F.; Wei Q.; Sacchi D.; Sia S. K. Angew. Chem., Int. Ed. Engl. 2010, 49, 1971–1975. [DOI] [PubMed] [Google Scholar]
- Patke S.; Boggara M.; Maheshwari R.; Srivastava S. K.; Arha M.; Douaisi M.; Martin J. T.; Harvey I. B.; Brier M.; Rosen T.; Mogridge J.; Kane R. S. Angew. Chem., Int. Ed. Engl. 2014, 53, 8037–8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck J. J.; Danner D. J.; Landgren R. M.; Rainbolt T. K.; Roberts D. S. Biomacromolecules 2012, 13, 1996–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig P. O.; Alzogaray V.; Goldbaum F. A. Biomacromolecules 2012, 13, 1112–1121. [DOI] [PubMed] [Google Scholar]
- Levine P. M.; Imberg K.; Garabedian M. J.; Kirshenbaum K. J. Am. Chem. Soc. 2012, 134, 6912–6915. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Lis J. T.; Shi H. Nucleic Acid Ther. 2013, 23, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad K. M.; Xiao Y.; Soh H. T. Nucleic Acids Res. 2012, 40, 11777–11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Eckert M. a; Ali M. M.; Liu L.; Kang D.-K.; Chang E.; Pone E. J.; Sender L. S.; Fruman D. a; Zhao W. ChemBioChem 2014, 15, 1268–1273. [DOI] [PubMed] [Google Scholar]
- Englund E. a; Wang D.; Fujigaki H.; Sakai H.; Micklitsch C. M.; Ghirlando R.; Martin-Manso G.; Pendrak M. L.; Roberts D. D.; Durell S. R.; Appella D. H. Nat. Commun. 2012, 3, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De M.; Ghosh P. S.; Rotello V. M. Adv. Mater. 2008, 20, 4225–4241. [Google Scholar]
- Salata O. V. J. Nanobiotechnol. 2004, 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanraj V. J.; Chen Y. Trop. J. Pharm. Res. 2006, 5, 561–573. [Google Scholar]
- Yildiz I.; Shukla S.; Steinmetz N. F. Curr. Opin. Biotechnol. 2011, 22, 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovlid M. L.; Steinmetz N. F.; Laufer B.; Lau J. L.; Kuzelka J.; Wang Q.; Hyypiä T.; Nemerow G. R.; Kessler H.; Manchester M.; Finn M. G. Nanoscale 2012, 4, 3698–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon J. O.; Kim S.; Choi E.; Shin K.; Cha K.; So I.-S.; Kim S.-J.; Jun E.; Kim D.; Ahn H. J.; Lee B.-H.; Lee S.-H.; Kim I.-S. ACS Nano 2013, 7, 7462–7471. [DOI] [PubMed] [Google Scholar]
- Gujrati V.; Kim S.; Kim S.-H.; Min J. J.; Choy H. E.; Kim S. C.; Jon S. ACS Nano 2014, 8, 1525–1537. [DOI] [PubMed] [Google Scholar]
- Löwik D. W. P. M.; Leunissen E. H. P.; van den Heuvel M.; Hansen M. B.; van Hest J. C. M. Chem. Soc. Rev. 2010, 39, 3394–3412. [DOI] [PubMed] [Google Scholar]
- Yu X.; Zou Y.; Horte S.; Janzen J.; Kizhakkedathu J. N.; Brooks D. E. Biomacromolecules 2013, 14, 2611–2621. [DOI] [PubMed] [Google Scholar]
- Hassouneh W.; Fischer K.; MacEwan S. R.; Branscheid R.; Fu C. L.; Liu R.; Schmidt M.; Chilkoti A. Biomacromolecules 2012, 13, 1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Veracoechea F. J.; Frenkel D. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 10963–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubacheva G. V.; Curk T.; Mognetti B. M.; Auzély-Velty R.; Frenkel D.; Richter R. P. J. Am. Chem. Soc. 2014, 136, 1722–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S.-H.; Yilmaz M. D.; Reinhoudt D. N.; Velders A. H.; Huskens J. Angew. Chem., Int. Ed. Engl. 2013, 52, 714–719. [DOI] [PubMed] [Google Scholar]
- Numata J.; Juneja A.; Diestler D. J.; Knapp E.-W. J. Phys. Chem. B 2012, 116, 2595–2604. [DOI] [PubMed] [Google Scholar]
- Kane R. S. Langmuir 2010, 26, 8636–8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.; Dong R.; Chen J.; Chen F.; Jiang W.; Zhou Y.; Zhu X.; Yan D. Biomacromolecules 2014, 15, 1828–1836. [DOI] [PubMed] [Google Scholar]
- Yang L.; Meng L.; Zhang X.; Chen Y.; Zhu G.; Liu H.; Xiong X.; Sefah K.; Tan W. J. Am. Chem. Soc. 2011, 133, 13380–13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Ali M. M.; Eckert M. a; Kang D.-K.; Chen Y. Y.; Sender L. S.; Fruman D. a; Zhao W. Biomaterials 2013, 34, 9728–9735. [DOI] [PubMed] [Google Scholar]
- Thomas T. P.; Huang B.; Choi S. K.; Silpe J. E.; Kotlyar A.; Desai A. M.; Zong H.; Gam J.; Joice M.; Baker J. R. Mol. Pharmaceutics 2012, 9, 2669–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A.; Raczka E.; Piehler L.; Lee I.; Myc A.; Majoros I.; Patri A. K.; Thomas T.; Mulé J.; Baker J. R. Pharm. Res. 2002, 19, 1310–1316. [DOI] [PubMed] [Google Scholar]
- Kukowska-Latallo J. F.; Candido K. A.; Cao Z.; Nigavekar S. S.; Majoros I. J.; Thomas T. P.; Balogh L. P.; Khan M. K.; Baker J. R. Cancer Res. 2005, 65, 5317–5324. [DOI] [PubMed] [Google Scholar]
- Majoros I. J.; Thomas T. P.; Mehta C. B.; Baker J. R. J. Med. Chem. 2005, 48, 5892–5899. [DOI] [PubMed] [Google Scholar]
- Thomas T. P.; Majoros I. J.; Kotlyar A.; Kukowska-Latallo J. F.; Bielinska A.; Myc A.; Baker J. R. J. Med. Chem. 2005, 48, 3729–3735. [DOI] [PubMed] [Google Scholar]
- Majoros I. J.; Williams C. R.; Becker A.; Baker J. R. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabez N.; Lynch R. M.; Xu L.; Gillies R. J.; Chassaing G.; Lavielle S.; Hruby V. J. J. Med. Chem. 2011, 54, 7375–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josan J. S.; Handl H. L.; Sankaranarayanan R.; Xu L.; Lynch R. M.; Vagner J.; Mash E. A.; Hruby V. J.; Gillies R. J. Bioconjugate Chem. 2011, 22, 1270–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Tian S.; Petros R. a; Napier M. E.; Desimone J. M. J. Am. Chem. Soc. 2010, 132, 11306–11313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchilin V. P. Nat. Rev. Drug Discovery 2005, 4, 145–160. [DOI] [PubMed] [Google Scholar]
- Mann A. P.; Bhavane R. C.; Somasunderam A.; Montalvo-Ortiz B. L.; Ghaghada K. B.; Volk D.; Nieves-Alicea R.; Suh K. S.; Ferrari M.; Annapragada A.; Gorenstein D. G.; Tanaka T. Oncotarget 2010, 2, 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz N. F.; Cho C.-F.; Ablack A.; Lewis J. D.; Marianne M. Nanomedicine (London) 2012, 6, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokorski J. K.; Hovlid M. L.; Finn M. G. ChemBioChem 2011, 12, 2441–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y.; Dalhaimer P.; Cai S.; Tsai R.; Tewari M.; Minko T.; Discher D. E. Nat. Nanotechnol. 2007, 2, 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltra N. S.; Swift J.; Mahmud A.; Rajagopal K.; Loverde S. M.; Discher D. E. J. Mater. Chem. B 2013, 1, 5177. [DOI] [PubMed] [Google Scholar]
- Shuvaev V. V.; Ilies M. a; Simone E.; Zaitsev S.; Kim Y.; Cai S.; Mahmud A.; Dziubla T.; Muro S.; Discher D. E.; Muzykantov V. R. ACS Nano 2011, 5, 6991–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine D. P. Clin. Infect. Dis. 2006, 42Suppl 1S5–12. [DOI] [PubMed] [Google Scholar]
- Courvalin P. Clin. Infect. Dis. 2006, 42Suppl 1S25–34. [DOI] [PubMed] [Google Scholar]
- Choi S. K.; Myc A.; Silpe J. E.; Sumit M.; Wong P. T.; McCarthy K.; Desai A. M.; Thomas T. P.; Kotlyar A.; Holl M. M. B.; Orr B. G.; Baker J. R. ACS Nano 2013, 7, 214–228. [DOI] [PubMed] [Google Scholar]
- Joshi A.; Vance D.; Rai P.; Thiyagarajan A.; Kane R. S. Chemistry 2008, 14, 7738–7747. [DOI] [PubMed] [Google Scholar]
- Vance D.; Shah M.; Joshi A.; Kane R. S. Biotechnol. Bioeng. 2008, 101, 429–434. [DOI] [PubMed] [Google Scholar]
- Jacobson J. M.; Kitov P. I.; Bundle D. R. Carbohydr. Res. 2013, 378, 4–14. [DOI] [PubMed] [Google Scholar]
- Tran H.-A.; Kitov P. I.; Paszkiewicz E.; Sadowska J. M.; Bundle D. R. Org. Biomol. Chem. 2011, 9, 3658–3671. [DOI] [PubMed] [Google Scholar]
- Maheshwari R.; Levenson E. A.; Kiick K. L. Macromol. Biosci. 2010, 10, 68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro-Viana R.; Sánchez-Navarro M.; Luczkowiak J.; Koeppe J. R.; Delgado R.; Rojo J.; Davis B. G. Nat. Commun. 2012, 3, 1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suami T. Pure Appl. Chem. 1987, 59, 1509–1520. [Google Scholar]
- Luczkowiak J.; Sattin S.; Sutkevičiu̅tė I.; Reina J. J.; Sánchez-Navarro M.; Thépaut M.; Martínez-Prats L.; Daghetti A.; Fieschi F.; Delgado R.; Bernardi A.; Rojo J. Bioconjugate Chem. 2011, 22, 1354–1365. [DOI] [PubMed] [Google Scholar]
- Rademacher C.; Guiard J.; Kitov P. I.; Fiege B.; Dalton K. P.; Parra F.; Bundle D. R.; Peters T. Chemistry 2011, 17, 7442–7453. [DOI] [PubMed] [Google Scholar]
- Mammen M.; Dahmann G.; Whitesides G. M. J. Med. Chem. 1995, 38, 4179–4190. [DOI] [PubMed] [Google Scholar]
- Mochalova L. V.; Tuzikov A. B.; Marinina V. P.; Gambaryan A. S.; Byramova N. E.; Bovin N. V.; Matrosovich M. N. Antiviral Res. 1994, 23, 179–190. [DOI] [PubMed] [Google Scholar]
- Choi S.-K.; Mammen M.; Whitesides G. M. J. Am. Chem. Soc. 1997, 119, 4103–4111. [Google Scholar]
- Papp I.; Sieben C.; Sisson A. L.; Kostka J.; Böttcher C.; Ludwig K.; Herrmann A.; Haag R. ChemBioChem 2011, 12, 887–895. [DOI] [PubMed] [Google Scholar]
- Martínez-Avila O.; Bedoya L. M.; Marradi M.; Clavel C.; Alcamí J.; Penadés S. ChemBioChem 2009, 10, 1806–1809. [DOI] [PubMed] [Google Scholar]
- Becer C. R.; Gibson M. I.; Geng J.; Ilyas R.; Wallis R.; Mitchell D. A.; Haddleton D. M.; Uni V.; Leicester L. E. J. Am. Chem. Soc. 2010, 132, 15130–15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay J. I.; Lai B. E.; Myszka D. G.; Mahalingam A.; Langheinrich K.; Katz D. F.; Kiser P. F.; Carolina N. Mol. Pharmaceutics 2010, 7, 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S.; Gavrilyuk J.; Burton D. R.; Barbas C. F. ACS Med. Chem. Lett. 2014, 5, 133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial M.; Root M. J.; Klok H.-A. Biomacromolecules 2012, 13, 1438–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo S. Biochim. Biophys. Acta, Biomembr. 2003, 1614, 36–50. [DOI] [PubMed] [Google Scholar]
- Rosa Borges A.; Wieczorek L.; Johnson B.; Benesi A. J.; Brown B. K.; Kensinger R. D.; Krebs F. C.; Wigdahl B.; Blumenthal R.; Puri A.; McCutchan F. E.; Birx D. L.; Polonis V. R.; Schengrund C.-L. Virology 2010, 408, 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli S. J.; Tsai M.; Piliponsky A. M. Nature 2008, 454, 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sil D.; Lee J. B.; Luo D.; Holowka D.; Baird B. ACS Chem. Biol. 2007, 2, 674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handlogten M. W.; Kiziltepe T.; Moustakas D. T.; Bilgiçer B. Chem. Biol. 2011, 18, 1179–1188. [DOI] [PubMed] [Google Scholar]
- Paar J. M.; Harris N. T.; Holowka D.; Baird B. J. Immunol. 2002, 169, 856–864. [DOI] [PubMed] [Google Scholar]
- Baird E. J.; Holowka D.; Coates G. W.; Baird B. Biochemistry 2003, 42, 12739–12748. [DOI] [PubMed] [Google Scholar]
- Huang Y.-F.; Liu H.; Xiong X.; Chen Y.; Tan W. J. Am. Chem. Soc. 2009, 131, 17328–17334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handlogten M. W.; Kiziltepe T.; Alves N. J.; Bilgicer B. ACS Chem. Biol. 2012, 7, 1796–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handlogten M. W.; Kiziltepe T.; Bilgicer B. Biochem. J. 2013, 449, 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handlogten M. W.; Kiziltepe T.; Serezani A. P.; Kaplan M. H.; Bilgicer B. Nat. Chem. Biol. 2013, 9, 789–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney A. H.; Puffer E. B.; Pontrello J. K.; Yang Z.-Q.; Kiessling L. L. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2500–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puffer E. B.; Pontrello J. K.; Hollenbeck J. J.; Kink J. a; Kiessling L. L. ACS Chem. Biol. 2007, 2, 252–262. [DOI] [PubMed] [Google Scholar]
- Wang L.; Huang P.; Fang H.; Xia M.; Zhong W.; McNeal M. M.; Jiang X.; Tan M. Biomaterials 2013, 34, 4480–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Cao D.; Wei C.; Meng X.-J.; Jiang X.; Tan M. Vaccine 2014, 32, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo M.; Wei C.-J.; Yassine H. M.; McTamney P. M.; Boyington J. C.; Whittle J. R. R.; Rao S. S.; Kong W.-P.; Wang L.; Nabel G. J. Nature 2013, 499, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W.; Kim B. Y. S.; Rutka J. T.; Chan W. C. W. Nat. Nanotechnol. 2008, 3, 145–150. [DOI] [PubMed] [Google Scholar]
- Maheshwari G.; Brown G.; Lauffenburger D. a; Wells A.; Griffith L. G. J. Cell Sci. 2000, 113, 1677–1686. [DOI] [PubMed] [Google Scholar]
- Holler N.; Tardivel A.; Hertig S.; Gaide O.; Tinel A.; Deperthes D.; Calderara S.; Schulthess T.; Engel J.; Schneider P.; Kovacsovics-bankowski M.; Martinon F. Mol. Cell. Biol. 2003, 23, 1428–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grochmal A.; Ferrero E.; Milanesi L.; Tomas S. J. Am. Chem. Soc. 2013, 135, 10172–10177. [DOI] [PubMed] [Google Scholar]
- Conway A.; Vazin T.; Spelke D. P.; Rode N. a; Healy K. E.; Kane R. S.; Schaffer D. V. Nat. Nanotechnol. 2013, 8, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazin T.; Ashton R. S.; Conway A.; Rode N. a; Lee S. M.; Bravo V.; Healy K. E.; Kane R. S.; Schaffer D. V. Biomaterials 2014, 35, 941–948. [DOI] [PubMed] [Google Scholar]
- Lee L. A.; Muhammad S. M.; Nguyen Q. L.; Sitasuwan P.; Horvath G.; Wang Q. Mol. Pharmaceutics 2012, 9, 2121–2125. [DOI] [PubMed] [Google Scholar]
- Tongers J.; Newcomb C. J.; Marquardt K.; Bauersachs J.; Losordo D. W.; Stupp S. I.; Webber M. J. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9220–9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E.; Patterson G. H.; Sougrat R.; Lindwasser O. W.; Olenych S.; Bonifacino J. S.; Davidson M. W.; Lippincott-Schwartz J.; Hess H. F. Science 2006, 313, 1642–1645. [DOI] [PubMed] [Google Scholar]
- Rust michael J.; Bates M.; Zhuang X. Nat. Methods 2006, 3, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponader D.; Ma P.; Aretz J.; Pussak D.; Ninnemann N. M.; Schmidt S.; Seeberger P. H.; Rademacher C.; Nienhaus G. U.; Hartmann L. J. Am. Chem. Soc. 2014, 136, 2008–2016. [DOI] [PubMed] [Google Scholar]
- Hall J.; Karplus P. A.; Barbar E. J. Biol. Chem. 2009, 284, 33115–33121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrot A.; Ro W.; Imberty A.; Audfray A. ACS Chem. Biol. 2013, 8, 1918–1924. [DOI] [PubMed] [Google Scholar]
- Neuman K. C.; Nagy A. Nat. Methods 2008, 5, 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieben C.; Kappel C.; Zhu R.; Wozniak A.; Rankl C.; Hinterdorfer P.; Grubmüller H.; Herrmann A. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 13626–13631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-casado A.; Dam H. H.; Yilmaz M. D.; Florea D.; Jonkheijm P.; Huskens J. J. Am. Chem. Soc. 2011, 133, 10849–10857. [DOI] [PubMed] [Google Scholar]
- Homola J. Chem. Rev. 2008, 108, 462–493. [DOI] [PubMed] [Google Scholar]
- Marx K. A. Biomacromolecules 2003, 4, 1099–1120. [DOI] [PubMed] [Google Scholar]
- Waldmann M.; Jirmann R.; Hoelscher K.; Wienke M.; Niemeyer F. C.; Rehders D.; Meyer B. J. Am. Chem. Soc. 2014, 136, 783–788. [DOI] [PubMed] [Google Scholar]
- Perl A.; Reinhoudt D. N.; Huskens J. Adv. Mater. 2009, 21, 2257–2268. [Google Scholar]