

Abstract
microRNAs (miRNAs) are a class of endogenous small RNAs (sRNAs) that play pivotal roles in plant development, abiotic stress response, and pathogen response. miRNAs have been extensively studied in plants, but rarely in Nicotiana benthamiana, despite its wide use in plant virology studies, particularly for studying N protein–tobacco mosaic virus (TMV) interactions. We report an efficient method using high-throughput sequencing and bioinformatics to identify genome-wide miRNAs in N. benthamiana. A total of 30 conserved miRNA families and 113 novel miRNAs belonging to 93 families were identified. Some miRNAs were clustered on chromosomes, and some were embedded in host gene introns. The predicted miRNA targets were involved in diverse biological processes, such as metabolism, signaling, and responses to stimuli. miRNA expression profiling revealed that most of them were differentially expressed during N-mediated immunity to TMV. This study provides a framework for further analysis of miRNA functions in plant immunity.
Keywords: miRNA, high-throughput sequencing, tobacco mosaic virus (TMV), immunity, N. benthamiana
Introduction
MicroRNA (miRNA) is a pivotal category of small RNA (sRNA) used in the regulation of gene expression in eukaryotes.1 miRNAs are approximately 21 nt endogenous non-coding RNAs that negatively regulate gene expression at the post-transcriptional level, by either repressing gene translation or cleaving target mRNAs.2 Unlike animals, plants use Dicer-like (DCL) proteins to generate stem-loop precursor mRNA (pre-miRNA) and process the miRNA:miRNA star (miRNA:miRNA*) duplex with two-nucleotide 3′ overhangs,3 which is then transported from the nucleus into the cytoplasm by HASTY (HST).4 Once 2’O-methylated by Hua En hancer 1 (HEN1), the mature miRNA strand is predominantly incorporated into argonaute-1 (AGO1) or argonaute-10 (AGO10) containing RNA-induced silencing complexes (RISCs) that inhibit gene expression by perfect or near-perfect complement to target transcripts.5,6 Many miRNA sequences are highly conserved within the same kingdom,7 whereas others are species specific. These non-conserved miRNAs are difficult to identify by conventional methods. However, recently established high-throughput sequencing technologies together with powerful bioinformatics tools have allowed efficient identification of not only conserved miRNAs but also low-abundance miRNAs in several plant species.8–10
In plants, miRNAs are involved in diverse processes such as development11,12 and responses to nutrient,13 and environmental stresses.14 They also play critical roles in resistance to bacterial pathogens and viruses. For example, Arabidopsis treatment with flg22, a flagellin-derived peptide, increases the transcriptional level of miR393, which then negatively regulates auxin receptors TIR1, AFB2, and AFB3 in bacterial resistance mechanisms.15 In Arabidopsis, miR160a, miR398b, and miR773 participate in plant innate immunity against Pseudomonas syringae by regulating Pathogen-associated molecular pattern (PAMP)-induced callose deposition.16 In diverse plant species, miR482/2118 superfamily members target the P-loop motif coding sequence of resistance genes with nucleotide binding site (NBS) and leucine-rich repeat (LRR) motifs, which leads to RNA-dependent RNA polymerase 6 (RDR6)-dependent mRNA degradation and production of secondary small interfering RNAs (siRNAs).17 Similarly, nta-miR6019 and nta-miR6020 from tobacco guide the cleavage of the mRNA of the immune receptor N’s TIR domain, which also leads to RDR6- and DCL4- dependent production of secondary siRNAs.18 In accordance with the function of miRNAs in plant immunity, genes required for miRNA biogenesis are also required for resistance against bacterial pathogens. For example, both HEN1 and DCL1 are required for PAMP-triggered immunity (PTI).19
The tobacco N gene belongs to the TIR-NB-LRR class of resistance (R) genes that confer resistance to tobacco mosaic virus (TMV).20 When TMV attacks tobacco cells, p50, the TMV replicase fragment, is recognized by N protein through direct interaction. This triggers a series of signal transduction cascades, which initiate a hypersensitive response (HR), inhibit TMV spread, and induce systemic acquired resistance (SAR). Interestingly, N protein’s function is temperature sensitive and reversible.21 At temperature above 28 °C, N-mediated HR is restricted and TMV spreads throughout the plants. When temperature is below 28 °C, N protein reactivated, resulting in HR in TMV-containing tissues. In recent decades, many proteins have been identified by virus-induced gene silencing (VIGS) technology as participating in N-mediated signaling pathways. Like other TIR-NB-LRR proteins, N protein requires enhanced disease susceptibility 1 (EDS1) for its function.22 The jasmonic acid (JA) and ethylene signaling pathways have been implicated in the resistance response to TMV through their respective hormone receptors, COI1 and CTR1. N protein occurs in a large complex with Rar1/SGT1, COP9 signalosome (CSN), and HSP90, suggesting that ubiquitin-mediated protein degradation and molecular chaperones play key roles in the N-mediated signaling pathway.22–24 Two MAPK cascades, MEK1 MAPKK and NRK1 MAPK, function downstream of the recognition step. The transcription factors WRKY1–3 and MYB1 might function downstream of the MAPK cascades.25
Although miRNAs have been implicated in plant immunity, whether miRNAs are involved in the N-mediated resistance pathway is still unknown. To address this question, we constructed three sRNA libraries of TMV-infected Nicotiana benthamiana plants from the selected time points after N gene activation. Through library sequencing and analysis, we identified 30 families of conserved miRNAs and 93 families of N. benthamiana-specific miRNAs. Furthermore, we identified numerous candidate miRNAs and their putative targets that may participate in regulating N-mediated resistance to TMV. This is the first large-scale survey of miRNAs in N. benthamiana, and has revealed putative miRNAs and targets that participate in the N-mediated resistance pathway.
Results
High-throughput sequencing of sRNAs in N. benthamiana
To probe miRNA regulation of N gene-mediated resistance to TMV, we deep sequenced (Solexa-Illumina) sRNAs from TMV-infected N. benthamiana plants containing the transgenic N gene22 at zero, two, and eight hours after transfer from a five-day 32 °C treatment to normal growth conditions. We obtained 11,597,524 (zero hour), 10,492,893 (two hours), and 11,125,715 (eight hours) reads. After removing 3′ and 5′ adaptors and low-quality reads, we obtained 11,200,906 (zero hour), 10,129,898 (two hours), and 11,125,715 (eight hours) high-quality reads, ranging in size from 10 to 30 nt (Table 1). These high-quality reads were then used to determine the sRNA length distribution. sRNA lengths varied but were similarly abundant between samples. The most abundant size class was 19–24 nt sRNA, accounting for 77.31% (zero hour), 77.79% (two hours), and 83.38% (eight hours) of the sRNAs (Fig. 1). Of the major specific sRNA lengths, the 21 and 24 nt sRNAs were similarly abundant in all three samples and significantly more abundant than other lengths (P < 0.01, Table S8 and Fig. S2).
Table 1.
Data summary of high-throughput sequencing of three small RNA libraries.
| SAMPLE | CATEGORY | READS NUMBER (RATIO) | |
|---|---|---|---|
| 0 hr | Total reads | 11597524 | |
| High quality reads | 11200906 (100%) | ||
| Smaller than 18 nt reads | 1720092 (15.36%) | ||
| Clean reads | 9405702 (83.97%) | 100% | |
| rRNA | 157435 | 1.67% | |
| snRNA | 757 | 0.01% | |
| snoRNA | 480 | 0.01% | |
| tRNA | 140883 | 1.50% | |
| unannotated | 9106147 | 96.82% | |
| 2 hr | Total reads | 10492893 | |
| High quality reads | 10129898 (100%) | ||
| Smaller than 18 nt reads | 1381982 (13.64%) | ||
| Clean reads | 8651639 (85.41%) | 100% | |
| rRNA | 539667 | 6.24% | |
| snRNA | 658 | 0.01% | |
| snoRNA | 978 | 0.01% | |
| tRNA | 422319 | 4.88% | |
| unannotated | 7688017 | 88.86% | |
| 8 hr | Total reads | 11125715 | |
| High quality reads | 10850589 (100%) | ||
| Smaller than 18 nt reads | 950866 (8.76%) | ||
| Clean reads | 9827853 (90.57%) | 100% | |
| rRNA | 413850 | 4.21% | |
| snRNA | 857 | 0.01% | |
| snoRNA | 931 | 0.01% | |
| tRNA | 352970 | 3.59% | |
| unannotated | 9059245 | 92.18% |
Figure 1.
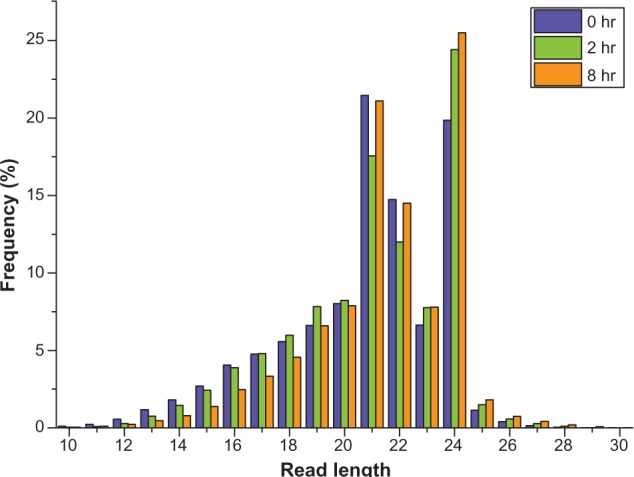
The length distribution of sRNAs in zero-hr, two-hr, and eight-hr libraries (hr, hour).
To identify putative miRNA in the pool of sRNA reads, we first removed other sRNA categories (rRNA, snRNA, snoRNA, tRNA) from our analysis. We identified the other sRNA categories by comparing the cleaned reads (see Materials and Methods) to entries in annotated sRNA databases of GenBank (http://www.ncbi.nlm.nih.gov/genbank/) and Rfam (http://rfam.sanger.ac.uk). The remaining unannotated reads were mapped to the N. benthamiana genome (version 0.4.4). Next, all mapped reads were analyzed to identify candidate miRNAs. Although we excluded the other sRNA categories from further analysis, we note here that rRNA levels clearly increased from zero hour (1.67%) to two hours (6.24%), but had decreased slightly by eight hours (4.21%). A similar pattern was also found for tRNA, indicating that many functional genes are expressed immediately after N gene activation and their expression peaked around two hours after the transfer to normal growth conditions.
Conserved N. benthamiana miRNAs
To identify conserved N. benthamiana miRNAs, we used a computational protocol similar to Mackowiak et al.26 with modifications for plant miRNA identification27 to align excised mapped reads to Nicotiana tabacum miRNAs and miRNA*s deposited in miRBase and a recent database reported by Frazier et al.28 We identified 95 miRNAs previously described in N. tabacum. With the remaining excised precursors, we used other plants’ miRNAs in miRBase as a reference and identified 17 N. benthamiana miRNAs to be similar to other plant species’ miRNAs. In total, 112 conserved miRNAs were identified, 100 of which were expressed in at least one of our sRNA libraries (97, 96, and 98 miRNA genes in the zero-, two-, and eight-hour samples, respectively; Table S1). The miRNA names, mature sequences, star sequences, the corresponding read numbers, and reference miRNAs from other plants with the same seed and pre-miRNAs’ position on the scaffolds are presented in Table S1. Furthermore, the partner miRNA* was identified for over 90% (102/112) of novel N. benthamiana miRNA genes. We also detected 16 sequences within the loop structure of miRNA genes. miRNA* and loop RNA are generally short lived, indicating that the high-throughput sequencing technology was very sensitive for identifying miRNA.
We found nearly equal numbers of reads from two arms of stem-loop precursors for 5 of the 112 known miRNAs (nbt-miR169a, nbt-miR160e, nbt-miR398, nbt-miR396b, and nbt-miR396a) and even more reads of the star strand than the miRNA strand annotated by miRBase for 3 miRNAs (nbt-miR319a, nbt-miR319b, nbt-miR482b) (Table S1). These results indicate that the biogenesis of mature miRNA is highly complex in N. benthamiana.
Mature plant miRNAs preferentially have a U at the first position from the 5′-end according to a previous study.6 We constructed position-weight-matrices (PWMs) in WebLogo29 for all conserved 18–22 nt mature N. benthamiana miRNA sequences. The results confirmed the reported findings. The extreme 5′-end of the miRNAs we examined had a 60% U bias based on the graphed nucleotide composition per position (Fig. 2A). There were also other positions that showed biased base conservations. For example, position 3 had more G, positions 5 and 15 more A/U, and position 11 more G/C than random expectations (Fig. 2A).
Figure 2.
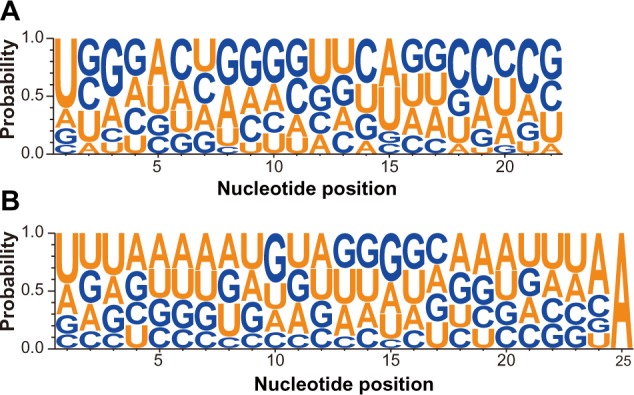
Position-specific nucleotide preference in known miRNA (A) and novel miRNA (B) in N. benthamiana.
We categorized the known miRNAs into 30 families according to their mature sequence identity (Table 2). The largest family, nbt-miR166, has 20 members, followed by nbt-miR156 with 10 members. Members of these two miRNA families match their Arabidopsis family member counterparts nearly perfectly, suggesting that the families are evolutionarily conserved and originated before the divergence of the two dicot branches.
Table 2.
Summary of conserved N. benthamiana miRNA families and number of member in each family.
| miRNA FAMILY | NUMBER OF MEMBER IN THE FAMILY |
|---|---|
| miR166 | 20 |
| miR156 | 10 |
| miR160 | 6 |
| miR167 | 6 |
| miR172 | 6 |
| miR169 | 5 |
| miR171 | 5 |
| miR396 | 5 |
| miR168 | 4 |
| miR390 | 4 |
| miR393 | 4 |
| miR399 | 4 |
| miR170 | 3 |
| miR394 | 3 |
| miR397 | 3 |
| miR403 | 3 |
| miR162 | 2 |
| miR164 | 2 |
| miR319 | 2 |
| miR479 | 2 |
| miR482 | 2 |
| miR6019 | 2 |
| miR7122 | 2 |
| miR159 | 1 |
| miR398 | 1 |
| miR1446 | 1 |
| miR5225 | 1 |
| miR6147 | 1 |
| miR6149 | 1 |
| miR6151 | 1 |
Novel N. benthamiana miRNAs
We identified novel N. benthamiana miRNAs with a computational protocol similar to that of Sebastian et al.26 with modifications for plant miRNA identification27 (the same program used for predicting conserved miRNAs) using a probabilistic method to score the compatibility of the miRNA position and the frequency of sRNA within the secondary structure of the miRNA precursors.30 A total of 113 unique sequences were identified as potential miRNA genes with a true possibility of over 71% (Table S2). Since we excluded miRNAs that had high similarity with the miRNA of the reference plants, these miRNAs are believed to be N. benthamiana specific.
The miRNA* read was present in over 79% (90/113) of the novel miRNAs in at least one of the libraries, similar to the ratio observed for conserved miRNAs. In all, 43 (38.1%) miRNA reads mapped to the loop structure, a ratio much higher than that of conserved miRNAs (14.3%). Interestingly, most reads of 32 novel miRNAs mapped to the loop rather than the star region of the pre-miRNA. For these 32 novel miRNA candidate genes, there were fewer miRNA* sequences than the corresponding mature miRNA sequences. Like conserved miRNAs, novel miRNAs’ mature sequence length was mostly 21 nt (62), followed by 22 nt (21) (Table 3). However, longer mature sequences with up to 24–25 nt were also present (Table 3).
Table 3.
Comparison of mature miRNA length between conserved and novel miRNA in N. benthamiana.
| MATURE miRNA LENGTH | CONSERVED | NOVEL |
|---|---|---|
| 18 | 6 | 3 |
| 19 | 4 | 3 |
| 20 | 11 | 4 |
| 21 | 80 | 62 |
| 22 | 11 | 21 |
| 23 | 0 | 5 |
| 24 | 0 | 14 |
| 25 | 0 | 1 |
We also inspected the nucleotide bias of the novel miRNA mature sequences by WebLogo.29 As expected, U was mostly preferred at the first position, albeit with a lower frequency (50%) than that of conserved miRNAs. Unlike conserved miRNAs, there was no obvious nucleotide bias at the other sites, indicating that the novel miRNAs in N. benthamiana are highly variable (Fig. 2).
Based on sequence similarity (identity >90%), the 113 novel N. benthamiana miRNAs were grouped into 93 families. The largest families were miRN3 and miRN10 with four members. Only 15 families had more than one member (Table S3). Of the 112 novel miRNAs, 50 had a very high confidence score (over 100) with a corresponding predicted true possibility higher than 94% (Table S2).
Clustered N. benthamiana miRNAs
Clustered miRNAs have been reported in both animal and plant genomes,31,32 but have not yet been described in tobacco. Since assembled chromosome data are not available for N. benthamiana, we mapped pre-miRNAs onto N. benthamiana genome scaffolds and contigs. We found 11 pre-miRNAs from five families distributed in five clusters (Table S4). After filtering out candidates who did not meet high stringency criteria, we identified two pre-miRNA clusters containing two nbt-miR399 and three nbt-miRN41 members (Fig. 3). The two nbt-miRN41 family members (nbt-miRN41a and nbt-miRN41b) on the scaffold Niben044Scf00014276 are separated by less than 400 bp. The closeness of nbt-miRN41a and nbt-miRN41b suggests that the two miRNAs are transcribed as a single primary transcript. However, the other clustered miRNAs are separated by much longer distances of 6–8 kb. Interestingly, members of the clustered pre-miRNAs belong to the same miRNA family. Moreover, both miRNA clusters are located in intergenic regions according to the current genome annotation.
Figure 3.
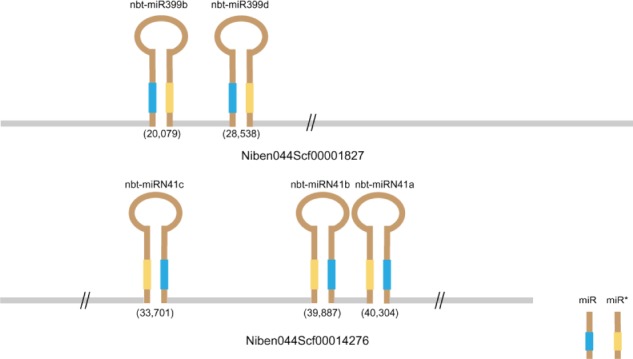
Clustered miRNAs in N. benthamiana.
Intronic miRNAs in N. benthamiana
Intronic miRNAs are a type of miRNAs located in the introns of host genes and were first identified in fruit flies and worms, and then in mammals. In animals, about 80% of the miRNAs are embedded in gene introns.33 Recently, they have also been detected in the genomes of plants, including rice, Arabidopsis, and Populus.34–37 However, their possible presence in the Solanaceae has not been previously explored. Therefore, we exploited our sRNA data to address this possibility. By mapping pre-miRNAs onto N. benthamiana intron sequences, we identified eight intronic miRNAs with high confidence in the genome sequence assembly. Information including the structures of genes that harbor intronic miRNAs, the positions in host genes, transcriptional directions, and folding structures of the pre-miRNAs is presented in Figure 4.
Figure 4.

Intronic miRNAs in N. benthamiana.
Conserved and novel miRNA expressions during N gene-mediated immunity to TMV
To test this hypothesis, we investigated the expression profiles of all the miRNAs at different time points of the N gene activation process using the quantifier module of the miRDeep226 algorithm. To increase the quantification’s fidelity, miRNAs with less than 100 total reads were excluded. Conserved and novel miRNAs were quantified separately, and each family member was quantified individually.
Most of the conserved miRNAs were differentially expressed; however, few displayed a dramatic change in pattern across the sampling time points (Fig. 5A). Most of the novel N. benthamiana-specific miRNAs were also differentially regulated during N gene activation (Fig. 5B). The dynamic N. benthamiana miRNA expression during N gene activation suggests a complex regulation of miRNAs in N gene-activated defense against TMV and indicates that the miRNAs identified in our study may be involved in this crucial immune pathway.
Figure 5.
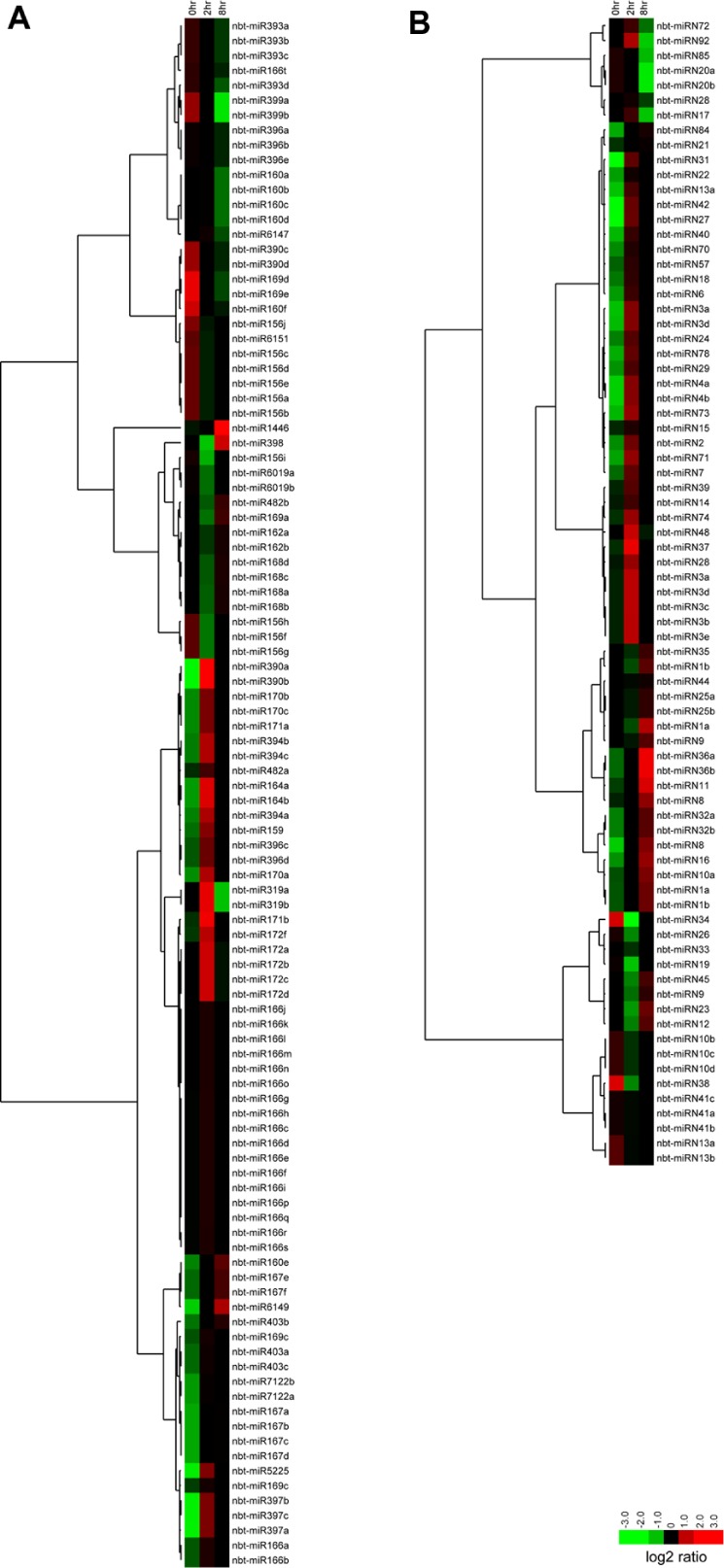
Expression pattern of known miRNAs (A) and novel miRNAs (B) at different time points after N gene was activated in N gene transgenic plants.
Prediction of miRNAs’ candidate target genes
miRNAs associate with their target transcripts by base-pair complementarity, which ultimately leads to modulation of target gene expression by mRNA cleavage or translation inhibition.38 Thus, predicting potential targets helps to infer a miRNA’s function. We computationally predicted potential N. benthamiana targets using predicted cDNAs from the N. benthamiana genome v0.4.4 (http://solgenomics.net/) of the plant miRNA target analysis tools on the psRNATarget server (http://plantgrn.noble.org/psRNATarget/).39
The predicted cDNAs contain about 48,342 non-overlapping gene models with annotations and comprise the longest representative transcripts. Using the default parameters’ setting, we identified 315 unique potential targets for 98 conserved miRNAs from 28 families and 609 unique potential targets for 104 novel miRNAs from 85 families. The miRNA ID, matched sequence, corresponding target transcript ID, inhibition target, and annotation of conserved and novel miRNAs are presented in Tables S5 and S6, respectively.
GO analysis of predicted miRNA target functions
A total of 924 putative target transcripts of both conserved and novel miRNAs were assigned GO terms based on a BLAST search of transcripts with known functions using the Blast2GO program (false discovery rate (FDR) cutoff of P < 0.05).40 Each transcript was assigned a molecular function and biological process. The molecular function of most transcripts fell into either the binding (50.2%) or catalytic activity (31.8%) categories (Fig. 6A). The two predominant binding activities were protein binding (35.1%) and nucleic acid binding (30.7%) (Table S7). These results suggest that to a large extent N. benthamiana miRNAs regulate transcription by modulating transcription factors. Among the GO biological processes assigned to the putative target transcripts, cellular (27.6%) and metabolic processes (24.8%) were the largest categories.
Figure 6.
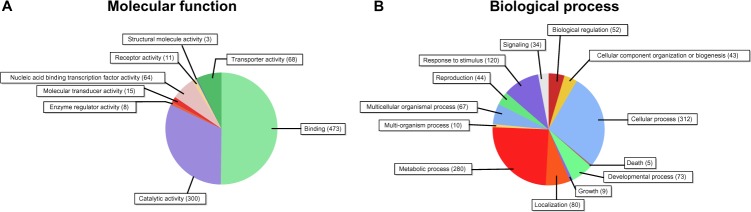
GO analysis of predicted target transcripts of miRNAs in N. benthamiana. (A) The pie diagram illustrating the significant numbers of predicted N. benthamiana miRNA targets within the (A) molecular function category and (B) biological process category.
Discussion
Characterization of N. benthamiana miRNAs
N. benthamiana is widely used as a host in plant-virus studies. Its susceptibility to a large number of pathogens, such as bacteria, fungi, and oomycetes, demonstrates its utility as a model in plant-pathogen research. To date, 21,516 mature miRNAs have been identified in plant species and deposited in miRBase (release 18).41 However, no N. benthamiana miRNA has been annotated in this database. Next-generation technologies have been instrumental in finding conserved as well as novel miRNAs in Arabidopsis, rice, Populus, and several non-model species.10,42–47 Through high-throughput sequencing, we identified 30 miRNA families conserved in N. benthamiana and other species. Most (95) of the conserved miRNAs (112 in total) are found in N. tabacum (Table S1) and show high homology of mature miRNA sequences, suggesting these two species share many common miRNA pathway features and might have only recently split in evolutionary history. We also identified 93 novel families, possibly N. benthamiana-specific miRNA families, which showed no sequence conservation with miRNAs from other plant species in the miRBase. By comparing miRNAs between N. tabacum and N. benthamiana, we found that only 28 miRNA families existed in N. tabacum, whereas 98 miRNAs are specific to N. benthamiana (Table S7). The differences in miRNAs between these two close species may account for their difference in response to TMV, and further studies are needed to prove this hypothesis. The abundance of novel miRNAs was lower than that of conserved miRNAs. Possibly, as previously proposed,48 conserved miRNAs are responsible for regulating basic cellular and developmental processes common to many eukaryotes, while species-specific miRNAs are involved in regulating unique pathways. The conserved N. benthamiana miRNAs tended to form multimember families (Table 2), whereas no novel miRNA family contained more than four members (only 2 families contained four members and 11 families contained two members; Table S3). This is consistent with observations from other species, such as Arabidopsis49 and Populus,50 and supports the hypothesis that conserved miRNA families have expanded by duplication.51–53 However, the exact frequency of birth and death needs to be further investigated by comparison on the basis of genomes of distantly related species. In accordance with observations of genomes of other plant species,49 the majority of both conserved and novel N. benthamiana 21 nt mature miRNA sequences contained a 5′ U, whereas 5′ A was overrepresented in 24 nt mature miRNA sequences (Fig. S1).
When miRNA was discovered, numerous observations suggested that only one strand of the miRNA duplex could be the effector sRNA and that the other strand, termed miRNA*, is degraded. However, traditional miRNA cloning methods and recently developed high-throughput sequencing technologies have increasingly revealed equal or close to equal miRNA/miRNA* ratios in vivo. In mouse, both strands of miR-30c and miR-142 have been cloned.54 In fruit fly, there are even more miRNA genes that yield close to equal miRNA*/miRNA ratios in vivo, most of which are relatively abundant in the total RNA transcriptome.55 Recently, nine miRNAs (ptc-miR160f, ptc-miR169b, ptc-miR169l, ptc-miR171h, ptc-miR171m, ptc-miR172h, ptc-miR393a, ptc-miR393b, and ptc-miR403c) with high miRNA* levels have been found in Populus euphratica from searches of cDNA libraries prepared from plants under drought stress and controls.35 Here, we identified five miRNA (nbt-miR169a, nbt-miR160e, nbt-miR398, nbt-miR396b, nbt-miR396a) genes that yielded nearly equal numbers of miRNA* and miRNA reads. Interestingly, two of these miRNAs (nbt-miR169a and nbt-miR160e) are similar to some of the abovementioned P. euphratica miRNAs with high miRNA* levels (ptc-miR160f, ptc-miR169b, and ptc-miR169l). We also identified three miRNA genes (miR319a, miR319b, and miR482b) with slightly more reads for the miRNA* than for the annotated miRNA. Since the sRNAs we measured were at the steady stage, these miRNA* sequences may be involved in particular biological processes in cells. In Drosophila, miRNA* preferentially associates with AGO2; thus, independent sorting of miRNA/miRNA* strands is a general character of Drosophila miRNA genes.55 In Arabidopsis, miR393* also binds AGO2, thereby downregulating a Golgi-localized SNARE gene (MEMB12) by translational inhibition.56 Because miR393 is bound by AGO1, a possible mechanism of independent sorting of duplex strands via distinct AGOs is suggested. Whether the abundant miRNA*s identified here are also bound by AGO2 is unknown and requires further investigation by coimmunoprecipitation with different AGOs.
Polycistronic miRNA
miRNAs are often clustered together in animal and plant genomes. They may share a common primary mRNA (pri-miRNA), a precursor subsequently excised by DCL1 into different pre-miRNAs. Clustered miRNAs with the same transcription start site are referred to as polycistronic miRNA. Polycistronic miRNA precursors are more abundant in animals (25–45% of total miRNAs) than in plants (10–20% of total miRNAs).57 In N. benthamiana, we identified two potentially polycistronic miRNA precursors, one containing known miRNAs and the other containing novel miRNAs. The proportion of potential polycistronic miRNA precursors (<1% of total miRNA genes) appears to be much lower than in other investigated plants, but this may be because of the incomplete sequencing of the N. benthamiana genome. We also identified another six potential polycistronic miRNA genes (Table S4), which were discarded because of consecutive Ns (unsequenced nucleotide positions) between individual miRNA loci. In animals, clustered miRNA genes are often heterogeneous. For example the miR-17 cluster consists of sequences encoding miR-17, miR-18, miR-19a, miR-19b, miR-20, miR-25, miR-92, miR-93, miR-106a, and miR-106b.67 However, the opinion that plant miRNA gene clusters generally comprise homologous members6,58,59 is consistent with our discovery. The clustered plant miRNAs may have been caused by tandem duplication and suggests a dosage effect of miRNA expression.
miRNA target prediction
We have identified over 100 known miRNAs, some of which are conserved across several model plant species, including Arabidopsis, Populus, and rice. Despite our stringent target prediction criteria, most of the targets of conserved N. benthamiana miRNAs were conserved with targets in other plant species and favored genes encoding transcription factors. For example, nbt-miR156 targets SPL transcription factors60; nbt-miR159 targets MYB domain containing transcription factors61; nbt-miR160 targets the ARF gene family62; nbt-miR165/166 targets the homeodomain-leucine zipper (HDZip) gene family63; and nbt-miR172 targets AP2-like transcription factors.64 These miRNAs are classified as highly conserved in plants.6 However, for moderately conserved miRNAs,6 only three (nbt-miR164, nbt-miR169, and nbt-miR397) out of eight N. benthamiana miRNAs target genes from the same family (Table S5). The targeting observed in N. benthamiana of conserved genes by conserved miRNA also occurs in other plants and even animals.52 For example, miR165/166 is conserved in all plant lineages, including mosses, monocots, and dicots, and the binding site of their targets, which encode the HD-Zip family transcription factors, is also conserved in these taxa. The conservation between miRNAs and their targets implies that regulatory networks involving miRNA-target interactions may have evolved over a very long time and play a pivotal role in key processes during the plant life cycle.
Dynamic miRNA expression programs during N-mediated resistance TMV
Most of the miRNAs we identified, from both conserved and novel miRNA pools, showed dynamic expression patterns during the TMV response, implying that miRNAs are involved in N-mediated resistance to TMV. This observation is in agreement with miRNA expression after bacterial infection in Arabidopsis.65 Since the predicted targets of these miRNAs have diverse functions, such as binding, catalytic activity, transporter activity, and transcription activity (Fig. 6A); and they are also involved in many biological processes, such as cellular process, metabolic process, developmental process, and stimuli process (Fig. 6B); the miRNAs’ dynamic expression might regulate gene expression systematically at different layers in N-mediated resistance pathway to TMV.
Usually, miRNAs from the same family have similar expression patterns. Interestingly, we identified a miRNA, nbt-miR160e, which displayed an expression pattern distinct from other members of its family. During TMV infection, nbt-miR160a/b/c/d was downregulated, while levels of nbt-miR160e increased (Fig. 5). This distinct expression pattern suggests that nbt-miR160e functions differently from the other members in the resistance pathway. Although miRNAs from the same family have the same or similar mature sequences, and therefore the same or similar target genes, their genomic contexts are different, which might explain the distinct expression patterns of different members. Similar observations for multicopy miRNAs from rice and Arabidopsis have been made.66
To date, few miRNAs have been shown to regulate plant immunity. miR393 targets TIR/AFB F-box genes, thereby downregulating auxin signaling and contributing to resistance to bacteria DC3000.15 However, we did not observe any significant changes in nbt-miR393 expression during N gene activation upon infection, suggesting that nbt-miR393 is not involved in the N gene pathway during N. benthamiana’s immunity response to TMV. miR160a enhances PTI, while overexpression of miR398b negatively regulates PTI in Arabidopsis.16 We found that both nbt-miR160e and nbt-miR398 increased rapidly within eight hours of N gene activation in N. benthamiana, indicating that these miRNAs positively regulate immunity to TMV in the N gene pathway. Accordingly, family member nta-miR6019 has been shown to cleave N gene transcripts, thereby attenuating N-mediated resistance to TMV.18 We also found that expression of the N receptor inhibitor nbt-miR6019a/b was inhibited two hours after N gene activation, suggesting a tight control between miRNAs and the immune receptor-mediated resistance pathway. This result also supports that our miRNA identification and expression profile analyses are reliable.
Materials and Methods
Plant material and growth conditions
Transgenic N-expressing N. benthamiana plants22 were grown in soil in a controlled climate chamber providing 16 hours light/8 hours dark cycles at 22–25 °C. For high-temperature treatment, sets of plants were transferred to a chamber providing 16 hours light/8 hours dark cycles at 32 °C. After two days of pre-treatment under these conditions, the plants were rub-inoculated with TMV-GFP and immediately moved back to high-temperature conditions for another five days. Leaves with full GFP fluorescence were collected at zero, two, and eight hours after the transfer to 22–25 °C.
Inoculation of TMV-GFP
N. benthamiana leaves were infiltrated with Agrobacterium carrying the TMV-GFP T-DNA construct. At five days post-infiltration (dpi), TMV-GFP infected leaves were homogenized and rub-inoculated onto the leaves of tested plants.
sRNA library construction and high-throughput sequencing
Total RNA was extracted with TRIzol (Invitrogen) following the manufacturer’s guide for the plant material. The RNA quality and quantity were determined with an Agilent 2100 Bioanalyzer (Agilent). The RNA was separated by PAGE, and then 16–30 nt sRNA was purified and ligated to 5′ and 3′ RNA adaptors. A reverse transcription reaction was performed with several cycles of PCR, and products were sequenced by Solexa technology.
Bioinformatics analysis of high-throughput sequencing data
The raw Solexa sequencing data were preprocessed by filtering out low-quality reads, trimming adaptors and contaminants formed by adaptors, and removing reads less than 18 nt (both siRNA and miRNA are longer than 18 nt in plants). The clean reads were then compared with entries in the available sRNA databases, Rfam (http://rfam.sanger.ac.uk, Release 9.1) and GenBank (http://www.genbank. com). All the reads that mapped to rRNA, tRNA, snRNA, and snoRNA entries in these two databases were annotated and removed. The remaining reads were first mapped to the N. benthamiana genome (Niben.genome.v0.4.4.scaffolds.nrcontigs) by miRDeep2’s mapper module.30 The arf file and reads file obtained from the mapping procedure together with the genome file were used to identify novel and known miRNAs. Before identification of miRNA using miRDeep2module, we modified several places of PERL script of the module to perform plant miRNA identification as Wen et al did.27 For conserved miRNA identification, we used mature miRNAs and precursors of miRNAs of the phylogenetically close N. tabacum as the first reference. N. tabacum mature miRNAs and their precursors were obtained from two sources: miRBase (http://www.mirbase.org) and reported miRNAs predicted using Expressed Sequence Tag (EST) sequences.28 Then we used all plant-derived mature miRNAs (from miRBase, http://www.mirbase.org) except N. tabacum as the second reference. After excluding reads mapped to the conserved miR-NAs, the remaining reads were subjected to novel miRNA identification. We grouped mature miRNAs (both conserved and novel) with identical and near identical (>90% identity) sequences into the same family. miRNA expression across distinct samples was profiled using the quantifier module of miRDeep2.30
For intronic miRNA identification, first, we obtained the 5′ and 3′ sites’ information in the genome of each miRNA’s precursor; second, we compared their location with the corresponding gene information in the gff file of the genome. If miRNA’s precursor’s 5′ and 3′ sites lay in an intron, we defined the miRNA as intronic miRNA. In addition, only genes with length more than 500 bp and intron length less than 10 kb were considered.
Supplementary Material
Supplementary Table 1. conserved miRNAs.
Supplementary Table 2. novel miRNAs.
Supplementary Table 3. number of member in each novel miRNA family.
Supplementary Table 4. clustered miRNAs.
Supplementary Table 5. target prediction of conserved miRNAs.
Supplementary Table 6. target prediction of novel miRNAs.
Supplementary Table 7. Nicotiana tabacum-specific and Nicotiana benthamiana-specific miRNAs.
Supplementary Table 8. comparison of length distribution of sRNA.
Supplementary Figure 1. nucleotide preference of 24nt novel miRNAs.
Supplementary Figure 2. read length comparison by ANOVA.
Acknowledgments
We thank Prof. Yule Liu’s suggestions on experimental design and critical reading of the manuscript. We also thank Jiapei Yuan’s help on bioinformatics analysis.
Footnotes
Author Contributions
Conceived and designed the experiments: KY, YT. Analyzed the data: KY. Wrote the first draft of the manuscript: KY. Contributed to the writing of the manuscript: KY, JZ. Agree with the manuscript results and conclusions: KY, YT, JZ. Jointly developed the structure and arguments for the paper: KY, JZ. Made critical revisions and approved final version: KY, YT, JZ. All authors reviewed and approved final version.
ACADEMIC EDITOR: Jike Cui, Associate Editor
FUNDING: This work was supported by the National Postdoc Fund of China (Grant No. 2011M500296) and the National Natural Science Foundation of China (Grant Nos. 30930060, 31071169, and 31270182). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
REFERENCES
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Kurihara Y, Watanabe Y. Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci USA. 2004;101(34):12753–8. doi: 10.1073/pnas.0403115101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cuperus JT, Fahlgren N, Carrington JC. Evolution and functional diversification of MIRNA genes. Plant Cell. 2011;23(2):431–42. doi: 10.1105/tpc.110.082784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park MY, Wu G, Gonzalez-Sulser A, Vaucheret H, Poethig RS. Nuclear processing and export of microRNAs in Arabidopsis. Proc Natl Acad Sci USA. 2005;102(10):3691–6. doi: 10.1073/pnas.0405570102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell. 2009;136(4):669–87. doi: 10.1016/j.cell.2009.01.046. [DOI] [PubMed] [Google Scholar]
- 6.Zhang B, Pan X, Cannon CH, Cobb GP, Anderson TA. Conservation and divergence of plant microRNA genes. Plant J. 2006;46(2):243–59. doi: 10.1111/j.1365-313X.2006.02697.x. [DOI] [PubMed] [Google Scholar]
- 7.Weber MJ. New human and mouse microRNA genes found by homology search. FEBS J. 2005;272(1):59–73. doi: 10.1111/j.1432-1033.2004.04389.x. [DOI] [PubMed] [Google Scholar]
- 8.Fahlgren N, Howell MD, Kasschau KD, et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One. 2007;2(2):e219. doi: 10.1371/journal.pone.0000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sunkar R, Zhou X, Zheng Y, Zhang W, Zhu JK. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008;8(1):25. doi: 10.1186/1471-2229-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pantaleo V, Szittya G, Moxon S, et al. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010;62(6):960–76. doi: 10.1111/j.0960-7412.2010.04208.x. [DOI] [PubMed] [Google Scholar]
- 11.Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol. 2006;57:19–53. doi: 10.1146/annurev.arplant.57.032905.105218. [DOI] [PubMed] [Google Scholar]
- 12.Mallory AC, Vaucheret H. Functions of microRNAs and related small RNAs in plants. Nat Genet. 2006;38:S31–6. doi: 10.1038/ng1791. [DOI] [PubMed] [Google Scholar]
- 13.Chiou T. The role of microRNAs in sensing nutrient stress. Plant Cell Environ. 2007;30(3):323–32. doi: 10.1111/j.1365-3040.2007.01643.x. [DOI] [PubMed] [Google Scholar]
- 14.Phillips JR, Dalmay T, Bartels D. The role of small RNAs in abiotic stress. FEBS Lett. 2007;581(19):3592–7. doi: 10.1016/j.febslet.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 15.Navarro L, Dunoyer P, Jay F, et al. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Sci Signal. 2006;312(5772):436. doi: 10.1126/science.1126088. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Zhang Q, Zhang J, Wu L, Qi Y, Zhou JM. Identification of microRNAs involved in pathogen-associated molecular pattern-triggered plant innate immunity. Plant Physiol. 2010;152(4):2222–31. doi: 10.1104/pp.109.151803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shivaprasad PV, Chen H-M, Patel K, Bond DM, Santos BACM, Baulcombe DC. A microRNA superfamily regulates nucleotide binding site leucine-rich repeats and other mRNAs. Plant Cell. 2012;24(3):859–74. doi: 10.1105/tpc.111.095380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li F, Pignatta D, Bendix C, et al. MicroRNA regulation of plant innate immune receptors. Proc Natl Acad Sci USA. 2012;109(5):1790–5. doi: 10.1073/pnas.1118282109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navarro L, Jay F, Nomura K, He SY, Voinnet O. Suppression of the microRNA pathway by bacterial effector proteins. Sci Signal. 2008;321(5891):964. doi: 10.1126/science.1159505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitham S, Dinesh-Kumar SP, Choi D, Hehl R, Corr C, Baker B. The product of the tobacco mosaic virus resistance gene N: similarity to toll and the interleukin-1 receptor. Cell. 1994;78(6):1101–15. doi: 10.1016/0092-8674(94)90283-6. [DOI] [PubMed] [Google Scholar]
- 21.Dinesh-Kumar SP, Whitham S, Choi D, Hehl R, Corr C, Baker B. Transposon tagging of tobacco mosaic virus resistance gene N: its possible role in the TMV-N-mediated signal transduction pathway. Proc Natl Acad Sci USA. 1995;92(10):4175–80. doi: 10.1073/pnas.92.10.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Schiff M, Marathe R, Dinesh-Kumar SP. Tobacco Rar1, EDS1 and NPR1/NIM1 like genes are required for N-mediated resistance to tobacco mosaic virus. Plant J. 2002;30(4):415–29. doi: 10.1046/j.1365-313x.2002.01297.x. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Burch-Smith T, Schiff M, Feng S, Dinesh-Kumar SP. Molecular chaperone Hsp90 associates with resistance protein N and its signaling proteins SGT1 and Rar1 to modulate an innate immune response in plants. J Biol Chem. 2004;279(3):2101–8. doi: 10.1074/jbc.M310029200. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Schiff M, Serino G, Deng X-W, Dinesh-Kumar SP. Role of SCF ubiquitin-ligase and the COP9 signalosome in the N gene mediated resistance response to Tobacco mosaic virus. Plant Cell. 2002;14(7):1483–96. doi: 10.1105/tpc.002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Schiff M, Dinesh-Kumar SP. Involvement of MEK1 MAPKK, NTF6 MAPK, WRKY/MYB transcription factors, COI1 and CTR1 in N-mediated resistance to tobacco mosaic virus. Plant J. 2004;38(5):800–9. doi: 10.1111/j.1365-313X.2004.02085.x. [DOI] [PubMed] [Google Scholar]
- 26.Mackowiak SD. Identification of novel and known miRNAs in deep sequencing data with miRDeep2. Curr Protoc Bioinformatics. 2011;12:12.10. doi: 10.1002/0471250953.bi1210s36. [DOI] [PubMed] [Google Scholar]
- 27.Wen M, Shen Y, Shi S, Tang T. miREvo: an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinformatics. 2012;13(1):140. doi: 10.1186/1471-2105-13-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frazier TP, Xie F, Freistaedter A, Burklew CE, Zhang B. Identification and characterization of microRNAs and their target genes in tobacco (Nicotiana tabacum) Planta. 2010;232(6):1289–308. doi: 10.1007/s00425-010-1255-1. [DOI] [PubMed] [Google Scholar]
- 29.Crooks GE, Hon G, Chandonia J-M, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedländer MR, Chen W, Adamidi C, et al. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008;26(4):407–15. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 31.Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132(21):4653–62. doi: 10.1242/dev.02073. [DOI] [PubMed] [Google Scholar]
- 32.Zhang L, Chia JM, Kumari S, et al. A genome-wide characterization of microRNA genes in maize. PLoS Genet. 2009;5(11):e1000716. doi: 10.1371/journal.pgen.1000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14(10a):1902–10. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Q-H, Spriggs A, Matthew L, et al. A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res. 2008;18(9):1456–65. doi: 10.1101/gr.075572.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joshi PK, Gupta D, Nandal UK, Khan Y, Mukherjee SK, Sanan-Mishra N. Identification of mirtrons in rice using MirtronPred: a tool for predicting plant mirtrons. Genomics. 2012;99:370–5. doi: 10.1016/j.ygeno.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Li B, Qin Y, Duan H, Yin W, Xia X. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J Exp Bot. 2011;62(11):3765–79. doi: 10.1093/jxb/err051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meng Y, Shao C. Large-scale identification of mirtrons in Arabidopsisand rice. PLoS One. 2012;7(2):e31163. doi: 10.1371/journal.pone.0031163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pillai RS, Bhattacharyya SN, Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007;17(3):118–26. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 39.Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39(suppl 2):W155–9. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conesa A, Götz S, García-Gómez JM, Terol J, Talön M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 41.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39(suppl 1):D152–7. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L, Ren Y, Zhang Y, Xu J, Zhang Z, Wang Y. Genome-wide profiling of novel and conserved PopulusmicroRNAs involved in pathogen stress response by deep sequencing. Planta. 2012;235(5):873–83. doi: 10.1007/s00425-011-1548-z. [DOI] [PubMed] [Google Scholar]
- 43.Martínez G, Forment J, Llave C, Pallás V, Gómez G. High-throughput sequencing, characterization and detection of new and conserved cucumber miRNAs. PLoS One. 2012;6(5):e19523. doi: 10.1371/journal.pone.0019523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sunkar R, Girke T, Zhu JK. Identification and characterization of endogenous small interfering RNAs from rice. Nucleic Acids Res. 2005;33(14):4443–54. doi: 10.1093/nar/gki758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16(8):2001. doi: 10.1105/tpc.104.022830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X-J, Reyes JL, Chua N-H, Gaasterland T. Prediction and identification of Arabidopsis thaliana microRNAs and their mRNA targets. Genome Biol. 2004;5(9):R65. doi: 10.1186/gb-2004-5-9-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yao Y, Guo G, Ni Z, et al. Cloning and characterization of microRNAs from wheat (Triticum aestivum L.) Genome Biol. 2007;8(6):R96. doi: 10.1186/gb-2007-8-6-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glazov EA, Cottee PA, Barris WC, Moore RJ, Dalrymple BP, Tizard ML. A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach. Genome Res. 2008;18(6):957–64. doi: 10.1101/gr.074740.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fahlgren N, Jogdeo S, Kasschau KD, et al. MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell. 2010;22(4):1074–89. doi: 10.1105/tpc.110.073999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barakat A, Wall PK, Diloreto S, Depamphilis CW, Carlson JE. Conservation and divergence of microRNAs in Populus. BMC Genomics. 2007;8(1):481. doi: 10.1186/1471-2164-8-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Axtell MJ, Bowman JL. Evolution of plant microRNAs and their targets. Trends Plant Sci. 2008;13(7):343–9. doi: 10.1016/j.tplants.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 52.Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8(2):93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- 53.Maher C, Stein L, Ware D. Evolution of ArabidopsismicroRNA families through duplication events. Genome Res. 2006;16(4):510–9. doi: 10.1101/gr.4680506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12(9):735–9. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 55.Okamura K, Liu N, Lai EC. Distinct mechanisms for microRNA strand selection by Drosophila argonautes. Mol Cell. 2009;36(3):431. doi: 10.1016/j.molcel.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang X, Zhao H, Gao S, et al. Arabidopsis argonaute 2 regulates innate immunity via miRNA393*-mediated silencing of a Golgi-localized SNARE gene MEMB12. Mol Cell. 2011;42(3):356. doi: 10.1016/j.molcel.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Griffiths-Jones S. miRBase: microRNA sequences and annotation. Curr Protoc Bioinformatics. 2010;12:12.9.1–10. doi: 10.1002/0471250953.bi1209s29. [DOI] [PubMed] [Google Scholar]
- 58.Guddeti S, Zhang DC, Li AL, et al. Molecular evolution of the rice miR395 gene family. Cell Res. 2005;15(8):631–8. doi: 10.1038/sj.cr.7290333. [DOI] [PubMed] [Google Scholar]
- 59.Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell. 2004;14(6):787–99. doi: 10.1016/j.molcel.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 60.Wang J-W, Czech B, Weigel D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell. 2009;138(4):738–49. doi: 10.1016/j.cell.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 61.Reyes JL, Chua N. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsisseed germination. Plant J. 2007;49(4):592–606. doi: 10.1111/j.1365-313X.2006.02980.x. [DOI] [PubMed] [Google Scholar]
- 62.Wang J-W, Wang L-J, Mao Y-B, Cai W-J, Xue H-W, Chen XY. Control of root cap formation by microRNA-targeted auxin response factors in Arabidopsis. Plant Cell. 2005;17(8):2204–16. doi: 10.1105/tpc.105.033076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Emery JF, Floyd SK, Alvarez J, et al. Radial patterning of Arabidopsisshoots by class III HD-ZIP and KANADI genes. Curr Biol. 2003;13(20):1768. doi: 10.1016/j.cub.2003.09.035. [DOI] [PubMed] [Google Scholar]
- 64.Chen X. A microRNA as a translational repressor of APETALA2 in Arabidopsisflower development. Science. 2004;303(5666):2022. doi: 10.1126/science.1088060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang W, Gao S, Zhou X, et al. Bacteria-responsive microRNAs regulate plant innate immunity by modulating plant hormone networks. Plant Mol Biol. 2011;75(1–2):93–105. doi: 10.1007/s11103-010-9710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang D, Yin C, Yu A, et al. Duplication and expression analysis of multicopy miRNA gene family members in Arabidopsisand rice. Cell Res. 2006;16(5):507–18. doi: 10.1038/sj.cr.7310062. [DOI] [PubMed] [Google Scholar]
- 67.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839–43. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. conserved miRNAs.
Supplementary Table 2. novel miRNAs.
Supplementary Table 3. number of member in each novel miRNA family.
Supplementary Table 4. clustered miRNAs.
Supplementary Table 5. target prediction of conserved miRNAs.
Supplementary Table 6. target prediction of novel miRNAs.
Supplementary Table 7. Nicotiana tabacum-specific and Nicotiana benthamiana-specific miRNAs.
Supplementary Table 8. comparison of length distribution of sRNA.
Supplementary Figure 1. nucleotide preference of 24nt novel miRNAs.
Supplementary Figure 2. read length comparison by ANOVA.